

Abstract
The conserved AAA‐ATPase Msp1 is embedded in the outer mitochondrial membrane and removes mislocalized tail‐anchored (TA) proteins upon dysfunction of the guided entry of tail‐anchored (GET) pathway. It remains unclear how Msp1 recognizes its substrates. Here, we extensively characterize Msp1 and its substrates, including the mitochondrially targeted Pex15Δ30, and full‐length Pex15, which mislocalizes to mitochondria upon dysfunction of Pex19 but not the GET pathway. Moreover, we identify two new substrates, Frt1 and Ysy6. Our results suggest that mislocalized TA proteins expose hydrophobic surfaces in the cytoplasm and are recognized by Msp1 through conserved hydrophobic residues. Introducing a hydrophobic patch into mitochondrial TA proteins transforms them into Msp1 substrates. In addition, Pex15Δ30 and Frt1 contain basic inter‐membrane space (IMS) residues critical for their mitochondrial mistargeting. Remarkably, Msp1 recognizes this feature through the acidic D12 residue in its IMS domain. This dual‐recognition mechanism involving interactions at the cytoplasmic and IMS domains of Msp1 and substrates greatly facilitates substrate recognition and is required by Msp1 to safeguard mitochondrial functions.
Keywords: AAA+ ATPase, ATAD1, mitochondrial protein quality control, Msp1, tail‐anchored proteins
Subject Categories: Membrane & Intracellular Transport, Protein Biosynthesis & Quality Control
Introduction
Mitochondria are essential organelles that play pivotal roles in energy supply, metabolism, and signaling processes such as cell death. Mitochondrial proteostasis is critical for mitochondrial fitness and is maintained by AAA‐proteases 1, 2, the ubiquitin‐Cdc48‐proteasome pathway 3, 4, 5, and an AAA‐ATPase Msp1 embedded in outer mitochondrial membrane (OMM) that clears mislocalized tail‐anchored (TA) proteins 6, 7.
TA proteins are post‐translationally targeted to the ER, peroxisome, and mitochondria 8. The best understood TA protein targeting pathway is the guided entry of tail‐anchored (GET) pathway: Newly synthesized TA protein is recognized at the transmembrane (TM) segment by the chaperone Sgt2, then delivered to the ATPase Get3, and finally transferred to the Get1/2 insertase complex at ER membrane 9, 10. In get mutant cells, many GET‐dependent TA proteins accumulate as aggregates in the cytoplasm, and a subset of them are mistargeted to mitochondria 11. OMM contains important TA proteins, including the fission receptor Fis1 12, the ER‐mitochondria encounter structure component Gem1 13, and subunits of the TOM import complex including Tom5, Tom6, Tom7, and Tom22 14. It remains unclear that how TA proteins are targeted to mitochondria as none of the known mitochondrial import machineries is required for their biogenesis 15, 16.
Msp1 is an evolutionarily conserved AAA‐ATPase (ATAD1 in human) that dually localizes to mitochondria and peroxisome. It removes TA proteins mistargeted to mitochondria in get mutant cells to safeguard mitochondrial function. Synthetic mutations of MSP1 and GET genes cause the accumulation of mistargeted TA proteins on mitochondria, resulting in severe mitochondrial defects and poor respiratory growth 6, 7. Peroxisomal Msp1 clears excessive TA protein Pex15 that fails to assemble into complex with its binding partner Pex3 17. ATAD1 knockout mice and patients carry ATAD1 mutations show severe mitochondrial damages 6, 18. It is interesting and essential to understand how Msp1 detects and distinguishes its substrates from mitochondrial TA proteins.
Msp1 belongs to the meiotic clade of AAA proteins that contains an N‐domain followed by an AAA‐ATPase domain 19. Other members of this clade include Vps4, Spastin, Katanin, and Fidgetin. Studies of these four enzymes suggest common features that they exist as monomers/dimers, directly bind substrates through the N‐terminal microtubule interacting and trafficking (MIT) domain, and assemble into hexamer upon substrate engagement 20. Msp1 alone is sufficient to dislocate TA protein from liposome as shown in an in vitro assay 21, indicating Msp1 can directly recognize substrates. But as the only transmembrane protein in this clade, Msp1 possesses a different N‐domain that contains a TM segment and lacks homology to those of other members 19. Furthermore, epichromosomally expressed Msp1 N‐domain mutants can rescue msp1Δ cells 21, arguing against a role of Msp1 N‐domain in substrate recognition. Msp1 also interacts with Cis1, a stress‐responsive protein that facilitates the Msp1‐dependent removal of mitochondrial precursor proteins clogged in the TOM complex 22. Whether Cis1 is involved in the removal of mistargeted TA proteins remains unclear. In this study, we combine genetic, biochemical, and imaging approaches to address the molecular mechanisms of substrate recognition by Msp1.
Results
Identification of Msp1 N‐domain residues critical for GFP‐Pex15Δ30 binding
Msp1 function was determined by monitoring the degradation of its model substrate GFP‐Pex15Δ30, in which the last 30 amino acids of Pex15 were truncated to make it constitutively targeted to mitochondria 7. Cis1 knockout did not affect GFP‐Pex15Δ30 degradation (Appendix Fig S1). We thus focused on Msp1 itself. Msp1 consists of an N‐domain of 98 amino acids, and a C‐terminal AAA‐domain highly analogous to other AAA‐ATPases (Fig 1A). We rescued msp1Δ cells with wild‐type (WT) Msp1 or Msp1TOM70(N), in which the N‐terminal 32 residues of Msp1 were replaced with those of Tom70 (Fig EV1A). When expressed by epichromosomal vector under the control of Msp1 promoter, Msp1TOM70(N) restored GFP‐Pex15Δ30 degradation and rescued the growth defect of get3Δ msp1Δ cells under respiratory growth condition (SCEG; Fig EV1B and C), consistent with previous results 21. In contrast, when expressed by knockin, Msp1Tom70(N) did not rescue these defects (Fig EV1B and C). Epichromosomal expression produced three times more proteins than the knockin method (Fig EV1D), suggesting that Msp1 N‐domain contains key residues facilitating substrate degradation, and the loss of these residues requires compensation by Msp1 overexpression. Furthermore, epichromosomal expression resulted in heterogeneous expression (Fig EV1E). We thus utilized the knockin method to analyze Msp1 N‐domain mutants.
Figure 1. Identification of residues in Msp1 N‐domain essential for GFP‐Pex15Δ30 binding.
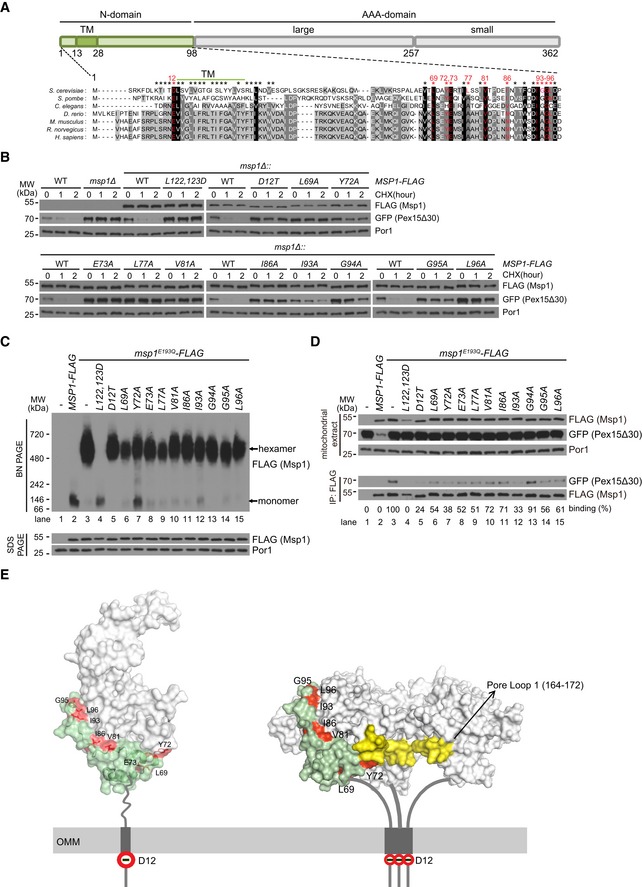
- Alignment of Msp1 N‐domain sequence (amino acids 1–98) with those of its orthologs. Residues marked by asterisks were tested for their roles in GFP‐Pex15Δ30 degradation. The critical residues are highlighted in red.
- Degradation of GFP‐Pex15Δ30 in strains expressing WT or mutant forms of Msp1‐FLAG. Cells were grown in synthetic glucose media (SCD) to log phase, treated with cycloheximide (CHX) to stop protein synthesis, and collected at the indicated time points. L122, 123D mutation disrupts Msp1 hexamer assembly and thus served as a control for Msp1 loss of function. Anti‐Por1 blots were shown as the loading controls.
- Hexamer assembly by WT and mutant forms of Msp1‐FLAG. Mitochondria‐enriched fraction was isolated from the indicated knockin strains, lysed with 1% digitonin. Lysates (10 μg) were analyzed by blue‐native (BN) PAGE or SDS–PAGE.
- Interaction of GFP‐Pex15Δ30 with WT and mutant forms of Msp1‐FLAG. Mitochondria were isolated, lysed with 1% digitonin, and subject to anti‐FLAG immunoprecipitation (IP). Mitochondrial extracts (10 μg) and immunoprecipitates (100 μl in total, load 1 μl for FLAG and 10 μl for GFP blot) were analyzed by Western blot. The intensity ratio of GFP‐Pex15Δ30/Msp1‐FLAG was calculated using ImageJ software and normalized to the value of positive control (GFP‐Pex15∆30/Msp1E193Q‐FLAG).
- The positions of conserved critical residues for substrate binding in the monomeric (PDB ID: 5W0T) and hexameric structures of Msp1 21. Three subunits of the hexamer were removed to visualize pore loop 1 (yellow). The AAA‐domain is in light gray and part of the N‐domain (amino acids 58–98) is in light green. Critical residues are highlighted in red. OMM: outer mitochondrial membrane.
Figure EV1. N‐domain is essential for Msp1 function under endogenous expression level (associated with Fig 1).
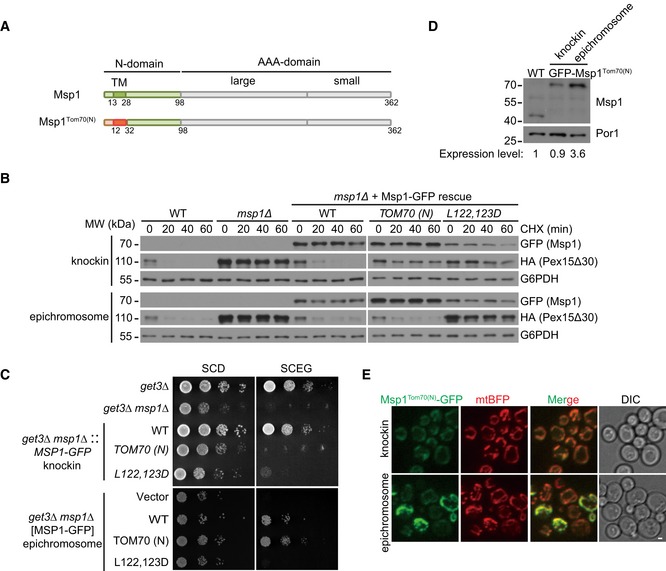
- Domain structure of Msp1 and Msp1Tom70(N). Msp1Tom70(N) was constructed by replacing the first 32 amino acids of Msp1 with that of Tom70.
- Msp1Tom70(N)‐GFP, when expressed at endogenous level by the knockin method, failed to restore mRFP‐HA‐Pex15Δ30 degradation. mRFP‐6xHA‐tagged Pex15Δ30 was expressed from a centromeric plasmid under the control of TEF1 promoter. The gene cassettes expressing WT or mutant forms of Msp1 under the control of MSP1 endogenous promoter were either integrated at the endogenous locus (knockin) or cloned into a centromeric plasmid (epichromosome). The indicated strains were grown in synthetic glucose media to log phase and then treated with CHX and collected at the indicated time points.
- Msp1Tom70(N)‐GFP, when expressed at endogenous level by the knockin method, failed to rescue the growth defect of get3Δ msp1Δ cells. The indicated strains constructed similarly as in (B) were grown in glucose media to log phase and then spotted on SCD or SCEG plates in a 10‐fold serial dilution, and then incubated for 2–5 days.
- The relative protein level of chromosomally and epichromosomally expressed Msp1TOM70(N)‐GFP. The intensity ratios of Msp1/Por1 were calculated using ImageJ software, and the value in WT was set to 1.
- Heterogeneous levels of Msp1TOM70(N)‐GFP expressed from a centromeric plasmid. Z projections and DIC images are shown. Scale bar represents 1 μm.
Alignment of the N‐domain sequences of Msp1 and its orthologs revealed conserved residues (Fig 1A). We performed mutagenesis scan of 39 residues within and flanking the TM domain and in the conserved C‐terminal region (marked by asterisks in Fig 1A). The L122, 123D mutant disrupts Msp1 hexamer assembly 21 and thus served as a control for Msp1 loss of function. We identified 11 residues whose mutations did not affect Msp1 localization (Fig EV2A) and stability (Fig 1B) but impaired GFP‐Pex15Δ30 degradation (Figs 1B and EV2B). The untagged Msp1 V81A and I93A mutants also impaired substrate degradation (Fig EV2C). We further examined whether Msp1 stability and folding were affected by the mutants. The thermostability of endogenous Msp1 was not affected by V81A and I93A mutations (Appendix Fig S2A–D). We then generated recombinant proteins of Msp1 cytoplasmic domain. WT Msp1 and its V81A, I86A, I93A, and G95A mutants had similar sensitivity to limited trypsin digestion (Appendix Fig S2E). These results indicate the N‐domain mutants did not alter Msp1 structure. These residues are highlighted in red (Fig 1A) and most of them are evolutionarily conserved. Notably, D12 is lost in the Msp1TOM70(N) mutant.
Figure EV2. The localization of Msp1 N‐domain mutants and their effects on GFP‐Pex15Δ30 degradation (associated with Fig 1).
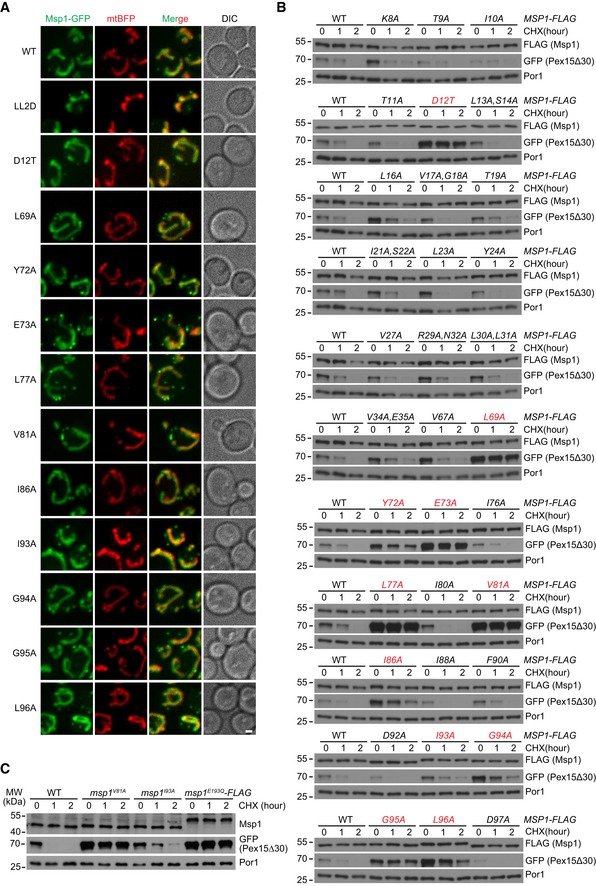
- The normal localization of Msp1 N‐domain mutants. Z projections and DIC images are shown. Mitochondrial Msp1‐GFP colocalizes with mtBFP. Peroxisomal Msp1 appears as extra‐mitochondrial dots. Scale bar represents 1 μm.
- Degradation of GFP‐Pex15Δ30 in WT and Msp1 N‐domain mutant cells. Mutants displaying defective degradation are highlighted in red.
- Degradation of GFP‐Pex15Δ30 in untagged Msp1 N‐domain mutant cells. msp1 E193Q ‐FLAG strain is used as a control for Msp1 loss of function.
The E193Q walker B mutant of Msp1 has normal ATP binding but loses ATP hydrolysis activity. It forms a hexamer and constitutively binds substrates and thus functions as a substrate‐trap mutant 6, 7, 21. When assayed by blue‐native gel, Msp1‐FLAG existed as monomers and Msp1E193Q‐FLAG as hexamers (Fig 1C, lanes 2 and 3), a typical feature of meiotic clade of AAA‐ATPases 20. Msp1L122,123D,E193Q served as a control to disrupt hexamer assembly. Most mutations did not affect the oligomerization of Msp1E193Q‐FLAG except Y72A, which caused a slight increase of monomeric forms (Fig 1C, lane 7). We then analyzed substrate binding with Msp1. As expected, Msp1E193Q‐FLAG pulled down GFP‐Pex15Δ30, whereas Msp1‐FLAG did not (Fig 1D, lanes 2 and 3). Nearly, all the mutations except G94A impaired GFP‐Pex15Δ30 pulldown by Msp1E193Q‐FLAG (Fig 1D, lanes 5–15). Therefore, most of the residues are required for substrate binding.
We highlighted all the conserved residues critical for substrate binding on Msp1 monomeric (PDB ID: 5W0T) and hexameric structures 21 (Fig 1E): D12 is a negatively charged residue in the inter‐membrane space (IMS) and the other residues are in the cytoplasm. Notably, most cytoplasmic critical residues are in the folded region and are hydrophobic residues, except E73 and G95. Y72, V81, I86, I93, and L96 are in contact with the AAA‐domain (light gray), most possibly to avoid exposing hydrophobic surfaces to the cytoplasm. Hydrophobic residues L69 and Y72, especially the latter, are close to pore loop 1, which is critical for substrate translocation into the pore 21.
Characterization of Msp1 and GFP‐Pex15Δ30 interaction by in vivo site‐specific photo‐crosslinking
Msp1 N‐domain may interact with substrates through the IMS and cytoplasmic regions because critical residues are present in both regions. We thus characterized the interaction between Msp1E193Q‐FLAG and GFP‐3xHA‐Pex15Δ30 by the in vivo site‐specific photo‐crosslinking method 23, 24. Photo‐activatable amino acid p‐benzoyl‐L‐phenylalanine (BPA) was introduced at a position specified by an amber codon in the target protein. Upon UV irradiation of living cells, BPA photo‐crosslinks with direct interacting proteins. By sampling multiple positions, the interaction surface consisting of residues positive for BPA photo‐crosslinking can be mapped. We and others have utilized the method to characterize mitochondrial complexes 25, 26, 27.
Substrate‐trap mutants of AAA‐ATPase can engage substrates into the central pore formed by the ATPase hexamer 28, which contains three conserved pore loops critical for substrate translocation (Fig 2A) 21. BPA incorporated at Msp1E193Q pore loops crosslinked with GFP‐3xHA‐Pex15Δ30 (Fig 2B, crosslinked residues are color‐labeled in Fig 2A), indicating Msp1 AAA‐domain works in a similar manner as other AAA‐ATPases.
Figure 2. Characterization of GFP‐3xHA‐Pex15Δ30 and Msp1E193Q‐FLAG interaction by in vivo site‐specific photo‐crosslinking.
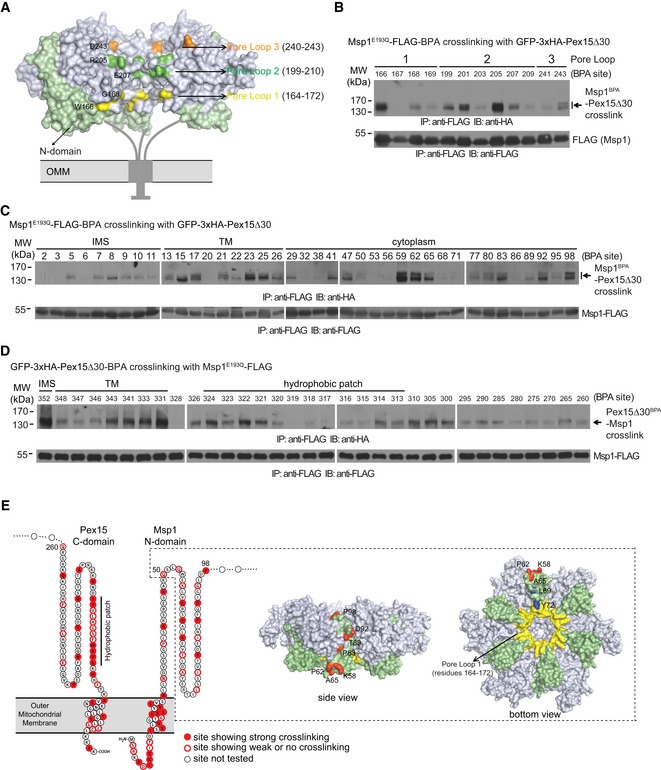
-
AA hexameric model of Msp1 with pore loop residues positive for crosslinking highlighted by colors.
-
B, CIn vivo photo‐crosslinking of BPA incorporated in pore loops (B) and N‐domain (C) of Msp1E193Q‐FLAG with GFP‐3xHA‐Pex15Δ30.
-
DIn vivo photo‐crosslinking of BPA incorporated in GFP‐3xHA‐Pex15Δ30 (amino acids 260–352) with Msp1E193Q‐FLAG. Experiments in (B–D) were performed as described in Materials and Methods. Immunoprecipitates (100 μl in total, load 1 μl for FLAG, and 40–50 μl for HA blot) were analyzed by Western blot. IMS, inter‐membrane space.
-
ECartoon summary of photo‐crosslinking results. The sites showing strong (filled red) and weak or no (red circle) crosslinking are highlighted in the topology model. The boxed region highlights the position of N‐domain (amino acids 50–98, shown in green) in the hexameric model of Msp1. N‐domain residues positive for crosslinking (red), hydrophobic residues (L69 and Y72, blue) critical for substrate binding, and pore loop 1 (yellow) are highlighted.
We then incorporated BPA into Msp1E193Q N‐domain (Fig 2C). We excluded most critical residues of Msp1 N‐domain from BPA incorporation because incorporating BPA into these residue positions is similar to alanine mutation and may disrupt substrate interaction and cause false‐negative results. We also incorporated BPA into residues 260–352 of Pex15Δ30 (Fig 2D), which contains all the critical elements for recognition by Msp1 (see Fig 3 for evidence). As summarized in Fig 2E, two features and indications can be obtained: (i) Direct interaction between Msp1 N‐domain and Pex15Δ30 extensively occurs in their IMS, TM, and cytoplasmic regions. (ii) The folded region of Msp1 N‐domain (amino acids 50–98, shown in green in the structural models) lines along the surface of Msp1 hexamer to positions near pore loop 1 and has direct interactions with Pex15Δ30 (crosslinked residues shown in red in the structural models). This spatial organization may facilitate substrate transfer from N‐domain into the central pore.
Figure 3. The IMS tail charge and cytoplasmic hydrophobic patch of Pex15Δ30 are required for its recognition and removal by Msp1.
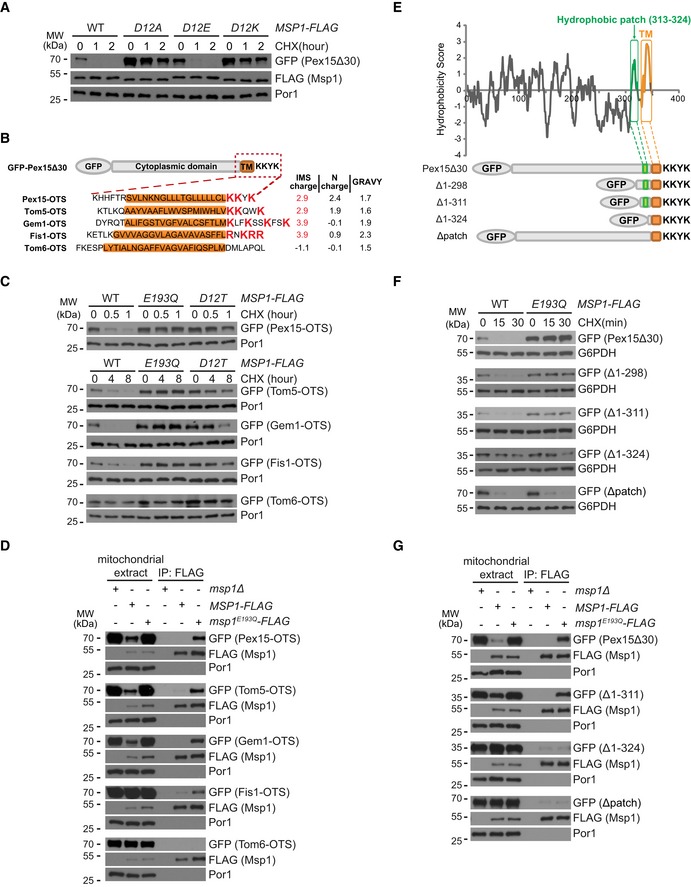
- Degradation of GFP‐Pex15Δ30 by Msp1 D12 mutants.
- Schematic illustration of the Outer mitochondrial membrane Targeting Sequences (OTSs) of GFP‐Pex15Δ30 and mitochondrial TA proteins. The GRAVY (Grand Average of Hydropathicity) values of TM domain (highlighted in orange) and the charges of the flanking sequences are shown on the right.
- Degradation of GFP‐Pex15Δ30 and its chimeric variants by Msp1.
- Interaction of GFP‐Pex15Δ30 and its chimeric variants with Msp1. Experiments were performed as in Fig 1D.
- Hydrophobicity plot of Pex15Δ30 using the Kyte–Doolittle scale. The hydrophobic patch and the TM domain are highlighted in green and orange, respectively. Truncated forms of Pex15Δ30 are shown below.
- Degradation of GFP‐Pex15Δ30 and its truncation mutants.
- Interaction of GFP‐Pex15Δ30 and its truncation mutants with Msp1. Cells were analyzed as in (D).
Positively charged residues in IMS tail of GFP‐Pex15Δ30 correlate with its recognition and removal by Msp1
The negatively charged D12 residue of Msp1 is critical for substrate binding (Fig 1D). Msp1 D12E mutation did not affect, whereas D12A and D12K mutations impaired substrate degradation (Fig 3A), indicating the negative charge of D12 is critical for its function. D12 may interact with the IMS tail of substrates through electrostatic interactions considering the presence of three positively charged K residues in Pex15Δ30 IMS tail (Fig 3B). We mutate these K residues, but even mutating one K to H mistargeted GFP‐Pex15Δ30 to the cytoplasm (Fig EV3A). This result is consistent with previous observations that positively charged residues in IMS tail are critical for the mitochondrial targeting of many TA proteins 8, 29, 30.
Figure EV3. Localization of WT or mutant forms of GFP‐Pex15Δ30, GFP‐Gem1, and GFP‐Fis1 (associated with Figs 3 and 4).
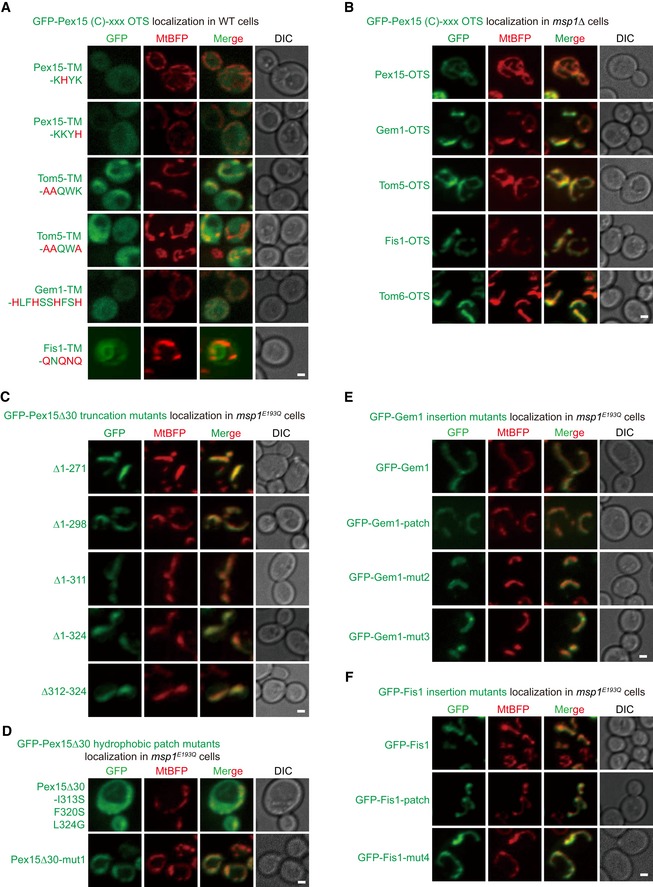
- Localization of GFP‐Pex15Δ30 carrying the indicated Outer mitochondrial membrane Targeting Sequence (OTS). Mutations in the IMS tail are highlighted in red.
- Localization of GFP‐Pex15Δ30 carrying indicated OTS in msp1Δ cells.
- Localization of GFP‐Pex15Δ30 truncation mutants in msp1 E193Q cells.
- Localization of GFP‐Pex15Δ30 hydrophobic patch mutants in msp1 E193Q cells.
- Localization of GFP‐Gem1 and its insertion mutants in msp1 E193Q cells.
- Localization of GFP‐Fis1 and its insertion mutants in msp1 E193Q cells.
Outer mitochondrial membrane TA proteins Tom5, Gem1, and Fis1 carry IMS sequences rich in positively charged residues, but Tom6 does not (Fig 3B). We thus replaced the OMM targeting sequence (OTS) of GFP‐Pex15Δ30 with that of OMM TA proteins. The chimeric proteins correctly localized to mitochondria (Fig EV3B). OTSs with positively charged residues (Tom5, Gem1, and Fis1) supported the degradation of chimeric proteins by Msp1 (Fig 3C). Similar to GFP‐Pex15Δ30, the degradation was impaired by the Msp1D12T mutation (Fig 3C). In contrast, the chimeric protein with Tom6 OTS was stable (Fig 3C). Consistent with the degradation results, OTSs of Tom5, Gem1, and Fis1 but not Tom6 supported substrate interaction with Msp1 (Fig 3D). We also tried to reduce positive charges in Tom5, Gem1, and Fis1 IMS tails. But again all the mutants tested were mislocalized (Fig EV3A). Taken together, these results support the idea that positively charged residues in substrate IMS tail facilitate their interaction with Msp1, very likely through electrostatic interactions with Msp1 D12 residue.
A hydrophobic patch in GFP‐Pex15Δ30 cytoplasmic domain is essential for its recognition and removal by Msp1
The enrichment of hydrophobic critical residues in Msp1 N‐domain suggests hydrophobic interactions might occur between Msp1 and substrate cytoplasmic domains. Hydrophobicity analysis revealed a hydrophobic patch near Pex15 TM segment (Fig 3E). The hydrophobic patch is rich in residue positions showing crosslinking with Msp1 (Fig 2E). Serial truncations showed that a minimum sequence (Δ1–311) consisting of the hydrophobic patch, TM segment, and IMS tail is sufficient for interaction with and removal by Msp1 (Fig 3F and G). Further deletion of the hydrophobic patch (Δ1–324) rendered the protein stable and unrecognized by Msp1 (Fig 3F and G). Deleting the hydrophobic patch alone (Δpatch) abolished the interaction between GFP‐Pex15Δ30 and Msp1 (Fig 3G), and caused GFP‐Pex15Δ30 degradation by an Msp1‐independent mechanism (Fig 3F). All the GFP‐Pex15Δ30 truncation mutants correctly localized to mitochondria (Fig EV3C). These results demonstrate that the hydrophobic patch of GFP‐Pex15Δ30 is essential for its recognition and removal by Msp1.
In the patch, I313, F320, and L324 likely form a hydrophobic core, with potential contribution from flanking L316 and A317 (Fig 4A). To determine whether this hydrophobic core is required for GFP‐Pex15Δ30 recognition by Msp1, we mutated three residues into non‐hydrophobic ones (I313S, F320S, and L324G). Surprisingly, this mutant form mislocalized to the cytoplasm (Fig EV3D), suggesting hydrophobic residues in the patch are critical for mitochondrial targeting. We then mutated these residues to A (Fig 4B, mut1), which is of medium hydrophobicity and correctly localized to mitochondria (Fig EV3D). GFP‐Pex15Δ30‐mut1 degradation was partially delayed in msp1 E193Q cells and mainly degraded by an Msp1‐independent mechanism (Fig 4B), similar as the degradation of the Δpatch mutant (Fig 3F). We screened a panel of mutants and found that temperature‐sensitive inactivation of the ATPase subunits of proteasome Cim3 delayed GFP‐Pex15Δ30‐mut1 degradation. Inactivating Cim3 in msp1Δ cells efficiently blocked GFP‐Pex15Δ30‐mut1 degradation (Fig 4C). GFP‐Pex15Δ30‐mut1 accumulated on mitochondria in Msp1 and Cim3 mutant strains (Fig 4D), allowing us to examine its interaction with Msp1. In comparison with GFP‐Pex15Δ30, GFP‐Pex15Δ30‐mut1 had significantly weaker interaction with Msp1E193Q‐FLAG (Fig 4E, lane 3 vs. 6) and the weak interaction was not enhanced by inactivating Cim3 (Fig 4E, lane 6 vs. 9). Collectively, these results suggest that the hydrophobic core in the patch of GFP‐Pex15Δ30 is crucial for its recognition by Msp1.
Figure 4. Patch hydrophobicity of Pex15Δ30 is critical for its interaction with Msp1 and insertion of the patch into mitochondrial TA proteins Fis1 and Gem1 transforms them into Msp1 substrates.
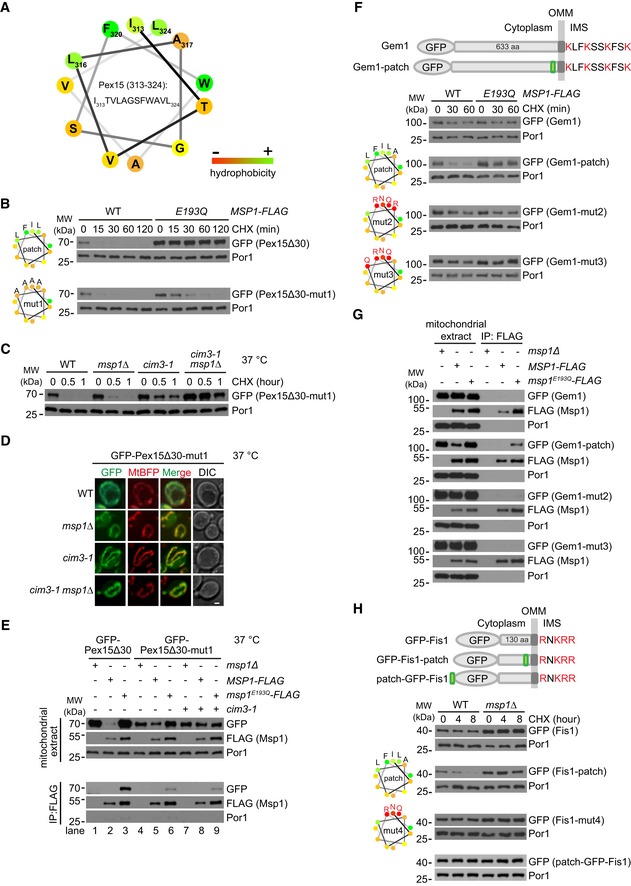
- Helical wheel plot of the hydrophobic patch of Pex15 (amino acids 313–324). Residues are color‐coded according to the hydrophobicity scores.
- Degradation of GFP‐Pex15Δ30 with WT or mutant hydrophobic patch in the indicated strains. Shown on the left are helical wheel plots of WT and mutant (mut1) hydrophobic patches of Pex15. Residue hydrophobicity is color‐coded as in (A). The mut1 form carries I313A, L316A, F320A, and L324A mutations.
- Degradation of GFP‐Pex15Δ30‐mut1 in the indicated mutants. Cim3‐1 is a temperature‐sensitive mutant of Cim3. Cells were cultured at 37°C for 1 h to inactivate Cim3 and then treated with CHX.
- Localization of GFP‐Pex15Δ30‐mut1 to mitochondria in the indicated strains. Cells were cultured at 37°C for 1 h before imaging. Scale bar represents 1 μm.
- Interaction of WT and mutant GFP‐Pex15Δ30 with Msp1‐ and Msp1E193Q‐FLAG. Cells were cultured at 37°C for 1 h before mitochondria purification. Mitochondrial extracts were subject to anti‐FLAG IP and analyzed as in Fig 1D.
- Insertion of Pex15 hydrophobic patch but not its hydrophilic mutants transforms Gem1 into Msp1 substrate. The patch was inserted between V628 and D629 of Gem1 (before Gem1 OTS) as shown in the upper panel. The mut2 form carries I313N, A317R, F320R, and L324Q mutations. The mut3 form carries I313N, L316Q, F320R, L324Q mutations.
- Interaction of GFP‐Gem1 and its insertion mutants with Msp1‐ and Msp1E193Q‐FLAG. Mitochondrial extracts were subject to anti‐FLAG IP and analyzed as in Fig 1D.
- Insertion of Pex15 hydrophobic patch before Fis1 OTS but not at the N‐terminus transforms Fis1 into Msp1 substrate. The insertion was placed between Q125 and K126 of Fis1 (before Fis1 OTS) or at the N‐terminus. The mut4 form carries I313N, F320R, L324Q mutations.
Insertion of the hydrophobic patch into OMM TA proteins transforms them into Msp1 substrates
The OMM TA proteins Gem1 and Fis1 contain positively charged IMS residues but are not degraded by Msp1. A possible reason is that these authentic OMM TA proteins do not expose hydrophobic surfaces in their cytoplasmic domains to be recognized by Msp1. We thus introduced the hydrophobic patch into these proteins. GFP‐Gem1 is stable in WT cells, but inserting the hydrophobic patch right before its OTS caused Gem1 degradation by Msp1 (GFP‐Gem1‐patch in Fig 4F). Patches with reduced hydrophobicity (mut2 and mut3) could not transform Gem1 into Msp1 substrate (Fig 4F). Consistently, Gem1 with WT but not mutant hydrophobic patch interacted with Msp1 (Fig 4G). Similarly, inserting the hydrophobic patch but not its mutant form (mut4) right before Fis1 OTS turned Fis1 into Msp1 substrate (Fig 4H). All the Gem1 and Fis1 mutants correctly localized to mitochondria (Fig EV3E and F). Msp1 N‐domain is close to membrane, which may impose spatial constraints on substrate recognition. Hydrophobic patch inserted at the N‐terminus of GFP (patch‐GFP‐Fis1) could not convert Fis1 into Msp1 substrate (Fig 4H), suggesting the existence of spatial constraints.
Pex15 is mistargeted to mitochondria and removed by Msp1 in pex19Δ cells
We then examined the turnover of full‐length GFP‐Pex15. The reported GET substrates (Pex15, Gos1, and Ubc6) that mislocalize to mitochondria in get mutant cells were identified by overexpression 6, 7, 11. Epichromosomal overexpression of GFP‐Pex15 under the control of TEF1 promoter did partially accumulate on mitochondria in get3Δ, msp1Δ, and get3Δ msp1Δ cells (Fig EV4). However, when chromosomally expressed by its promoter, GFP‐Pex15 did not localize to mitochondria (Fig 5A) but remained peroxisomal (Fig 5B). Pex19 binds the TM segment of peroxisomal TM proteins and targets them to peroxisome 31, 32, 33, 34, 35. GFP‐Pex15 disappeared in pex19Δ cells (Fig 5C), indicating Pex15 is mistargeted and degraded. Mitochondrial accumulation of GFP‐Pex15 was detected in part of pex19Δ msp1Δ cells (~ 32%), and greatly enhanced in pex19Δ get3Δ msp1Δ cells (~ 86%; Fig 5C and D). These results suggest that Pex19 mediates the sorting of endogenous Pex15. Pex15 is likely mistargeted to both the ER and mitochondria in pex19Δ cells, and mostly to mitochondria in pex19Δ get3Δ cells. Msp1 knockout saves mitochondrial Pex15. We then rescued pex19Δ msp1Δ and pex19Δ get3Δ msp1Δ cells with Msp1 N‐domain mutants. The hydrophobic residues are required for removing GFP‐Pex15, but D12 is largely not required (Fig 5D). These results indicate that Msp1 hydrophobic residues and D12 play key and facilitatory roles in substrate recognition, respectively. The facilitatory role of D12 is no longer rate‐limiting when substrates are expressed at low level (chromosomally expression by endogenous promoter) or when Msp1 mutant without D12 is overexpressed (Fig EV1B and C).
Figure EV4. Localization of GFP‐Pex15 overexpressed by the TEF1 promoter in the indicated strains (associated with Fig 5).
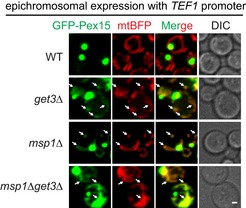
Arrows point to mitochondrial mislocalization of GFP‐Pex15. Scale bar represents 1 μm.
Figure 5. Pex15 is mislocalized to mitochondria and removed by Msp1 in pex19∆ cells.
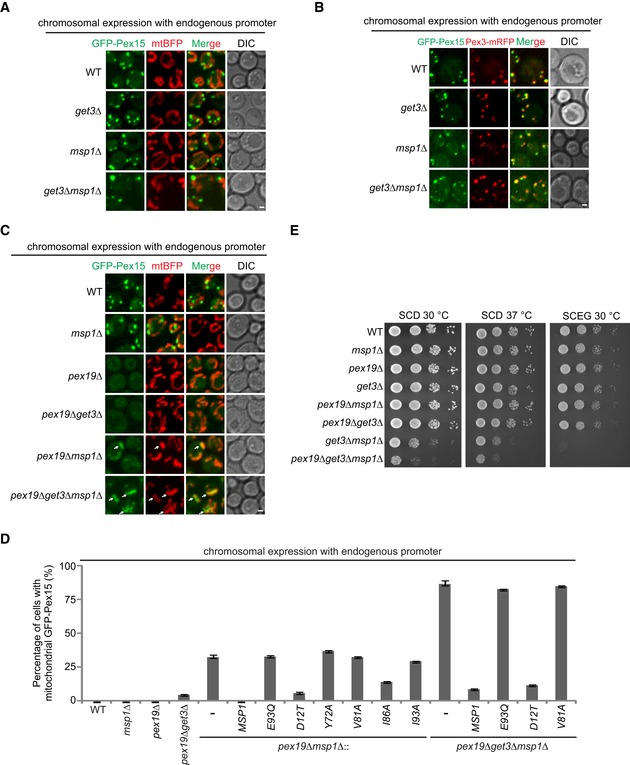
- Chromosomally expressed GFP‐Pex15 driven by endogenous promoter did not mislocalize to mitochondria in GET3 and MSP1 mutant strains.
- GFP‐Pex15 expressed as in (A) localizes to peroxisome in GET and MSP1 mutant strains. Pex3 was chromosomally tagged with mRFP to visualize peroxisomes.
- Localization of GFP‐Pex15 expressed as in (A) in the indicated stains. Arrows point to mislocalized GFP‐Pex15 on mitochondria.
- The percentage of cells with mitochondrial GFP‐Pex15 in the indicated strains. Data values represent means ± SEM from three independent experiments, with at least 100 cells counted in each experiment.
- The indicated strains were grown in glucose media to log phase and then spotted on SCD or SCEG plates in a 10‐fold serial dilution, and then incubated at indicated temperatures for 2–5 days.
Knocking out PEX19 did not affect cell growth in WT, msp1Δ, and get3Δ cells, but aggravated the growth defect of get3Δ msp1Δ cells under fermentable growth condition (SCD; Fig 5E), suggesting the mislocalization of Pex19 substrates to mitochondria contributes to mitochondrial dysfunction.
Identification of Msp1 substrates mistargeted to mitochondria upon dysfunction of the GET pathway
Similar as Pex15, when GFP‐tagged Gos1 and Ubc6 were expressed at near physiological level, they weakly mislocalized to mitochondria in get3Δ and get3Δ msp1Δ cells (< 5%; Fig 6A). We thus performed an unbiased screen of TA proteins to find out GET substrates that can mislocalize to mitochondria and be cleared by Msp1.
Figure 6. Identification of TA proteins that are mislocalized to mitochondria and removed by Msp1 in get3Δ cells.
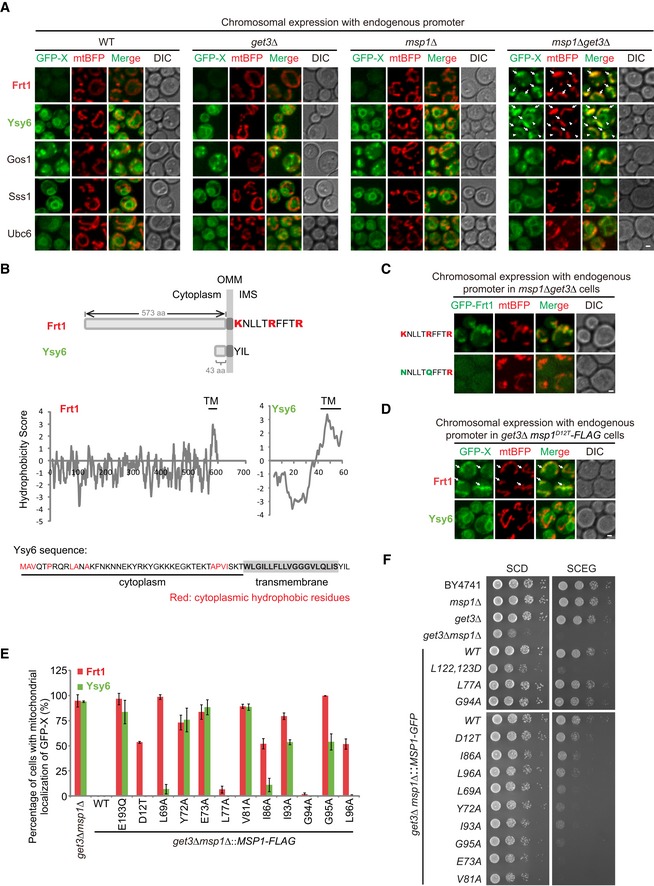
- Localization of GFP‐tagged TA proteins in the indicated strains. Arrows point to the mitochondrial GFP‐Frt1 and GFP‐Ysy6 signals. Arrowheads point to GFP‐Ysy6 signals at the ER.
- Schematic illustration of the IMS tail and sequence hydrophobicity of Frt1 and Ysy6. The amino acid sequence of Ysy6 is shown with the cytoplasmic hydrophobic residues highlighted in red.
- Localization of GFP‐Frt1 with WT or mutant IMS tail in msp1Δ get3Δ cells.
- Localization of GFP‐Frt1 and GFP‐Ysy6 in get3Δ msp1 D12T‐FLAG mutant cells. Arrows point to mitochondrial GFP‐Frt1.
- The percentage of cells with mitochondrial GFP‐Frt1 or GFP‐Ysy6 in get3Δ cells expressing WT or mutant forms of Msp1‐FLAG. Data values represent means ± SEM from three independent experiments, with at least 50 cells counted in each experiment.
- The indicated strains were grown in glucose media to log phase and then spotted on SCD or SCEG plates in a 10‐fold serial dilution, and then incubated at 30°C for 2–5 days.
In addition to Pex15, there are 48 non‐mitochondrial TA proteins encoded by the yeast genome (Appendix Table S1) 36. We epichromosomally overexpressed all of them in get3Δ msp1Δ cells and observed that 34 TA proteins did not localize to mitochondria (Fig EV5A), the expression of six TA proteins was undetectable (Fig EV5B), and eight TA proteins (Frt1, Ysy6, Gos1, Ubc6, Sss1, Far10, Sps2, and Vps64) accumulated on mitochondria (Fig EV5C). Tail charge and TM hydrophobicity as indicated by TM GRAVY score have been suggested as important determinants of TA protein sorting 8, 30, 37. The TA proteins with mitochondrial mislocalization had medium TM GRAVY score (1.7–2.6) and zero or positive tail charge. But these two parameters are not predictive because many TA proteins in this range did not mislocalize to mitochondria (Fig EV5D).
Figure EV5. Imaging analysis of non‐mitochondrial TA proteins in msp1Δ get3Δ cells (associated with Fig 6).
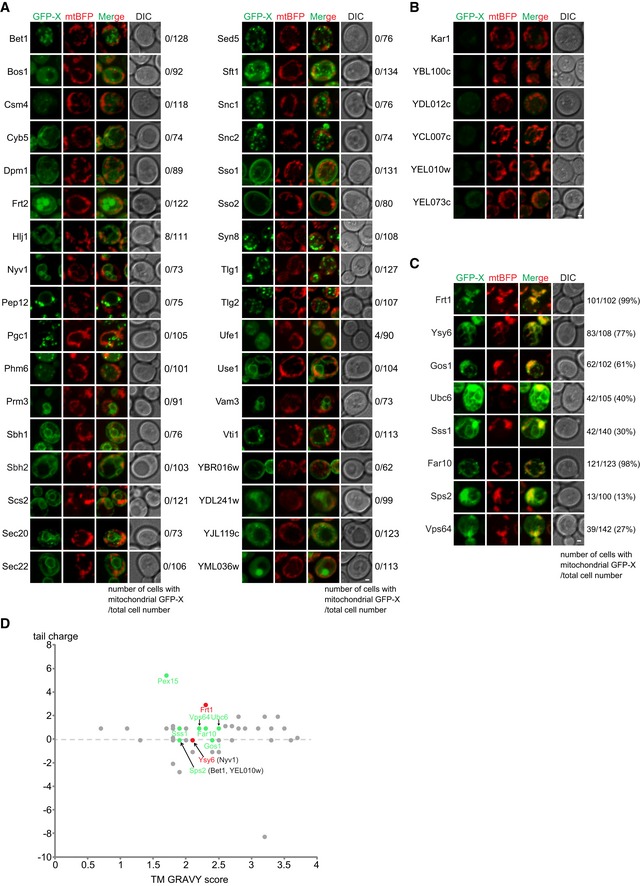
-
A–CGFP‐tagged TA proteins were expressed from a centromeric plasmid under the control of TEF1 promoter in msp1Δ get3Δ cells. (A) TA proteins with non‐mitochondrial localization. (B) TA proteins with undetectable GFP signals. (C) TA proteins with mitochondrial localization. Z projections and DIC images are shown. Scale bars represent 1 μm.
-
DScatter plots depicting tail charge (y‐axis) and TM GRAVY (x‐axis) for each non‐mitochondrial TA protein in yeast. Their mitochondrial mislocalization in msp1Δget3Δ cells is color‐encoded: Gray means no mitochondrial mislocalization; green indicates mitochondrial mislocalization when epichromosomally overexpressed; red indicates mitochondrial mislocalization under both over‐ and physiological expression levels. Proteins in the bracelets are non‐mislocalized proteins having overlapping scores with mislocalized ones.
We continued to analyze the eight TA proteins by chromosomal expression with their endogenous promoters. As shown in Fig 6A, GFP‐Frt1 was undetectable in WT, get3Δ, and msp1Δ cells, but strongly accumulated on mitochondria (pointed by arrows) in about 95% of get3Δ msp1Δ cells. GFP‐Ysy6 localized to the ER in WT, get3Δ, and msp1Δ cells, but dually localized to the ER (pointed by arrowheads) and mitochondria (pointed by arrows) in about 95% of get3Δ msp1Δ cells, indicating that in get3Δ cells, GFP‐Ysy6 is partially mistargeted to mitochondria and removed by Msp1. GFP‐tagged Gos1 and Ubc6 exhibited very weak mitochondrial localization in a small number of cells (< 5%). GFP‐Sss1 did not localize to mitochondria in all the cells examined. GFP‐tagged Far10, Sps2, and Vps64 were undetectable. In summary, we identified two TA proteins Frt1 and Ysy6 that prominently mislocalize to mitochondria in get3Δ cells and get cleared by Msp1. Most TA proteins did not mislocalize to mitochondria in get3Δ cells probably because there are additional complexes to target TA proteins to the ER, such as the SND 38 and the EMC 39 complexes.
Evaluation of Msp1 N‐domain critical residues in the removal of GET pathway substrates
Frt1 IMS tail contains three positively charged residues but Ysy6 IMS tail has none (Fig 6B). Like Pex15Δ30, the positively charged IMS tail is critical for the mitochondrial mislocalization of Frt1 (Fig 6C). If Msp1 D12 residue only contributes to interaction with substrates and has no additional roles for Msp1 activity, then the D12T mutation should result in the mitochondrial accumulation of GFP‐Frt1 but not GFP‐Ysy6 in get3Δ cells. Indeed, in get3Δ msp1 D12T cells, about 50% of cells showed mitochondrial accumulation of GFP‐Frt1, and mitochondrial signals of GFP‐Ysy6 were not detected (Fig 6D and E).
Hydrophobicity analysis of Frt1 and Ysy6 cytoplasmic domains did not reveal a hydrophobic patch near their TM segments (Fig 6B). Ysy6 has a short hydrophilic cytoplasmic domain with distributed hydrophobic residues. We then examined whether Msp1 N‐domain critical residues, especially hydrophobic ones, are still required for substrate clearance (Fig 6E). L77 and G94 mutations had no or very minor effects on substrate removal. L69, I86, and L96 had a more prominent role in GFP‐Frt1 removal. Y72, E73, V81, I93, and G95 were generally required for the removal of both substrates. These results indicate that although a localized hydrophobic patch was not found in Frt1 and Ysy6, Msp1 may still recognize them through hydrophobic interactions in the cytoplasm. Consistent with imaging results, under respiratory growth conditions (SCEG), Msp1 L77A and G94A mutations had no effects on the growth of get3Δ cells, D12T, I86A, and L96A mutations partially compromised the growth of get3Δ cells, and Y72A, E73A, V81A, I93A, and G95A mutations strongly inhibited the growth of get3Δ cells (Fig 6F).
Discussion
Our extensive characterizations of Msp1 and its substrates demonstrate that Msp1 recognizes exposed hydrophobic surfaces of mistargeted TA proteins. An important consequence of protein mislocalization is the loss of their binding partners. This is true for Pex15, which complexes with Pex3 at peroxisome 17, for Frt1, which interacts with Frt2 at the ER 40, and may also apply to the poorly characterized protein Ysy6. Losing binding partners may cause structural deformation and instability to expose hydrophobic surfaces. The crystal structure of Msp1 monomer 21 shows that the critical hydrophobic residues of N‐domain for substrate binding are packed against the large AAA‐domain to form a closed conformation (Fig 1E). N‐domain may exist in a dynamic equilibrium of closed and open conformations to allow Msp1 to sample substrate surfaces and avoid prolonged exposure of its own hydrophobic surfaces. When hydrophobic surfaces of substrates are detected, Msp1 may undergo conformational rearrangements to assemble active hexamers capable of ATP hydrolysis and substrate dislocation (Fig 7).
Figure 7. A working model for Msp1‐mediated clearance of mislocalized TA proteins.
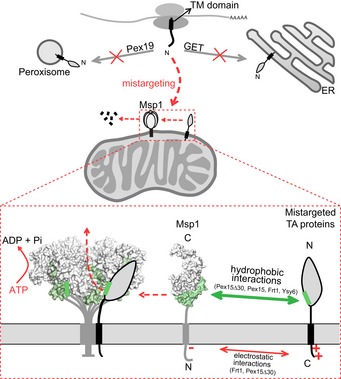
Newly expressed TA proteins are targeted to peroxisome and the ER by the Pex19 and GET pathways, respectively. Disrupting these pathways mistargets TA proteins to mitochondria. Mistargeted TA proteins expose hydrophobic surfaces in the cytoplasm and are recognized by Msp1 N‐domain hydrophobic residues. The interaction is further facilitated by IMS electrostatic interactions when substrates contain positively charged IMS tails. Substrate‐engaged Msp1 assembles into hexamer to dislocate substrates through ATP hydrolysis.
An unexpected mechanism of substrate recognition by Msp1 is the involvement of a second layer of interaction with substrates Pex15Δ30 and Frt1 at the IMS side, very likely through electrostatic interactions between positively charged residues of substrates and the negatively charged D12 residue of Msp1 (Fig 7). Positively charged residues in IMS tail are critical for the targeting of mitochondrial and peroxisomal TA proteins 8, 29, and also required for mistargeting of Pex15Δ30 and Frt1 to mitochondria (Figs EV3A and 6C). Recognizing this property enables Msp1 to survey a large number of mitochondrial and peroxisomal TA proteins. When dual interactions in the IMS and cytoplasmic domains occur, the affinity between Msp1 and substrates will be substantially enhanced to enable efficient substrate removal and thus enable Msp1 to safeguard mitochondrial function at its physiological expression level, as suggested by the suboptimal growth of get3Δ msp1 D12T cells (Fig 6F).
These results together highlight a crucial role of Msp1 N‐domain in substrate recognition, a feature of the meiotic clade of AAA‐ATPases 20. Considering that Msp1 recognizes misfolded domains with exposed hydrophobic surfaces, it may have a more general role in the quality control of OMM and peroxisomal proteins beyond mistargeted TA proteins. Consistent with this, Msp1 was recently found to remove mitochondrial precursor proteins clogged in the TOM import channel 22.
Materials and Methods
Culture media
Media used in this study included standard YP and synthetic minimal media supplemented with 2% glucose (D), 2% lactate(L), 2% galactose (Gal), or 3% ethanol and glycerol (EG). The synthetic media contained either complete supplements or the appropriate amino acid dropout mixture. ClonNAT (100 μg/l, WERNER BioAgents) was added to media for plasmid maintenance as needed. Yeast strains were grown at 30°C if not otherwise indicated.
Yeast strains and plasmids
The yeast strains used in this study are listed in Appendix Table S2. Strain transformation was performed using the lithium acetate method, selected on appropriate media, and confirmed by PCR and gene expression as needed 41, 42, 43. The strains expressing Msp1 variants from the endogenous loci were generated by PCR‐based homologous recombination to replace the original selection cassette in msp1Δ strain with cassettes containing sequences encoding WT or mutant forms of MSP1 and a different selection marker. The msp1Δget3Δ strains epichromosomally expressing Msp1 variants were generated using the systematic genetic analysis (SGA) technique 44: (i) msp1Δ strain (in BY4741 background) was mated with get3Δ strain (in Y7092 background). (ii) The resulting diploid strain was transformed with an empty vector or a centromeric plasmid expressing WT or mutant forms of Msp1 under the control of its endogenous promoter. (iii) The resulting strain was cultured in sporulation media and then subject to a series of selection processes for the isolation of the desired haploid strains.
The plasmids used in this study (Appendix Table S3) were generated using the Gibson‐based assembly method 45. Mutations were created by Quick Change site‐directed mutagenesis (Stratagene).
Antibodies and chemicals
The following antibodies were from Sigma: G6PDH produced in rabbit (A9521), HA‐peroxidase (H6533), and FLAG M2 produced in mouse (F1804). The antibody for Por1 produced in mouse (459500) was from Life Technologies. The antibody for GFP produced in mouse (11814460001) was from Roche. A synthetic peptide (amino acid residues CTLDAYERTILSSIV of Msp1) was used to generate Msp1 antibody.
Yeast extract, peptone, and yeast nitrogen base without amino acids were from BD Biosciences. Yeast complete supplement mixture was from MP Biomedicals. Yeast amino acid dropout supplements were from Clontech. Cycloheximide (used at 50 μg/ml) was from Amresco. Benzoyl‐phenylalanine (BPA) was from Bachem. Other chemicals or reagents were from Sigma if not otherwise indicated.
Yeast whole cell extract preparation and thermostability measurement
Cell pellets were resuspended in 300 μl yeast lysis buffer (50 mM NaCl, 50 mM NaF, 100 mM Tris–HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, 1% Triton X‐100, 10% glycerol, 14 mM 2‐mercaptoethanol, 2 mM PMSF, 5 μM pepstatin A, 10 μM Leupeptin). After adding ~ 80 μl of glass beads, cells were lysed via three rounds of bead‐beating (40 s beating followed by 1 min of cooling on ice).
Cell extracts were divided into equal volume (50 μl) and heated at the indicated temperatures for 3 min on a PCR machine followed by cooling down to 4°C on ice. Heated cell lysates were centrifuged at 18,407 g for 10 min at 4°C to collect supernatants. Equal volume of supernatants was analyzed by Western blot. Band intensity was measured by ImageJ software.
Purification of recombinant proteins and limited trypsin digestion
Msp1 (33–345) was amplified by PCR from plasmid and subcloned into a pET28a derivative encoding an N‐terminal 6xHis‐Smt3 tag. Site‐specific mutagenesis was performed by Quick Change PCR. All plasmids were confirmed by DNA sequencing.
Plasmids were transformed into Rosetta (DE3). Cells were grown at 37°C until an OD600 of 0.6–1.0. Cultures were induced with 1 mM IPTG (Sigma) and grown at 18°C for 16–18 h. Cells were harvested by centrifugation, and resuspended in lysis buffer (20 mM Tris–HCl pH 8.0, 500 mM NaCl, 1 mM PMSF, 10 mM imidazole), and lysed by sonication. The supernatant was isolated by centrifugation for 1 h at 4°C at 38,759 g. The supernatant was filtered with 0.22‐μm filter and purified by Ni‐NAT column. Ni‐NTA resin was washed with 10 column volumes (CV) of lysis buffer and 10 CV of wash buffer (lysis buffer with 30 mM imidazole), then eluted with lysis buffer containing 250 mM imidazole. After Ni‐NTA purification, protein concentrations were determined by Bradford assay. Recombinant proteins were treated with 1/100 SUMO Protease 1 (Ulp1) for 16 h at 4°C to cleave off the 6xHis‐Smt3 tag, and reloaded to Ni‐NAT column to remove the tag. Recombinant proteins were digested with 1/500 (by concentration) trypsin at 37°C for the indicated times and then subjected to SDS–PAGE and Coomassie Blue staining.
Isolation of the mitochondria‐enriched fraction and immunoprecipitation
Mitochondria were isolated following a previously described method 46, with some modifications. Briefly, about 200 ml of cells grown in synthetic glucose media to late log phase (OD ~ 3) was collected. Cells were washed once with water and incubated in TD buffer [10 mM DTT, 100 mM Tris‐SO4 (pH 9.4)] for 15 min at 30°C. Cells were then washed once with SP buffer [1.2 M sorbitol, 20 mM potassium phosphate (pH 7.4)] and treated with Zymolyase 20T/100T (MP Biomedicals) for 40 min at 30°C to generate spheroplasts. After two times of washes with SP buffer, the spheroplasts were resuspended in SHE buffer [0.6 M sorbitol, 20 mM HEPES‐KOH (pH 7.4), 1 mM EGTA (pH 8), 2 mM MgCl2] supplemented with protease inhibitors and homogenized by a French press (EmulsiFlex‐C3, AVESTIN Inc.) at pressures in the range of 1,000 to 1,500 psi. The mitochondria‐enriched fraction was obtained by differential centrifugation, flash frozen by liquid nitrogen, and stored at −80°C until use.
Crude mitochondria were solubilized with 1% digitonin buffer [50 mM HEPES‐KOH (pH 7.4), 50 mM KOAc, 2 mM Mg(OAc)2, 1 mM CaCl2, 200 mM sorbitol, 1 mM NaF, 1% (w/v) digitonin] supplemented with 1 mM DTT, 2 mM ATP, and protease inhibitors for 45–60 min at 4°C. The lysates were cleared by centrifugation at 17,000 g for 10 min at 4°C. The supernatant was then mixed with anti‐FLAG agarose beads (Sigma) and incubated at 4°C for 5–6 h. The agarose beads were then washed five times with 0.1% digitonin buffer and eluted overnight with FLAG peptide (ChinaPeptides Co. Ltd.) at 4°C.
Site‐specific in vivo photo‐crosslinking and immunoprecipitation
Experiments were performed following a previously described method 23 with specific modifications as follows. For GFP‐3xHA‐Pex15Δ30‐BPA, strains in W303 background were transformed with two 2‐μ plasmids: One expresses GFP‐3xHA‐Pex15Δ30 with an amber (TAG) codon at a specific site under the control of the repressible GAL1 promoter; and the other one expresses an amber suppressor tRNA and a modified aminoacyl‐tRNA synthetase that specifically charges the amber suppressor tRNA with the photo‐reactive unnatural amino acid BPA. For Msp1‐BPA, a gene cassette expressing Msp1 with TAG codon under the control of GAL1 promoter was integrated into his3 locus. The resulting strain was transformed with the same plasmid to express the tRNA and tRNA ligase. Strains were grown in selective glucose media to late log phase and then switched to BPA (0.2 mM)‐containing selective media supplemented with 2% lactate and 0.05% galactose for Msp1‐BPA or 2% galactose for Pex15Δ30‐BPA for 16–18 h. About 80 OD600 units of cells in late log phase were subject to UV irradiation for 15 min. To prepare whole cell extracts, cells were resuspended in 0.1 M NaOH and incubated at room temperature for 5–10 min. Cell pellets were then resuspended in SDS buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 2% (w/v) SDS, 4% (v/v) BME] and boiled at 98°C for 10 min. The lysates were cleared by centrifugation at 17,000 g for 10 min. The supernatant was then diluted with 15 volumes of Triton buffer (50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% (v/v) Triton), mixed with anti‐FLAG agarose beads, and incubated at 4°C for 5–6 h. The agarose beads were then washed four times with Triton buffer and eluted overnight with FLAG peptide at 4°C.
Microscopy
Yeast cells were grown in synthetic glucose media to log phase, concentrated, and immobilized on microscope slides. Fluorescent images were captured at 25°C using a 100× objective (CFI Plan Apochromat Lambda; NA 1.45; Nikon) with immersion oil (type NF) on an inverted fluorescence microscope (Eclipse Ti‐E; Nikon) with a spinning‐disk confocal scanner unit (UltraView; PerkinElmer) with 488 [emission filter 525 (W50)] and 405 [emission filter 445 (W60) and 615 (W70)] lasers. Z‐stack images with 0.5 μm increments were acquired with Volocity software (PerkinElmer) and processed with Volocity and ImageJ software.
Sequence, structure, and bioinformatics analysis
Sequence alignment was performed using ClustalW and processed by GeneDoc software. The structure of monomeric or hexameric form of Msp1 was analyzed and processed using PyMOL. The GRAVY (Grand Average of Hydropathicity) values of TM domains and the hydrophobicity plot of Pex15Δ30 were calculated using the ProtParam and ProtScale server, respectively, from ExPASy. The charges of TM domain flanking sequences were calculated using the Protein Calculator v3.4 (http://protcalc.sourceforge.net). The helical wheel plots were generated using the helical wheel projections server (http://rzlab.ucr.edu/scripts/wheel/wheel.cgi). Protein topology model was generated using the Protter server (http://wlab.ethz.ch/protter/#;).
Author contributions
HJ and XW conceived the project, supervised the study, and wrote the manuscript. LL generated substrates and their mutations and analyzed their localization and turnover. LL performed thermostability analysis of Msp1 and its mutants and generated recombinant proteins and performed limited trypsin digestion. JZ generated Msp1 mutants, analyzed their effects on substrate degradation, and performed immunoprecipitation and blue‐native gel experiments. JZ and LL performed the in vivo site‐specific photo‐crosslinking experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Dr. Charlie Boone (University of Toronto, Canada) for providing the Y7092 strain. The research is supported by National Natural Science Foundation of China (Grant No. 31871346), the National Basic Research Program of China 973 (program 2012CB837503), and Beijing Municipal Science and Technology Commission. X.W. is supported by Beijing Postdoctoral Research Foundation and China Postdoctoral Science Foundation.
EMBO Reports (2019) 20: e46989
Contributor Information
Xi Wu, Email: xi.wu@beigene.com.
Hui Jiang, Email: jianghui@nibs.ac.cn.
References
- 1. Gerdes F, Tatsuta T, Langer T (2012) Mitochondrial AAA proteases–towards a molecular understanding of membrane‐bound proteolytic machines. Biochim Biophys Acta 1823: 49–55 [DOI] [PubMed] [Google Scholar]
- 2. Baker MJ, Tatsuta T, Langer T (2011) Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol 3: a007559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karbowski M, Youle RJ (2011) Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 23: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bragoszewski P, Turek M, Chacinska A (2017) Control of mitochondrial biogenesis and function by the ubiquitin‐proteasome system. Open Biol 7: 170007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu X, Li L, Jiang H (2016) Doa1 targets ubiquitinated substrates for mitochondria‐associated degradation. J Cell Biol 213: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen YC, Umanah GK, Dephoure N, Andrabi SA, Gygi SP, Dawson TM, Dawson VL, Rutter J (2014) Msp1/ATAD1 maintains mitochondrial function by facilitating the degradation of mislocalized tail‐anchored proteins. EMBO J 33: 1548–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Okreglak V, Walter P (2014) The conserved AAA‐ATPase Msp1 confers organelle specificity to tail‐anchored proteins. Proc Natl Acad Sci USA 111: 8019–8024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borgese N, Colombo S, Pedrazzini E (2003) The tale of tail‐anchored proteins: coming from the cytosol and looking for a membrane. J Cell Biol 161: 1013–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hegde RS, Keenan RJ (2011) Tail‐anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol 12: 787–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Denic V, Dotsch V, Sinning I (2013) Endoplasmic reticulum targeting and insertion of tail‐anchored membrane proteins by the GET pathway. Cold Spring Harb Perspect Biol 5: a013334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, Weissman JS (2008) The GET complex mediates insertion of tail‐anchored proteins into the ER membrane. Cell 134: 634–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mozdy AD, McCaffery JM, Shaw JM (2000) Dnm1p GTPase‐mediated mitochondrial fission is a multi‐step process requiring the novel integral membrane component Fis1p. J Cell Biol 151: 367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kornmann B, Osman C, Walter P (2011) The conserved GTPase Gem1 regulates endoplasmic reticulum‐mitochondria connections. Proc Natl Acad Sci USA 108: 14151–14156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wiedemann N, Pfanner N (2017) Mitochondrial machineries for protein import and assembly. Annu Rev Biochem 86: 685–714 [DOI] [PubMed] [Google Scholar]
- 15. Kemper C, Habib SJ, Engl G, Heckmeyer P, Dimmer KS, Rapaport D (2008) Integration of tail‐anchored proteins into the mitochondrial outer membrane does not require any known import components. J Cell Sci 121: 1990–1998 [DOI] [PubMed] [Google Scholar]
- 16. Setoguchi K, Otera H, Mihara K (2006) Cytosolic factor‐ and TOM‐independent import of C‐tail‐anchored mitochondrial outer membrane proteins. EMBO J 25: 5635–5647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weir NR, Kamber RA, Martenson JS, Denic V (2017) The AAA protein Msp1 mediates clearance of excess tail‐anchored proteins from the peroxisomal membrane. Elife 6: e28507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Piard J, Umanah GKE, Harms FL, Abalde‐Atristain L, Amram D, Chang M, Chen R, Alawi M, Salpietro V, Rees MI et al (2018) A homozygous ATAD1 mutation impairs postsynaptic AMPA receptor trafficking and causes a lethal encephalopathy. Brain 141: 651–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frickey T, Lupas AN (2004) Phylogenetic analysis of AAA proteins. J Struct Biol 146: 2–10 [DOI] [PubMed] [Google Scholar]
- 20. Monroe N, Hill CP (2016) Meiotic clade AAA ATPases: protein polymer disassembly machines. J Mol Biol 428: 1897–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wohlever ML, Mateja A, McGilvray PT, Day KJ, Keenan RJ (2017) Msp1 is a membrane protein dislocase for tail‐anchored proteins. Mol Cell 67: 194–202 e196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weidberg H, Amon A (2018) MitoCPR‐A surveillance pathway that protects mitochondria in response to protein import stress. Science 360: eaan4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shiota T, Nishikawa S, Endo T (2013) Analyses of protein‐protein interactions by in vivo photocrosslinking in budding yeast. Methods Mol Biol 1033: 207–217 [DOI] [PubMed] [Google Scholar]
- 24. Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG (2003) An expanded eukaryotic genetic code. Science 301: 964–967 [DOI] [PubMed] [Google Scholar]
- 25. Shiota T, Imai K, Qiu J, Hewitt VL, Tan K, Shen HH, Sakiyama N, Fukasawa Y, Hayat S, Kamiya M et al (2015) Molecular architecture of the active mitochondrial protein gate. Science 349: 1544–1548 [DOI] [PubMed] [Google Scholar]
- 26. Shiota T, Mabuchi H, Tanaka‐Yamano S, Yamano K, Endo T (2011) In vivo protein‐interaction mapping of a mitochondrial translocator protein Tom22 at work. Proc Natl Acad Sci USA 108: 15179–15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu X, Li L, Jiang H (2018) Mitochondrial inner‐membrane protease Yme1 degrades outer‐membrane proteins Tom22 and Om45. J Cell Biol 217: 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin A, Baker TA, Sauer RT (2008) Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor‐dependent recognition of ssrA‐tagged substrates. Mol Cell 29: 441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rapaport D (2003) Finding the right organelle. Targeting signals in mitochondrial outer‐membrane proteins. EMBO Rep 4: 948–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Costello JL, Castro IG, Camões F, Schrader TA, McNeall D, Yang J, Giannopoulou EA, Gomes S, Pogenberg V, Bonekamp NA et al (2017) Predicting the targeting of tail‐anchored proteins to subcellular compartments in mammalian cells. J Cell Sci 130: 1675–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rottensteiner H, Kramer A, Lorenzen S, Stein K, Landgraf C, Volkmer‐Engert R, Erdmann R (2004) Peroxisomal membrane proteins contain common Pex19p‐binding sites that are an integral part of their targeting signals. Mol Biol Cell 15: 3406–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hettema EH, Girzalsky W, van Den Berg M, Erdmann R, Distel B (2000) Saccharomyces cerevisiae pex3p and pex19p are required for proper localization and stability of peroxisomal membrane proteins. EMBO J 19: 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones JM, Morrell JC, Gould SJ (2004) PEX19 is a predominantly cytosolic chaperone and import receptor for class 1 peroxisomal membrane proteins. J Cell Biol 164: 57–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Halbach A, Landgraf C, Lorenzen S, Rosenkranz K, Volkmer‐Engert R, Erdmann R, Rottensteiner H (2006) Targeting of the tail‐anchored peroxisomal membrane proteins PEX26 and PEX15 occurs through C‐terminal PEX19‐binding sites. J Cell Sci 119: 2508–2517 [DOI] [PubMed] [Google Scholar]
- 35. Yagita Y, Hiromasa T, Fujiki Y (2013) Tail‐anchored PEX26 targets peroxisomes via a PEX19‐dependent and TRC40‐independent class I pathway. J Cell Biol 200: 651–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burri L, Lithgow T (2004) A complete set of SNAREs in yeast. Traffic 5: 45–52 [DOI] [PubMed] [Google Scholar]
- 37. Rao M, Okreglak V, Chio US, Cho H, Walter P, Shan SO (2016) Multiple selection filters ensure accurate tail‐anchored membrane protein targeting. Elife 5: e21301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aviram N, Ast T, Costa EA, Arakel EC, Chuartzman SG, Jan CH, Haßdenteufel S, Dudek J, Jung M, Schorr S et al (2016) The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature 540: 134–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guna A, Volkmar N, Christianson JC, Hegde RS (2018) The ER membrane protein complex is a transmembrane domain insertase. Science 359: 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heath VL, Shaw SL, Roy S, Cyert MS (2004) Hph1p and Hph2p, novel components of calcineurin‐mediated stress responses in Saccharomyces cerevisiae . Eukaryot Cell 3: 695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- 42. Voth WP, Richards JD, Shaw JM, Stillman DJ (2001) Yeast vectors for integration at the HO locus. Nucleic Acids Res 29: E59–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno‐Borchart A, Doenges G, Schwob E, Schiebel E et al (2004) A versatile toolbox for PCR‐based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962 [DOI] [PubMed] [Google Scholar]
- 44. Tong AH, Boone C (2006) Synthetic genetic array analysis in Saccharomyces cerevisiae . Methods Mol Biol 313: 171–192 [DOI] [PubMed] [Google Scholar]
- 45. Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- 46. Diekert K, de Kroon AI, Kispal G, Lill R (2001) Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae . Methods Cell Biol 65: 37–51 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File