

Abstract
AChE and BuChE are druggable targets for the discovery of anti-Alzheimer’s disease drugs, while dual-inhibition of these two targets seems to be more effective. In this study, we synthesised a series of novel isoflavone derivatives based on our hit compound G from in silico high-throughput screening and then tested their activities by in vitro AChE and BuChE bioassays. Most of the isoflavone derivatives displayed moderate inhibition against both AChE and BuChE. Among them, compound 16 was identified as a potent AChE/BuChE dual-targeted inhibitor (IC50: 4.60 μM for AChE; 5.92 μM for BuChE). Molecular modelling study indicated compound 16 may possess better pharmacokinetic properties, e.g. absorption, blood–brain barrier penetration and CYP2D6 binding. Taken together, our study has identified compound 16 as an excellent lead compound for the treatment of Alzheimer’s disease.
Keywords: Alzheimer’s disease, isoflavone derivatives, AChE/BuChE dual-targeted inhibitor, molecular modelling
Introduction
Alzheimer’s disease (AD) is one of the most common neurodegenerative diseases and accounts for more than 80% of dementia cases worldwide in elderly people1,2. It is in particular characterised by a forebrain cholinergic neuronal loss, a progressive cognitive decline and neuropsychiatric cholinergic disturbances, which seems to be closely related to pathological formation of β-amyloid3. Studies have demonstrated that memory impairment in patients with dementia is due to the selective and irreversible deficiency in the cholinergic functions4. Cholinesterase is the enzyme which catalyses the hydrolysis of acetylcholine, and thus inhibition of cholinesterase can be an effective way for the treatment of AD5–7.
Two types of the cholinesterase, namely, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) were discovered in the nervous system8. However, AChE has a 1013 fold higher acetylcholine hydrolytic activity than BuChE does under the same condition9,10. Many AChE inhibitors were discovered, of which donepezil11 and galantamine12 (cf. Figure 1) have been approved by Food and Drug Administration (FDA). Nevertheless, the clinical use demonstrated they have modest memorial and cognitive improving functions for patients with AD. Specifically, they only provide a symptomatic and palliative pharmacological effect instead of curing AD, and their effects would “wear off” after a certain period of treatment time13,14. Evidences suggested that additional inhibition of brain BuChE may become an important therapeutic strategy for AD treatment. It was reported that the BuChE had a key role that it could partly compensate for the action of AChE15. In addition, studies indicated that a balanced inhibition of both AChE and BuChE may be beneficial for treating the cognitive deficits16,17. Such multifunctional cholinestarase inhibitors have been identified so far and were showing positive clinical outcome for the treatment of AD18,19. For instance, the AChE/BuChE dual-targeted inhibitor, i.e. a “blockbuster drug” rivastigmin (cf. Figure 1) displayed a more potent effect than donepezil20.
Figure 1.

Structures of currently marketed cholinesterase inhibitors.
In order to identify new classes of AChE/BuChE dual-targeted inhibitors, in silico high-throughput screening (HTS) was performed to screen our in-house chemical library that contained more than 30,000 compounds. On the top-ranked compound list, an isoflavone derivative G (cf. Figure 2) attracted our attention as it displayed favourable binding modes with both AChE and BuChE (cf. Supplementary Figure S1). To the best of our knowledge, the scaffolds of flavonoids21,22 and its closely related ones such as homoisolavonoids23,24, trihydroxyflavone25,26 were privileged structures that showed beneficial effects on neurological disorders, e.g. anti-inflammation, metal chelation, neuroprotection, Aβ fibril formation inhibition and free radical scavenging effect. In addition, a recent study revealed a few compounds structurally similar to G were AChE inhibitors21. These evidences prompted us to test the activity of G against both AChE and BuChE. Interestingly, the compound G showed a dual-targeting effect, with an IC50 of 1.47 μM for AChE and 3.37 μM for BuChE. In this study, we explored its preliminary structure-activity relationship (SAR) by synthesising a series of novel isoflavone derivatives and testing their inhibitory effects on both AChE and BuChE. Then we carried out molecular docking to predict binding modes of isoflavone derivatives to AChE and BuChE. Moreover, we predicted the pharmacokinetic profile of those dual-targeting isoflavone derivatives, i.e. absorption, distribution, metabolism and excretion (ADME) properties.
Figure 2.

Chemical Structure of the hit compound G.
Materials and methods
Chemistry
All melting points (mps) were measured by a Melting Point YRT-3 apparatus (Tianjin precision apparatus factory, China) and were uncorrected. NMR spectra were performed using 300 or 400 MHz spectrometers (Varian Mercury, USA) with TMS as an internal standard. High resolution mass spectra were determined by Thermo Scientific Exactive Plus mass spectrometry with ESI method (Thermo, USA). Some compounds were purified by medium-pressure preparative column chromatography (Shimadzu, Japan). The purity of all these synthesised compounds was determined by HPLC analysis (Waters, USA). All the materials were obtained from commercial suppliers and used without purification, unless otherwise specified. Yields were not optimised. TLC analysis was carried out on silica gel plates GF254 (Yantai chemical research institute, China). Column chromatography was performed on silica gel (200–300 mush; Qingdao Marine Chemical Inc.).
General procedure for the preparation of intermediates (a1–a4)
Formononetin (5 g, 18.6 mmol), finely grounded anhydrous K2CO3 (30 g, 270 mmol) and 1,2-dibromoethane (17.2 ml, 199 mmol) or 1,3-dibromopropane (20.1 ml, 199 mmol) or 1,4-dibromobutane (24 ml, 198 mmol) were added into 500 ml acetone solution. The mixture was refluxed for 10 h. The residues were then added into 100 ml water and stirred for 1 h and filtrated by suction. After that, the pastry was washed by 5 ml water for three times. The solid was finally dried in vacuum at 50 °C to give the desired product without further purification.
7-(2-Bromoethoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (a1)
The compound was obtained as white solid in 93.4% yield. Purity 98.5% (by HPLC); mp 177.7–179.6 °C; 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 8.8 Hz, 1H), 7.92 (s, 1H), 7.50 (d, J = 8.8 Hz, 2H), 7.02–6.96 (m, 3H), 6.86 (d, J = 2.0 Hz, 1H), 4.39 (t, J = 6.4 Hz, 2H), 3.84 (s, 3H), 3.69 (t, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 175.9, 162.4, 159.8, 157.9, 152.2, 130.2, 128.2, 125.1, 124.2, 119.1, 114.7, 114.1, 101.2, 68.3, 55.5, 28.4. ESI-MS m/z 375.38 [M + H]+.
7-(3-Bromopropoxy)-3–(4-methoxyphenyl)-4H-chromen-4-one (a2)
The compound was obtained as white solid in 91.5% yield. Purity 99.2% (by HPLC); mp 121.5–123.6 °C; 1H NMR (400 MHz, CDCl3) δ 8.22 (d, J = 8.8 Hz, 1H), 7.92 (s, 1H), 7.50 (d, J = 8.8 Hz, 2H), 7.02–6.91 (m, 3H), 6.87 (d, J = 2.4 Hz, 1H), 4.22 (t, J = 6.0 Hz, 2H), 3.84 (s, 3H), 3.63 (t, J = 6.0 Hz, 2H), 2.38 (m, J = 6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 176.0, 163.1, 159.7, 158.0, 152.2, 130.3, 128.0, 125.1, 124.3, 118.7, 114.8, 114.1, 100.9, 66.1, 55.5, 32.1, 29.7. ESI-MS m/z 389.34 [M + H]+.
7-(4-Bromobutoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (a3)
The compound was obtained as white solid in 88.6% yield. Purity 98.7% (by HPLC); mp 152.8–154.0 °C; 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.50 (d, J = 8.8 Hz, 2H), 6.99–6.96 (m, 3H), 6.84 (d, J = 2.4 Hz, 1H), 4.10 (t, J = 6.0 Hz, 2H), 3.84 (s, 3H), 3.51 (t, J = 6.0 Hz, 2H), 2.14–1.98 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 175.8, 163.2, 159.6, 157.9, 152.0, 130.1, 127.8, 124.9, 124.2, 118.5, 114.7, 114.0, 100.6, 67.6, 55.3, 33.2, 29.3, 27.6. ESI-MS m/z 403.31 [M + H]+.
7-(2-Bromoethoxy)-5-hydroxy-3-(4-methoxyphenyl)-4H-chromen-4-one (a4)
The procedure is the same as above method, however, the formononetin was replaced by 5-hydroxyl formononetin. The compound was obtained as white solid in 94.4% yield. Purity 99.6% (by HPLC); mp 157.1–159.2 °C; 1H NMR (400 MHz, CDCl3) δ 12.87 (s, 1H), 7.88 (s, 1H), 7.46 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.40 (dd, J = 2.0, 15.2 Hz, 2H), 4.36 (t, J = 6.0 Hz, 2H), 3.85 (s, 3H), 3.66 (t, J = 6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 181.0, 164.0, 163.1, 160.0, 158.0, 152.9, 130.2, 123.9, 123.0, 114.3, 106.8, 98.7, 93.2, 68.3, 55.5, 28.4. ESI-MS m/z 391.38[M + H]+.
7-(2-Bromoethoxy)-3-(4-(2-bromoethoxy)phenyl)-4H-chromen-4-one (a5)
The procedure for the preparation of a5 was the same with that for a1–a4, except that the formononetin was replaced with daidzein and the final product was purified by silico column and eluted by CH2Cl2. The compound obtained as white solid in 26.3% yield. mp 170.2–172.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 9.2 Hz, 1H), 7.93 (s, 1H), 7.50 (d, J =8.4 Hz, 2H), 7.03–6.97 (m, 3H), 6.87 (d, J = 1.6 Hz, 1H), 4.40 (t, J = 6.4 Hz, 2H), 4.33 (d, J =6.4 Hz, 2H), 3.69 (t, J = 6.4 Hz, 2H), 3.66 (t, J = 6.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 174.6, 162.2, 157.7, 157.3, 153.6, 130.1, 127.1, 124.6, 123.3, 117.9, 115.0, 114.4, 101.4, 68.5, 67.8, 31.4, 31.0. ESI-MS (m/z): 467.44 [M + H]+.
General procedure for the synthesis of compounds 1–15
Intermediate a1 (1.87 g, 5 mmol) or a2 (1.88 g, 5 mmol) or a3 (2.02 g, 5 mmol) or a4 (100 mg, 0.26 mmol) were dissolved in 150 ml acetonitrile or 70 ml DMF. Then K2CO3 (13.8 g, 100 mmol) and various substituted amino-group (16.4 mmol) were added. The mixture was stirred and refluxed for 3 h and monitored by TLC. When the reaction was finished, the reaction solution was poured into 600 ml water and stirred for 5 min. After that, the mixture was filtrated by suction and the pastry was washed by water, then, dried by vacuum at 50 °C to yield the desired products with chromatography, recrystallisation, or without any further purification.
5-Hydroxy-3-(4-mehtoxyphenyl)-7-(2-(piperidin-1-yl)ethoxy)-4H-chromen-4-one (1)
The compound was obtained as white solid in 85.7% yield. Purity 99.3% (by HPLC); mp 81.3–83.1 °C. 1H NMR (400 MHz, CDCl3) δ: 12.83 (s, 1H), 7.86 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 6.40 (d, J = 13.2 Hz, 2H), 4.20 (brs, 2H), 3.84 (s, 3H), 2.83 (brs, 2H), 2.56 (brs, 4H), 1.65 (brs, 4H), 1.47 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 181.0, 164.9, 162.8, 159.9, 158.1, 152.8, 130.2, 123.8, 123.1, 114.2, 106.4, 983.9, 93.1, 66.8, 57.7, 55.5, 55.2, 26.0, 24.2. HRMS: calcd for C23H26NO5[M + H]+, 396.1811, found: 396.1807.
3-(4-Methoxyphenyl)-7-(3-(piperidin-1-yl)propoxy)-4H-chromen-4-one (2)
The compound was obtained as white solid in 7% yield. Purity 98.8% (by HPLC); mp 152.8–154.0 °C; 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.50 (d, J = 8.8 Hz, 2H), 6.99–6.96 (m, 3H), 6.86 (d, J = 2.0 Hz, 1H), 4.12 (t, J = 6.4 Hz, 2H), 3.84 (s, 3H), 2.50 (t, J = 7.2 Hz, 2H), 2.42 (brs, 4H), 2.07–2.00 (m, 2H), 1.64–1.58 (m, 4H), 1.48–1.44 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 175.9, 163.5, 159.6, 158.0, 152.0, 130.1, 127.7, 124.8, 124.3, 118.3, 114.9, 114.0, 100.6, 67.2, 55.7, 55.3, 54.7, 26.6, 26.0, 24.4. HRMS: calcd for C24H28NO4[M + H]+, 394.2018, found: 394.2017.
3-(4-Methoxyphenyl)-7-(4-(piperidin-1-yl)butoxy)-4-H-chromen-4-one (3)
The compound was obtained as white solid in 93.6% yield. Purity 99.4% (by HPLC); mp 152.8–154.0 °C; 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.51 (d, J = 8.8 Hz, 2H), 6.98–6.96 (m, 3H), 6.83 (d, J = 2.0 Hz, 1H), 4.08 (t, J = 6.4 Hz, 2H), 3.84 (s, 3H), 2.44 (brs, 6H), 1.90–1.83 (m, 2H), 1.78–1.73 (m, 2H), 1.65–1.63 (m, 4H), 1.46 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ 175.9, 163.4, 159.6, 158.0, 152.0, 130.1, 127.8, 124.9, 124.3, 118.3, 114.8, 114.0, 100.6, 68.4, 58.8, 55.4, 54.5, 27.1, 25.7, 24.2, 23.1. HRMS: calcd for C25H30NO4[M + H]+, 408.2175, found: 408.21738.
7-(2-(Ethyl(methyl)amino)ethoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (4)
The compound was obtained as pale yellow solid in 40% yield. Purity 99.2% (by HPLC); mp 110.3–112.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.49 (d, J = 8.8 Hz, 2H), 7.01–6.95 (m, 3H), 6.86 (d, J = 2.0 Hz, 1H), 4.18 (t, J = 6.0 Hz, 2H), 3.84 (s, 3H), 2.87 (t, J = 6.0 Hz, 2H), 2.58 (q, J = 7.2 Hz, 2H), 2.38 (s, 3H), 1.12 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 176.0, 163.3, 159.7, 158.0, 152.2, 130.3, 127.9, 125.0, 124.4, 118.6, 115.0, 114.1, 100.9, 67.0, 55.6, 55.5, 52.1, 42.4, 12.3. HRMS: calcd for C21H24NO4 [M + H]+, 354.1705, found: 354.1701.
7-(3-(Ethyl(methyl)amino)propoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (5)
The compound was obtained as white solid in 26.4% yield. Purity 98.7% (by HPLC); mp 100.7–101.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.50 (d, J = 8.8 Hz, 2H), 6.99–6.96 (m, 3H), 6.86 (d, J = 2.0 Hz, 1H), 4.12 (t, J = 6.4 Hz, 2H), 3.84 (s, 3H), 2.56 (t, J = 6.8 Hz, 2H), 2.47 (q, J = 7.2 Hz, 2H), 2.27 (s, 3H), 2.05–1.99 (m, 2H), 1.08(t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 176.0, 163.6, 159.7, 158.1, 152.2, 130.3, 127.9, 125.0, 124.4, 118.5, 115.0, 114.1, 100.8, 67.1, 55.5, 53.7, 51.6, 41.8, 27.1, 12.3. HRMS: calcd for C22H26NO4[M + H]+, 368.1862, found: 368.1859.
7-(4-(Ethyl(methyl)amino)butoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (6)
The final compound was obtained as white solid in 11.7% yield purified by medium-pressure preparative column chromatography method (CH2Cl2: CH3OH =97: 3). Purity 99.1% (by HPLC); mp 128.3–139.9 °C; 1H-NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.8 Hz, 1H), 7.88 (s, 1H), 7.48 (d, J = 8.8 Hz, 2H), 6.96–6.94 (m, 3H), 6.81 (d, J = 2.0 Hz, 1H), 4.05 (t, J = 6.0 Hz, 2H), 3.81 (s, 3H), 2.48–2.41 (m, 4H), 2.24 (s, 3H), 1.88–1.81 (m, 2H), 1.71–1.64 (m, 2H), 1.07 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 175.9, 163.5, 159.6, 158.0, 152.1, 130.2, 127.7, 124.9, 124.3, 118.3, 114.9, 114.0, 100.6, 68.5, 56.8, 55.4, 51.5, 41.5, 27.0, 23.8, 12.2. HRMS: calcd for C23H28NO4 [M + H]+, 382.2018, found: 382.2018.
7-(2-((2-(5-Methoxy-1H-indol-3-yl)ethyl)amino)ethoxy)-3-(4-methoxyphenyl)-4H-chr-omen-4-one (7)
The compound was obtained as pale yellow solid in 10.5% yield. Purity 99.6% (by HPLC); mp 236.0 –238.3 °C; 1H NMR (400 MHz, CD3OD) δ 8.14 (d, J = 0.8 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 8.8 Hz, 2H), 7.20 (d, J = 8.8 Hz, 1H), 7.05 (s, 1H), 7.01 (d, J = 2.4 Hz, 1H), 6.96 (d, J = 8.8 Hz, 2H), 6.89–6.85 (m, 2H), 6.72 (dd, J = 8.8, 2.4 Hz, 1H), 4.13 (t, J = 4.2 Hz, 2H), 3.81 (s, 3H), 3.75 (s, 3H), 3.03 (t, J = 4.2 Hz, 2H), 2.97 (t, J = 3.6 Hz, 4H); 13C NMR (100 MHz, CD3OD) δ 177.9, 164.8, 161.1, 159.5, 154.9, 154.9, 133.5, 131.3, 128.9, 128.2, 125.9, 125.4, 124.4, 119.2, 116.2, 114.8, 113.0, 112.8, 112.6, 101.9, 101.2, 68.3, 56.2, 55.7, 50.3, 48.7, 26.0. HRMS: calcd for C29H29N2O5 [M + H]+, 485.2076, found: 485.2077.
7-(3-((2-(5-Methoxy-1H-indol-3-yl)ethyl)amino)propoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (8)
The compound was obtained as yellow solid in 51.8% yield. Purity 98.7% (by HPLC); mp 108.8–109.5 °C; 1H NMR (400 MHz, CD3OD) δ 8.17 (d, J = 0.8 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.47 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.4 Hz, 1H), 7.06–7.05 (m, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 2.0 Hz, 1H), 6.82 (dd, J = 8.8, 2.0 Hz, 1H), 6.75 (dd, J = 8.8, 2.0 Hz, 1H), 4.08 (t, J = 6.0 Hz, 2H), 3.82 (s, 3H), 3.80 (s, 3H), 2.98 (s, 4H), 2.87 (t, J = 6.8 Hz, 2H), 2.05–1.99 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 178.0, 165.0, 161.2, 159.6, 155.0, 154.9, 133.5, 131.4, 128.9, 128.2, 125.9, 125.4, 124.4, 119.1, 116.4, 114.9, 113.1, 112.7, 112.7, 101.7, 101.3, 68.4, 56.3, 55.7, 50.7, 47.5, 29.3, 25.8. HRMS: calcd for C30H31N2O5 [M + H]+, 499.2233, found: 499.2231.
7-(4((2-(5-Methoxy-1H-indol-3-yl)ethyl)amino)butoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (9)
The compound was purified by silica column (CH2Cl2: CH3OH= 400: 9) and obtained as yellow solid in 26.1% yield. Purity 99.2% (by HPLC); mp 76.8–79.5 °C; 1H NMR (400 MHz, CD3OD) δ 8.14 (s, 1H), 8.06 (d, J = 6.8 Hz, 1H), 7.45 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.8 Hz, 1H), 7.04 (s, 2H), 6.97–6.95 (m, 3H), 6.75 (dd, J = 8.8, 2.4 Hz, 1H), 4.03 (t, J = 6.0 Hz, 2H), 3.80 (s, 3H), 3.80 (s, 3H), 2.98 (brs, 4H), 2.70 (t, J = 7.2 Hz, 2H), 1.82–1.75 (m, 2H), 1.71–1.64 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 178.0, 165.3, 161.2, 159.7, 155.0, 154.9, 133.5, 131.4, 128.9, 128.2, 125.9, 125.4, 124.3, 119.0, 116.4, 114.9, 113.0, 113.0, 112.7, 101.8, 101.3, 69.6, 56.3, 55.7, 50.7, 49.9, 27.7, 26.8, 25.9. HRMS calcd for C31H33N2O5 [M + H]+, 513.2389, found: 513.2380.
7-(2-(Benzyl(methyl)amino)ethoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (10)
The compound was obtained as white solid in 76.1% yield. Purity 98.8% (by HPLC); mp 135.0–136.6 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.42 (s, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.32–7.24 (m, 5H), 7.18 (s, 1H), 7.08 (d, J = 8.8 Hz, 1H), 7.00 (d, J = 8.4 Hz, 2H), 4.26 (t, J = 5.6 Hz, 2H), 3.79 (s, 3H), 3.59 (s, 2H), 2.79 (t, J = 5.6 Hz, 2H), 2.25(s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 174.6, 162.9, 159.0, 157.4, 153.5, 138.9, 130.1, 128.1, 126.9, 126.9, 124.1, 123.4, 117.6, 115.1, 113.6, 101.1, 66.8, 61.6, 55.1, 54.9, 42.3. HRMS: calcd for C26H26NO4[M + H]+, 416.1862, found: 416.1855.
7-(2-(Dibenzylamino)ethoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (11)
The crude product was recrystallised by ethyl acetate. The final product was obtained as white solid in 49.9% yield. Purity 98.4% (by HPLC); mp 118.9–120.0 °C; 1H NMR (400 MHz, DMSO-d6) δ: 8.42 (s, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 7.6 Hz, 4H), 7.32 (d, J = 7.6 Hz, 4H), 7.23 (t, J = 7.2 Hz, 2H), 7.09 (s, 1H), 7.05–6.99 (m, 3H), 4.25 (t, J = 5.6 Hz, 2H), 3.79 (s, 3H), 3.68 (s, 4H), 2.83 (t, J = 5.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 174.6, 162.8, 159.0, 157.4, 153.4, 139.2, 130.0, 128.5, 128.2, 126.9, 126.9, 124.1, 123.3, 117.5, 115.1, 113.6, 101.0, 66.7, 57.9, 55.1, 51.2. HRMS: calcd for C32H30NO4 [M + H]+, 492.2175, found: 492.2169.
3-(4-Methoxyphenyl)-7-(2-(prop-2-yn-1-ylamino)ethoxy)-4H-chromen-4-one (12)
The compound was obtained as yellow solid in 54.1% yield. Purity 99.4% (by HPLC); mp 127.8–130.7 °C; 1H NMR (400 MHz, CD3COCD3) δ 8.22 (s, 1H), 8.11 (d, J = 8.4 Hz, 1H), 7.57 (d, J = 7.6 Hz, 2H), 7.09–6.97 (m, 4H), 4.28 (t, J = 4.8 Hz, 2H), 3.31 (s, 3H), 3.49 (s, 2H), 3.12 (t, J = 4.8 Hz, 2H), 2.66 (s, 1H); 13C NMR (100 MHz, CD3COCD3) δ 175.6, 164.4, 160.5, 158.8, 153.6, 131.0, 128.1, 125.4, 125.1, 119.2, 115.7, 114.4, 101.7, 83.2, 72.7, 69.4, 55.6, 47.7, 38.5. HRMS: calcd for C21H30NO4[M + H]+, 350.1392, found: 350.1383.
7-(2-(((3 R,5 S,7r)-3,5-dimethyladamantan-1-yl)amino)ethoxy)-3-(4-methoxyphenyl)-4H-chromen-4-one (13)
The compound was obtained as pale yellow solid in 41% yield. Purity 98.9% (by HPLC); mp 127.5–129.5 °C; 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.49 (d, J = 8.8 Hz, 2H), 7.00–6.96 (m, 3H), 6.84 (d, J = 1.6 Hz, 1H), 4.15 (t, J = 5.6 Hz, 2H), 3.84 (s, 3H), 3.04 (t, J = 5.6 Hz, 2H), 2.18–2.15 (m, 1H), 1.52 (d, J = 2.0 Hz, 2H), 1.35–1.27 (m, 9H), 2.21 (dd, J = 12.4, 7.2 Hz, 2H), 0.86 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 176.0, 163.4, 159.7, 158.0, 152.2, 130.3, 127.9, 125.0, 124.4, 118.6, 115.0, 114.1, 100.8, 69.5, 55.5, 52.4, 51.1, 49.2, 43.1, 41.4, 39.8, 32.6, 30.5, 30.4. HRMS calcd for C30H36NO4 [M + H]+, 474.2644, found: 474.2638.
3-(4-Hydroxyphenyl)-7-(2-(piperidin-1-yl)ethoxy)-4H-chromen-4-one.HBr (14)
The product from the prior step (569 mg, 1.5 mmol) was dissolved into 10 ml 40% methanol solution of HBr and refluxed at 80 °C for 3 h. After cooling down to 0 °C, the reactant was filtrated by suction and washed by methanol (3 ml) for three times. Then, the liquid was removed by vacuum and the residues were recrystallised by methanol. The compound was obtained as white solid in 15.8% yield. Purity 99.3% (by HPLC); mp 241.2–246.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.39 (s, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.40 (d, J = 8.0 Hz, 2H), 7.21 (s, 1H), 7.10 (d, J = 8.8 Hz, 1H), 6.82 (d, J = 8.0 Hz, 2H), 4.34 (t, J = 4.2 Hz, 2H), 3.03 (brs, 2H), 2.77 (brs, 4H), 1.64–1.58 (m, 4H), 1.44 (brs, 2H); 13C NMR (100 MHz, D2O/CD3OD) δ 178.7, 163.4, 159.2, 157.4, 155.5, 131.5, 128.2, 125.6, 124.2, 119.1, 116.4, 116.4, 102.4, 63.3, 56.4, 54.7, 23.7, 22.1. HRMS: calcd for C22H24NO4 [M − Br]+, 366.1705, found: 366.1694.
3-(4-Hyroxyphenyl)-7-(2-(pyrrolidin-1-yl)ethoxy)-4H-chromen-4-one .HBr (15)
The procedure for the synthesis of compound 15 was the same as that of 14. The compound was obtained as white solid in 15.8% yield. Purity 98.9% (by HPLC); mp 234.4–236.8 °C; 1H NMR (400 MHz, D2O/CD3OD) δ 8.15(s, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.8 Hz, 1H), 7.07 (s, 1H), 6.93 (d, J = 8.4 Hz, 2H), 4.38 (t, J = 4.4 Hz, 2H), 3.62 (t, J = 4.4 Hz, 2H), 3.45 (brs, 4H), 2.14 (brs, 4H); 13C NMR (100 MHz, D2O/CD3OD) δ 178.4, 163.4, 159.0, 157.4, 155.3, 131.4, 128.2, 125.4, 124.1, 119.0, 116.3, 116.3, 102.3, 64.6, 55.6, 54.5, 23.7. HRMS: calcd for C21H22NO4 [M − Br]+, 352.1543, found: 352.1541.
7-(2-(Piperidin-1-yl)ethoxy)-3-(4-(2-(piperidin-1-yl)ethoxy)phenyl)-4H-chrom en-4-one (16)
Intermediate a5 (932 mg, 2 mmol) was dissolved in 50 ml acetonitrile. Then K2CO3 (4.60 g, 33.2 mmol) and piperidine (0.31 ml, 3.13 mmol) were added. The mixture was refluxed and stirred overnight. When the reaction ended, the reaction mixture was poured into 200 ml water and stirred for 30 min. After that, the mixture was filtrated by suction and the pastry was washed by water (6 ml) for three times. The crude product was purified by 200–300 mush silica column (CH2Cl2:CH3OH =20:3). The purified compound was obtained as white solid in 13.6% yield. Purity 99.6% (by HPLC); mp 148.7–152.2 °C; 1H NMR (400 MHz, CDCl3) δ 8.19(d, J = 9.2 Hz, 1H), 7.91 (s, 1H), 7.48 (d, J = 8.8 Hz, 2H), 7.00–6.96 (m, 3H), 6.86 (d, J = 2.4 Hz, 1H), 4.20 (t, J = 6.0 Hz, 2H), 4.15 (t, J = 6.0 Hz, 2H), 2.84–2.79 (m, 4H), 2.53 (brs, 8H), 1.65–1.59 (m, 8H), 1.46 (brs, 4H); 13C NMR (100 MHz, CDCl3) δ 176.0, 163.3, 158.9, 158.0, 152.2, 130.2, 127.9, 125.0, 124.4, 118.6, 115.1, 114.8, 100.9, 66.9, 66.1, 58.0, 57.8, 55.3, 55.2, 26.1, 26.0, 24.3, 24.3. HR MS: calcd for C29H37N2O4 [M + H]+, 477.2753, found: 477.2743.
3-(4-Methoxyphenyl)-7-(2-oxo-2-(piperidin-1-yl)ethoxy)-4H-chromen4-one (17)
The piperidine (4 ml, 80 mmol) which was diluted in THF (5 ml) was added dropwise into the chloroacetyl chloride (3.1 ml, 40 mmol) dissolved in THF (10 ml) and stirred for 10 h at room temperature, the mixture was condensed by reduced pressure to dryness and the residue was dissolved in 50 ml acetone. Then, formononetin (1 g, 3.73 mmol) and K2CO3 (4.60 g, 33.2 mmol) were added into the solution. The reactant was refluxed overnight. When the reaction finished, the reaction solution was poured into 200 ml water, stirred and filtrated by suction. The pastry was washed by water and purified by 200–300 mush silica column (CH2Cl2:CH3OH =100:1) .The compound was obtained as pale yellow solid in 45.3% yield. Purity 99.0% (by HPLC); mp 144.8–145.8 °C; 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.8 Hz, 1H), 7.87 (s, 1H), 7.45 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 1H), 6.93–6.89 (m, 3H), 4.75 (s, 2H), 3.79 (s, 3H), 3.53 (brs, 2H), 3.42 (brs, 2H), 1.62–1.51 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 175.7, 165.0, 162.3, 159.5, 157.7, 152.2, 130.1, 127.9, 124.8, 124.1, 118.9, 114.6, 113.9, 101.4, 67.5, 55.3, 46.3, 43.2, 26.5, 25.5, 24.3. HRMS calcd for C25H30NO5 [M + H]+, 424.2124, found: 424.2124.
3-(4-Methoxyphenyl)-7-(3-oxo-3-(piperidin-1-yl)propoxy)-4H-chromen-4-one (18)
K2CO3 (7.5 g, 54.3 mmol), 3-bromopropionic acid (6 g, 39.2 mmol) and formononetin (2 g, 7.4 mmol) were dissolved in 100 ml acetone and refluxed for 2 days. After that, the reactant was condensed by reduced pressure to dryness and then the residue was poured into 400 ml water. In order to precipitate the product, the solution should be acidified with HCl (1 N) to pH 5. Then, the precipitated white solid was filtrated by suction and washed with water (5 ml) for three times. 3-((3-(4-Methoxyphenyl)-4H-chromogen 4-one-7-yl)oxy)propanoic acid crude product (2.465 g) was obtained.
The obtained crude product was dissolved in 50 ml DMF, then, N,N-diisopropylethylamine (1.02 ml, 5.85 mmol), piperidine (0.5 ml, 5.1 mmol) and hydrochloro-1-(3-dimethylaminopropyl)-3-ethylcarbodiimide salt were added and stirred overnight at room temperature. When the reaction finished, the reactant was poured into 200 ml water and extracted with CH2Cl2, washed with saturated K2CO3 and brine respectively. The organic phase was taken, dried with Na2SO4, filtrated by suction and the filtrate was condensed by reduced pressure. The residues were purified by silica column (petroleum ether:ethyl acetate =2:1). The final compound was obtained as pale yellow solid in 26.9% yield. Purity 99.5% (by HPLC); mp 161.0–163.0 °C; 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.50 (d, J = 8.4 Hz, 2H), 6.96 (brd, J = 8.4 Hz, 3H), 6.90 (s, 1H), 4.42 (t, J = 6.0 Hz, 2H), 3.84 (s, 3H), 3.54 (brd, 4H), 2.89 (t, J = 6.0 Hz, 2H), 1.70–1.60 (m, 6H); 13C NMR (100 MHz, DMSO-d6) δ 174.6, 167.6, 162.9, 159.0, 157.4, 153.5, 130.1, 126.9, 124.1, 123.3, 117.5, 115.0, 113.6, 101.0, 65.1, 55.1, 45.9, 42.0, 31.8, 26.0, 25.3, 24.0. HRMS: calcd for C24H26NO5 [M + H]+, 408.1811, found: 408.1805.
2-((3-(4-Methoxyphenyl)-4-oxo-4H-chromen-7-yl)oxy)ethyl piperidine-1-carboxylate (19)
Intermediate a1 (0.748 mg, 2 mmol), K2CO3 (4.60 g, 33.3 mmol) was dissolved in DMF (70 ml), then, piperidine (0.302 ml, 3.30 mmol) was poured into the reactant liquid, stirred and refluxed for 3 h. When the reaction finished, the mixture was poured into water (300 ml), stirred for 30 min and filtrated by suction. The pastry was washed by water (5 ml) for three times and purified by 200–300 mush silica column eluted by CH2Cl2. The compound was obtained as pale yellow solid in 12.6% yield. Purity 99.1% (by HPLC); mp 104.5–109.4 °C. 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.8 Hz, 1H), 7.90 (s, 1H), 7.48 (d, J = 8.8 Hz, 2H), 7.00 (dd, J = 8.8, 2.0 Hz, 1H), 6.95 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 2.0 Hz, 1H), 4.46 (t, J = 6.0 Hz, 2H), 4.27 (t, J = 6.0 Hz, 2H), 3.82 (s, 3H), 3.41 (brs, 4H), 1.56–1.51 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 175.1, 163.0, 159.5, 157.8, 155.0, 152.1, 130.1, 127.8, 124.8, 124.2, 118.6, 114.8, 113.9, 100.9, 67.0, 63.1, 55.3, 44.9, 25.6, 24.3. HRMS: calcd for C24H26NO6 [M + H]+, 424.1760, found: 424.1751.
2-((3-(4-Methoxyphenyl)-4-oxo-4H-chromen-7-yl)oxy)ethyl prop-2-yn-1-ylc-arbamate (20)
The procedure was the same as above. However, the piperidine was replaced with prop-2-yn-1-amine (0.226 ml, 3.30 mmol). The final compound was obtained as pale yellow solid in 18.7% yield. Purity 99.4% (by HPLC); mp 155.1–157.4 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.42 (s 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.78 (t, J = 5.6 Hz, 1H), 7.53 (d, J = 8.8, 2.0 Hz, 2H), 7.20 (d, J = 2.0 Hz, 1H), 7.10 (dd, J = 8.8, 2.0 Hz, 1H), 7.01–6.97 (m, 2H), 4.37–4.32 (m, 4H), 3.79 (s, 5H), 3.10 (t, J = 2.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 174.6, 162.6, 159.0, 157.4, 155.8, 153.5, 130.1, 127.0, 124.0, 123.4, 117.7, 115.0, 113.6, 101.2, 81.3, 73.0, 67.1, 62.5, 55.1, 29.8. HRMS: calcd for C22H20NO6 [M + H]+, 394.1291, found: 394.1286.
AChE/BuChE bioassay
To measure in vitro AChE/BuChE activity, modified Ellman's method27–30 was performed using a 96-well plate reader (BioTek ELx808). AChE (0.5 U/mg) was extracted from rat cortex while BuChE (3.4 U/mg) was obtained from human plasma. Each well contained 50 μl potassium phosphate buffer (KH2PO4/K2HPO4, 0.1 M, pH 8.0), 25 μl test compounds and 25 μl enzyme. Notably, the test compounds were firstly dissolved in the mixture of 50% methanol and 50% DMSO, and eventually diluted so that the final concentration of both solvents was less than 1% in the assay. They were pre-incubated for 60 min at 37 °C, and then 125 μl 5,5′-dithiobis-2-nitrobenzoic acid (DNTB, 3 mM in buffer) was added. Characterisation of the hydrolysis of acetylthiocholine iodide or butyrylthiocholine chloride catalysed by AChE/BuChE was performed spectrometrically at 412 nm, followed by the addition of substrate (acetylthiocholine iodide or butyrylthiocholine chloride 3 mM in water, respectively). The activity was determined by measuring the increase in absorbance at 412 nm after 15 min. For those compounds with inhibition rate >50% at the concentration of 50 μM, the IC50 values were further determined. A control experiment was performed under the same condition without any inhibitor and the blank contained buffer, water, DTNB and substrate. The described method was also performed for BuChE bioassay. In the bioassay, donepezil (an AChE inhibitor) and tetraisopropyl pyrophosphoramide (Iso-OMPA, a BuChE inhibitor) were used as positive controls of AChE and BuChE, respectively.
Molecular docking
In order to understand the protein-ligand interactions between the synthesised isoflavone derivatives and AChE/BuChE, the most potent inhibitor as an example was docked against AChE and BuChE. The ligand structure was built and prepared by DS 2016 (Discovery Studio version 2016, San Diego, CA, USA). Hydrogen atoms were added to the structure and its ionisation states at pH 7.3 to 7.5 were generated. Besides, its lowest-energy conformation was generated prior to docking. The X-ray structures of AChE (PDB ID: 5EIH)31 and BuChE (PDB ID: 4BDS)32 were retrieved from the Protein Data Bank. Both of them were high-resolution structures of protein-ligand complexes and their organisms were consistent with those of the enzymes used for bioassay. After stripping the cognate ligand from each complex, molecular docking was carried out by GOLD 3.0.1 in which the target protein was kept rigid, while the ligands were left flexible to explore the conformational space inside the binding site. After that, the docking poses of the ligand were visually inspected.
In silico prediction of pharmacokinetic properties
Pharmacokinetic properties, in particular blood–brain barrier penetration (BBB), are quite important for the drugs for the treatment of AD. Therefore, the pharmacokinetic properties of AChE/BuChE inhibitors were predicted by the “ADMET Descriptors” module implemented in DS2016. The pharmacokinetic properties available in this module were aqueous solubility, BBB, CYP2D6 binding, hepatotoxicity, intestinal absorption and plasma protein binding.
Results and discussion
Chemistry
In total, 20 isoflavone derivatives (cf. Table 1) were synthesised and their chemical structures were confirmed by melting point, HR MS, 1H NMR and 13C NMR.
Table 1.
Chemical structures of synthesised isoflavone derivatives.
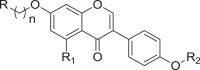 | ||||
|---|---|---|---|---|
| Compound | n | R1 | R2 | R |
| 1 | 2 | OH | CH3 | 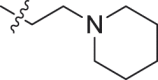 |
| 2 | 3 | H | CH3 | 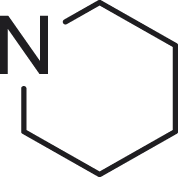 |
| 3 | 4 | H | CH3 | 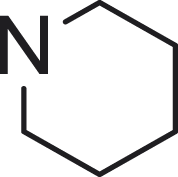 |
| 4 | 2 | H | CH3 | 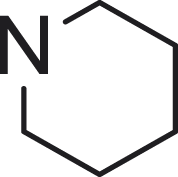 |
| 5 | 3 | H | CH3 | 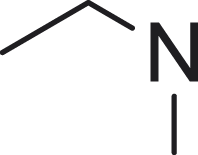 |
| 6 | 4 | H | CH3 | 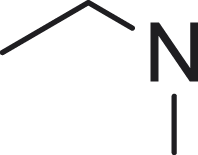 |
| 7 | 2 | H | CH3 | 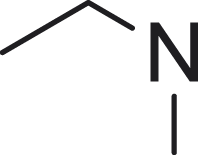 |
| 8 | 3 | H | CH3 | 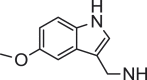 |
| 9 | 4 | H | CH3 | 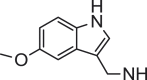 |
| 10 | 2 | H | CH3 | 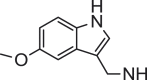 |
| 11 | 3 | H | CH3 | 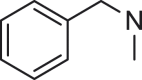 |
| 12 | 4 | H | CH3 | 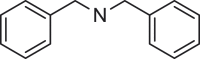 |
| 13 | 2 | H | CH3 | 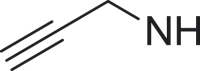 |
| 14 | 2 | H | H | 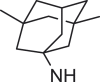 |
| 15 | 2 | H | H | 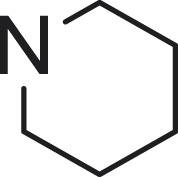 |
| 16 | 2 | H | 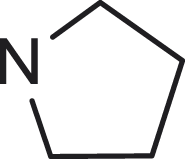 |
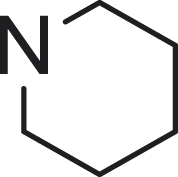 |
| 17 | 1 | H | CH3 | 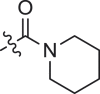 |
| 18 | 2 | H | CH3 | 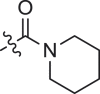 |
| 19 | 2 | H | CH3 | 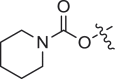 |
| 20 | 2 | H | CH3 | 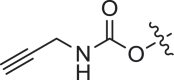 |
The synthetic route of new isoflavone derivatives was designed and depicted in Scheme 1 according to earlier reports33,34. Briefly, the bromo-substituted intermediates a1–a5 were prepared through commercially available isoflavones and dibromoalkane in acetone in the presence of potassium carbonate. Reaction between a1–a5 and corresponding amines in acetonitrile afforded compounds 1–16. At first, we synthesised compounds 1–9 and evaluated their biological activity. Structure-activity relationship (SAR) of these compounds demonstrated the optimal length of the linker between the amino group and isoflavone core structure seems to be 2. Previous reports indicated that various amino groups can form a cation–pi interaction with the aromatic amino acid residues (i.e. Trp84 in AChE and Try82 in BuChE) located in the catalysis active site of the cholinesterases35. This observation prompted us to design and synthesise compounds 10–20 by substitution of different amino groups.
Scheme 1.
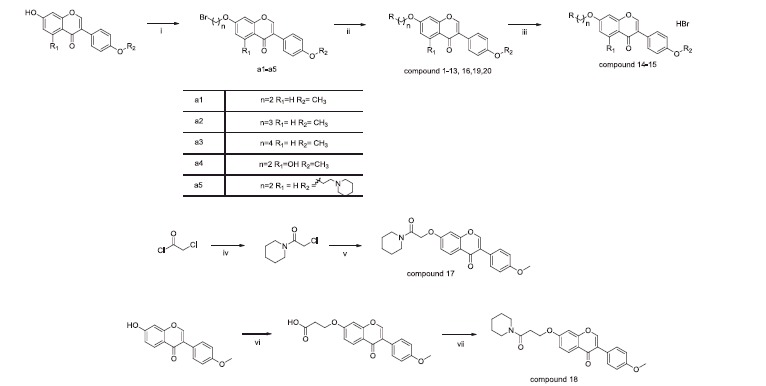
The general procedure for the synthesis of compounds 1–20. (i): K2CO3, acetone, Br(CH2)nBr, 60 °C; (ii): RH (amines), K2CO3, DMF/Acetonitrile, 100 °C, 3 h; (iii): 40% HBr, 120 °C, 3 h; (iv): piperidine, THF r.t. 10 h; (v): formononetin, K2CO3, acetone; (vi) K2CO3, 3-bromopropionic acid, acetone, 60 °C, 2 days; (vii) DMF, N, N-diisopropylethylamine, piperidine, 12 h, r.t.
It is worth noting that carbamate substituted compounds 19 and 20 can be produced by the reaction of amines and bromo-substituted isoflavone in the presence of DMF and K2CO3 rather than the reported reaction of acyl chloride with hydroxyl-substituted isoflavone36 in acetonitrile. Interestingly, compound 17 can be synthesised by step 1 and step 2 as shown in Scheme 1, however this synthetic route was not suitable for compound 18 because of the poor reactive activity of 3-chloro-1-(piperidin-1-yl)propan-1-one which has a longer distance between reactive site and amide compared to 2-chloro-1-(piperidin-1-yl) ethan-1-one. Therefore, the intermediate 3-((3-(4-methoxyphenyl)-4H-chromogen-4-one-7-yl)oxy)propanoic acid was obtained by the reaction of formononetin with 3-bromopropionic acid. Piperidine was then added to the intermediate to produce the final compound 18.
In vitro inhibition of AChE and BuChE
Both in vitro AChE and BuChE inhibitory activity were evaluated for all the synthesised isoflavone derivatives. The results are shown in Table 2. To be noted donepezil and Iso-OMPA were used as the positive drugs.
Table 2.
In vitro inhibition of AChE and BuChE for compounds 1–20.
| Inhibition % at 50 μM |
IC50 (μM) |
|||
|---|---|---|---|---|
| Name | AChE | BuChE | AChE | BuChE |
| G | n.d.a | n.d. | 1.47 | 3.37 |
| 1 | 71.10 | 95.74 | 10.3 | 4.12 |
| 2 | 28.34 | 97.00 | n.d. | 23.2 |
| 3 | 85.65 | 98.11 | 46.39 | 11.1 |
| 4 | 58.66 | 84.13 | 100.4 | 141.3 |
| 5 | 25.38 | 91.32 | n.d. | 103.6 |
| 6 | 63.76 | 94.77 | 106.6 | 68.08 |
| 7 | 3.68 | 45.74 | n.d. | n.d. |
| 8 | 2.42 | 60.63 | n.d. | 15.6 |
| 9 | 0.72 | 37.92 | n.d. | n.d. |
| 10 | 3.34 | 2.72 | n.d. | n.d. |
| 11 | −6.27 | −18.48 | n.d. | n.d. |
| 12 | 5.47 | −16.87 | n.d. | n.d. |
| 13 | 17.61 | −6.62 | n.d. | n.d. |
| 14 | 79.38 | 99.86 | 9.75 | 7.66 |
| 15 | 75.22 | 99.88 | 57.74 | 7.19 |
| 16 | 100.34 | 99.74 | 4.60 | 5.92 |
| 17 | 0.54 | −2.83 | n.d. | n.d. |
| 18 | −2.36 | 5.59 | n.d. | n.d. |
| 19 | −11.56 | 53.03 | n.d. | 9.59 |
| 20 | −4.84 | −5.49 | n.d. | n.d. |
| Donepezil | 64.82 | n.d. | 1.05 | n.d. |
| Iso-OMPA | n.d. | 94.28 | n.d. | 5.78 |
n.d.: not determined.
As shown in Table 2, five isoflavone derivatives, i.e. 1, 3, 14, 15 and 16, showed both AChE and BuChE inhibitory effect. Notably, compound 16 displayed equivalent inhibitory activity to two single-targeting drugs (Donepezil and Iso-OMPA) as well as our hit compound G.
Based on the chemical structures and their biological activity, it is clear that AChE and BuChE inhibitory activities changed markedly because of the change of amino substitutes. Compounds would manifest inhibitory effect when C7 was substituted by piperidine or N-methylethanamide. In the meanwhile, the length of the side chain also played a significant role in maintaining the inhibitory activity. Generally, compounds with side chains of two or four carbon atoms can display a dual-targeting inhibition whereas those with three carbon atoms selectively inhibited BuChE. Besides, compounds with a side chain of four carbon atoms showed stronger inhibition for BuChE than those with two carbon atoms. However, ester-substituted compounds displayed no AChE inhibitory effect. The alternative isoflavone core changed the inhibition level for both AChE and BuChE. C5 hydroxyl substitution improved the inhibitory effect. When C4′ was substituted for 2-piperidineethoxyl group, the most potent AChE inhibitor 16 was obtained. All the information provided us with clues for further lead optimisation.
Binding modes of isoflavone derivatives
As compound 16 was the most potent AChE/BuChE dual-targeted inhibitor, it was selected as an example and used in the docking simulation. As shown in Figure 3, compound 16 was able to bind both AChE and BuChE. Regarding AChE, the piperidine moiety in C4’ position occupied the “entrance cavity” by forming π–cation interaction with the key amino acid residues TRP286 and TYR341, while the isoflavone core structure was well accommodated in the catalytic cleft through π–π T-shaped interactions with PHE297/TYR341 and hydrogen bonds with TYR124/TYR337. Furthermore, the π–cation interaction between the piperidine ring in C7 position with TRP86, the salt bridge with GLU202 and the hydrogen bond with HIS447 also contributed to the stabilisation of protein–ligand interaction.
Figure 3.

(a) 2D schematic diagram of potential interactions between compound 16 and AChE. (b) The predicted binding mode of compound 16 with AChE.
Protein–ligand interactions between compound 16 with BuChE were similar to that for AChE (cf. Figure 4). It deserved to mention that the piperidine moiety in C7 position formed a hydrogen bond with the key amino acid residue TYR332 which can also interact with the isoflavone structure through π–π stacking and π–π T-shape. Protonated amino group in C4 position can stabilise the complex through forming a salt bridge with both TRP82.
Figure 4.

(a) 2D schematic diagram of potential interactions between compound 16 and BuChE. (b) The predicted binding mode of compound 16 with BuChE.
Predicted pharmacokinetic properties
As shown in Table 3, all the AChE/BuChE dual-targeted inhibitors reported in this study appeared to have poor solubility in aqueous media whereas possess good absorption. Like compound G, compounds 3, 14 and 16 were predicted to penetrate the BBB, which was a favoured property for drugs to treat neurodegenerative diseases, e.g. AD. Encouragingly, compound 16 may not bind to CYP2D6, which was different from other compounds, including the hit compound G. This unique feature would be beneficial for ensuring the efficacy of compound 16 and avoiding the side-effect. These data prove our hit-to-lead optimisation strategy seems to be also effective in optimising pharmacokinetic property.
Table 3.
Predicted pharmacokinetic properties of compounds 1, 3, 14, 15 and 16.
| Compound | AlogP98 | PSA-2D | Solubility level | Absorption level | BBB level | PPB | CYP2D6 |
|---|---|---|---|---|---|---|---|
| 1 | 3.647 | 68.259 | 2 | 0 | 2 | True | True |
| 3 | 4.532 | 47.443 | 2 | 0 | 1 | True | True |
| 14 | 3.664 | 59.328 | 2 | 0 | 1 | True | True |
| 15 | 3.208 | 59.328 | 2 | 0 | 2 | True | True |
| 16 | 4.945 | 50.796 | 2 | 0 | 1 | True | False |
| G | 4.267 | 47.443 | 2 | 0 | 1 | True | True |
AlogP98: Lipophilicity descriptor; PSA-2D: Polar surface area; AlogP98: Lipophilicity descriptor; PSA-2D: Polar surface area; Solubility Level: (0, Good; 1, Moderate; 2, Poor; 3, Very poor); Absorption Level: (0, Good; 1, Moderate; 2, Poor; 3, Very poor); BBB Level: (0, very high blood–brain barrier penetration; 1, high; 2, medium; 3, low).
Conclusion
In this study, we report the synthesis of a series of novel isoflavone derivatives based on our hit compound G identified by in silico HTS. The in vitro AChE/BuChE bioassay has shown 5 out of 20 isoflavone derivatives were AChE/BuChE dual-targeted inhibitors. SAR analysis has demonstrated the length of the side chain and the type of amino-substituted group played an essential role in AChE inhibition, whereas both factors had little effect on the BuChE inhibition. Among these derivatives, compound 16 possessed the greatest AChE inhibition as well as strong BuChE inhibitory activity. The molecular docking study demonstrated that π–cation and π–π interactions were essential for compound 16 to display its dual-targeting effect. In addition, the in silico prediction of pharmacokinetic properties has indicated 1) compounds 3, 14 and 16 were able to penetrate the BBB and 2) compound 16 may be more effective and less toxic due to no binding to CYP2D6. Taken together, compound 16 may warrant further development as a potential drug-like AChE/BuChE dual-targeted inhibitor for the treatment of AD.
Supplementary Material
Funding Statement
This work was partly supported by National Major Scientific and Technological Special Project for Significant New Drugs Development [2012ZX09301002-001, 2013ZX09402103 and 2014ZX09507003-002] and National Natural Science Foundation of China [81603027].
Disclosure statement
The authors declare no conflict of interest in this work.
References
- 1.Kumar A, Singh A.. A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol Rep 2015;67:195–203. [DOI] [PubMed] [Google Scholar]
- 2.Anand R, Gill KD, Mahdi AA.. Therapeutics of Alzheimer's disease: past, present and future. Neuropharmacology 2014;76 Pt A:27–50. [DOI] [PubMed] [Google Scholar]
- 3.Geula C, Mesulam MM.. Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis Assoc Disord 1995;9 Suppl 2:23–8. [DOI] [PubMed] [Google Scholar]
- 4.Overk CR, Felder CC, Tu Y, et al. Cortical M1 receptor concentration increases without a concomitant change in function in Alzheimer's disease. J Chem Neuroanat 2010;40:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Q-S, Holloway HW, Flippen-Anderson JL, et al. Methyl analogues of the experimental alzheimer drug phenserine: synthesis and structure/activity relationships for acetyl- and butyrylcholinesterase inhibitory action. J Med Chem 2001;44:4062–71. [DOI] [PubMed] [Google Scholar]
- 6.Silva T, Reis J, Teixeira J, Borges JF.. Alzheimer's disease, enzyme targets and drug discovery struggles: from natural products to drug prototypes. Ageing Res Rev 2014;15:116–45. [DOI] [PubMed] [Google Scholar]
- 7.Pepeu G, Giovannini MG.. Cholinesterase inhibitors and memory. Chem Biol Interact 2010;187:403–8. [DOI] [PubMed] [Google Scholar]
- 8.AnandSingh PB.A review on cholinesterase inhibitors for Alzheimer's disease. Arch Pharm Res 2013;36:375–99. [DOI] [PubMed] [Google Scholar]
- 9.Houghton PJ, Ren Y, Howes MJ.. Acetylcholinesterase inhibitors from plants and fungi. Nat Prod Rep 2006;23:181–99. [DOI] [PubMed] [Google Scholar]
- 10.Munoz FJ, Inestrosa NC.. Neurotoxicity of acetylcholinesterase amyloid beta-peptide aggregates is dependent on the type of abeta peptide and the ache concentration present in the complexes. FEBS Lett. 1999;450:205–9. [DOI] [PubMed] [Google Scholar]
- 11.Sugimoto H. Donepezil hydrochloride: a treatment drug for Alzheimer's disease. Chem Rec 2001; 1: 63–73. [DOI] [PubMed] [Google Scholar]
- 12.Zarotsky V, Sramek JJ, Cutler NR.. Galantamine hydrobromide: an agent for Alzheimer's disease. Am J Health Syst Pharm 2003;60:446–52. [DOI] [PubMed] [Google Scholar]
- 13.CastroMartinez AA.Targeting beta-amyloid pathogenesis through acetylcholinesterase inhibitors. Curr Pharm Des 2006;12:4377–87. [DOI] [PubMed] [Google Scholar]
- 14.Birks J.Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev 2006;1:CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darvesh S, Hopkins D, Geula AC.. Neurobiology of butyrylcholinesterase. Nat Rev Neurosci 2003;4:131–8. [DOI] [PubMed] [Google Scholar]
- 16.Greig NH, Utsuki T, Yu Q-S, et al. A new therapeutic target in Alzheimer's disease treatment: attention to butyrylcholinesterase. Curr Med Res Opin 2001;17:159–65. [DOI] [PubMed] [Google Scholar]
- 17.Recanatini M, Cavalli A.. Acetylcholinesterase inhibitors in the context of therapeutic strategies to combat Alzheimer’s disease. Expert Opin Ther Pat 2005;12:1853–65. [Google Scholar]
- 18.Ismaili L, Refouvelet B, Benchekroun M, et al. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer's disease. Prog Neurobiol 2017;151:4–34. [DOI] [PubMed] [Google Scholar]
- 19.Guzior N, Wieckowska A, Panek D, Malawska DB.. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr Med Chem 2015;22:373–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bacalhau P, San Juan AA, Goth A, et al. Insights into (S)-rivastigmine inhibition of butyrylcholinesterase (Buche): molecular docking and saturation transfer difference NMR (STD-NMR). Bioorg Chem 2016; 67:105–9. [DOI] [PubMed] [Google Scholar]
- 21.Singh M, Silakari O.. Design, synthesis and biological evaluation of novel 2-phenyl-1-benzopyran-4-one derivatives as potential poly-functional anti-Alzheimer's agents. RSC Adv 2016;6:108411–22. [Google Scholar]
- 22.Li SY, Wang XB, Xie SS, et al. Multifunctional tacrine-flavonoid hybrids with cholinergic, beta-amyloid-reducing, and metal chelating properties for the treatment of Alzheimer's disease. Eur J Med Chem 2013;69:632–46. [DOI] [PubMed] [Google Scholar]
- 23.Sun Y, Chen J, Chen X, et al. Inhibition of cholinesterase and monoamine oxidase-B activity by tacrine-homoisoflavonoid hybrids. Bioorg Med Chem 2013;21:7406–17. [DOI] [PubMed] [Google Scholar]
- 24.Desideri N, Bolasco A, Fioravanti R, et al. Homoisoflavonoids: natural scaffolds with potent and selective monoamine oxidase-B inhibition properties. J Med Chem 2011;54:2155–64. [DOI] [PubMed] [Google Scholar]
- 25.Liao S, Deng H, Huang S, et al. Design, synthesis and evaluation of novel 5,6,7-trimethoxyflavone-6-chlorotacrine hybrids as potential multifunctional agents for the treatment of Alzheimer's disease. Bioorg Med Chem Lett 2015;25:1541–5. [DOI] [PubMed] [Google Scholar]
- 26.Sang Z, Qiang X, Li Y, et al. Design, synthesis and evaluation of scutellarein-O-alkylamines as multifunctional agents for the treatment of Alzheimer's disease. Eur J Med Chem 2015;94:348–66. [DOI] [PubMed] [Google Scholar]
- 27.Ellman GL, Courtney KD, Andres V, Featherstone RM.. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961;7:88–95. [DOI] [PubMed] [Google Scholar]
- 28.Kim DH, Hung TM, Bae KH, et al. Gomisin A improves scopolamine-induced memory impairment in mice. Eur J Pharmacol 2006; 542: 129–35. [DOI] [PubMed] [Google Scholar]
- 29.Lima JA, Costa RS, Epifanio RA, et al. Geissospermum vellosii stembark: anticholinesterase activity and improvement of scopolamine-induced memory deficits. Pharmacol Biochem Behav 2009;92:508–13. [DOI] [PubMed] [Google Scholar]
- 30.Kurt BZ, Gazioglu I, Basile L, et al. Potential of aryl-urea-benzofuranylthiazoles hybrids as multitasking agents in Alzheimer's disease. Eur J Med Chem 2015;102:80–92. [DOI] [PubMed] [Google Scholar]
- 31.Bourne Y, Sharpless KB, Taylor P, Marchot PP.. Steric and dynamic parameters influencing in situ cycloadditions to form triazole inhibitors with crystalline acetylcholinesterase. J Am Chem Soc 2016;138:1611–21. [DOI] [PubMed] [Google Scholar]
- 32.Nachon F, Carletti E, Ronco C, et al. Crystal structures of human cholinesterases in complex with huprine w and tacrine: elements of specificity for anti-Alzheimer's drugs targeting acetyl- and butyryl-cholinesterase. Biochem J 2013;453:393–9. [DOI] [PubMed] [Google Scholar]
- 33.Delcanale M, Amari G, Armani E, et al. Novel basic isoflavones as inhibitors of bone resorption. Helv Chim Acta 2001;84:2417–29. [Google Scholar]
- 34.Seguin H, Gardette D, Moreau M-F, et al. A general method for the synthesis of N, N-dialkylaminobutylamines. Synth Commun 1998;28:4257–72. [Google Scholar]
- 35.Overk CR, Felder CC, Tu Y, et al. Cortical M1 receptor concentration increases without a concomitant change in function in Alzheimer’s disease. J Chem Neuroanat 2010;40:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inesi A, Mucciante L, Rossi VL.. A convenient method for the synthesis of carbamate esters from amines and tetraethylammonium hydrogen carbonate. J Organ Chem 1998;63:1337–8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.