

Abstract
Nephron formation continues throughout kidney morphogenesis in both mice and humans. Lineage tracing studies in mice identified a self‐renewing Six2‐expressing nephron progenitor population able to give rise to the full complement of nephrons throughout kidney morphogenesis. To investigate the origin of nephrons within human pluripotent stem cell‐derived kidney organoids, we performed a similar fate‐mapping analysis of the SIX2‐expressing lineage in induced pluripotent stem cell (iPSC)‐derived kidney organoids to explore the feasibility of investigating lineage relationships in differentiating iPSCs in vitro. Using CRISPR/Cas9 gene‐edited lineage reporter lines, we show that SIX2‐expressing cells give rise to nephron epithelial cell types but not to presumptive ureteric epithelium. The use of an inducible (CreERT2) line revealed a declining capacity for SIX2+ cells to contribute to nephron formation over time, but retention of nephron‐forming capacity if provided an exogenous WNT signal. Hence, while human iPSC‐derived kidney tissue appears to maintain lineage relationships previously identified in developing mouse kidney, unlike the developing kidney in vivo, kidney organoids lack a nephron progenitor niche capable of both self‐renewal and ongoing nephrogenesis.
Keywords: CRSIPR/Cas9, fate mapping, kidney organoid, lineage tracing, pluripotent stem cell
Subject Categories: Development & Differentiation, Stem Cells
Introduction
Our understanding of kidney morphogenesis is predominantly based on studies performed in model organisms. Gene‐knockout and fate‐mapping studies, performed largely in mice, have uncovered many of the fundamental molecular processes underlying kidney development, homeostasis, and disease 1. While inferences can be made using a well‐characterized mammalian model, our capacity to validate the true relevance of lineage relationships or key genetic pathways in human development is hampered by the scarcity of human fetal tissue for experimental purposes. Recent analyses of human fetal kidney have provided important insights and highlighted several differences between humans and mice at both the immunohistochemical and transcriptional levels 2, 3, 4. However, the examination of humans fetal tissue is ethically constrained, provides only snapshots at a fixed developmental time‐point, and does not represent an ideal platform for evaluating whether differences between human and model organisms convey functional relevance.
The capacity to create a model of the developing human kidney in vitro may provide a unique opportunity to better understand nephrogenesis at the molecular level in a human context. This will depend, however, on the accuracy with which these models of human kidney organogenesis recapitulate normal development. Several protocols have now been described for the directed differentiation of human pluripotent stem cells (hPSCs) to kidney organoids 5. We have established a protocol that generates complex multicellular kidney organoids containing patterning and segmenting nephrons and endothelial, perivascular, and stromal cells 6, 7. After day 25 of differentiation, a single organoid contains up to 100 nephrons, each beginning to show functional maturation with podocyte foot process formation and albumin uptake in proximal tubules. Moreover, kidney organoids exhibit a transcriptional profile that is remarkably similar to first‐trimester human fetal kidney 7. Combined with an ability to generate developing kidney tissue in vitro, improvements in the speed and accuracy with which it is possible to edit the genome of hPSCs using CRISPR/Cas9 technology 8 provide a unique opportunity to interrogate the molecular and cellular basis of morphogenesis within kidney organoids to better understand these model systems.
During kidney morphogenesis in the mouse, new nephrons form throughout development from a mesenchymal population located at the periphery of the developing kidney 9, 10. Marked by expression of Six2 and Cited1, these cells exist in close association with the tips of the branching ureteric epithelium. Nephron formation involves a mesenchymal‐to‐epithelial transition to form a renal vesicle with this process being accompanied by downregulation of Six2 11, 12. In a seminal fate‐mapping study performed in mice, Kobayashi et al demonstrated that Six2‐expressing cells represent a multipotent self‐renewing nephron progenitor population that, after undergoing mesenchymal‐to‐epithelial transition, gives rise to all cells of the murine nephron, but not to those within the branching ureteric epithelium 9, 10, which is instead derived from a distinct ureteric progenitor population 13.
In this study, we chose to perform a similar fate‐mapping analysis of the SIX2‐expressing lineage in iPSC‐derived kidney organoids. Using iPSCs harboring Cre recombinase within the endogenous SIX2 locus, in addition to a ubiquitously expressed loxP‐flanked fluorescence cassette, we demonstrate that SIX2‐expressing cells can indeed contribute to nephron formation. While SIX2‐derived cells formed proximal nephron segments, they were absent from the GATA3+CDH1+ epithelium, consistent with a ureteric epithelium identity for the latter. Importantly, the use of an inducible CreERT2 lineage reporter showed a loss of contribution to new nephrons across time within organoids, highlighting an absence of a nephrogenic zone in such models. Despite this, SIX2+ cells could contribute to new nephrons late in organoid culture if provided an exogenous WNT signal, suggesting that they retain a nephron progenitor identity across time even in the absence of a ureteric tip. These findings not only improve our understanding of nephrogenesis within kidney organoids, but provide a proof of concept that organoid and gene‐editing technologies can be combined to interrogate and dissect human lineage relationships in vitro.
Results
SIX2 expression persists throughout kidney organoid differentiation
We first generated a SIX2 fluorescent reporter iPSC line to evaluate and monitor SIX2‐expressing cells within differentiating kidney organoids in real time. Here, CRISPR/Cas9 was used to knock‐in EGFP at the 3′ end of the SIX2 coding region, linked by the T2A self‐cleaving peptide (Fig 1A). Several clonally derived iPSC lines with either heterozygous or homozygous insertion of the EGFP reporter were established using a previously described method that combines reprogramming and gene editing together in a single step 8, 14. Both heterozygous (SIX2EGFP/+) and homozygous (SIX2EGFP/EGFP) clones, distinguished by PCR analysis (Fig EV1A), were used in subsequent differentiation experiments. Kidney organoids were generated using our previously described protocol 6 (Fig 1B) with two modifications: (i) TeSR‐E6 was used instead of APEL as the base differentiation medium, and (ii) a 3D bioprinter was used to generate day 7 aggregates via extrusion bioprinting onto Transwell filters instead of manual transfer with a handheld pipette. This modified protocol enables the generation of large numbers of highly reproducible organoids that are equivalent at the level of morphology, component cell types, and gene expression to those previously reported via manual generation [preprint: 15]. Reporter gene expression in SIX2EGFP organoids was monitored routinely by flow cytometry and fluorescent microscopy and was first detected at approximately days 8–10 of differentiation, consistent with previous reports 13, 16, 17. Notably, reporter gene expression was steadily maintained after this time‐point, until the cessation of our differentiation experiments at day 25 (Fig 1C and D). Co‐localization of EGFP and SIX2 was confirmed by immunofluorescence (Fig 1E), with EGFP‐ and SIX2‐expressing cells restricted largely to the interstitial/mesenchymal compartment (Fig EV1B), as anticipated. Co‐localization of EGFP and SIX2 was also confirmed by RT–PCR analysis of sorted EGFP‐expressing and non‐expressing fractions (Fig EV1C). Organoids derived from either SIX2EGFP/+ or SIX2EGFP/EGFP iPSCs exhibited highly similar dynamics with respect to reporter gene expression and nephron formation, although reporter gene intensity was greater in SIX2EGFP/EGFP kidney organoids (Fig EV1D). EGFP expression was also dependent on CHIR99021 concentration and duration during the first stage of differentiation (Fig EV1E), consistent with previous studies showing that increased WNT signaling during hPSC differentiation induces a more posterior intermediate mesoderm 7. Organoids generated from cultures treated with < 4 μM CHIR99021 failed to induce SIX2 expression or any recognizable epithelial structures, whereas differentiations performed with 8 μM CHIR for ≥ 4 days contained the highest fraction of SIX2‐expressing cells (> 50%). Cultures treated with 6–8 μM CHIR99021 for 4 days formed organoids with the most balanced profile with respect to relative abundance of NEPHRIN+ podocytes, LTL+ proximal tubules, ECAD+ distal tubule structures, and putative GATA3+/CDH1+ collecting duct epithelium, as determined by whole‐mount immunostaining (Fig 1F). This condition was used for all subsequent differentiations.
Figure 1. Analysis of SIX2‐expressing cells during kidney organoid development.
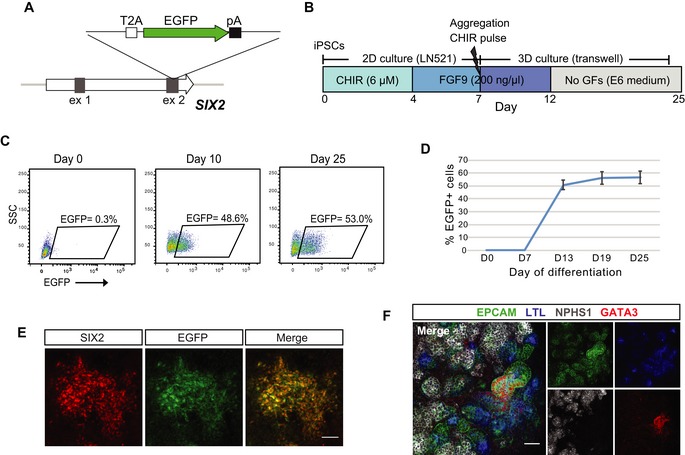
- Schematic diagram of the targeting strategy for generation of SIX2 reporter iPSCs. The EGFP gene was inserted just upstream of the SIX2 stop codon, linked via the self‐cleaving T2A sequence.
- Outline of the kidney differentiation protocol used throughout this study.
- Flow cytometry analysis of kidney organoids derived from SIX2EGFP/EGFP iPSCs.
- Flow cytometry analysis of SIX2EGFP/EGFP kidney organoids shows EGFP/SIX2 expression emerges after day 7 of differentiation and persists thereafter. Data represent mean ± SD, n = 3.
- Immunostaining of a day 12 SIX2EGFP/EGFP kidney organoid confirming co‐localization of SIX2 and EGFP.
- Immunofluorescence of SIX2EGFP/EGFP organoid demonstrating expression and correct localization of nephron segment‐specific markers for proximal tubules (EPCAM+/LTL+; blue/green), distal tubules (EPCAM+; green), collecting duct (EPCAM+/GATA3+; green/red), and glomeruli (NPHS1+; gray).
Figure EV1. Characterization of SIX2EGFP iPSCs and SIX2EGFP kidney organoids.

- PCR analysis of SIX2 reporter iPSCs to confirm correct insertion, or heterozygous versus homozygous insertion of the EGFP reporter. ODNs used for each analysis are marked.
- Immunofluorescence of SIX2EGFP/EGFP organoid demonstrating localization of endogenous SIX2 protein (green) within the interstitial but not epithelial compartment. Low‐magnification and high‐magnification (inset) images are shown. Scale bar = 100 μm.
- RT–PCR analysis of sorted fractions from SIX2EGFP organoids shows highly enriched SIX2 expression in the EGFP+ but not EGFP− cells. Scale bar = 50 μm.
- Flow cytometry analysis of SIX2EGFP/+ (heterozygous) versus SIX2EGFP/EGFP (homozygous) kidney organoids at day 13 of differentiation.
- SIX2 expression (measured as number of EGFP+ cells) positively correlates with CHIR99021 concentration and duration during the first stage of differentiation.
Single‐cell RNAseq of kidney organoids reveals widespread SIX2‐expression
To examine SIX2‐expressing cells in greater depth during the process of kidney organoid differentiation, single‐cell transcriptome profiling of day 18 and 25 organoids was performed using the 10× Chromium platform. Twenty cell populations emerged from guided clustering analyses using Seurat 18, with several of these pertaining to different nephron segments and cells at various stages of nephrogenesis (Fig 2A, Dataset EV1). Multiple stromal populations were also identified, which expressed collagens COL1A1 and COL3A1, as well as kidney stromal markers DCN and CXCL12. We note the apparent absence of an endothelial cluster in this dataset, despite clear evidence for this cell type in previous studies 19, 20, 21, including in bioprinted organoids [preprint: 15].
Figure 2. Analysis of SIX2 expression in kidney organoids by single‐cell RNAseq.

- tSNE plot representing the merged datasets from single‐cell RNAseq analysis of day 18 (1,865 cells) and day 25 (3,500 cells) organoids. Twenty clusters were identified.
- A distinct SIX2‐expressing cluster was identified as a nephron progenitor‐like population based on expression of other known markers, including CITED1, DAPL1, EYA1, and SALL1.
- SIX2 marks a diverse range of cell types within human kidney organoids with SIX2 transcripts detected in numerous clusters in both day 18 and day 25 organoids.
- Graphical representation showing the cell populations that are enriched or depleted in day 18 versus day 25 kidney organoids.
With respect to SIX2‐expressing cells, a distinct population (cluster 9) exhibited strong congruence with human fetal nephron progenitors, as determined by co‐expression of several other previously described nephron progenitor markers, including CITED1, DAPL1, EYA1, and SALL1 3, 13, 16 (Fig 2B). Notably, SIX2 expression was not solely restricted to the putative nephron progenitor cluster, with SIX2 transcripts detected in scattered cells within several additional clusters, including a subset of the renal stroma, and within an “off‐target” population that displayed a muscle‐like transcriptional profile (Fig 2C). Interestingly, this SIX2‐expressing muscle‐like population was clearly evident in day 25 organoids, but largely absent in earlier day 18 organoids (Fig 2C and D). Compared to day 18, day 25 organoids also showed an overall reduction in both the SIX2‐expressing nephron progenitor and “early committed nephron” clusters (Fig 2D). Taken together, these findings reveal that SIX2 is not confined to the presumptive nephron progenitor compartment within human kidney organoids, but also marks several additional distinct cell types, including renal stroma and a muscle‐like population which becomes more prevalent as the differentiation proceeds.
Generation of a dual‐fluorescence cassette for human fate‐mapping studies
To facilitate human fate‐mapping experiments in iPSC‐derived organoids, we generated a dual‐fluorescence gene‐targeting cassette comprising a loxP‐flanked EGFP and adjacent mCherry reporter that can be incorporated into the 3′ end of the endogenous GAPDH coding region (Fig 3A). We have previously shown that this targeting strategy facilitates ubiquitous and consistent transgene expression in hPSCs, both prior to and following differentiation into various different cell types 22. We first evaluated the functionality of our fluorescence cassette in the human embryonic stem cell line, H9. Following CRISPR/Cas9‐mediated knock‐in of our targeting cassette, correctly edited cells were identified based on expression of the EGFP reporter (Fig 3A). To validate the Cre‐mediated color switching capacity of our dual‐fluorescence cassette, EGFP‐expressing clones were isolated, expanded, and subsequently transfected with mRNA encoding Cre recombinase. This resulted in the rapid induction of mCherry expression and a corresponding loss of EGFP expression within 8 h post‐transfection in > 95% of cells (Fig 3B). Kidney organoids could also be successfully derived from EGFP or mCherry‐expressing H9 cells, representing cells before and after exposure to Cre recombinase, respectively. We observed maintenance of appropriate reporter gene expression in all component cell types as determined by live fluorescent microscopy and flow cytometry (Fig 3C and D).
Figure 3. Generation and characterization of a dual fluorescent reporter construct for downstream lineage tracing experiments.

-
ASchematic diagram of the targeting strategy. A loxP‐flanked EGFP and adjacent mCherry reporter were inserted downstream of the endogenous GAPDH coding region, linked via a self‐cleaving T2A sequence. In the absence of Cre recombinase, cells constitutively express EGFP.
-
BThe loxP‐flanked EGFP gene is deleted following Cre‐mediated recombination. Shortly after (< 8 h) the transient transfection of mRNA encoding Cre recombinase, GAPDHdual hPSCs permanently switch from EGFP to mCherry reporter gene expression.
-
C, DReporter gene expression is maintained in all cell types within kidney organoids generated from GAPDHdual hPSCs before and after exposure to Cre, as detected by fluorescent microscopy (C) and flow cytometry (D).
Tracing the fate of SIX2+ cells in human kidney organoids
To examine the fate of SIX2‐expressing cells in hPSC‐derived kidney organoids, we used CRISPR/Cas9‐mediated gene editing to insert the Cre recombinase gene into the endogenous SIX2 locus using the targeting strategy described earlier for generation of SIX2EGFP iPSCs. Clonally derived iPSCs with homozygous insertion of Cre recombinase were established using our one‐step reprogramming/gene‐editing protocol 8 which were subsequently used for knock‐in of the dual‐fluorescence cassette into the GAPDH locus as described above (Fig 4A). Correctly targeted EGFP‐expressing colonies, hereafter referred to as SIX2Cre/Cre:GAPDHdual iPSCs, were identified by fluorescent microscopy, isolated, and expanded for downstream differentiation experiments. Kidney organoids were generated from SIX2Cre/Cre:GAPDHdual iPSCs and monitored by flow cytometry for reporter gene expression. Cells expressing mCherry could be detected at approximately day 10 of differentiation, coinciding with activation of endogenous SIX2 and reporter expression within kidney organoids derived from the SIX2EGFP iPSCs described earlier (Figs 4B and 1C). As differentiation progressed, a steady increase in mCherry‐expressing cells and a corresponding loss of EGFP‐expressing cells were also observed (Fig 4B). Live mCherry+/EGFP− cells could also be detected in kidney organoids by fluorescent microscopy, some of which appeared to be localized within epithelial structures (Fig 4C). Whole‐mount immunofluorescence of day 25 SIX2Cre/Cre:GAPDHdual organoids was performed to determine the precise location of mCherry‐expressing cells within specific cellular compartments, using markers specific to nephrons (WT1, NPHS1, LTL, CDH1, EPCAM), presumptive ureteric epithelium (GATA3, CDH1), renal interstitium (MEIS1), and endothelium (CD31). SIX2‐expressing cells were seen to contribute to nephron formation, as evidenced by the appearance of mCherry+ cells within LTL+ proximal tubules, EPCAM+/LTL− distal tubules, and NPHS1+ podocytes (Fig 4D). Consistent with our scRNAseq analysis, interstitial cells co‐expressing MEIS1 and mCherry were also clearly evident, indicating that SIX2 + cells give rise to at least a subset of the renal stroma (Fig 4D). Conversely, CD31+/mCherry+ cells were not observed, indicating that endothelial cells within kidney organoids are not derived from SIX2 + cells (Fig 4D). Notably, mCherry+ cells were excluded from GATA3+/CDH1+ structures (Fig 4E), which is consistent with a ureteric epithelium‐like identity for this population of cells, as previous fate‐mapping analyses in mice demonstrate SIX2 + cells do not give rise to the ureteric epithelium 10. Using image analysis software, we detected < 1% of mCherry+ cells within GATA3+/CDH1+ epithelium, with most of these events observed at the proximal end of GATA3+/CDH1+ structures (Fig 4F). Taken together, our findings are consistent with previous studies performed in mice, which show SIX2 + cells can give rise to all cells of the nephron but not the collecting duct network, which is instead derived from a more anterior ureteric progenitor population 10, 13 (Fig 4G).
Figure 4. Fate mapping of the SIX2 population in hPSC‐derived kidney organoids.

- Schematic diagram of the targeting strategy used for generation of SIX2Cre/Cre:GAPDHdual iPSCs.
- Flow cytometry analysis of kidney organoids derived from SIX2Cre/Cre:GAPDHdual iPSCs showing induction of mCherry and corresponding loss of EGFP expression.
- Low (upper panel)‐ and high‐magnification (lower panel) images showing mCherry+ cells detected by live fluorescent microscopy in SIX2Cre/Cre:GAPDHdual kidney organoids.
- Immunostaining of SIX2Cre/Cre:GAPDHdual kidney organoids shows localization of mCherry cells within proximal (LTL+/EPCAM+), distal (LTL−/EPCAM+), and glomerular (NPHS1+) nephron segments and within renal stroma (MEIS1+) but not within the CD31+ vasculature.
- mCherry+ cells were excluded from the presumptive GATA3+/ECAD+ collecting duct epithelium.
- Plot showing exclusion of mCherry+ cells from GATA3+/ECAD+ epithelium as determined by image analysis software.
- Model depicting the separation of nephron and collecting duct lineages during kidney morphogenesis.
A SIX2‐expressing population gives rise to nephrons early in organoid culture but retains nephron‐forming capacity across organoid differentiation
In the developing kidney in vivo, new nephrons arise throughout fetal development, arising from a self‐renewing Six2 + nephron progenitor population present around the tips of the ureteric epithelium 10. This process continues until approximately week 36 in humans 23, 24 and the first few days after birth in mice 25, at which point all NPCs have committed to nephron formation. To determine the duration of nephrogenesis across the period of kidney organoid culture, we generated SIX2 knock‐in iPSCs using the tamoxifen‐inducible Cre recombinase, CreERT2 26. The dual‐fluorescence cassette was subsequently inserted into the endogenous GAPDH locus of a clonally derived iPSC line harboring a homozygous insertion of CreERT2 (Fig 5A). Day 12 kidney organoids derived from SIX2CreERT2/CreERT2:GAPDHDual iPSCs were cultured in the presence of 4‐hydroxytamoxifen (4‐OHT) for 1 h to induce Cre recombination. Activation of the mCherry reporter could be detected by flow cytometry and fluorescent microscopy within 24 h post‐treatment (Fig 5B). A dose‐dependent trend was also noted, with the number of mCherry+ cells positively correlating with 4‐OHT concentration as anticipated (Fig 5C). Importantly, no mCherry+ cells were observed in the absence of 4‐OHT treatment.
Figure 5. Temporal and limited labeling of the SIX2 lineage using the inducible Cre recombinase, CreERT2.

- Schematic diagram of the targeting strategy used for generation of SIX2CreERT2/CreERT2:GAPDHdual iPSCs.
- mCherry+ cells detected by live fluorescent microscopy in SIX2CreERT2/CreERT2:GAPDHdual kidney organoid following 4‐OHT induction at day 10 of differentiation. Epithelial mCherry+ cells are indicated (white arrow). Scale bar = 50 μm.
- The number of mCherry+ cells in kidney SIX2CreERT2/CreERT2:GAPDHdual organoids positively correlates with 4‐OHT concentration. Data represent mean ± SD, n = 3.
To determine if SIX2 + cells could contribute to nephron formation throughout organoid development, we staggered the labeling of SIX2‐expressing cells by initiating 4‐OHT treatment (1 μM) at 2‐day intervals between days 10 and 18 of differentiation (Fig 6A). Organoids were dissociated at day 25, stained with a directly conjugated EPCAM antibody, and analyzed by flow cytometry to determine the percentage of mCherry+ cells localized within epithelial structures. Using this assay, we observed significantly fewer EPCAM+ cells within the mCherry+ fraction of organoids induced at day 18 compared to those induced at day 10 (Figs 6B and EV2A). While a negative correlation between the time of 4‐OHT treatment and EPCAM+/mCherry+ cells was observed, there was no correlation between time of 4‐OHT treatment and the total number of mCherry+ cells (Fig EV2B) nor the overall fraction of EPCAM+ cells (Fig EV2C). Whole‐mount immunofluorescence of day 25 SIX2CreERT2/CreERT2:GAPDHdual organoids was also performed, where we observed mCherry+ cells both within EPCAM+ structures and the interstitial compartment, but not within the GATA3+/EPCAM+ epithelium (Fig 6C). In organoids induced early, at day 10, mCherry+ cells were easily detected in all nephron epithelial segments and EPCAM−/NEPHRIN+ podocytes (Fig 6D, early induction). Conversely, in organoids induced at later time‐points, mCherry+ cells were notably absent within nephron structures and predominantly restricted to the interstitium surrounding them (Fig 6D, late induction). Because our transcriptional profiling data revealed a SIX2‐expressing cell population with strong similarity to human nephron progenitors in day 18 kidney organoids (see Fig 2), we considered whether an additional CHIR pulse could promote SIX2 + cells in late (day 18) kidney organoids to undergo nephron commitment. In this set of experiments, kidney organoids induced with 4‐OHT at day 18 were subsequently subjected to a 1‐h CHIR pulse the following day. When the organoids were harvested 1 week later (day 26) and analyzed by whole‐mount immunofluorescence, we could clearly detect mCherry+ cells derived from the SIX2+ lineage in EPCAM+ epithelial structures and NEPHRIN+ podocytes, whereas mCherry+ cells were restricted to the interstitium in control organoids that had not undergone a late CHIR pulse. Collectively, these findings indicate that the capacity for SIX2 + cells to contribute to nephron formation is retained across organoid culture, but requires an exogenous differentiation signal such as can be provided via induction of WNT signaling.
Figure 6. Staggered labeling of SIX2 lineage shows declining nephrogenic potential of SIX2 + cells during kidney organoid differentiation.

- Outline of the strategy used to interrogate the nephrogenic potential of SIX2 + cells throughout kidney organoid differentiation.
- The percentage of mCherry+ cells localized within EPCAM+ epithelial structures negatively correlates with the time of 4‐OHT initiation, as determined by flow cytometric analysis. Data represent mean ± SD, n ≥ 3. Data from day 10 and 18 time‐points were obtained from two independent experiments. P‐value was calculated using unpaired two‐sided t‐test.
- Immunostaining of SIX2CreERT2/CreERT2:GAPDHdual kidney organoids shows mCherry+ cells localized within EPCAM+ structures (white arrow) and the interstitial compartment (yellow arrow), but not within the GATA3+/EPCAM+ epithelium.
- mCherry+ cells derived from the SIX2+ lineage were detected in all nephron epithelial segments when 4‐OHT induction was initiated at day 10 (early induction) but were largely restricted to the interstitium when induced at later time‐points (late induction).
- In late kidney organoids induced with 4‐OHT at day 18, mCherry+ cells derived from the SIX2+ lineage were detected in nephron segments when subjected to a late (day 19) CHIR pulse. mCherry+ cells were restricted to the interstitium in control organoids that had not undergone a late CHIR pulse.
Figure EV2. Flow cytometric analysis of SIX2CreERT2/CreERT2 kidney organoids.

-
ARepresentative plots for early (day 10) and late (day 18) 4‐OHT‐induced kidney organoids derived from SIX2CreERT2/CreERT2 iPSCs. The mCherry+ fraction was plotted separately to determine the percentage of mCherry+ cells localized within EPCAM+ epithelial structures, following immunostaining with a directly conjugated EPCAM antibody.
-
B, CNo correlation between the time of 4‐OHT treatment and the total number of mCherry+ cells (B) nor the overall fraction of EPCAM+ cells (C) was observed. Data represent mean ± SD, n ≥ 3.
Discussion
Organoids derived from hPSCs offer enormous utility in personalized disease modeling and drug testing platforms, while also providing promise for the development of autologous cellular therapies to treat/correct many inherited and acquired diseases. Organoid‐based cultures also represent a potential source of human tissue at developmental stages that are typically unavailable for research purposes. In combination with gene‐editing technologies, this could facilitate the study of gene function and cellular processes that govern human development in vitro. Genome engineering also facilitates studies aimed at characterizing the cell types produced in organoid‐based cultures and how well these compare with primary developing tissue, which is imperative for understanding and addressing limitations associated with hPSC‐derived tissue.
In this study, we use gene‐edited iPSCs to interrogate the SIX2 lineage in human kidney organoids and to examine how this compares with our existing understanding of mammalian nephrogenesis in vivo, which is largely based on studies performed in mice. We first used our previously described kidney organoid differentiation protocol and a SIX2 EGFP reporter line to monitor SIX2 + cells in developing kidney organoids. SIX2 + cells emerged soon after organoid formation and persisted until the termination of differentiation at day 25. Although this contrasts to other previously described differentiation protocols where a rapid loss of SIX2 + cells is observed soon after the formation of 3D kidney organoids 16, our findings are consistent with a recent study that employed a similar SIX2‐EGFP reporter iPSC line and kidney organoid protocol, where SIX2 expression also persisted until the termination of differentiation 27. Importantly, our findings do not support their hypothesis that the presence of these cells definitively represents a progenitor niche that may supply the organoid with more mature cells over time. Rather, we show that SIX2 is expressed in a variety of other cell types, and a lack of ongoing nephrogenesis across organoid culture. While single‐cell transcriptome profiling of organoids generated in this study did reveal a distinct SIX2 + population that exhibited strong congruence with human fetal nephron progenitors, SIX2 expression was also detected in several additional cell clusters. Interestingly, this included an “off‐target” muscle‐like population that was enriched in late but not early organoids. SIX2 expression was also detected in several clusters identified as “renal stroma”. Consistent with this observation, our fate‐mapping experiments revealed an obvious SIX2 + lineage‐derived MEIS1+ stromal population. Although this contrasts sharply with studies performed in mice, where a strict lineage boundary between nephron progenitors and the interstitial progenitor cells that give rise to the renal stroma has been shown to exist 28, 29, 30, recent single‐cell RNAseq analysis of human fetal kidney has revealed substantial overlap between these two progenitor populations, with co‐expression of SIX2, MEIS1, and FOXD1 detected within human nephron progenitors 3. Although difficult to determine definitively whether the co‐expression of nephron progenitor and stromal markers is an artifact of our organoid culture system, the findings from this previous study suggest that this may in fact be a true species difference. In mice, the origin of the endothelial population has not been regarded to be the SIX2‐expressing population and we did not see evidence for endothelial cells of SIX2 lineage in this study; however, endothelial cell types were low in abundance, and hence, the lineage relationship here remains equivocal and requires more investigation. Further studies are also warranted to determine which sub‐populations of the overall SIX2‐expressing population can in fact contribute to nephron formation in our organoid model.
The presence of cell clusters with a muscle signature that apparently increase in prevalence with time is difficult to interpret. We would note, however, the prevalent formation of ectopic muscle within Wilms’ tumors, a childhood renal neoplasia regarded as showing disrupted development 31. It has previously been suggested that Wilms’ tumor arises as a result of inappropriate differentiation of the nephron progenitors 31, 32, with this conclusion recently supported at the single‐cell level 33. It has also been suggested that muscle formation within Wilms’ tumors is associated with reductions in WT1 expression. Hence, it is possible that this off‐target population is arising from the initial nephron‐forming SIX2 population.
Using a lineage tracing iPSC reporter line, we demonstrate that SIX2‐expressing cells can contribute to all segments of the developing nephron but are excluded from the distal ends of the GATA3+/CDH1+ epithelial structures, suggestive of a presumptive ureteric epithelium population in kidney organoids. Of note, not all cells within each nephron had undergone Cre‐mediated color switching, with many nephrons comprised of both mCherry+ and EGFP+ cells. One explanation for this is that Cre recombination may not have been complete. Alternatively, there may be a genuine contribution of both SIX2‐expressing and non‐expressing cells within forming nephrons. More studies are required to investigate this further.
By using a tamoxifen‐inducible variant of Cre recombinase to stagger the labeling of SIX2 + cells during organoid differentiation, we noted a significantly reduced capacity for these cells to contribute to nephron formation over time. This suggests human kidney organoids, in contrast to fetal kidney in vivo, lack a true nephrogenic zone capable of sustained nephrogenesis. In mice, correct localization of nephron and stromal progenitors around the tips of the ureteric epithelium enables reciprocal inductive signals between these populations. This is required for continued branching of the ureteric epithelium and both nephron progenitor self‐renewal and commitment, which drives organogenesis throughout fetal development 12, 34. Interestingly, although SIX2 + cells in more mature (day 18) organoids did not contribute to nephron formation, these cells could in fact give rise to nephrons upon the addition of a late (day 19) CHIR pulse. This finding is consistent with the transcriptional profiling of day 18 organoids, where a SIX2‐expressing population with strong congruence with human nephron progenitors was identified. However, it suggests that this competent population ceases nephron formation in the absence of an exogenous differentiation signal. Hence, although we demonstrate clear evidence of nephron progenitor commitment in our organoid cultures, a self‐renewing niche comprised of a branching ureteric epithelium with corresponding tips and a distinct domain of nephron and stromal progenitors is not evident. This suggests that appropriate spatial organization and/or reciprocal interactions between nephron progenitors, ureteric epithelium, and possibly also the renal stroma are deficient.
Perhaps a more logical strategy for generating kidney organoids with a sustainable nephrogenic niche would be to derive homogenous populations of nephron and stromal progenitors, and ureteric epithelium in parallel which could be subsequently combined in 3D culture, in a spatial arrangement that more accurately depicts the developing organ in vivo. This strategy was partially recapitulated in a recent study which demonstrated a substantially improved higher‐order structure in kidney organoids derived from mouse PSCs, and was highlighted by an impressive capacity for the ureteric epithelium to undergo several rounds of branching morphogenesis 35. However, while a human branching ureteric epithelium was generated, there was not a successful reciprocal interaction between this epithelium and surrounding presumptive human metanephric mesenchyme, highlighting our limited understanding of the exact conditions required to recapitulate a self‐renewing nephron progenitor niche coupled with ongoing productive nephron formation in vitro.
In conclusion, our findings show that nephrons within kidney organoids arise from a SIX2‐expressing mesenchymal population, as anticipated from previous studies in mice, but also show the absence of ongoing nephrogenesis, likely due to the lack of a peripheral nephrogenic zone. However, at least a subset of SIX2‐expressing cells retain nephron‐forming capacity longer term and can form nephrons if induced to do so. As such, this represents a proof of concept that gene‐editing and organoid technologies can be combined to facilitate fate‐mapping studies in differentiating hPSCs, thereby providing a unique opportunity to investigate lineage relationships in real time and in a higher‐throughput and more cost‐effective manner compared with mammalian models. Additional fate‐mapping and CRISPR/Cas9‐mediated gene‐knockout studies in organoids may facilitate the development of more efficient and robust protocols to generate renal cell types for downstream applications. Indeed, such approaches may also provide deeper insight into human kidney development. While applied to kidney in this instance, such a lineage tracing approach is applicable in other organoid settings in which complex multicellular tissues are formed.
Materials and Methods
Cell lines
Human foreskin fibroblasts (ATCC ID: CRL‐2429) were cultured in DMEM (Thermo Fisher Scientific) supplemented with 15% fetal bovine serum (FBS; Hyclone) and 1X MEM Non‐Essential Amino Acids Solution (Thermo Fisher Scientific) at 37°C, 5% CO2, and 5% O2. All iPSC lines were maintained and expanded at 37°C, 5% CO2, and 5% O2 in Essential 8 medium (Thermo Fisher Scientific) on Matrigel‐coated plates with daily medium changes and passaged every 3–4 days with EDTA in 1X PBS as previously described 36. The genomic integrity of iPSCs was confirmed by molecular karyotyping using Infinium CoreExome‐24 v1.1 SNP arrays (Illumina), and expression of common pluripotency markers (TRA‐1‐81, SSEA‐4, CD9, OCT4) was confirmed by immunofluorescence and flow cytometry.
Kidney organoid production
The day prior to differentiation, cells were dissociated with TrypLE (Thermo Fisher Scientific), counted using a hemocytometer, and seeded onto Laminin 521‐coated 6‐well plates at a density of 50 × 103 cells per well in Essential 8 medium. Intermediate mesoderm induction was performed by culturing iPSCs in TeSR‐E6 medium (Stem Cell Technologies) containing 4–8 μM CHIR99021 (R&D Systems) for 4 days. On day 4, cells were switched to TeSR‐E6 medium supplemented with 200 ng/ml FGF9 (R&D Systems) and 1 μg/ml Heparin (Sigma‐Aldrich). On day 7, cells were dissociated with TrypLE, diluted fivefold with TeSR‐E6 medium, transferred to a 15‐ml conical tube, and centrifuged for 5 min at 300 × g to pellet cells. The supernatant was discarded, and cells were resuspended in residual medium and transferred directly into a syringe for bioprinting. Syringes containing the cell paste were loaded onto a NovoGen MMX Bioprinter, primed to ensure cell material was flowing, and user‐defined aliquots (5,000–100,000 cells per organoid) were deposited on 0.4‐μm Transwell polyester membranes in 6‐well plates (Corning). Following bioprinting, organoids were cultured for 1 h in the presence of 6 μM CHIR99021 in TeSR‐E6 medium in the basolateral compartment and subsequently cultured until day 12 in TeSR‐E6 medium supplemented with 200 ng/ml FGF9 and 1 μg/ml Heparin. From day 12 to day 25, organoids were grown in TeSR‐E6 medium without supplementation. Unless otherwise stated, kidney organoids were cultured until harvest at day 25. For induction of CreERT2 protein in kidney organoids derived from SIX2CreERT2 iPSCs, 4‐hydroxytamoxifen (Sigma‐Aldrich) dissolved in ethanol at a concentration of 100 μM was diluted to working concentration in TeSR‐E6. Induction medium (1 ml) was pipetted under the transwell, and individual drops from a 20‐μl pipette were carefully placed on top of the organoids on the filter to ensure complete coverage. Organoids were incubated at 37°C for 1 h. Induction medium was removed by washing three times with TeSR‐E6 medium every 10 min and then returned to the media they were in prior to induction.
Vector construction
The SIX2:EGFP vector (pDNR‐SIX2:EGFP) carries a targeting cassette encoding the T2A peptide and EGFP gene flanked by ~700 and ~450 bp of sequence corresponding to sequence immediately upstream and downstream of the SIX2 stop codon, respectively. Two gBlocks (Integrated DNA Technologies) encoding the targeting cassette were inserted into the AatII and EcoRI sites of the pDNR‐Dual (Clontech) plasmid vector. The pDNR‐SIX2:Cre and pDNR‐SIX2:CreERT2 targeting vectors were generated as described above but substituting the Cre recombinase and CreERT2 recombinase genes for EGFP, respectively. The GAPDH targeting vector encoding the dual‐fluorescence cassette (pGAPTrap‐loxEGFPloxCherry) was generated by inserting sequence encoding the T2A peptide, loxP‐flanked EGFP gene with SV40 polyA signal and adjacent mCherry gene with SV40 polyA signal into the SfiI and ClaI sites of the pGAPTrap‐mtagBFP2‐IRESMuro plasmid vector (after removal of the mtagBFP2‐IRESMuro cassette). A sgRNA plasmid specific to the 3′ end of the SIX2 coding region (pSMART‐sgRNA‐SIX2) was generated by annealing ODNs SIX2_sgRNA1a and SIX2_sgRNA1b followed by ligation into the BbsI sites of the pSMART‐sgRNA vector 37. A sgRNA plasmid specific to the 3′ end of the GAPDH coding region (pSMART‐sgRNA‐GAPDH) was generated by annealing ODNs GAPDH_sgRNA1a and GAPDH_sgRNA1b followed by ligation into the BbsI sites of the pSMART‐sgRNA vector. All plasmids were propagated in DH5‐alpha Escherichia coli (BIOLINE) and prepared for transfection using a Plasmid Maxi kit (QIAGEN). See Table 1 for list of ODNs and Table 2 for list of plasmids used in this study.
Table 1.
Oligonucleotides used in this study
| Name | Sequence |
|---|---|
| GAPDH_sgRNA1a | CACCGCTTCCTCTTGTGCTCTTGCT |
| GAPDH_sgRNA1b | AAACAGCAAGAGCACAAGAGGAAGC |
| SIX2_sgRNA1a | CACCGTCAGCCAACCTCGTGGACC |
| SIX2_sgRNA1b | AAACGGTCCACGAGGTTGGCTGAC |
| SIX2F | CATCTACCCAGCAAACCTGG |
| EGFPR | GTCCAGCTCGACCAGGATGG |
| SV40pAF | GCGACTCTAGATCATAATC |
| SIX2R | GAGTACAAGAGACTGGCAGG |
| CreR | GAGTTGATAGCTGGCTGGTG |
Table 2.
Plasmids used in this study
| Name | References | Identifier |
|---|---|---|
| pEP4 E02S ET2K | Yu et al 39 | Addgene plasmid #20927 |
| pEP4 E02S EN2L | Yu et al 39 | Addgene plasmid #20922 |
| pEP4 E02S EM2K | Yu et al 39 | Addgene plasmid #20923 |
| pSimple‐miR302/367 | Howden et al 14 | Addgene plasmid #98748 |
| pSP6‐EBNA2A+DBD | Howden et al 38 | Addgene plasmid #98749 |
| pDNR‐SpCas9‐Gem | Howden et al 14 | Addgene plasmid #98749 |
| pSMART‐sgRNA(Sp) | Howden et al 14 | Addgene plasmid #80427 |
| pGAPTrap‐mtagBFP2‐IRESMuro | Kao et al 22 | Addgene plasmid #82335 |
| pGAPTrap‐loxEGFPloxCherry | This study | TBC |
| pDNR‐Dual | Clontech | N/A (discontinued) |
| pDNR‐SIX2:EGFP | This study | TBC |
| pDNR‐SIX2:Cre | This study | TBC |
| pDNR‐SIX2:CreERT2 | This study | TBC |
Generation of knock‐in iPSCs
All SIX2 knock‐in iPSCs (EGFP, Cre, CreERT2) were derived from human foreskin fibroblasts (ATCC: CRL‐2429) using a previously described protocol that combines reprogramming and gene editing in one step 8. Episomal reprogramming plasmids (pEP4E02SET2K, pEP4E02SEN2L, pEP4E02SEM2K, and pSimple‐miR302/367), in vitro transcribed mRNA encoding the SpCas9‐Gem variant 37, the pSMART‐sgRNA‐SIX2 plasmid, and either the pDNR‐SIX2:EGFP, pDNR‐SIX2:Cre, or pDNR‐SIX2:CreERT2 targeting vectors were introduced into fibroblast using the Neon Transfection System as described below. In vitro transcribed mRNA encoding a truncated version of the EBNA1 protein was also included to enhance nuclear uptake of the reprogramming plasmids 36, 38. Genomic DNA was isolated from resulting iPSCs using the DNeasy Blood & Tissue Kit (QIAGEN) in accordance with the manufacturer's protocol, and PCR analysis was performed using GoTaq Green Master Mix (Promega) to identify correctly targeted clones. ODNs SIX2F and EGFPR flank the 5′ recombination junction of SIX2:EGFP knock‐in iPSCs, whereas SIX2F and CreR flank the 5′ recombination junction of SIX2:Cre and SIX2:CreERT2 knock‐ins. ODNs SV40pAF and SIX2R flank the 3′ recombination junction of SIX2:EGFP, SIX2:Cre, and SIX2:CreERT2 knock‐in iPSCs. Heterozygous and homozygous clones were distinguished using ODNs SIX2F and SIX2R. For knock‐in of the dual‐fluorescence cassette, the GAPDH targeting construct (pGAPTrap‐loxEGFPloxCherry) was co‐transfected with pSMART‐sgRNA‐GAPDH and mRNA encoding SpCas9‐Gem into hPSCs using the Neon Transfection System as described below. Correctly targeted (EGFP‐expressing) clones were identified by fluorescent microscopy.
In vitro transcription
Capped and polyadenylated in vitro transcribed mRNA encoding SpCas9‐Gem protein 37 was generated using the mMESSAGE mMACHINE T7 ULTRA transcription kit (Thermo Fisher Scientific) according to the manufacturer's recommendations. Plasmid template was linearized with PmeI endonuclease prior to transcription. LiCl was used to precipitate mRNA before resuspension. A truncated version of the EBNA1 protein 38, used to facilitate uptake of reprogramming plasmids, was transcribed using the mMESSAGE mMACHINE SP6 transcription kit (Thermo Fisher Scientific) according to the manufacturer's recommendations.
Cell transfection
Transfections were performed using the Neon Transfection System (Thermo Fisher Scientific). Human fibroblasts or iPSCs were harvested with TrypLE (Thermo Fisher) 2 days after passaging and resuspended in Buffer R at a final concentration of 1 × 107 cells/ml. Electroporation was performed in a 100‐μl tip using 1,400 V, 20 ms, and 2 pulses for human fibroblasts, or 1,100 V, 30 ms, and 1 pulse for human iPSCs. Following electroporation fibroblasts were transferred to 6‐well Matrigel‐coated plates containing DMEM + 15% FBS and switched to reprogramming medium (TeSR‐E7 + 100 μM sodium butyrate) after 3 days, with medium changes every other day. Electroporated human iPSCs were plated on 6‐well Matrigel‐coated plates containing Essential 8 medium with 5 μM Y‐27632 (Tocris).
Flow cytometry
Prior to analysis, single kidney organoids were dissociated with 0.2 ml of a 1:1 TrypLE/Accutase solution in 1.5‐ml tubes at 37°C for 15–25 min, with occasional mixing (flicking) until large clumps were no longer clearly visible. 1 ml of HBBS supplemented with 2% FBS was added to the cells before passing through a 40‐μM FACS tube cell strainer (Falcon). Flow cytometry was performed using a LSRFortessa Cell Analyzer (BD Biosciences). Data acquisition and analysis were performed using FACSDiva (BD) and FlowLogic software (Inivai). Gating was performed on live cells based on forward‐ and side‐scatter analysis.
Whole‐mount immunofluorescence
Organoids were transferred to 48‐well plates for fixation and immunofluorescence procedures. Fixation was performed using ice‐cold 2% paraformaldehyde (PFA; Sigma‐Aldrich) for 20 min followed by 15 min of washing in three changes of phosphate‐buffered saline (PBS). For immunofluorescence, blocking and antibody staining incubations were performed on a rocking platform for 3 h at room temperature or overnight at 4°C, respectively. Blocking solution consisted of 10% donkey serum with 0.3% Triton X‐100 (TX‐100; Sigma‐Aldrich) in PBS. See Table 3 for list of antibodies used in this study. Antibodies were diluted in 0.3% TX‐100/PBS. Primary antibodies were detected with Alexa Fluor‐conjugated fluorescent secondary antibodies (Invitrogen), diluted 1:500. Organoids were washed in at least three changes of PBS for a minimum of 1 h following primary and secondary antibody incubations. Imaging was performed in glass‐bottomed dishes (MatTek) with glycerol submersion using either the Zeiss LSM 780 or Dragonfly Spinning Disk confocal microscope.
Table 3.
Antibodies used in this study
| Name | Source | Identifier |
|---|---|---|
| Mouse monoclonal anti‐TRA‐1‐81 | BioLegend | Cat#330706 |
| Mouse monoclonal anti‐SSEA‐4 | BioLegend | Cat#330408 |
| Mouse monoclonal anti‐CD9 | BD Biosciences | Cat#555371 |
| Rabbit monoclonal anti‐OCT4 | Abcam | Cat#Ab181557 |
| Rabbit polyclonal anti‐RFP (and mCherry) | MBL Medical & Biological Laboratories | Cat#PM005 |
| Biotinylated Lotus Tetragonolobus Lectin | Vector Laboratories | Cat#B‐1325 |
| Rabbit polyclonal anti‐SIX1 | Cell Signaling | Cat#12891 |
| Rabbit polyclonal anti‐SIX2 | Proteintech Group | Cat#11562‐1‐AP |
| Mouse monoclonal anti‐MEIS1/2/3 | Active Motif | Cat#39795 |
| Mouse anti‐EPCAM (Alexa Fluor 488‐conjugated) | BioLegend | Cat#324210 |
| Mouse anti‐EPCAM (Alexa Fluor 647‐conjugated) | BioLegend | Cat#324212 |
| Mouse monoclonal anti‐GATA3 | Thermo Fisher Scientific | Cat#MA1‐028 |
| Goat polyclonal anti‐GATA3 | R&D Systems | Cat#AF2605 |
| Sheep polyclonal anti‐NEPHRIN | R&D Systems | Cat#AF4269 |
| Mouse monoclonal anti‐CD31 | BD Pharmingen | Cat#550274 |
| Mouse monoclonal anti‐E‐CADHERIN | BD Biosciences | Cat#610181 |
| Chicken polyclonal anti‐GFP | Abcam | Cat#Ab13970 |
Single‐cell transcriptional profiling and data analysis
Organoids were dissociated as described above (for flow cytometry) and passed through a 40‐μM FACS tube cell strainer. Following centrifugation at 300 g for 3 min, the supernatant was discarded and cells resuspended in 50 μl TeSR‐E6 medium. Viability and cell number were assessed, and samples were run across separate runs on a Chromium Chip Kit (10× Genomics). Libraries were prepared using Chromium Single Cell Library kit V2 (10× Genomics) and sequenced on an Illumina HiSeq with 100‐bp paired‐end reads. Cell Ranger (v1.3.1) was used to process and aggregate raw data from each of the samples returning a count matrix. Quality control and analysis was performed in R using the Seurat package (v2.3.1) 18. Cells with more than 125,000 UMIs, < 500 genes expressed, or more than 20% reads assigned to mitochondrial genes were filtered out. UMI counts, percentage of mitochondrial and ribosomal gene expression, and cell cycle phase identity were regressed out. Genes with less than two counts across the whole dataset were also filtered out. The final dataset had 5,365 cells and 22,105 identified genes. The two samples were merged using a canonical correlation analysis (CCA) using 1,429 genes with the highest dispersion present in both samples. The CCA subspaces were aligned, and the first 25 principal components based on these genes were used to build a graph, which was clustered at a resolution of 1.6. Data from the single‐cell transcriptional profiling have been deposited in the Gene Expression Omnibus under accession number GSE119561.
Author contributions
SEH and MHL conceived the study and wrote the manuscript; SEH performed gene‐editing experiments; SEH, JMV, SBW, and KST performed kidney differentiation experiments; JMV, SBW, and KST performed immunostaining; SBW performed scRNAseq analysis; and all authors assisted in manuscript preparation.
Conflict of interest
M.H.L. is an inventor on a patent associated with kidney organoid generation and has a research contract with and has consulted for Organovo Inc.
Supporting information
Expanded View Figures PDF
Dataset EV1
Review Process File
Acknowledgements
The Murdoch Children's Research Institute is supported by the Victorian Government's Operational Infrastructure Support Program. The MCRI Gene Editing Facility is supported by the Stafford Fox Foundation. MHL is a Senior Principal Research Fellow of the National Health and Medical Research Council, Australia (GNT1136085). This work was supported by the National Institutes of Health, USA (DK107344‐01), and the NHMRC (GNT1100970).
EMBO Reports (2019) 20: e47483
Contributor Information
Sara E Howden, Email: sara.howden@mcri.edu.au.
Melissa H Little, Email: melissa.little@mcri.edu.au.
References
- 1. Humphreys BD, DiRocco DP (2014) Lineage‐tracing methods and the kidney. Kidney Int 86: 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Brien LL, Guo Q, Lee Y, Tran T, Benazet JD, Whitney PH, Valouev A, McMahon AP (2016) Differential regulation of mouse and human nephron progenitors by the Six family of transcriptional regulators. Development 143: 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lindstrom NO, Guo J, Kim AD, Tran T, Guo Q, De Sena Brandine G, Ransick A, Parvez RK, Thornton ME, Basking L et al (2018) Conserved and divergent features of mesenchymal progenitor cell types within the cortical nephrogenic niche of the human and mouse kidney. J Am Soc Nephrol 29: 806–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lindstrom NO, McMahon JA, Guo J, Tran T, Guo Q, Rutledge E, Parvez RK, Saribekyan G, Schuler RE, Liao C et al (2018) Conserved and divergent features of human and mouse kidney organogenesis. J Am Soc Nephrol 29: 785–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morizane R, Miyoshi T, Bonventre JV (2017) Concise Review: kidney generation with human pluripotent stem cells. Stem Cells 35: 2209–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takasato M, Er PX, Chiu HS, Little MH (2016) Generation of kidney organoids from human pluripotent stem cells. Nat Protoc 11: 1681–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, Parton RG, Wolvetang EJ, Roost MS, Chuva de Sousa Lopes SM et al (2015) Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526: 564–568 [DOI] [PubMed] [Google Scholar]
- 8. Howden SE, Thomson JA, Little MH (2018) Simultaneous reprogramming and gene editing of human fibroblasts. Nat Protoc 13: 875–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyle S, Misfeldt A, Chandler KJ, Deal KK, Southard‐Smith EM, Mortlock DP, Baldwin HS, de Caestecker M (2008) Fate mapping using Cited1‐CreERT2 mice demonstrates that the cap mesenchyme contains self‐renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev Biol 313: 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP (2008) Six2 defines and regulates a multipotent self‐renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3: 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Short KM, Combes AN, Lefevre J, Ju AL, Georgas KM, Lamberton T, Cairncross O, Rumballe BA, McMahon AP, Hamilton NA et al (2014) Global quantification of tissue dynamics in the developing mouse kidney. Dev Cell 29: 188–202 [DOI] [PubMed] [Google Scholar]
- 12. Little MH, McMahon AP (2012) Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol 4: a008300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, Nishinakamura R (2014) Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14: 53–67 [DOI] [PubMed] [Google Scholar]
- 14. Howden SE, Maufort JP, Duffin BM, Elefanty AG, Stanley EG, Thomson JA (2015) Simultaneous reprogramming and gene correction of patient fibroblasts. Stem Cell Reports 5: 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Higgins JW, Chambon A, Bishard K, Hartung A, Arndt D, Brugnano J, Xuan Er P, Lawlor KT, Vanslambrouck JM, Wilson S et al (2018) Bioprinted pluripotent stem cell‐derived kidney organoids provide opportunities for high content screening. bioRxiv 10.1101/505396 [PREPRINT] [DOI] [Google Scholar]
- 16. Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV (2015) Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol 33: 1193–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, Parton RG, Wolvetang EJ, Roost MS, Lopes SM et al (2016) Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 536: 238 [DOI] [PubMed] [Google Scholar]
- 18. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36: 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Phipson B, Er PX, Combes AN, Forbes TA, Howden SE, Zappia L, Yen HJ, Lawlor KT, Hale LJ, Sun J et al (2019) Evaluation of variability in human kidney organoids. Nat Methods 16: 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Combes AN, Zappia L, Er PX, Oshlack A, Little MH (2019) Single‐cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med 11: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu H, Uchimura K, Donnelly EL, Kirita Y, Morris SA, Humphreys BD (2018) Comparative analysis and refinement of human PSC‐derived kidney organoid differentiation with single‐cell transcriptomics. Cell Stem Cell 23: 869–881 e868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kao T, Labonne T, Niclis JC, Chaurasia R, Lokmic Z, Qian E, Bruveris FF, Howden SE, Motazedian A, Schiesser JV et al (2016) GAPTrap: a simple expression system for pluripotent stem cells and their derivatives. Stem Cell Reports 7: 518–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hinchliffe SA, Sargent PH, Howard CV, Chan YF, van Velzen D (1991) Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Lab Invest 64: 777–784 [PubMed] [Google Scholar]
- 24. Ryan D, Sutherland MR, Flores TJ, Kent AL, Dahlstrom JE, Puelles VG, Bertram JF, McMahon AP, Little MH, Moore L et al (2018) Development of the human fetal kidney from mid to late gestation in male and female infants. EBioMedicine 27: 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rumballe BA, Georgas KM, Combes AN, Ju AL, Gilbert T, Little MH (2011) Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev Biol 360: 110–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D (1999) Temporally‐controlled site‐specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen‐inducible Cre‐ER(T) and Cre‐ER(T2) recombinases. Nucleic Acids Res 27: 4324–4327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Borestrom C, Jonebring A, Guo J, Palmgren H, Cederblad L, Forslow A, Svensson A, Soderberg M, Reznichenko A, Nystrom J et al (2018) A CRISP(e)R view on kidney organoids allows generation of an induced pluripotent stem cell‐derived kidney model for drug discovery. Kidney Int 94: 1099–1110 [DOI] [PubMed] [Google Scholar]
- 28. Kobayashi A, Mugford Joshua W, Krautzberger AM, Naiman N, Liao J, McMahon Andrew P (2014) Identification of a multipotent self‐renewing stromal progenitor population during mammalian kidney organogenesis. Stem Cell Reports 3: 650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mugford JW, Sipilä P, McMahon JA, McMahon AP (2008) Osr1 expression demarcates a multi‐potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1‐dependent nephron progenitor compartment within the mammalian kidney. Dev Biol 324: 88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Levinson R, Mendelsohn C (2003) Stromal progenitors are important for patterning epithelial and mesenchymal cell types in the embryonic kidney. Semin Cell Dev Biol 14: 225–231 [DOI] [PubMed] [Google Scholar]
- 31. Miyagawa K, Kent J, Schedl A, van Heyningen V, Hastie ND (1994) Wilms’ tumour–a case of disrupted development. J Cell Sci Suppl 18: 1–5 [DOI] [PubMed] [Google Scholar]
- 32. Schedl A (2007) Renal abnormalities and their developmental origin. Nat Rev Genet 8: 791–802 [DOI] [PubMed] [Google Scholar]
- 33. Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, Collord G, Botting RA, Popescu DM, Loudon KW et al (2018) Single‐cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361: 594–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Combes AN, Lefevre JG, Wilson S, Hamilton NA, Little MH (2016) Cap mesenchyme cell swarming during kidney development is influenced by attraction, repulsion, and adhesion to the ureteric tip. Dev Biol 418: 297–306 [DOI] [PubMed] [Google Scholar]
- 35. Taguchi A, Nishinakamura R (2017) Higher‐order kidney organogenesis from pluripotent stem cells. Cell Stem Cell 21: 730–746 e736 [DOI] [PubMed] [Google Scholar]
- 36. Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga‐Otto K, Howden SE, Diol NR, Propson NE et al (2011) Chemically defined conditions for human iPSC derivation and culture. Nat Methods 8: 424–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Howden SE, McColl B, Glaser A, Vadolas J, Petrou S, Little MH, Elefanty AG, Stanley EG (2016) A Cas9 variant for efficient generation of Indel‐free knockin or gene‐corrected human pluripotent stem cells. Stem Cell Reports 7: 508–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Howden SE, Wardan H, Voullaire L, McLenachan S, Williamson R, Ioannou P, Vadolas J (2006) Chromatin‐binding regions of EBNA1 protein facilitate the enhanced transfection of Epstein‐Barr virus‐based vectors. Hum Gene Ther 17: 833–844 [DOI] [PubMed] [Google Scholar]
- 39. Yu J, Hu K, Smuga‐Otto K, Tian S, Stewart R, Slukvin II, Thomson JA (2009) Human induced pluripotent stem cells free of vector and transgene sequences. Science 324: 797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Review Process File