

Abstract
Cardiac dysfunctions dramatically increase with age. Revealing a currently unknown contributor to cardiac ageing, we report the age‐dependent, cardiac‐specific accumulation of the lysosphingolipid sphinganine (dihydrosphingosine, DHS) as an evolutionarily conserved hallmark of the aged vertebrate heart. Mechanistically, the DHS‐derivative sphinganine‐1‐phosphate (DHS1P) directly inhibits HDAC1, causing an aberrant elevation in histone acetylation and transcription levels, leading to DNA damage. Accordingly, the pharmacological interventions, preventing (i) the accumulation of DHS1P using SPHK2 inhibitors, (ii) the aberrant increase in histone acetylation using histone acetyltransferase (HAT) inhibitors, (iii) the DHS1P‐dependent increase in transcription using an RNA polymerase II inhibitor, block DHS‐induced DNA damage in human cardiomyocytes. Importantly, an increase in DHS levels in the hearts of healthy young adult mice leads to an impairment in cardiac functionality indicated by a significant reduction in left ventricular fractional shortening and ejection fraction, mimicking the functional deterioration of aged hearts. These molecular and functional defects can be partially prevented in vivo using HAT inhibitors. Together, we report an evolutionarily conserved mechanism by which increased DHS levels drive the decline in cardiac health.
Keywords: dihydrosphingosine, DNA damage, genomic instability, histone modification, transcription
Subject Categories: Ageing; Chromatin, Epigenetics, Genomics & Functional Genomics; Metabolism
Introduction
A temporal decline in the functional and molecular integrity of an organism defines its ageing process. While multiple hallmarks of ageing have recently been enumerated, our understanding of their functional, molecular and temporal hierarchy remains incomplete. The significance of these hallmarks in the ageing process varies in a tissue‐/cell type‐specific manner. Considering the physiological differences between organs, it is conceivable that their susceptibility to these distinct triggers of ageing can differ. Therefore, it is important to delineate those mechanisms that influence the ageing process of specific organs to better understand organismal ageing. The heart is the first functional organ in a developing embryo. Once formed, cardiomyocytes are seldom replaced during the lifetime of an organism 1. Therefore, to sustain cardiac health, cardiomyocytes need to maintain their functional and molecular integrity with age 2. Still, our knowledge of the specific molecular processes and mechanisms affected by age in cardiomyocytes is far from complete.
Results
African turquoise killifish (Nothobranchius furzeri) as a model for cardiac ageing
To address this, we used a short‐lived vertebrate, the African turquoise killifish (N. furzeri) as a model for cardiac ageing (Fig EV1A and B) 3, 4. The killifish heart showed classical hallmarks of ageing including elevated senescence‐associated‐β‐galactosidase (SA‐β‐GAL) activity as well as accumulation of lipofuscin granules (Sudan Black B staining) as early as ~ 8 weeks post‐hatching (Fig EV1C and D).
Figure EV1. Killifish hearts show molecular, physiological and metabolic signatures of cardiac ageing.
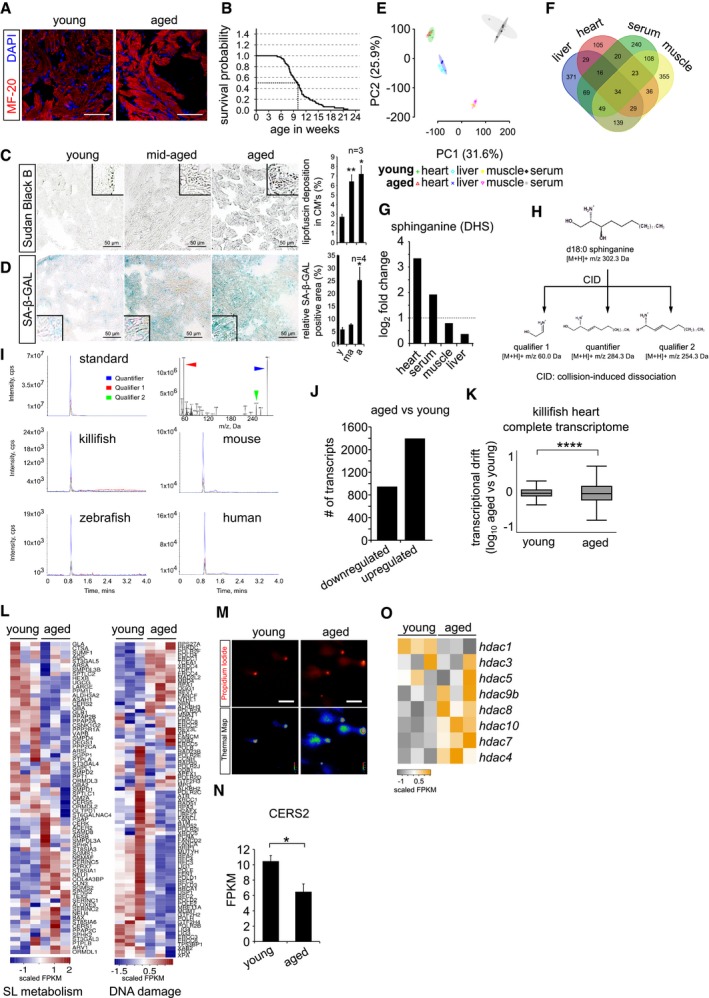
- Immunohistochemistry on 10 μm cryosections of young (6 weeks) and aged (12 weeks) killifish ventricular tissue with cardiac marker MF‐20. Cardiomyocyte sarcomeres are labelled with MF‐20 (red).
- Kaplan–Meier analysis representing the survival of GRZ‐AD killifish strain used in this study. The median lifespan of killifish in laboratory condition is ˜ 9.5 weeks, indicated as stripped line (n = 170).
- Lipofuscin staining by Sudan Black B on 5 μm paraffin sections of different ages of killifish heart. Lipofuscin granules are labelled in grey‐black colour. The bar graph on right represents the percentage area of the ventricular region acquired by lipofuscin granule with respect to total tissue area. Quantification was performed on a total of 12 sections per condition, covering independent regions of the ventricle. Number of animals = 3 per condition. Insets represent magnified view of the ventricles.
- Senescence detection based on SA‐beta‐galactosidase activity in ageing killifish hearts. Blue/cyan colour represents the positive regions in the ventricular sections. The bar graph on right represents percentage of SA‐β‐GAL‐stained area with respect to the total ventricle area. Quantification was performed on a total of 12 sections per condition, covering different regions of the ventricle. Number of animals = 4 per condition. Insets represent magnified view of the ventricle.
- Principal component analysis of all the metabolites detected in various tissue samples. Number of animals = 4 per condition.
- Venn diagram of all the detected metabolites by untargeted metabolomics. Number of animals = 4 per condition.
- Bar graph depiction of the relative abundance of sphinganine levels in various killifish tissues obtained from untargeted metabolomics n = 4, each condition. Log2 fold enrichment represents the relative abundance of sphinganine in aged tissues in comparison with young ones.
- Graphical illustration of collision‐induced dissociation of standard sphinganine d18:0, resulted in 2 qualifiers and 1 quantifier. Mass by charge ratios (m/z) of corresponding fragments are mentioned underneath.
- Spectral peaks of both qualifiers (in red and green) and a quantifier (in blue) obtained from dissociation of standard sphinganine and of aged heart tissues from killifish, zebrafish, mouse and humans.
- Bar graph depicting the number of up and downregulated genes in the killifish ventricles upon ageing (n = 3 animals per condition).
- Transcriptional drift variance of all the detected transcripts in young and aged killifish hearts. Data are represented as box‐plot, and Levene's test was used for the estimation of the statistical significance. Error bars represent drift‐variance. Box plot whiskers show 1.5 IQR of highest and lowest quartile. Horizontal line within the bars represent median of the underlying population.
- Heatmap representation of the hallmark genes of sphingolipid metabolism and DNA damage pathways shows differential expression in young and aged killifish hearts. Scale bar represents scaled FPKM (Fragments Per Kilobase of transcript per Million mapped reads), to graphically represent expression levels of the indicated genes, on a scale given scale.
- Micrographs of comet assay on isolated killifish cardiomyocytes from young and old individuals n = 4, each condition.
- Bar graph illustrating the absolute transcript expression of ceramide synthase 2 between young and aged killifish ventricles, n = 3 per condition. Expression levels are depicted in FPKM (Fragments Per Kilobase of transcript per Million mapped reads).
- Heatmap with scaled FPKM values of all detected histone deacetylase genes, which are differentially expressed in the killifish transcriptome. Scale bar represents scaled FPKM, to graphically represent expression levels of the indicated genes on a scale given scale.
Elevated sphinganine levels and loss of genomic integrity are an evolutionarily conserved hallmark of the aged vertebrate heart
Since the heart is one of the most metabolically active organs, we hypothesized that breakdown of its metabolic homeostasis would critically mediate cardiac ageing. To identify cardiac‐specific changes in the metabolome with age, we performed untargeted LC‐mass spectrometry‐based metabolome profiling of young and aged killifish hearts. General age‐associated changes were distinguished from heart‐specific ones by a side‐by‐side analysis of the liver, skeletal muscle and serum metabolomes (Fig 1A and Dataset EV1). Tissue types segregated on the basis of their metabolome, which allowed us to identify tissue‐specific and age‐dependent changes (Fig EV1E and F, and Dataset EV1). The lysosphingolipid sphinganine (dihydrosphingosine, DHS) showed significant and cardiac‐specific accumulation with age (Figs 1B and C, and EV1G). Independently, targeted mass spectrometry approach confirmed this age‐dependent accumulation of DHS across killifish, zebrafish, mouse and human hearts (Figs 1D, and EV1H and I). DHS, an intermediate in the sphingolipid biosynthesis pathway, is formed by the reduction of 3‐ketosphinganine by 3‐ketosphinganine reductase. Upon its synthesis, ceramide synthase (CERS) converts DHS to dihydroceramide, which is then converted to downstream ceramides by desaturation. This makes DHS a critical intermediate in the synthesis of multiple complex sphingoid bases and ceramide analogues.
Figure 1. Sphinganine accumulation accompanied by loss of genome integrity is an evolutionarily conserved hallmark of the aged vertebrate heart.
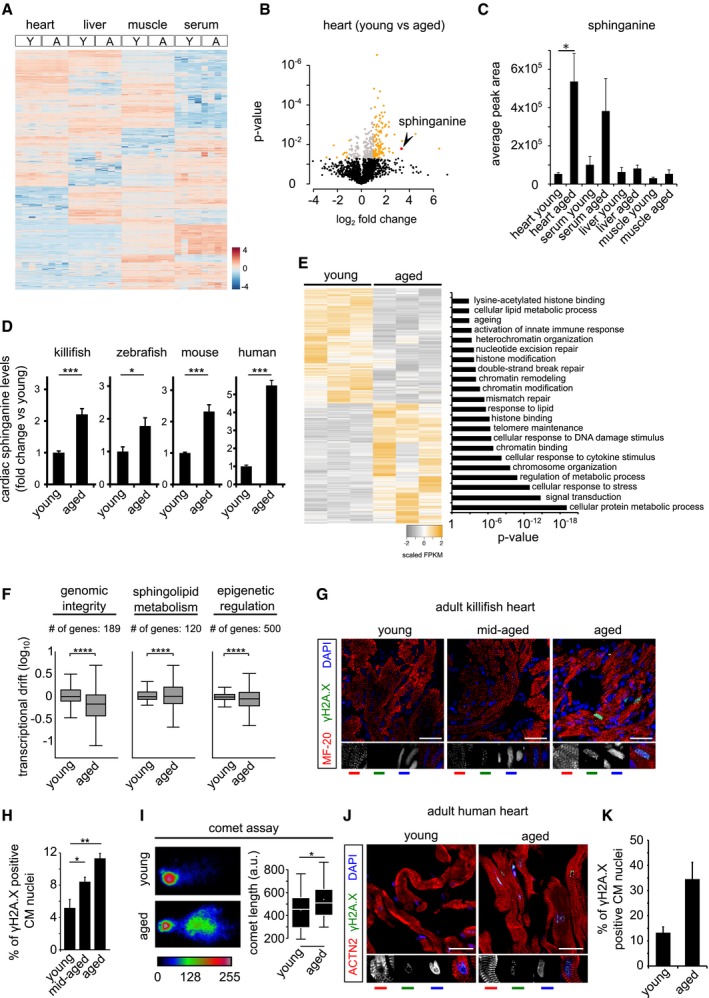
- Heatmap of differentially enriched metabolites in heart, liver, skeletal muscle and serum of young (Y) and aged (A) killifish (n = 4). Color scale represents scaled enrichment values.
- Ageing leads to significant cardiac‐specific accumulation of sphinganine. Volcano plot depicting the (de)enrichment profiles of all the identified metabolites in the heart, with sphinganine indicated by an arrowhead. Metabolites depicted in orange colour are the significantly altered metabolites with age in the heart (Log2 fold change ≥ 1 or ≤ −1; P‐value < 0.05); Student's t‐test was used to compute statistical significance.
- Bar graph depicting the relative abundance of sphinganine in indicated tissue from young and aged killifish.
- Targeted lipidomics confirm age‐dependent accumulation of sphinganine in killifish (n = 4; per condition, young = 6 weeks and aged = 12 weeks), zebrafish (n = 4, per condition, young = 7 months and aged = 26 months), mouse (n = 4 for young and n = 6 for aged samples, young = 21–23 weeks and aged = 131–143 weeks) and human hearts (n = 1 for young and 2 for aged, young = 30 years and aged = 61 and 64 years). In the case of human heart tissue, statistical significance was computed from technical replicates.
- Heatmap of differentially expressed transcripts (Log2 fold change ± 1, P‐value cut‐off < 0.05) between young and aged killifish hearts (n = 3). Significantly enriched relevant GO categories depicted as bar graph.
- Transcriptional drift variance of genes involved in genome integrity, epigenetic regulation and sphingolipid metabolism. Data are represented as box‐plot, and Levene's test was used for the estimation of the statistical significance. Error bars represent drift‐variance. Box plot whiskers show 1.5 IQR of highest and lowest quartile. Horizontal line within the bars represent median of the underlying population.
- γH2A.X staining of young, mid‐aged and aged killifish hearts show signs of DNA damage, increasing with age (n = 4, no. of sections per animal = 10 alternate sections, 10 μm thickness).
- Quantification of γH2A.X+ cardiomyocyte nuclei.
- The aged killifish hearts show significant signs of genome instability shown here by neutral comet assays. Representative images of single nuclei from young and old hearts along with comet length on the right (n = 4).
- γH2A.X staining of young and aged human ventricles shows signs of DNA damage accumulating with age (n = 2, no. of sections per sample = 5 alternate sections, 10 μm thickness).
- Quantification of γH2A.X+ cardiomyocyte nuclei is on the right.
To gain an unbiased understanding of age‐associated molecular changes accompanying the aberrant increase in DHS levels in the heart, we performed whole transcriptome analysis of young and aged killifish cardiac tissue (Fig EV1J and Dataset EV2). Gene Ontology analysis of differentially expressed genes revealed a significant enrichment for ageing hallmarks, including changes in chromatin organization, genomic integrity and metabolism (Fig 1E). Since deregulation of gene expression has been reported as an independent measure for identifying molecular processes breaking down with age, we performed a transcriptional drift‐variance analysis for genes curated in the aforementioned GO categories 5. Transcriptional drift‐variance analysis computes the variability in gene expression by estimating the stoichiometric co‐expression of genes within the same functional group. It assumes that the coordinated gene expression is essential for optimal tissue functionality and is ideal in young and declines with age. Genes involved in the maintenance of genomic and epigenomic integrity as well as in sphingolipid metabolism showed significant transcriptional drift suggesting aberrations in these processes with age in the heart (Figs 1F, and EV1K and L). Next, we confirmed the age‐associated loss of genomic integrity in the heart using two independent methods, by evaluating the formation of phosphorylated histone H2A‐positive (γH2A.X+) nuclear foci and by using a single‐cell electrophoresis assay (comet assay). A significant fraction of cardiomyocytes in aged killifish harboured γH2A.X+ nuclear foci in line with the longer “tail moments” in cells isolated from aged hearts (Figs 1G–I and EV1M). Importantly, aged human hearts show substantial accumulation of γH2A.X+ nuclear foci in cardiomyocytes, further indicating the relevance of this observation (Fig 1J and K). Collectively, our results point to DHS accumulation and loss of genome integrity as an evolutionarily conserved hallmark of aged vertebrate hearts.
Increase in intracellular sphinganine levels causes genomic instability in human cardiomyocytes
We next considered the possibility of a causal link between age‐associated accumulation of DHS and the loss of genomic stability. To ensure the cellular specificity of this phenomenon as well as to test its evolutionary significance and broad relevance, we interrogated this using human cardiomyocytes (hCMs). Therefore, we derived post‐mitotic human cardiomyocytes expressing ventricle‐specific maturation markers and synchronized calcium currents from pluripotent stem cells (Fig EV2A–D) 6. We manipulated DHS levels in these hCMs either by direct incubation with DHS or by increasing endogenous DHS levels through pharmacological inhibition of ceramide synthase (CERS) with Fumonisin B1 (FB1) 7. We confirmed that the exogenous exposure of hCMs to DHS leads to an increase in intracellular DHS levels by targeted mass spectrometry (Fig EV2E). Notably, our gene expression data from the killifish already revealed an age‐dependent decrease in CERS levels in the heart, consistent with the elevated DHS levels (Fig EV1N). A transient increase in intracellular DHS levels either by direct incubation with DHS or FB1 led to γH2A.X+ nuclear foci in ~ 50% of hCMs and to a significant increase in comet tail moment (Figs 2A–E and EV2F–I). Titration, as well as chronic exposure experiments, indicated that as little as 0.01 μM DHS suffices to induce DNA damage in cardiomyocytes (Fig EV2F and J). However, increased DHS levels triggered apoptosis rather than DNA damage in mitotic cells (human primary fibroblasts; Fig EV2K) 8. Importantly, DHS exposure does not induce apoptosis in hCMs (Fig EV2K). Taken together, these results suggest that the observed cardiac specificity might be due to the selective effect of DHS largely depending on the proliferative status of the cell type. This observation is further strengthened by the fact that DHS exposure to cancer cells (proliferating), both in vivo and in vitro, is known to cause apoptosis 9, 10, 11. It should be noted that we observed an age‐dependent increase in DHS levels only in the heart in comparison with the other tissue types measured (Fig 1D). Thus, increased cellular DHS levels lead to genomic instability in human cardiomyocytes, recapitulating the observation from aged killifish and human hearts.
Figure EV2. Elevated sphinganine levels induce DNA damage without causing apoptosis in cardiomyocytes.
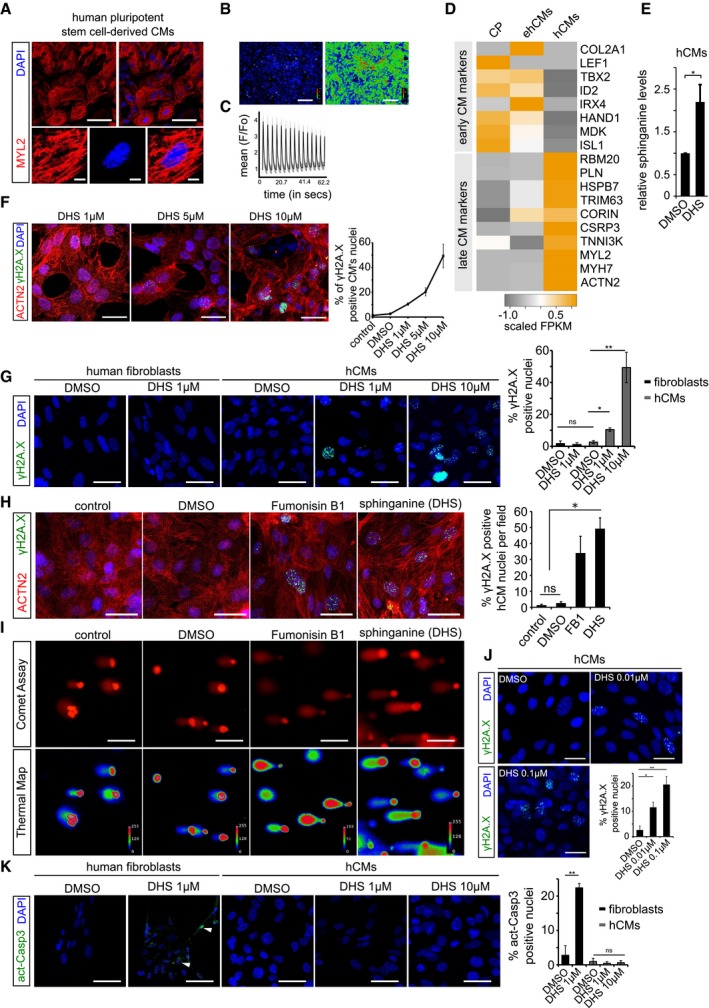
- Human pluripotent stem cell‐derived cardiomyocytes (hCMs) express ventricle‐specific marker MYL2 (red); insets show a magnified single cardiomyocyte.
- Calcium response in post‐mitotic hCMs. Left and right panel depicts basal and excitation states of hCMs, respectively.
- Graphical depiction of ratiometric measurement of fluorescence from different ROI (region of interest). Fluorescence intensities from the indicated ROIs (faint red circles) were measured (F) and normalized to basal fluorescence levels (Fo). The normalized data points (F/Fo) were plotted over time.
- Heatmap depicting expression profile of early and late cardiomyocytes markers during key time points of differentiation. CP (cardiac progenitors), ehCMs (early human cardiomyocytes) and hCMs (human cardiomyocytes). Scale bar represents scaled FPKM, to graphically represent expression levels of the indicated genes, on a scale of −1 to +1.
- Exogenous treatment of hCMs with 10 μM DHS leads to significant increase in intracellular DHS levels in comparison with DMSO control. Experiment was performed in biological triplicate.
- Dose‐dependent increase in γH2A.X+ nuclear foci in hCMs upon DHS exposure. hCMs were pre‐incubated for 3 days with either 1, 5 μM or 10 μM of DHS. The graph on the right represents quantifications from n = 3 experiments. ACTN2 is used to specifically label human cardiomyocytes (red).
- Elevated DHS levels induce DNA damage in hCMs but not in human primary fibroblasts shown here by immunostaining for γH2A.X (green). Bar graph on the right represents percentage of γH2A.X+ positive nuclei per condition. These experiments were performed in biological triplicate, and a total of ˜ 170 cells were quantified.
- Human cardiomyocytes were pre‐incubated for 3 days with either DMSO, 10 μM Fumonisin B1 or 10 μM sphinganine (DHS), and the induction of DNA damage was assayed by immunostaining for γH2A.X (green). Bar graph to the right depicts the percentage of γH2A.X positive nuclei in the indicated conditions. (n = 3 biological replicates; number of cells quantified per replicate = 150–200). ACTN2 is used to specifically label human cardiomyocytes (red).
- Representative micrographs depicting the results of comet assay in the indicated conditions.
- Chronic exposure of hCMs with lower concentrations of sphinganine (0.01 and 0.1 μM) for 7 days induces signs of DNA damage shown here by γH2A.X immunostaining. Bar graph represents percentage of positive nuclei per condition.
- Elevated DHS levels cause apoptosis in human primary fibroblasts but not in hCMs shown here by immunostaining with active caspase‐3 (green). White arrowheads indicate the active caspase‐3‐stained regions. Bar graph on the right represents percentage of active caspase‐3‐positive cells in the indicated conditions. Experiment was performed in biological triplicate, and a total of ˜ 150 cells were quantified per condition.
Figure 2. Elevated sphinganine level causes genomic and epigenomic aberrations leading to senescence and ageing signatures in human cardiomyocytes.
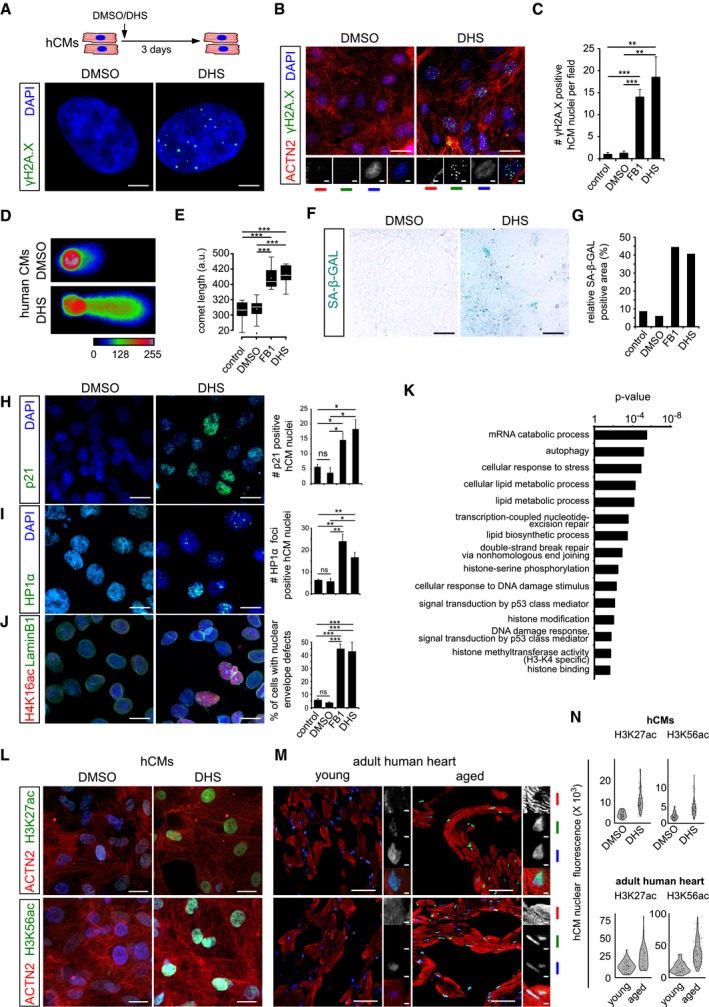
- Schematic representation depicting experimental design and the incubation duration used in these experiments. Sphinganine induces DNA damage in human cardiomyocytes depicted here by γH2A.X+ single nuclei.
- Micrographs depicting DHS‐induced DNA damage in human cardiomyocytes depicted here by γH2A.X+ nuclei in cardiac‐specific ACTN2+ cells (n = 3 biological replicates; no. of cells quantified = 147 per condition). ACTN2 is to specifically label human cardiomyocytes.
- Bar graph depicting the number of γH2A.X+ nuclei in the indicated conditions.
- Sphinganine induces DNA damage in human cardiomyocytes depicted here by neutral comet assay of DHS‐treated hCMs.
- Quantification of comet length in the indicated conditions. Data are represented as box‐plot, and the extremes of the error bars represent non‐outlier range and their length represents the variability within the data. Horizontal line within the bars represents median of the underlying population. Box plot whiskers show 1.5 IQR of highest and lowest quartile, outliers are included (dots). Extremes of the error bars represent non‐outlier range and their length represents the variability within the data. Horizontal line and white dot within the bars represent median and mean of the underlying population, respectively.
- Elevated DHS levels lead to ageing signatures in hCMs shown here by SA‐β‐GAL activity.
- Quantification of SA‐β‐GAL activity assay in the indicated conditions. (n = 3 biological replicates). Data are represented as bar graph depicting percentage mean of the SA‐β‐GAL labelled area.
- Elevated DHS levels lead to ageing signatures in hCMs shown here by p21 activation. Respective graphs depict the quantification of p21‐positive nuclei is on the right. (n = 3 biological replicates; number of cells quantified per replicate = 150–200).
- Elevated DHS levels lead to ageing signatures in hCMs shown here by HP1‐α+ nuclear foci. Respective graphs depict the quantification of HP1‐α‐positive nuclei is on the right. (n = 3 biological replicates; number of cells quantified per replicate = 120–200).
- Elevated DHS levels lead to ageing signatures in hCMs shown here by disruption of the nuclear envelope marked by Lamin B1 along with an increase in H4K16ac staining (n = 3 biological replicates; number of cells quantified per replicate = 100–150). Respective graphs depict the quantification of nuclear membrane defect is on the right.
- Proteome analysis confirms sphinganine‐induced disruption of (epi)genomic integrity (n = 3). Noteworthy, comparison of the proteomic profiles was performed between hCMs pretreated with DMSO and DHS.
- High sphinganine levels cause significant increase in H3K27ac and H3K56ac marks in hCMs. ACTN2 is used to specifically label human cardiomyocytes.
- Ageing leads to significant increase in H3K27ac and H3K56ac marks in human hearts. Insets on the right depict magnified view of cardiomyocyte nuclei. ACTN2 is used to specifically label human cardiomyocytes.
- Violin plot depicting the distributions of the greyscale nuclear intensity of the indicated markers. Quantification represents measurements of ˜ 150 single nuclei per condition, derived from three biological replicates.
Sphinganine‐mediated DNA damage causes ageing‐associated signatures in human cardiomyocytes
Next, we examined whether increased DHS levels were sufficient to recapitulate the changes in epigenome in hCMs, as observed in aged killifish and human hearts. We evaluated SA‐β‐GAL activity, p21 expression and the emergence of HP1α+ nuclear foci 12 upon directly or indirectly increasing DHS levels in hCMs via incubation with DHS or FB1. A transient increase in DHS levels led to an increase in SA‐β‐GAL activity as well as a significant increase in p21+ hCMs (Figs 2F–H and EV3A–D). Additionally, ~ 45% of hCMs acquired HP1α+ nuclear foci 12, another sign of senescence (Figs 2I and EV3E–G, and Dataset EV3). Our results demonstrate that DHS‐mediated DNA damage results in an induction of senescence and ageing‐associated signatures in hCMs. Since transcriptome analysis of killifish hearts suggested that genes regulating the epigenome are significantly affected by age in the heart (Fig 1E and F, and Dataset EV3), we next assessed the impact of DHS levels on chromatin modifications and nuclear membrane architecture by evaluating the levels and distribution of H4K16ac 13, 14 and Lamin B1 15 in hCMs. An increase in DHS levels caused a disruption of the nuclear envelope along with increased H4K16ac levels in ~ 40% of hCMs (Figs 2J and EV3H and I, Dataset EV3). To test whether an increase in DHS levels is sufficient to induce widespread age‐associated molecular changes in vitro in hCMs, we performed mass spectrometry‐based proteomics upon incubation with DHS. Gene Ontology analysis of significantly differentially enriched proteins (DMSO‐ vs DHS‐treated hCMs) revealed an impairment in DNA damage response, genome stability, cellular stress response and chromatin modifications, in line with the molecular changes observed in ageing killifish hearts (Figs 2K and EV3J, and Dataset EV4). More specifically, proteins directly involved in DNA damage response and histone deacetylation were deregulated (Fig EV3J and K). Noteworthy, examination of the changes in histone methylation levels revealed no significant difference upon DHS treatment (Fig EV3L and M). Deregulation in histone acetylation levels has previously been linked to ageing 16. Thus, our data imply that increased DHS levels are sufficient to recapitulate major hallmarks of ageing in human cardiomyocytes, including the loss of genomic integrity and the concomitant changes in the epigenome.
Figure EV3. Elevated sphinganine levels induce genomic instability and ageing signatures in human cardiomyocytes.
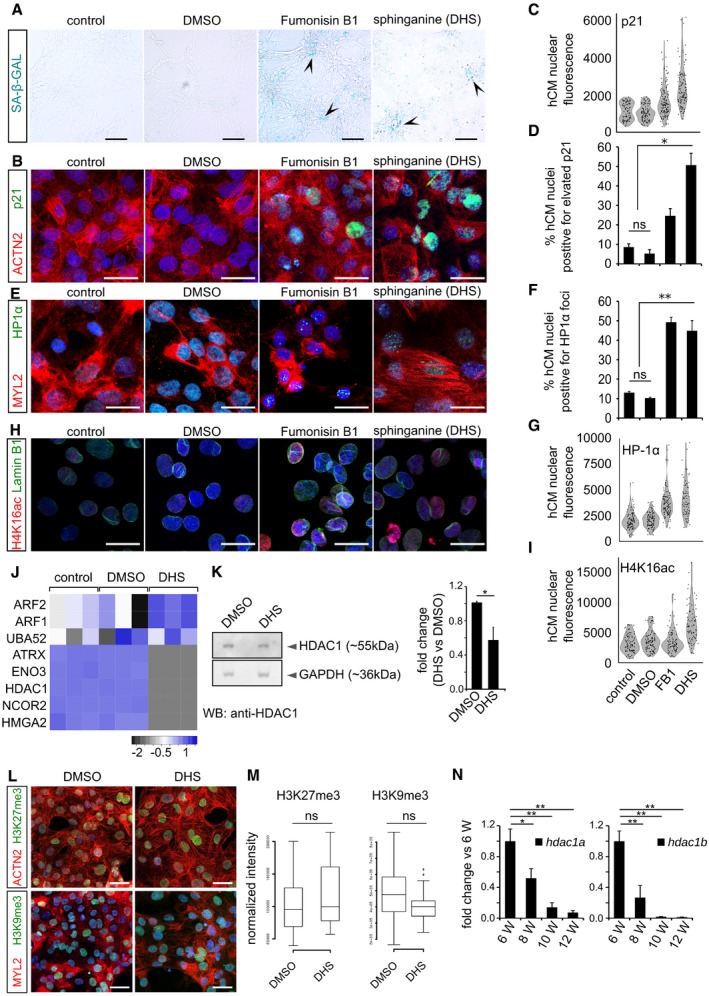
- Elevated DHS levels in hCMs induce signature of cellular senescence indicated here by representative micrographs from SA‐beta‐galactosidase staining (blue/cyan colour represents the positive regions). Arrowheads in the representative panels indicate the SA‐beta‐galactosidase‐stained regions.
- Elevated DHS levels in hCMs induce p21 expression indicated here by representative micrographs from p21 immunostaining (in green). ACTN2 is used to specifically label human cardiomyocytes.
- Violin plot depicting the distributions of the greyscale nuclear intensity of the indicated markers. n = 3 biological replicates; number of fields evaluated per condition = 12; total number of cells quantified per replicate = 150–200).
- Bar graph representing the percentage of hCMs nuclei harbouring elevated p21 levels. n = 3 biological replicates; number of fields evaluated per condition = 12; total number of cells quantified per replicate = 150–200).
- Elevated DHS levels lead to HP1‐α+ nuclear loci in hCMs. MYL2 is used to specifically label cardiomyocytes (red).
- Bar graph depicting the percentage positive nuclei HP1‐α foci in human cardiomyocytes (n = 3 biological replicates; number of fields evaluated per condition = 9; number of cells quantified per replicate = 120–200).
- Violin plot depicting the distributions of the greyscale nuclear intensity of the indicated markers (n = 3 biological replicates; number of fields evaluated per condition = 9; number of cells quantified per replicate = 120–200).
- DHS exposure leads to significant increase in H4K16ac (in red) levels accompanied by nuclear envelope defects (Lamin B1 immunostaining in green) of cardiomyocytes.
- Violin plot depicting the distributions of the greyscale nuclear intensity of the indicated marker (n = 3 biological replicates; number of cells quantified per replicate = 100–150).
- Heatmap depicting the relative enrichment/de‐enrichment of the indicated proteins as inferred from label‐free proteomics analysis.
- Western blot analysis revealed decreased levels of HDAC1 protein in the DHS‐treated human cardiomyocytes. Quantifications are on the right.
- DHS exposure of hCMs does not alter the levels of H3K27me3 and H3K9me3. ACTN2 or MYL2 is used to specifically label human cardiomyocytes.
- Box‐plot representing the normalized nuclear intensities of H3K27me3 and H3K9me3. Extremes of the error bars represent non‐outlier range, and their length represents the variability within the data. Horizontal line within the bars represents median of the underlying population. Box plot whiskers show 1.5 IQR of highest and lowest quartile, outliers are included (dots). Extremes of the error bars represent non‐outlier range and their length represents the variability within the data. Horizontal line within the bars represent median of the underlying population.
- Bar graphs representing the decline in the transcript levels of hdac1a (left) and hdac1b (right) in the killifish ventricles as inferred by RT–qPCR. X‐axis represents fish age in weeks (annotated as W).
HDAC1 inhibition by phosphorylated sphinganine derivatives cause aberrant increase in histone acetylation levels and transcription leading to DNA damage in cardiomyocytes
We next set out to delineate the mechanisms by which increased DHS levels induce senescence and ageing signatures in the heart. Recent evidence indicates that levels of intracellular metabolites can modulate the activity of chromatin modifiers 16. To examine whether intracellular DHS levels in hCMs can alter histone acetylation levels, we measured H3K27ac 17 (marking active transcription) and H3K56ac 18 (associated with DNA damage response). A substantial increase in both H3K27ac and H3K56ac in hCMs was observed upon DHS accumulation (Fig 2L and N). Consistent with this observation, we found that histone deacetylase, HDAC1 levels were significantly decreased in aged killifish hearts, confirmed here by RNA‐seq and qPCR assays (Figs EV1O and EV3N, and Dataset EV2) as well as in hCMs upon DHS exposure (Fig EV3J and K, and Dataset EV4). Additionally, aged human hearts displayed significant accumulation of H3K27ac and H3K56ac marks in line with epigenomic changes associated with active transcription and DNA damage in hCMs exposed to DHS (Fig 2M and N).
To establish whether this DHS‐induced increase in histone acetylation levels causes DNA damage in cardiomyocytes, we increased histone acetylation levels exogenously by pharmacologically inhibiting HDAC1/2 19. In order to nullify the potential pleiotropic effects caused by individual HDAC inhibitors, we performed these experiments using three independent HDAC inhibitors, namely romidepsin, MS‐275 and pyroxamide 20, 21. Our experiments revealed that HDAC inhibition using each of these inhibitors consistently caused significant DNA damage in hCMs, indicated by the accumulation of γH2AX+ nuclear foci (Fig 3A and B), suggesting that HDAC1/2 inhibition alone is sufficient to induce DNA damage in cardiomyocytes. These data are in agreement with previous evidence showing that tissue‐specific loss of HDAC1/2 is prenatally lethal in mice due to cardiac insufficiencies 19, 22. Since DHS and its derivatives are predominantly functional both in the nucleus and in the mitochondria 7, we next tested whether increased DHS levels could affect mitochondrial integrity and function in hCMs. There were no detectable changes in either mitochondrial distribution or membrane integrity upon treatment of hCMs with DHS (Fig EV4A–C). Furthermore, altered mitochondrial integrity/function could lead to increased reactive oxygen species (ROS) levels 23 and result in DNA damage. To understand whether the DHS‐induced DNA damage is ROS‐mediated, we incubated hCMs with DHS along with ROS scavengers. DHS caused DNA damage even in the presence of ROS scavengers (Fig EV4E). Additionally, DHS exposure did not increase the ROS levels in hCMs (Fig EV4D). These experiments demonstrate that DHS induced DNA damage in hCMs by a ROS‐independent mechanism. Together, this suggests that the DHS‐induced increase in DNA damage likely occurs via the direct inhibition of HDAC1/2.
Figure 3. DHS‐induced DNA damage in cardiomyocytes is caused by inhibition of histone deacetylases.
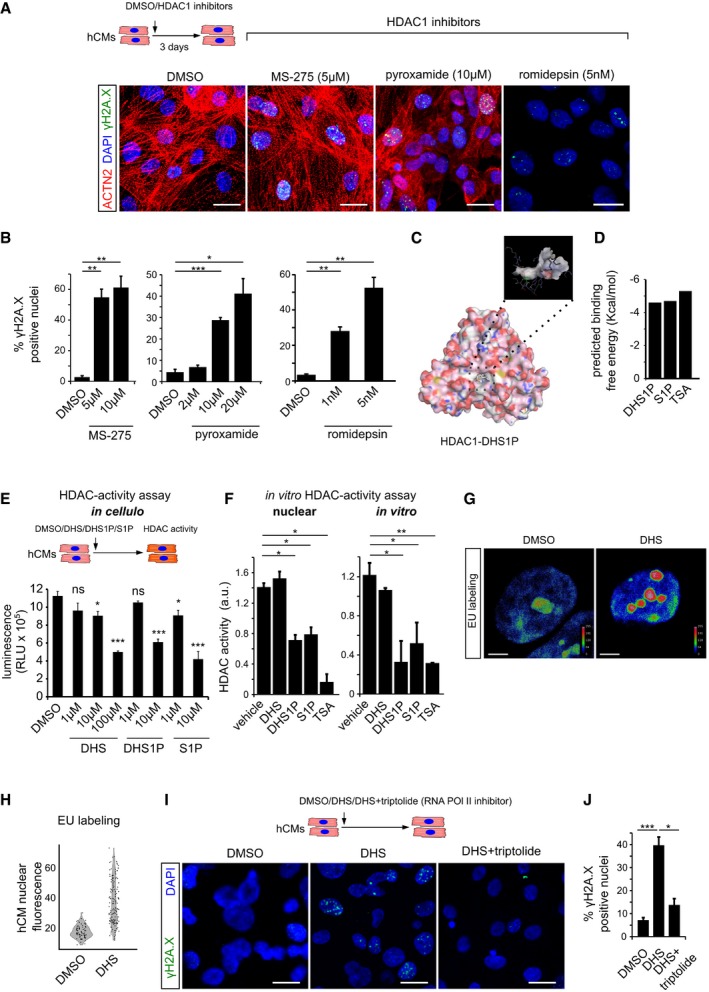
- Pharmacological inhibition of HDAC1 using romidepsin, MS‐275 and pyroxamide in human cardiomyocytes causes extensive DNA damage, shown here by γH2A.X staining. ACTN2 is used to specifically label human cardiomyocytes.
- Bar graphs depict quantification of γH2A.X+ hCM nuclei in the indicated conditions. Data represent measurements from ˜ 100 to 200 cells per condition, derived from three biological replicates.
- In silico simulation showed docking of sphinganine‐analogue DHS1P in the tubular active site of human HDAC1.
- Sphinganine derivatives DHS1P and S1P show comparable binding affinity to HDAC1, similar to the known HDAC inhibitor TSA.
- Sphinganine and its derivative DHS1P inhibits class 1 HDACs in the human cardiomyocytes as inferred from the in cellulo HDAC activity assay, shown here as bar graph. Data represent measurements from four biological replicates.
- In vitro HDAC activity assay revealed inhibition of nuclear HDACs and purified HDAC1 activity by DHS1P and S1P, shown here as bar graphs. Data represent measurements from three independent replicates.
- Representative micrographs depicting the increase in nascent transcripts upon DHS treatment of hCMs, measured by EU labelling assay. Micrographs are depicted as a thermal map derived from greyscale images. Scale represents the relative EU labelling intensities within the nucleus, ranging from red colour (higher intensity) to blue colour (lower intensity).
- Quantitative assessment of transcription levels measured by EU labelling assay upon treatment with DHS on hCMs. Quantification represents measurements of ˜ 80–120 single nuclei per condition, derived from three biological replicates.
- Representative micrographs of hCMs indicating the rescue of the DHS‐induced DNA damage by co‐incubation with RNA Pol II inhibitor, triptolide.
- Quantifications of γH2A.X+ CM nuclei represented as bar graph (n = 3 biological replicates).
Figure EV4. DHS‐induced genomic instability is a consequence of HDAC inhibition in human cardiomyocytes.
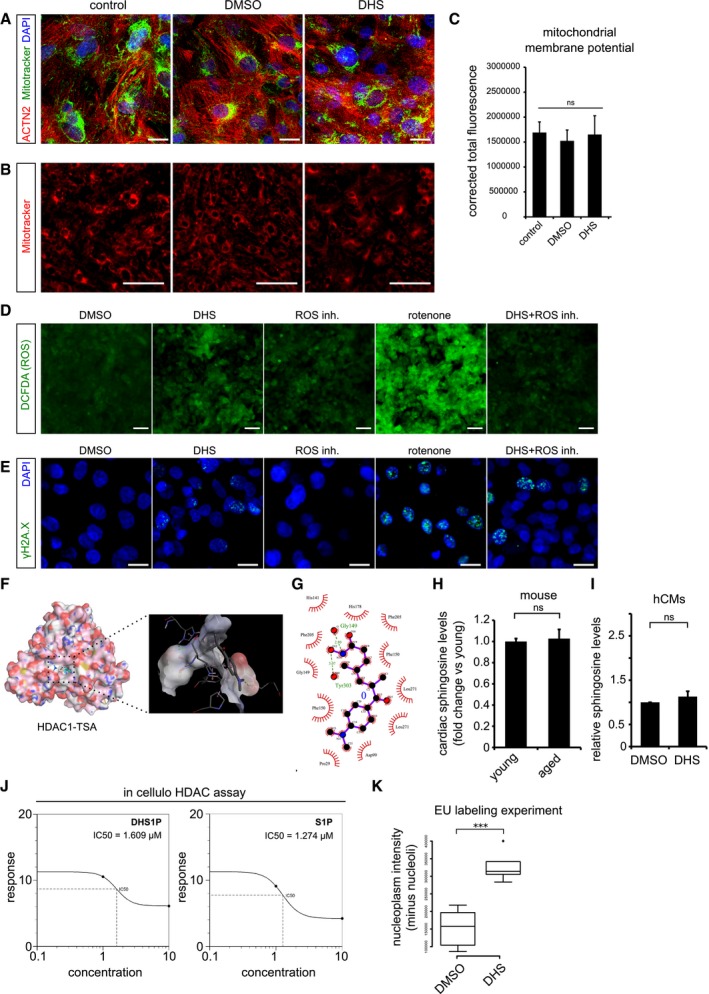
- Elevated DHS levels in hCMs do not affect mitochondrial network and its membrane potential. Co‐staining of mitochondria by MitoTracker (in green) along with the cardiac‐specific marker ACTN2 (in red).
- Micrographs of live cell imaging of hCMs stained with MitoTracker (in red).
- Bar graph representing the corrected total fluorescence mean ± SEM, from live cardiomyocytes stained with MitoTracker n = 3, each condition.
- Elevated DHS levels do not increase the ROS levels in human cardiomyocytes. Measurement of reactive oxygen species (ROS) in hCMs pre‐incubated for 72 h with DMSO, DHS, ROS inhibitors, rotenone and DHS + ROS inhibitors.
- Elevated DHS levels induce ROS‐independent DNA damage as shown by immunostaining with γH2A.X (in green).
- Molecular docking of human HDAC1 protein with Trichostatin TSA, a pan HDAC inhibitor, suggests potential binding within the known active site pocket (spotted square and the magnified view of the active site is to the right). TSA and HDAC1 are represented in ball & stick and spacefill models, respectively.
- Ligplot representing the two‐dimensional interaction between the HDAC1 residues with TSA at the active site.
- Cardiac sphingosine levels remain unchanged with age in vivo in mice. Bar graph depicting the relative sphingosine (Spo) levels in aged mouse hearts in comparison with the young ones.
- Exogenous treatment of hCMs with 10 μM DHS did not alter sphingosine (Spo) levels, depicted as a bar graph in comparison with DMSO control. Experiment was performed in biological triplicate.
- Concentration curves depicting the comparable IC50 values of DHS1P and S1P from in cellulo HDAC assay.
- Box‐plot depicting the normalized nuclear (without nucleolus) intensities of nascent transcript, measured by EU labelling. Extremes of the error bars represent non‐outlier range, and their length represents the variability within the data. Horizontal line within the bars represents median of the underlying population. Box plot whiskers show 1.5 IQR of highest and lowest quartile, outliers are included (dots). Extremes of the error bars represent non‐outlier range and their length represents the variability within the data. Horizontal line within the bars represent median of the underlying population, respectively.
To test this, we asked whether DHS (or its derivatives) could bind human HDAC1. To address this, firstly we performed docking simulations using DHS and its downstream derivatives, sphinganine‐1‐phosphate (DHS1P), sphingosine (SPO) and sphingosine 1‐phosphate (S1P) on the human HDAC1 crystal structure (4BKX) 24. In these simulation experiments, we have used TSA as a positive control, a well‐characterized HDAC inhibitor, which directly binds to the active site of HDAC proteins 24. The root‐mean‐square deviation (RMSD) values (which is an indicator of stability in the simulated interactions) for DHS and SPO were above the threshold limit (< 0.5 Å), suggesting a non‐specific and unreliable binding conformation. Importantly, DHS1P showed potential to bind directly to the tubular active site pocket of human HDAC1, similar to TSA and with a comparable free energy (Figs 3C and D, and EV4F and G). As previously reported, the downstream metabolite of SPO, S1P, also displayed similar potential in binding capacity (Fig 3D) 25. However, SPO levels remain unchanged in the heart with age (Fig EV4H) as well as upon exposure of hCMs to DHS (Fig EV4I). This suggests that DHS and not SPO could be the major contributing factor to cardiac genomic instability. Secondly, we enquired whether an increase in DHS levels in hCMs leads to HDAC inhibition using the same experimental conditions that resulted in DNA damage. DHS and its immediate downstream derivatives DHS1P and S1P caused significant inhibition of HDACs revealed by in cellulo HDAC activity assay with comparable IC50 values (for class 1 HDACs; Figs 3E and EV4J). Thirdly, we assessed the ability of DHS and its derivatives inhibit HDAC activity in nuclear extracts. In line with the previous observations, DHS1P showed significant ability to inhibit nuclear HDAC activity (Fig 3F). Finally, our experiments measuring the ability of DHS and its derivatives to inhibit the activity of purified human HDAC1 further consolidated our observation that indeed DHS1P can inhibit HDAC1 activity directly (Fig 3F).
In agreement with the DHS‐dependent increase in histone acetylation levels, hCMs showed significant increase in transcription, as shown here by EU labelling of nascent RNA (Fig 3G and H). Moreover, the observed increase in nascent transcripts upon treatment with DHS in cardiomyocytes is global and not restricted to specific regions in the nucleus (Fig EV4K). This increase in transcriptional output can be a major contributor to the accumulation of DNA damage as previously suggested 16, 26. To test this, we co‐treated hCMs with DHS and the RNA P II inhibitor triptolide. Inhibition of transcription by triptolide was sufficient to block the DHS‐induced DNA damage. This indicates that the DHS‐mediated DNA damage is due to the aberrant increase in nascent transcription caused by the inhibition of HDAC activity (Fig 3I and J). Collectively, these results indicate that the age‐dependent increase in DHS leads to an inhibition of HDAC activity, resulting in an increase of transcription‐dependent loss of genomic integrity in hCMs.
SPHK2 inhibition prevents sphinganine‐induced DNA damage in human cardiomyocytes
DHS is converted in the nucleus to DHS1P by sphingosine kinase 2 (SPHK2) 7, which also mediates the synthesis of its downstream analogue S1P. Since DHS1P and S1P are the primary mediators of HDAC inhibition, we hypothesized that inhibition of SPHK2 would prevent DHS‐induced DNA damage in hCMs. Strikingly, treatment with the SPHK2 inhibitors, ABC294640 and SLM6031434 27, 28, led to ~ 80% reduction (from > 40 to < 10%) in DHS‐induced DNA damage visualized here by γH2A.X+ nuclear foci in hCMs, while histone acetylation was restored to control levels (Fig 4A–F and Appendix Fig S1A–E). Thus, partial inhibition of SPHK2 could constitute a potential strategy to alleviate the DHS‐induced DNA damage in the cardiomyocytes. Together, this confirmed that HDAC1/2 inhibition by DHS derivatives is the primary cause of the observed genomic instability in hCMs.
Figure 4. SPHK2 and HAT inhibitors prevent the DHS‐induced cardiac DNA damage in vitro .
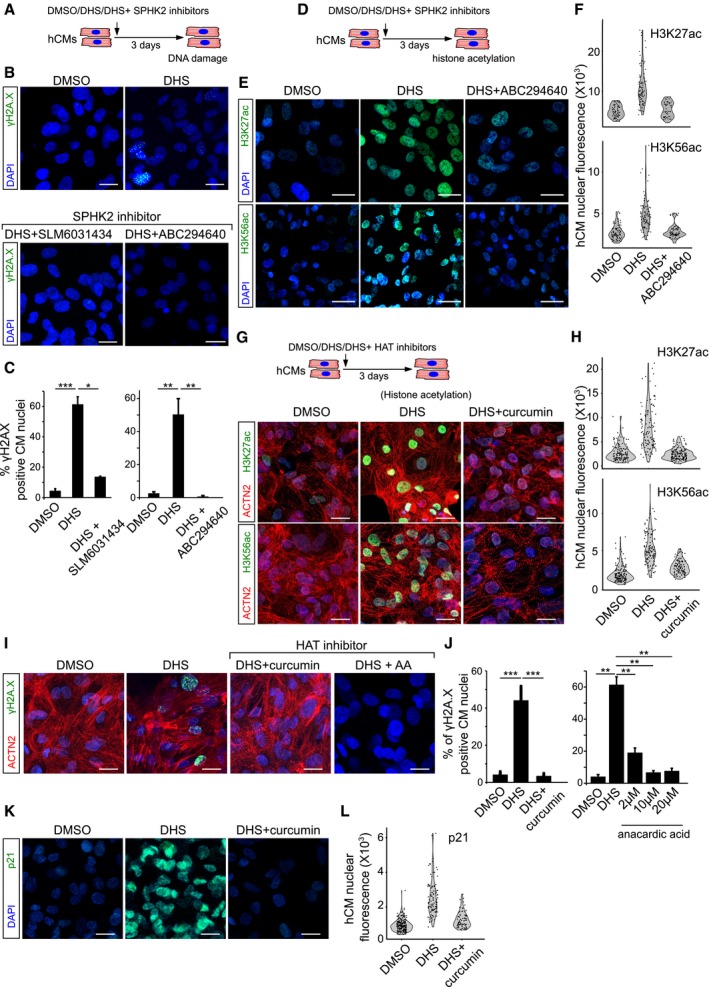
- Schematic representation of the experimental set‐up.
- SPHK2 inhibition using ABC294640 and SLM6031434 prevents DHS‐induced DNA damage as shown here by staining for γH2A.X (green) in hCMs.
- Bar graphs representing the percentage of γH2A.X+ hCMs in the indicated conditions. Data represent measurements from ˜ 200 cells per condition, derived from three biological replicates.
- Schematic representation of the experimental set‐up.
- Treatment of hCMs with ABC294640, a specific SPHK2 inhibitor, prevents DHS‐induced epigenetic aberrations in hCMs visualized here by H3K56ac and H3K27ac staining (˜ 100 cells quantified/condition).
- Violin plots depicting the distributions of the greyscale nuclear intensity of the indicated markers. Quantification represents measurements of ˜ 100–150 single nuclei per condition, derived from three biological replicates.
- HAT inhibition by curcumin prevents the aberrant increase in H3K56ac and H3K27ac levels.
- Violin plots depicting the distributions of the greyscale nuclear intensity of the indicated markers. Quantification represents measurements of ˜ 100–150 single nuclei per condition, derived from three biological replicates.
- HAT inhibition prevents DHS‐induced DNA damage as shown here by staining for γH2A.X (green) on human cardiomyocytes.
- Quantification of γH2A.X+ hCMs nuclei represented as percentage bar graph.
- Curcumin‐mediated HAT inhibition abrogates the DHS‐induced upregulation of p21.
- Violin plots depicting the distributions of the greyscale nuclear intensity of p21 stained nuclei. Quantification represents measurements of ˜ 100–150 single nuclei per condition, derived from three biological replicates.
Restoring histone acetylation levels in cardiomyocytes by HAT inhibition using curcumin prevents the DHS‐induced DNA damage in vitro and in vivo
Next, we hypothesized that if the DHS‐induced cardiac ageing phenomenon operates through the inhibition of HDACs, we would be able to prevent this by exogenously decreasing the histone acetylation levels. We tested this by independently exposing DHS‐treated hCMs to histone acetyltransferase (HAT) inhibitors, curcumin and anacardic acid (AA) 29. As expected, HAT inhibition prevented the aberrant increase in histone acetylation levels (Fig 4G and H). Importantly, HAT inhibition abolished the DHS‐induced DNA damage visualized here by the accumulation of γH2A.X+ nuclear foci and the activation of p21 (Fig 4I–L and Dataset EV3).
Curcumin, while having well‐documented health benefits as well as proven HAT inhibitory activity 18, has been prone to pleiotropic effects. To further confirm whether the prevention of DNA damage by curcumin in our system acts primarily through its HAT inhibitory activity, we investigated its effect on DNA damage caused by a histone acetylation‐independent mechanism. We induced DNA damage in cardiomyocytes using the topoisomerase II inhibitor etoposide. Curcumin failed to inhibit DNA damage caused by topoisomerase II inhibition (Appendix Fig S1F and G). Taken together, this indicates that the prevention of DNA damage in hCMs by curcumin upon DHS treatment acts via its HAT inhibitory activity, which is further supported by the concomitant restoration of histone acetylation levels (Fig 4G–J).
Next, to evaluate the global molecular changes caused by elevated DHS levels in cardiomyocytes in an unbiased manner, we performed microarray analysis of hCMs upon DHS treatment alone and upon co‐treatment with curcumin, in comparison with corresponding DMSO controls. Gene Ontology analysis of differentially expressed genes revealed significant enrichment for expected categories, including histone modifications, transcriptional regulation, senescence and DNA damage (Fig 5A and Appendix Fig S1H–K, and [Link], [Link]). The differentially enriched genes were involved in clinical phenotype‐related Gene Ontologies including age‐associated cardiovascular disorders (Fig 5B). Furthermore, in comparison with the DHS‐treated samples, co‐treatment with curcumin activates major DNA repair pathways, indicating that curcumin treatment rescues the DNA repair defects upon DHS treatment (Fig 5C–E). This observation is in line with the near lack of DNA damage upon co‐treatment of cardiomyocytes with DHS and curcumin (Fig 4I and J).
Figure 5. Sphinganine exposure activates molecular signatures of genome instability and senescence in human cardiomyocytes while concomitant HAT inhibition amends the genotoxicity.
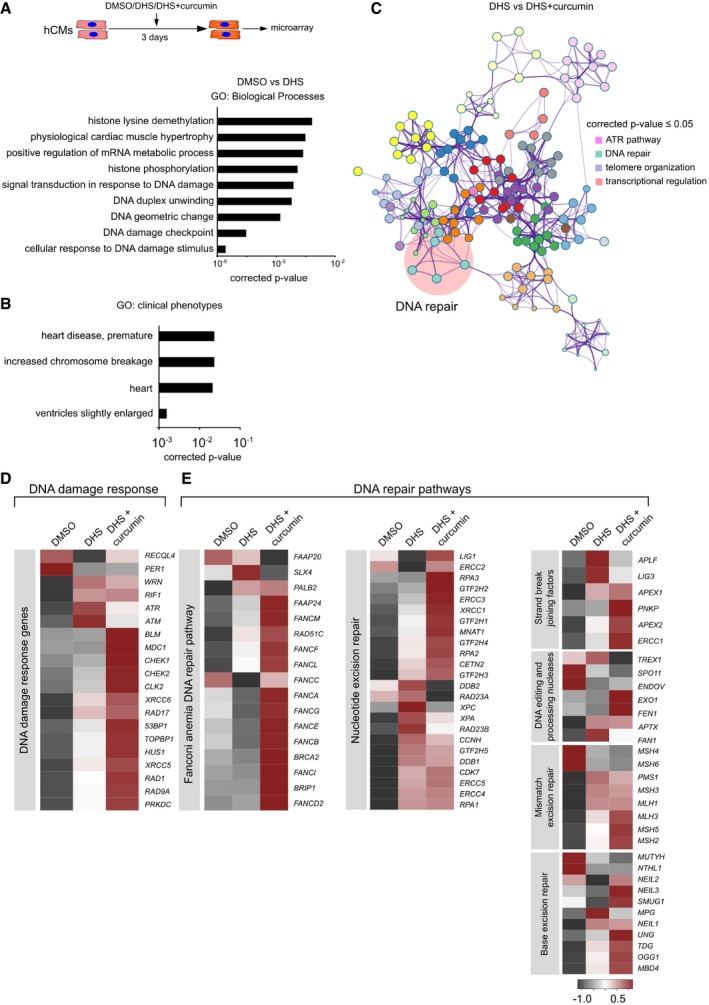
- hCMs were treated for 3 days with DMSO, DHS and DHS + curcumin (co‐incubation), represented in the scheme. After incubation, cells were harvested for RNA isolation and differentially expressed genes were identified by microarray analysis. Bar graph depicting Gene Ontology (GO) enrichment status for the differentially expressed transcripts upon DHS treatment. Hypergeometric test was used for determining the P‐value. Experiment was performed in biological triplicate for DHS and DHS + Curcumin condition, whereas two biological replicates were used for DMSO condition.
- Bar graph representing GO enrichment levels for clinical phenotypes. Hypergeometric test was used for determining the P‐value.
- Co‐treatment of curcumin with DHS leads to the activation of DNA repair pathways as shown here by functional enrichment analysis on the differentially expressed transcripts using Metascape. Hypergeometric test was used for determining the P‐value.
- Heatmap depicting the expression profile of the markers of DNA damage response in the indicated conditions.
- Co‐treatment of curcumin activates DNA repair pathways as depicted here by heatmaps. Color scale represents scaled expression values.
To understand the molecular and physiological effects of increased DHS levels on the mammalian heart in vivo, we exposed young adult mice (~ 12 weeks old) for five consecutive days to FB1 alongside indicated controls by intraperitoneal administration (Fig 6A). Pharmacological inhibition of ceramide synthase (CERS) by FB1 resulted in a substantial increase of endogenous cardiac DHS levels (Appendix Fig S2E, upper panel). This in vivo increase in cardiac DHS levels resulted in significant elevation of histone acetylation and DNA damage indicated by γH2A.X+ nuclear foci and activation of phospho‐ATM in cardiomyocytes (Fig 6B–E, and Appendix Fig S2A and B), whereas no DNA damage was observed in kidney and liver (Appendix Fig S2C and D). This suggests a causal link between elevated DHS levels and DNA damage observed in the heart in vivo.
Figure 6. In vivo recapitulation of the molecular defects caused by elevated DHS levels leads to severe decline of cardiac functionality which can be prevented by HAT inhibition.
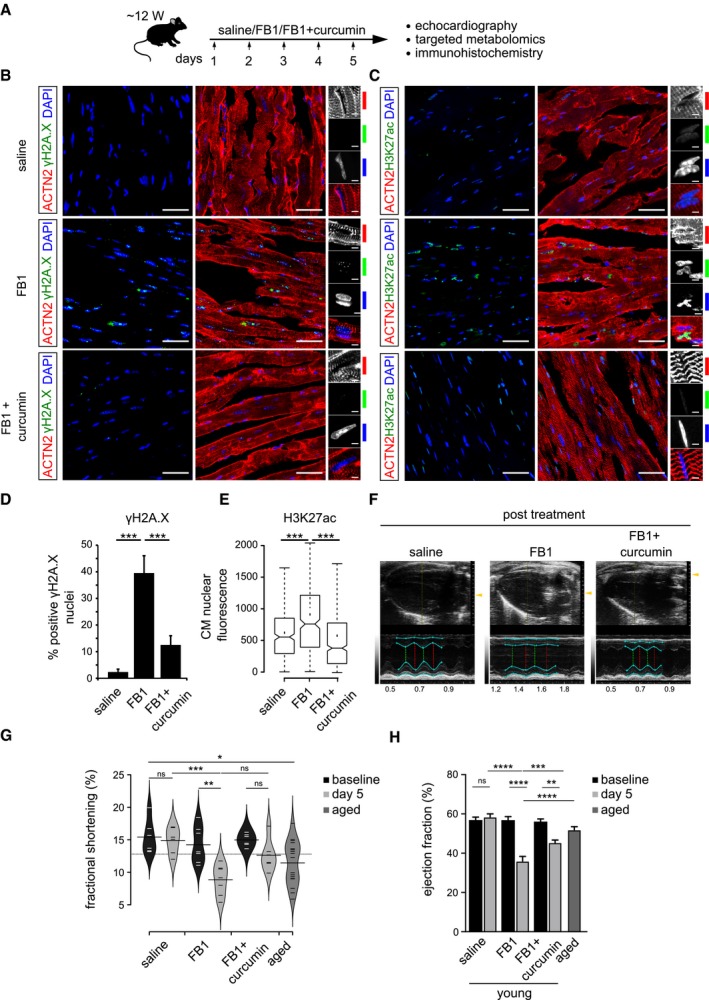
- Schematic representation of the experimental set‐up employed in the in vivo mouse experiments (n = 6 animals per condition). 12‐week‐old C57BL/6J mice were injected i.p. once daily with PBS, FB1 (6.75 mg/kg) or FB1 and curcumin (6.75 and 50 mg/kg, respectively) for five consecutive days (n = 6 animals per condition).
- Representative micrographs depicting γH2A.X staining on the ventricle sections for the indicated cohorts. Insets on the right depict magnified view of cardiomyocyte nuclei (n = 6 animals per condition). ACTN2 is used to label mouse cardiomyocytes.
- Representative micrographs depicting H3K27ac staining on the ventricle sections for the indicated cohorts (n = 6 animals per condition). ACTN2 is used to label mouse cardiomyocytes.
- Quantification of γH2A.X+ CMs nuclei represented as percentage bar graph. Data are represented as bar graph depicting percentage mean of the γH2A.X+ CMs population. For quantification, five randomized micrographs, representing independent regions of the ventricles from six different animals per condition, were used (n = 6 animals per condition).
- Box‐plot depicting the distributions of the corrected total nuclear fluorescence of the indicated cohorts. Data depict corrected total nuclear fluorescence estimated from greyscaled immunostained micrographs of the ventricular region from the mice treated with the indicated compounds. Extremes of the error bars represent non‐outlier range, and their length represents the variability within the data. Horizontal line with the bar represents median of the underlying population (n = 6 animals per condition). Box plot whiskers show 1.5 IQR of highest and lowest quartile. Extremes of the error bars represent non‐outlier range and their length represents the variability within the data. Horizontal line and dot within the bars represent median and mean of the underlying population, respectively. Notch represents confidence interval around the median.
- Representative images depicting ventricular view from the echocardiography of the indicated cohorts. Cyan lines indicate the margins of epicardium and endocardium, the green dashed lines indicate the minimum diameter and the red lines measures the maximum diameter during a heart cycle. The smaller the relative difference between red and green line, the worse is systolic heart function.
- Bean‐plot depicting the quantification of fractional shortening in the indicated groups (n = 6 animals per condition). Horizontal lines depict the mean of the underlying distributions.
- Bar graph depicting the quantification of ejection fraction (in percentage) in the indicated groups (n = 6 animals per condition).
Finally, to understand the physiological impact of increased DHS level on cardiac function, we performed echocardiography on the indicated cohorts both before (Appendix Fig S2E, lower panel) and after the treatment in comparison with aged mice (~ 2.5 years old; Fig 6F). An increase in DHS levels led to significant impairment in cardiac function as revealed by the decrease in left ventricular fractional shortening, comparable to that of aged mice (Fig 6G and Movies EV1, EV2, EV3, EV4, EV5 and EV6) and ejection fraction (Fig 6H). It has been reported that ageing leads to progressive decrease in ventricular fractional shortening and ejection fractions 30, in line with our data from old mice (Fig 6G and H). Further demonstrating a causal link, our data indicate that increasing DHS levels significantly contribute to this age‐dependent deterioration of cardiac function.
Denoting the evolutionary significance of our observations, exposing young adult zebrafish (~ 7 months old) to 1 μM of DHS (5 h/day) for 3 days led to significant accumulation of γH2A.X+ nuclear foci in cardiomyocytes in vivo (Appendix Fig S2F–H).
Remarkably, in line with our observation in hCMs, curcumin treatment prevented aberrant histone acetylation and DNA damage caused by elevated DHS levels in vivo. As a result, this leads to significant alleviation of cardiac functional defects caused by the increase in DHS levels. Together, our data imply that curcumin has potent cardio‐protective properties, blocking the molecular and physiological damage caused by DHS accumulation in vivo.
Discussion
The age‐dependent decline in structural and functional integrity of the heart possesses a major risk factor for various cardiovascular diseases. Recent evidences indicate that alterations in the cardiac epigenome are a major contributor to its functional and physiological decline 31, 32, 33, 34. This is supported by the global changes in histone modifications, both active (H3K9ac, H3K27ac, H3K4me3 and H3K36me3) and repressive marks (H3K27me3), reported in the failing human hearts 34, 35, while the cause of these epigenomic changes or the particular susceptibility of the heart to such alterations is currently not well understood. Here, we identified a specific metabolic mediator, whose age‐dependent accumulation in the heart causes genome instability by directly inhibiting histone deacetylases. This phenomenon of age‐dependent accumulation of DHS is evolutionarily conserved among vertebrates. Furthermore, DHS‐mediated inhibition of HDACs leads to an increase in transcription‐dependent accumulation of DNA damage. Loss of transcriptional control has been proposed as a causal link to cardiac ageing 36. However, its mechanistic basis is at the best speculative. Our study provides the mechanistic link to this observation, by which the DHS‐mediated HDAC inhibition causes the age‐dependent loss of transcriptional control in the heart. Additionally, we provide for the first time the direct in vivo causal link between the increasing cardiac DHS levels and the observed loss in genomic integrity, negatively impacting cardiac function. Furthermore, our data suggest that the age‐associated loss of cardiac function could be at least partially contributed by the accumulation of DHS and can be prevented by curcumin (HAT inhibitor). Collectively, these data illustrate the significance of lysosphingolipid‐mediated loss of genomic integrity in the cardiac ageing process.
Materials and Methods
Antibodies and animal handling
Primary antibodies used were anti‐MYL2 antibody (rabbit IgG; 1:500; catalog no. 10906‐1‐AP, Proteintech), anti‐ACTN2 antibody (mouse IgG; 1:500; catalog no. A7811, Sigma), anti‐SPHK2 antibody (rabbit IgG; 1:250; catalog no. GTX105152, GeneTex), anti‐HP1α (mouse IgG1;1:500; catalog no. 39977; Active Motif), anti‐MF20 (supernatant, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), anti‐γH2A.X (rabbit IgG; 1:500; catalog no. 9718; Cell Signaling), anti‐HDAC1 (rabbit IgG; 1:1,000; catalog no. ab7028, Abcam), anti‐tubulin antibody (mouse IgG; 1:1,000; catalog no. MS‐581‐P1ABX, ThermoFisher Scientific), anti‐Phospho‐ATM (Ser1981) (rabbit IgG; 1:250; catalog no. 5883S, Cell Signaling), anti‐p21 (mouse IgG, 1:500; catalog no. ab184640, Abcam), anti‐cleaved caspase‐3 (rabbit IgG; 1:500; catalog no. 9661; Cell Signaling); anti‐H3K27ac (rabbit antibody IgG, 1:500; catalog no. 39133, Active Motif); anti‐H4K16ac (mouse monoclonal antibody IgG, 1:500; catalog no. 61529, Active Motif), anti‐H3K56ac (rabbit antibody IgG, 1:500; catalog no. ab71956, Abcam) and anti‐Lamin B1 (rabbit antibody IgG, 1:500; catalog no. ab16048, Abcam). Secondary antibodies used were donkey anti‐rabbit IgG conjugated to Alexa Fluor 488 (A21206, Invitrogen) and goat anti‐mouse conjugated to Alexa Fluor 568 (A‐11004, Invitrogen). Turquoise killifish (GRZ‐AD strain) were housed at 28°C in a central filtration recirculating system with a 12‐h light/dark cycle. Fish were fed twice a day with fresh bloodworms and brine shrimp nauplii. All turquoise killifish care and use were approved by the MPI committee on Research Animal Care (Permit number = 576.1.36.36.G28/13 Be).
Age information of killifish, zebrafish and human cohorts
In killifish experiments young, mid‐aged and aged referred to 6, 8 and 12 weeks (Fig 1C), 6 and 16 weeks (Fig 1E–G), 6 and 12 weeks (Fig 1H and I), 6, 8 and 12 weeks (Fig EV1C and D). For zebrafish experiments, young referred to 7 months old and aged referred to 26 months old. For mouse experiments, young referred to 21–23 weeks old and aged referred to 131–143 weeks old. Human ventricular samples were purchased from Cureline, Inc. A material transfer agreement (MTA) was signed between Cureline, Inc., and University of Cologne. All human samples were categorized as normal tissues by the Cureline, Inc. Samples were collected by autopsy from subjects with no cardiac history. Left ventricle from 30 years old and right and left ventricles from 61‐ and 64‐year‐old humans, respectively, were used in this study for metabolomics as well as histology.
Immunohistochemistry on cryosections
Hearts were pre‐incubated in freshly prepared 4% PFA in PBS (pre‐fixed tissue). Horizontal cryosections (8 μm) of the hearts were thaw‐mounted onto Superfrost Plus slide glasses (Thermo), incubated in acetone at −20°C for 15 min, washed several times in PBST and blocked in 5% normal goat serum in PBST (blocking solution) for 1 h at room temperature. For double labelling, the slides were overnight incubated at 4°C with both the primary antibodies from two different species, washed three times in PBST to remove unbound primary antibodies and incubated for 2 h at room temperature with the two secondary antibodies. Slides were further washed three times in PBST. The slides were washed in PBS and mounted with VectaShield containing DAPI (Vector). Noteworthy, in order to avoid regional bias, five micrographs were taken from each ventricle section covering left area, right area, two areas in the middle and one micrograph capturing the tip of the ventricle. In the majority of experiments, double‐blinded quantification was performed.
Sudan Black staining for lipofuscin
Fresh solution of Sudan Black was prepared by dissolving 0.7 g of Sudan Black B in 100 ml of 70% ethanol. Solution was stirred overnight at room temperature and was post‐filtered through filter glass with medium porosity. The resulting dye was tightly capped to prevent evaporation induced dye saturation. Killifish hearts were dissected from different ages and fixed in 4% PFA overnight. Next day, hearts were dehydrated in series of ethanol and waxed. Five‐micrometre‐thin sections were made by using microtome (Thermo Scientific) and were mounted on the glass slides. Slides were incubated overnight at 37°C. The tissue samples were dewaxed with xylene and dehydrated till 70% ethanol. A drop of freshly prepared SBB solution was applied to the tissue with the help of syringe. The staining was constantly observed under the light microscope. After 2–8 min post‐SBB exposure, the slides were dipped in 50% ethanol for two to three times to remove dirt and residual SBB solution. Immediately, slides were transferred to distilled water and mounted with 40% glycerol/TBS mounting solution.
RNA‐Seq data analysis
Poly‐A enriched RNA‐seq libraries (2 × 75nt; stranded) were prepared from ventricles of three 6‐week‐old and three 12‐week‐old animals. Adapter sequences were removed and quality of clipped RNA‐seq reads was assessed using Flexbar 2.5. Trimmed reads were mapped to the killifish genome (NotFur1_genome_draft) with a splice‐aware aligner (STAR 2.5.0a) 37 including all available splice site annotation (NotFur1_protein_coding_gene_models_15‐07‐2014.gtf). All mapping results were analysed with the cufflinks software suite (version 2.2.1). Briefly, initial gene abundance estimates were computed with cuffquant and contrasted between the groups using cuffdiff.
RT–qPCR
RT–qPCR was performed using SYBR green master mix on CFX 384 Real Time System (Bio‐Rad). Briefly, RNA was isolated from the ventricles of 6‐week‐old, 8‐week‐old, 10‐week‐old and 12‐week‐old killifish. Each set of primers was validated before the final experiment. The used primer sequences are as follows:
hdac1a: forward: caaacggatagcgtgtgatg; reverse: tttggctcgcttgactttct
hdac1b: forward: gcttcaacgtcaccatcaaa; reverse: cacgttgcgaatggtgtatc
rpl37a: forward: ccagcacgcgaaatatacct; reverse: cagccactgtcttcttgcag
Drift‐variance calculations
Transcriptional drift describes how the relative expression levels of genes involved in the same biological function change with age 5. The underlying assumption is that the stoichiometric co‐expression of genes within the same functional group matters for functionality and is optimal in young adult animals, the time when mortality in nearly all organisms is lowest. The variable drift‐variance dv describes how closely the co‐expression of a group of genes resembles the co‐expression levels observed in young organisms, with dv = 0 indicating identical expression. Chronic increases in drift variance across a specific function are likely to lead to functional decline as it signifies alteration in co‐expression away from levels observed in young organisms. Transcriptional drift (td) is the change in transcript level of a gene at a given age from its level in young animals (“young reference”). The gene expression levels of sample 1 (age = 6 weeks)* was used as a young reference set. For any gene x, transcriptional drift (td) is defined as (equation 1).
To evaluate changes in co‐expression, we calculated the drift‐variance (dv) (equation 2) over a group of n genes with transcriptional drift‐values ranging from tdi = 1 to tdn.
Thus, if genes maintain a youthful co‐expression pattern, drift‐variance stays relatively small. If large fractions of genes within a GO or an entire transcriptome change expression in opposing directions, the drift‐variance increases, suggesting a loss of youthful co‐expression patterns and probable functional decline.
*Note: While the exact P‐values varied slightly, the results were the same no matter whether we used sample 1, 2 or 3 (age = 6 weeks) as a young reference sample.
Detection of senescent cells
Senescent cells were detected using the Senescence detection kit (Abcam catalog no. ab65351) according to the manufacturer's suggestions. In brief, immediately after dissecting killifish hearts, they were embedded in Tissue‐Tek and frozen immediately. Eight‐micrometre‐thin sections were made and mounted on poly‐L‐lysine‐coated slides. Slides were emerged in 1× PBS for 5 min and fixed in fixative solution (provided by supplier) for another 5 min. Staining solution was removed and slides were washed three times, 5 min each with 1× PBS. Slides were then incubated overnight with staining solution mix at 28°C for killifish samples and 37°C for human cardiomyocytes. Success of the staining was observed under the bright‐field microscope, and the reaction was stopped with two times washes with 1× PBS.
Maintenance of human pluripotent stem cells
HuES6 cells (genotype:female) [Boris Greber Lab, Max Planck Institute for Molecular Medicine (Muenster, Germany)], derived from inner cell mass of blastocysts, were used for the in vitro differentiation experiments. Authenticated cell lines were provided by the indicated providers. Human pluripotent stem cells were maintained on Matrigel‐coated 6‐well dishes in FTDA medium 38. For passaging, cells were washed with PBS and dissociated with Accutase (ThermoFisher, A1110501), supplemented with 10 μM ROCK inhibitor (Y‐27632, Stemgent), for 10 min at 37°C. Accutase was blocked with F12/DMEM (Life Technologies), and the desired number of cells was centrifuged for 2 min at 300 × g at room temperature. The cell pellet was resolved in 2 ml FTDA medium, supplemented with 10 μM Y‐27632 and pipetted onto fresh Matrigel‐coated plates followed by 24‐h incubation at 37°C and 5% CO2. After 24 h, medium was changed to 2.5 ml fresh FTDA medium. Medium change was repeated daily with increasing FTDA volumes, until cells reached confluency.
Cardiomyocyte differentiation
Cardiomyocyte differentiation was performed as previously described 6. Briefly, confluent hPSCs were dissociated into single cells with Accutase at 37°C for 10 min. Accutase was stopped with two volumes of F12/DMEM. Cells were counted, and 230,000 cells per square cm (450,000 for one well of a 24‐well plate) were centrifuged for 2 min at 300 × g at room temperature. The cell pellet was resolved in ITS medium, containing 1.5 μM CHIR (Axon, #1386) and 1.5 ng/ml BMP4, and seeded on Matrigel‐coated 24‐well plates. To ensure equal distribution and attachment of cells, plates were moved crosswise, tapped several times and left outside for 20 min before transferring them to the incubator. After 24 h, medium was changed to TS medium. After 48 h, medium was changed to TS medium supplemented with 10 μM canonical Wnt‐Inhibitor IWP‐2 (Santa Cruz #sc‐252928) for 48 h. After 48 h, medium was changed to fresh TS until beating cells were observed at days 8–10. Medium was then changed to Knockout DMEM supplemented with 2% FCS, l‐glutamine and penicillin/streptomycin until cells were used for downstream analysis.
Treatment of hCMs with chemical compounds and their experimental conditions
In order to negate any potential pleiotropic effects of the used inhibitors, we have used more than one specific inhibitor for all the targets. Furthermore, in order to estimate the best working concentration of the used chemical compounds, titration experiments were performed. For all the tested chemicals, final used concentrations were within the range or well below the IC50 values of each inhibitor. Unless mentioned, human cardiomyocytes were treated with the final concentrations of the indicated chemicals and the incubation duration was 72 h at 37°C in 5% CO2. In summary, unless indicated in the figure or their respective legends, the standard final concentrations of the indicated compounds are as follows: DHS (10 μM), triptolide (20 nM), curcumin (10 μM), anacardic acid (10 μM), MS‐275 (5 μM), pyroxamide (10 μM) and romidepsin (5 nM).
For triptolide treatment, a final concentration of 20 nM of triptolide was used. In these experiments, hCMs were pretreated with sphinganine (with or without 20 nM triptolide) for 3 days. Post‐exposure, the media were removed and the standard immunocytochemistry protocol was used for staining.
For the rescue experiments, hCMs were treated with the inhibitors for sphingosine kinase (SPHK2): ABC294640 and SLM6031434, histone acetyltransferases (HATs): curcumin and anacardic acid and histone deacetylase type 1‐specific inhibitors (HDAC1): MS‐275, pyroxamide and romidepsin. Final concentrations used were indicated in the respective figures. In these experiments, hCMs were pretreated with sphinganine (with or without rescue compounds) for 3 days. Post‐exposure, the media were removed and the standard immunocytochemistry protocol was used for staining.
For the etoposide experiments, hCMs were treated with etoposide (Sigma, catalog no: E1383) alone or in co‐incubation with curcumin. Cells were treated with 10 μM final concentration of etoposide (with and without curcumin). Three different concentrations of curcumin were used: 10, 30 and 60 μM. In order to negate the temporal parameter for rescue, we have tested two different time points (24 and 72 h). Post‐exposure, the media were removed and the standard immunocytochemistry protocol was used for staining.
For human fibroblasts, cells with 30–40% confluency were treated with 1 μM final concentration of sphinganine for 24 h at 37°C in 5% CO2, immediately fixed with 4% PFA and used for immunocytochemistry. Longer incubation periods (3 days) resulted in vast cell death.
For the chronic exposure experiments, hCMs were treated with 0.01 and 0.1 μM of sphinganine for 7 days was immediately fixed with 4% PFA and used for immunocytochemistry.
Microarray data analysis
Human cardiomyocytes were treated with DMSO, sphinganine (10 μM) and co‐treatment with sphinganine and curcumin (10 μM each) for 72 h at 37°C in 5% CO2. Post‐treatment, the media were removed and the cells were harvested immediately with cold TRIzol for the RNA isolation. RNA isolation was performed using the standard TRIzol‐based protocol. Quantity and the purity of the isolated RNA were accessed by NanoDrop™. Microarray was performed on GeneChip™ Human Transcriptome Array 2.0, and the data analysis was performed using Transcriptome Analysis Console (TAC) Software (Thermo Fisher Scientific). This experiment was performed in biological duplicates for DMSO‐treated hCMs and in biological triplicates for DHS‐ and DHS + curcumin‐treated hCMs. Gene Ontology analysis was performed using Metascape and FunRich.
Exposure of sphinganine on young adult zebrafish
Young zebrafish (~ 7–8 months old) were exposed to 1 μM sphinganine in fish tank water for 5 h/day for three consecutive days. For the rescue experiments with curcumin, fishes of the same age group were treated with a mixture of 1 μM concentration of each DHS and curcumin. Three days post‐treatment, fishes were sacrificed and tissues were harvested for immunostainings. Noteworthy, ventricle sections from the DHS‐ treated zebrafish were hard to retain on glass slides, presumably due to altered lipid content. We therefore took thicker cryosections (15 μm). To ensure that the observed DNA damage is indeed present in the nucleus of the cardiomyocytes, line‐scan approach was used on the images from confocal z‐stack.
Isolation of single cardiomyocytes from killifish
Cardiomyocytes from adult killifish ventricles were isolated as described 39. Briefly, hearts were dissected and immediately transferred to perfusion buffer on ice. Perfusion buffer was subsequently replaced by digestion buffer, and the tubes were incubated at 37°C for 2 h at 750 rpm. After digestion, the suspended cells were incubated with a series of different SB buffers and finally plated on 6‐well plates and cultured at 28°C in 5% CO2.
Comet assay
Alkaline lysis‐based comet assay was performed on killifish and human cardiomyocytes (hCMs) as discussed 40. Briefly, isolated/dissociated cardiomyocytes were mixed with 1% low‐gelling‐temperature agarose. The resulting solution was immediately poured on slides pre‐coated with agarose. Immediately, the slides were submerged in covered dish containing A1 lysis buffer for overnight at 4°C. Next day, the slides were submerged in A2 buffer for 20 min, three times each at room temperature. Electrophoresis was conducted at 12 V for 30 min. Post‐electrophoresis, the slides were incubated with the recommended concentration of propidium iodide for 20 min and immediately photographed.
Immunocytochemistry on hCMs/fibroblasts
The cells in the chamber slides were washed 1 time with 1× PBS and fixed with 4% PFA for 10 min at room temperature. Post‐fixation, the PFA was removed and the cells were washed three times with PBS. Cells in each chamber were treated with 1% Triton X‐100 (in PBS) for permeabilization. Cells were then incubated with the blocking solution, containing 2% BSA and 2% glycine in PBST. After blocking, the blocking solution was removed and cells were washed one time with PBST. Cells were incubated with either single or double primary antibodies (different host species) in 0.5% BSA in PBST overnight at 4°C. The cells were then washed three times with PBST and incubated with secondary antibody/antibodies for 2 h at room temperature. The cells were washed in PBS and mounted with VectaShield containing DAPI (Vector).
Image analysis
All images, unless mentioned, were captured as z‐stack using confocal microscopy. Confocal microscopy was performed in three frames for tricolour stainings (red, green and blue) and two frames for two colour stainings (red/green and blue), for which the intensities of only the red and green channels were kept constant across samples for later quantification. Quantification of the signal intensities was performed on greyscale images acquired after maximum intensity projections on z‐stacks. Quantification of disruption of nuclear envelope was performed using Fiji (ImageJ). Micrographs from confocal microscopy (z‐stacks) were first merged with maximum intensity projections. Dapi channel was removed from each image, to better visualize the Lamin B1 signals. Single‐channel micrographs with Lamin B1 stainings were digitally enhanced to manually observe the nuclear defect if any. Nuclei with ruptured nuclear envelope were considered as positive. For each condition, more than 200 nuclei were quantified from randomized micrographs.
HDAC activity assay
Nuclear extract HDAC activity was determined using fluorimetric assay as described by the manufacturer (Abcam; catalog no: ab156064). In brief, HDAC reaction was initiated by mixing substrate (X‐X‐X‐Lys(Ac)‐MCA), HDAC buffer along with the nuclear extract, incubated for 20 min at room temperature, and developer was added to each well. Samples were incubated for another 10–15 min at room temperature, and fluorescence readings were taken at excitation/emission at 355/460 nm. For the activity assay with the purified human HDAC1 protein (Abcam; catalog no: ab101661), similar steps were followed. The concentration of purified protein used was 200 ng per well. The final concentration of each of the tested sphingolipids was 10 μM. All controls were taken as recommended by the manufacturer.
In cellulo HDAC activity assay
In cellulo HDAC activity assay was performed using HDAC‐Glo™ I/II Assay (Promega). Briefly, human cardiomyocytes were treated with different concentrations of DHS and DHS1P using the standard exposure method in the 96‐well plate. After incubation, the media were removed and the downstream steps were performed as written in the manufacturer's instructions.
Assessment of nascent transcripts in pretreated hCMs by EU labelling
Assessment of the cellular transcription was done by 5‐ethynyl uridine (EU) labelling procedure. All steps performed and reagent concentrations used were as indicated in the manufacturer's protocol (Catalog no.: C10329, C10330).
In vivo assessment of the cardiac function by echocardiography
Twelve‐week‐old C57BL/6J mice were injected i.p. once daily with PBS, FB1 (6.75 mg/kg) or FB1 and curcumin (6.75 and 50 mg/kg, respectively) for five consecutive days. Under isoflurane anaesthesia, echocardiography was performed on 12‐week‐old C57BL/6J WT mice, along with the ~ 124‐week‐old mice using a Vevo 3100 Preclinical Imaging System and an MX550D ultrasonic transducer. Images were analysed with VevoLab software (all FUJIFILM VisualSonics, Toronto, Canada). Left ventricular ejection fraction (EF) and fractional shortening (FS) were calculated from parasternal long‐axis B‐mode both before (baseline) and after (day 5) the treatment. These parameters were also calculated for aged mice for the intergroup comparison.
Formula used to compute percentage ejection fraction and fractional shortening are as follows:
Mass spectrometry and sample preparation
For metabolomics studies, killifish tissue and serum samples were analysed by liquid chromatography–mass spectrometry (LC‐MS). Frozen killifish tissue samples, consisting of heart, liver and muscle, were transferred into a 2.0‐ml impact resistant tube containing 200 mg of 1 mm zirconium beads. To each tube, solvent volumes were adjusted so that 1,000 μl or 100 μl/mg of ice‐cold 80:20 methanol: water was added to liver and muscle, and heart, respectively. This was followed by three 15‐s homogenization cycles at 6,400 Hz in a Precellys 24® tissue homogenizer. Muscle and heart samples were cooled on dry ice and subject to one additional three 15‐s homogenization cycles. Samples were then placed in the −20°C freezer for 30 min to allow for precipitation of protein. Samples were thereafter vortexed for 30 s, centrifuged at 14,000 g × 10 min at 4°C, and supernatants were transferred to LC‐MS vials containing a 200‐μl glass insert. Injection volumes and sample concentrations were adjusted so that 50, 70 and 75 μg of heart, liver and muscle tissue were injected per run, respectively. Killifish serum samples were prepared for metabolomics analysis by adding 20 μl of ice‐cold solvent to the serum. Solvent ratios were modified for each sample to keep a ratio of 80:20 methanol: water. Samples were vortexed for 30 s and placed in the −20°C freezer for 30 min to facilitate protein precipitation. Samples were vortexed again for 30 s followed by being centrifuged at 14,000 g × 10 min at 4°C. The supernatants were collected and transferred to LC‐MS vials containing a 200‐μl glass insert. For each sample, the injection volume was adjusted so that equivalent to 0.45 μl of serum was injected for each sample. All samples were kept at 4°C in the autosampler compartment until they were injected for analysis. LC‐MS‐based metabolomics analysis was performed using a Thermo Q Exactive Orbitrap mass spectrometer coupled to a Thermo Vanquish UHPLC system. Chromatographic separation of metabolites was achieved using a Millipore (Sequant) Zic‐pHILIC 2.1 × 150 mm 5 μm column maintained at 25°C using a flow rate of 0.3 ml/min. Compounds were eluted via a 19‐min linear gradient starting from 90:10 acetonitrile:20 mM ammonium bicarbonate to 45:55 acetonitrile:20 mM ammonium bicarbonate. A Thermo Q Exactive Orbitrap mass spectrometer was operated in positive‐ion and negative‐ion modes using a heated electrospray ionization (HESI) source at 35,000 resolution, 100 ms ion trap time for MS1 and 17,500 resolution, 50 ms ion trap time for MS2 collection. Data were collected over a mass range of m/z 67–1,000, using a sheath gas flow rate of 40 units, auxiliary gas flow rate of 20 units, sweep gas flow rate of 2 units, spray voltage of 3.5 and 2.5 kV for positive‐ and negative‐ion modes (respectively), capillary inlet temperature of 275°C, auxiliary gas heater temperature of 350°C and an S‐lens RF level of 45. For MS2 collection, MS1 ions were isolated using a 1.0 m/z window and fragmented using normalized collision energy of 35. Fragmented ions were placed on dynamic exclusion for 30 s before being allowed to be fragmented again. Collected data were imported into the mzMine 2.20 software suite for analysis. Pure standards were used for identification of metabolites through manual inspection of spectral peaks and matching of retention time and MS1 accurate mass, with confirmation of identification through comparison to MS/MS fragmentation patterns. Results obtained were analysed online by MetaboAnalyst 41.
Targeted metabolomics
d18:0 Sphinganine levels were determined by liquid chromatography coupled to electrospray ionization tandem mass spectrometry (LC‐ESI‐MS/MS) using a procedure previously described with several modifications: Young and aged heart samples from killifish, zebrafish, mouse and humans were homogenized in 300 μl of water using the Precellys 24 Homogenisator (Peqlab, Erlangen, Germany) at 4,732 g for 30 s. The protein content of the homogenate was determined using the Direct Detect Infrared Spectrometer (Merck Millipore, Darmstadt, Germany). The homogenate was diluted to 1 ml with water, and 2 ml of methanol and 1 ml of chloroform were added. After addition of 75 pmol of internal standard (d17:0 Sphinganine, Avanti Polar Lipids, Alabaster, AL, USA), the mixture was sonicated for 5 min, and lipids were extracted overnight at 37°C. The solvent was evaporated in a stream of nitrogen. Interfering glycerolipids were degraded by alkaline hydrolysis with 2.5 ml of 100 mM sodium hydroxide in methanol for 2 at 37°C. After neutralization with 20 μl of acetic acid, the mixture was centrifuged (4,000 × g, 5 min, 4°C) to separate insoluble tissue debris and salts. The supernatant was collected and the residue re‐extracted with 1 ml of methanol/chloroform 2:1 (v/v). The supernatants were combined, and the solvent was evaporated in a stream of nitrogen. The residues were resolved in 200 μl of methanol/water/formic acid 58:41:1 (v/v/v) with 5 mM ammonium formate (solvent A) and 100 μl of methanol/formic acid 99:1 (v/v) with 5 mM ammonium formate (solvent B) and sonicated for 5 min. After centrifugation (13,200 × g, 5 min, 4°C), 70 μl of the clear supernatants was transferred to autoinjector vials. LC‐MS/MS analysis was performed using a Core‐Shell Kinetex C18 column (100 mm × 2.1 mm ID, 2.6 μm particle size, 100 Å pore size, Phenomenex) with detection using a QTRAP 6500 triple quadrupole/linear ion trap mass spectrometer (SCIEX). The LC (1260 Infinity Binary LC System, Agilent, Waldbronn, Germany) was operated at a flow rate of 400 μl/min. Prior to sample injection, the column was equilibrated for 1.0 min with 60% A/40% B. After injection of 50 μl of sample, 60% A/40% B was continued for 0.5 min, then linearly changed to 100% B in 1.8 min and held for 5.3 min, finally restored to 60% A/40% B by a 0.1‐min linear gradient and held for 0.5 min to re‐equilibrate the column. The total run time was 8.2 min. d18:0 Sphinganine was monitored in the positive‐ion mode using its specific multiple reaction monitoring (MRM) transitions m/z 302–284 Da (quantifier) and m/z 302–60 Da (qualifier 1) and m/z 302–254 Da (qualifier 2). For the detection of the internal standard d17:0 Sphinganine, the MRM transitions m/z 288–270 Da (quantifier) and m/z 288–60 Da (qualifier 1) and m/z 288–240 Da (qualifier 2) were used. The instrument settings for nebulizer gas (Gas 1), turbogas (Gas 2), curtain gas and collision gas were 50, 55, 40 psi and medium, respectively. The Turbo V ESI source temperature was 450°C, and the ionspray voltage was 5,500 V. For all MRM transitions, the values for declustering potential, entrance potential and cell exit potential were 120, 10 and 8 V, respectively. The collision energies ranged from 20 to 45 V. The LC chromatogram quantifier peaks of endogenous d18:0 Sphinganine and the internal standard d17:0 Sphinganine were integrated using the Analyst 1.6.2 software (AB SCIEX). Endogenous d18:0 Sphinganine was quantified by normalizing its quantifier peak area to that of the internal standard d17:0 Sphinganine. The calculated amounts of d18:0 Sphinganine were normalized to the protein content of the sample.
Proteomics and Western blot
Human cardiomyocytes were treated with DMSO and sphinganine (10 μM) for 3 days at 37°C in 5% CO2. Post‐treatment, cells were lysed in the lysis buffer. Label‐free mass spectrometry was performed in CECAD central proteomics facility. For Western blot, cells were pretreated for 72 h with: (i) DMSO (ii) DHS (10 μM) and (iii) DHS (10 μM) + curcumin (10 μM). Post‐treatment, the cells were lysed in the lysis buffer and the Western blotting was performed on 10% gel. After transfer and blocking, the PVDF membrane containing lysate was probed with HDAC1 antibody. Anti‐tubulin antibody was used as loading control.
Protein target, ligand structures and molecular docking
The crystal structure of Human HDAC1 in complex with the dimeric ELM2‐SANT domain of MTA1 from the NuRD complex was obtained from protein data bank (http://www.rcsb.org); structure id: 4BKX. HDAC1 monomer was obtained and saved as a separate pdb file, which was later used as a targeted receptor/protein. Mol2 files of the tested ligands were obtained from the Zinc database (http://zinc.docking.org/). The molecular docking simulation was performed using PyRX 42 consisting of AutoDock and AutoDock Vina with a Lamarckian genetic algorithm as scoring function. The grid parameters were selected in the active site of HDAC1 protein. Only the top hits of the ligand–protein interactions were selected with minimal free energy and lowest rmsd values (< 0.5 Å). PyMol and Discovery Studio Visualizer software were used for analysing the protein–ligand interactions. The two‐dimensional interaction plots were generated by LIGPLOT plus software 43.
Mitochondrial staining of hCMs
hCMs were incubated with MitoTracker red dye (1:5,000) dilution (stock concentration = 1 mM) for 15–30 min in incubator. For estimating the relative mitochondrial membrane potential, the cells were immediately subjected to fluorescent microscopy. For immunostaining with the cardiac marker, anti‐ACTN2, cells were immediately fixed with 4% PFA and the staining procedure was performed as discussed above.
Calcium imaging of hCMs
Calcium responses were measured in hCMs by using EarlyTox™ Cardiotoxicity Kit. All the steps were followed as recommended by the supplier. In brief, cardiomyocytes were incubated for 2 h with EarlyTox Cardiotoxicity Dye and were later analysed by live imaging with the fluorescent microscope. Captured videos were later analysed by ImageJ for calcium signals.
ROS detection assay
hCMs were treated with DMSO, DHS (10 μM), ROS inhibitors cocktail [mannitol 15 mM (Sigma) and catalase 200 U (Sigma)], rotenone (0.5 μM) and DHS (10 μM) plus ROS inhibitor cocktail. Cells were loaded with DCFDA dye, with final concentration of 1 μM and incubated for 30 min in the dark. Subsequently, the dye was removed and the cells were washed thrice with PBS and were supplemented with fresh media. ROS levels were estimated by fluorescence microscopy.
Microscopy and image processing
Leica SP8 confocal microscope was used to acquire fluorescent images with z‐stack. For the estimation of increase/decrease in fluorescent intensities, the optimal excitation intensities were kept constant. Bright‐field images were taken from Nikon Eclipse Ci and BZ‐9000 wide‐field fluorescence microscope (Keyence, Japan). Image processing was done by Fiji (ImageJ).
Statistical analysis
Statistical analysis was performed using unpaired two‐tailed Student's t‐test for comparison of two groups and analysis of variance (ANOVA) for analysing experiments consisting of three or more groups (PAST version 3.15). In all immunostaining assays, samples were randomly assigned and the blind evaluation was performed, which later decoded and statistical analysis was performed on them. To estimate, whether two intensity distributions were significantly different, we performed Kolmogorov–Smirnov, a measure of distribution differences, makes no assumption about the nature of the distributions, which is essential, because distributions analysed here are non‐Gaussian. Results of the Kolmogorov–Smirnov test were confirmed by Monte Carlo permutation analysis in Past 3.15. Graphical illustrations are plotted with R Stats packages. P‐value cut‐off used in this study is 0.05. *, **, *** and **** in the figures refer to P‐values ≤ 0.05, ≤ 0.01, ≤ 0.001 and ≤ 0.0001, respectively.
Author contributions
LK conceived the project and supervised the experiments. LK, DRV & GA designed all the experiments. GA, DB, WY, SF, J‐EM, NR, NEV, KAL & SBa performed the experiments. JD performed killifish husbandry and killifish tissue preparation for this study. JSM contributed to killifish husbandry GA, AP, DRV, MJ, MP, AA, SF, AS, JH, AT, CD & LK analysed and interpreted the data. SG, GA, LK, AK and SBr designed, performed and evaluated the in vivo mouse experiments. GA & LK drafted the illustrations. GA & LK prepared the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File
Acknowledgements
The Kurian lab is supported by the NRW Stem Cell Network Independent Group Leader Grant (Grant No: 3681000801 and 2681101801, CECAD (Grant No: 3624720211), German Heart Association (DZHK; Grant No: 3632009221), Else Kröner‐Fresenius‐Stiftung, Deutsche Forschungsgemeinschaft grant (KU 3511/4‐1), University of Cologne (Grant No: 3622801511), Koeln Fortune funding and Grants4Targets initiative by Bayer AG. The Valenzano Lab is funded by the Max Planck Institute for the Biology of Ageing. We acknowledge the central facilities at CECAD, CMMC and MPI Ageing for technical assistance. Jain Lab is funded by NIH R01ES027595, R03HL133720 and S10OD020025. Aguirre Lab is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number K01HL135464. We would like to thank Dr. Janine Altmueller, CGI‐Cologne, for assistance with whole transcriptome sequencing and all members of Valenzano Lab and Kurian Lab for intellectual contributions at various stages of this project. We thank Dr. Thomas Langer and Dr. Björn Schumacher for their valuable comment on the manuscript.
EMBO Reports (2019) 20: e47407
Contributor Information
Dario Riccardo Valenzano, Email: valenzano@age.mpg.de.
Leo Kurian, Email: leo.kurian@uni-koeln.de.
Data availability
Microarray data from this publication have been deposited to the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) and assigned the identifier E‐MTAB‐7530. Raw transcriptomics data have been deposited to the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) and assigned the identifier E‐MTAB‐7582. Processed proteomics data and microarray data are available in [Link], [Link].
References
- 1. Kikuchi K, Poss KD (2012) Cardiac regenerative capacity and mechanisms. Annu Rev Cell Dev Biol 28: 719–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguirre A, Sancho‐Martinez I, Izpisua Belmonte JC (2013) Reprogramming toward heart regeneration: stem cells and beyond. Cell Stem Cell 12: 275–284 [DOI] [PubMed] [Google Scholar]
- 3. Reichwald K, Petzold A, Koch P, Downie BR, Hartmann N, Pietsch S, Baumgart M, Chalopin D, Felder M, Bens M et al (2015) Insights into sex chromosome evolution and aging from the genome of a short‐lived fish. Cell 163: 1527–1538 [DOI] [PubMed] [Google Scholar]
- 4. Valenzano DR, Benayoun BA, Singh PP, Zhang E, Etter PD, Hu C‐K, Clément‐Ziza M, Willemsen D, Cui R, Harel I, et al (2015) The African turquoise killifish genome provides insights into evolution and genetic architecture of lifespan. Cell 163: 1539–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rangaraju S, Solis GM, Thompson RC, Gomez‐Amaro RL, Kurian L, Encalada SE, Niculescu AB, Salomon DR, Petrascheck M (2015) Suppression of transcriptional drift extends C. elegans lifespan by postponing the onset of mortality. Elife 4: e08833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rao J, Pfeiffer MJ, Frank S, Adachi K, Piccini I, Quaranta R, Araúzo‐Bravo M, Schwarz J, Schade D, Leidel S et al (2016) Stepwise clearance of repressive roadblocks drives cardiac induction in human ESCs. Cell Stem Cell 18: 341–353 [DOI] [PubMed] [Google Scholar]
- 7. Lucki NC, Sewer MB (2012) Nuclear sphingolipid metabolism. Annu Rev Physiol 74: 131–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hung WC, Chang HC, Chuang LY (1999) Activation of caspase‐3‐like proteases in apoptosis induced by sphingosine and other long‐chain bases in Hep3B hepatoma cells. Biochem J 338(Pt 1): 161–166 [PMC free article] [PubMed] [Google Scholar]
- 9. Ahn EH, Schroeder JJ (2010) Induction of apoptosis by sphingosine, sphinganine, and C(2)‐ceramide in human colon cancer cells, but not by C(2)‐dihydroceramide. Anticancer Res 30: 2881–2884 [PubMed] [Google Scholar]
- 10. Chumanevich AA, Poudyal D, Cui X, Davis T, Wood PA, Smith CD, Hofseth LJ (2010) Suppression of colitis‐driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis 31: 1787–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ahn EH, Schroeder JJ (2002) Sphingoid bases and ceramide induce apoptosis in HT‐29 and HCT‐116 human colon cancer cells. Exp Biol Med (Maywood) 227: 345–353 [DOI] [PubMed] [Google Scholar]
- 12. Zhang R, Chen W, Adams PD (2007) Molecular dissection of formation of senescence‐associated heterochromatin foci. Mol Cell Biol 27: 2343–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verdone L, Caserta M, Di Mauro E (2005) Role of histone acetylation in the control of gene expression. Biochem Cell Biol 83: 344–353 [DOI] [PubMed] [Google Scholar]
- 14. Krishnan V, Chow MZY, Wang Z, Zhang L, Liu B, Liu X, Zhou Z (2011) Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24‐deficient mice. Proc Natl Acad Sci USA 108: 12325–12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu G‐H, Barkho BZ, Ruiz S, Diep D, Qu J, Yang S‐L, Panopoulos AD, Suzuki K, Kurian L, Walsh C et al (2011) Recapitulation of premature ageing with iPSCs from Hutchinson‐Gilford progeria syndrome. Nature 472: 221–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peleg S, Feller C, Ladurner AG, Imhof A (2016) The metabolic impact on histone acetylation and transcription in ageing. Trends Biochem Sci 41: 700–711 [DOI] [PubMed] [Google Scholar]
- 17. Tie F, Banerjee R, Stratton CA, Prasad‐Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ (2009) CBP‐mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136: 3131–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vempati RK, Jayani RS, Notani D, Sengupta A, Galande S, Haldar D (2010) p300‐mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J Biol Chem 285: 28553–28564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Valdez BC, Brammer JE, Li Y, Murray D, Liu Y, Hosing C, Nieto Y, Champlin RE, Andersson BS (2015) Romidepsin targets multiple survival signaling pathways in malignant T cells. Blood Cancer J 5: e357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bracker TU, Sommer A, Fichtner I, Faus H, Haendler B, Hess‐Stumpp H (2009) Efficacy of MS‐275, a selective inhibitor of class I histone deacetylases, in human colon cancer models. Int J Oncol 35: 909–920 [DOI] [PubMed] [Google Scholar]
- 21. Noor Z, Afzal N, Rashid S (2015) Exploration of novel inhibitors for class I histone deacetylase isoforms by QSAR modeling and molecular dynamics simulation assays. PLoS One 10: e0139588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN (2007) Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 21: 1790–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. López‐Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153: 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L, Schwabe JWR (2013) Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell 51: 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S et al (2009) Regulation of histone acetylation in the nucleus by sphingosine‐1‐phosphate. Science 325: 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, Gromak N, Petermann E (2016) Increased global transcription activity as a mechanism of replication stress in cancer. Nat Commun 7: 13087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Britten CD, Garrett‐Mayer E, Chin SH, Shirai K, Ogretmen B, Bentz TA, Brisendine A, Anderton K, Cusack SL, Maines LW et al (2017) A phase I study of ABC294640, a first‐in‐class sphingosine kinase‐2 inhibitor, in patients with advanced solid tumors. Clin Cancer Res 23: 4642–4650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kharel Y, Morris EA, Congdon MD, Thorpe SB, Tomsig JL, Santos WL, Lynch KR (2015) Sphingosine kinase 2 inhibition and blood sphingosine 1‐phosphate levels. J Pharmacol Exp Ther 355: 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, Kundu TK (2004) Curcumin, a novel p300/CREB‐binding protein‐specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase‐dependent chromatin transcription. J Biol Chem 279: 51163–51171 [DOI] [PubMed] [Google Scholar]
- 30. Dai D‐F, Chen T, Johnson SC, Szeto H, Rabinovitch PS (2012) Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal 16: 1492–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Perrino C, Barabási A‐L, Condorelli G, Davidson SM, De Windt L, Dimmeler S, Engel FB, Hausenloy DJ, Hill JA, Van Laake LW et al (2017) Epigenomic and transcriptomic approaches in the post‐genomic era: path to novel targets for diagnosis and therapy of the ischaemic heart? Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc Res 113: 725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang W, Song M, Qu J, Liu G‐H (2018) Epigenetic modifications in cardiovascular aging and diseases. Circ Res 123: 773–786 [DOI] [PubMed] [Google Scholar]
- 33. Illi B, Ciarapica R, Capogrossi MC (2015) Chromatin methylation and cardiovascular aging. J Mol Cell Cardiol 83: 21–31 [DOI] [PubMed] [Google Scholar]
- 34. Papait R, Greco C, Kunderfranco P, Latronico MVG, Condorelli G (2013) Epigenetics: a new mechanism of regulation of heart failure? Basic Res Cardiol 108: 361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A, Viganò V, Stirparo GG, Latronico MVG, Hasenfuss G et al (2013) Genome‐wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci USA 110: 20164–20169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé MET, Calder RB, Chisholm GB, Pollock BH, Klein CA et al (2006) Increased cell‐to‐cell variation in gene expression in ageing mouse heart. Nature 441: 1011–1014 [DOI] [PubMed] [Google Scholar]
- 37. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frank S, Zhang M, Schöler HR, Greber B (2012) Small molecule‐assisted, line‐independent maintenance of human pluripotent stem cells in defined conditions. PLoS One 7: e41958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sander V, Suñe G, Jopling C, Morera C, Izpisua Belmonte JC (2013) Isolation and in vitro culture of primary cardiomyocytes from adult zebrafish hearts. Nat Protoc 8: 800–809 [DOI] [PubMed] [Google Scholar]
- 40. Olive PL, Banáth JP (2006) The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1: 23–29 [DOI] [PubMed] [Google Scholar]
- 41. Xia J, Sinelnikov IV, Han B, Wishart DS (2015) MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res 43: W251–W257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dallakyan S, Olson AJ (2015) Small‐molecule library screening by docking with PyRx. Methods Mol Biol 1263: 243‐250 [DOI] [PubMed] [Google Scholar]
- 43. Laskowski RA, Swindells MB (2011) LigPlot+: multiple ligand‐protein interaction diagrams for drug discovery. J Chem Inf Model 51: 2778‐2786 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File
Data Availability Statement
Microarray data from this publication have been deposited to the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) and assigned the identifier E‐MTAB‐7530. Raw transcriptomics data have been deposited to the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) and assigned the identifier E‐MTAB‐7582. Processed proteomics data and microarray data are available in [Link], [Link].