

Abstract
Cachexia is a wasting disorder that accompanies many chronic diseases including cancer and results from an imbalance of energy requirements and energy uptake. In cancer cachexia, tumor‐secreted factors and/or tumor–host interactions cause this imbalance, leading to loss of adipose tissue and skeletal and cardiac muscle, which weakens the body. In this review, we discuss how energy enters the body and is utilized by the different organs, including the gut, liver, adipose tissue, and muscle, and how these organs contribute to the energy wasting observed in cachexia. We also discuss futile cycles both between the organs and within the cells, which are often used to fine‐tune energy supply under physiologic conditions. Ultimately, understanding the complex interplay of pathologic energy‐wasting circuits in cachexia can bring us closer to identifying effective treatment strategies for this devastating wasting disease.
Keywords: adipose tissue, cachexia, inflammation, liver, skeletal muscle
Subject Categories: Cancer, Metabolism
Glossary
- AAV
adeno‐associated virus
- AIDS
acquired immune deficiency syndrome
- Akt/PKB
protein kinase B
- AMPK
adenosine monophosphate‐activated protein kinase
- Apc
adenomatous polyposis coli
- AT
adipose tissue
- Atg5
autophagy‐related 5
- ATGL
adipocyte triglyceride lipase
- ATP
adenosine triphosphate
- Atrogin 1
muscle atrophy F‐box/MAFbx
- BA
bile acid
- BMI
body mass index
- C26
Colon 26 cell line
- CD36
cluster of differentiation 36
- CEBP‐β
CCAAT enhancer binding protein beta
- eIF3f
eukaryotic translation initiation factor 3 subunit f
- ER
endoplasmic reticulum
- FITC‐dextran
Fluorescein isothiocyanate‐dextran
- G6Pase
glucose 6‐phosphatase
- G6P
glucose 6‐phosphate
- GAA
acid α‐glucosidase
- Gabarap
gamma‐aminobutyric acid receptor‐associated protein
- GIP
glucose‐dependent insulinotropic peptide
- GK
glucokinase
- GKRP
GK regulatory protein
- GLP‐1
glucagon‐like peptide 1
- GLUT
glucose transporter
- GTP
guanosine‐5′‐triphosphate
- HSL
hormone‐sensitive lipase
- IL
interleukin
- MAPK
mitogen‐activated protein kinase
- MGL
monoglyceride lipase
- MHC
myosin heavy chain
- mTOR
mammalian target of rapamycin
- MuRF1
muscle RING finger‐1
- MUSA1
muscle ubiquitin ligase of SCF complex in atrophy‐1
- myoD
myogenic differentiation 1
- NPC1L1
Niemann‐Pick C1‐Like 1
- PYY
peptide yy
- RIP140
receptor‐interacting protein 140
- S6K1
ribosomal S6 kinase
- SCFA
short‐chain fatty acids
- SGLT1
sodium/glucose cotransporter 1
- SNP
single nucleotide polymorphism
- SRB1
scavenger receptor class B type 1
- T3
triiodothyronine
- T4
inactive thyroxine
- TGR5
G protein‐coupled bile acid receptor 1
- TNFα
tumor necrosis factor alpha
- TRAF6
TNF receptor associated factor 6
- TSC22D4
TSC22 domain family member 4
- UCP1
uncoupling protein 1
- UPS
ubiquitin‐proteasome system
- ZAG
zinc‐α2‐glycoprotein
Introduction
A well‐balanced and controlled energy homeostasis is critical for human health. Excessive caloric intake and insufficient physical activity lead to a plus in energy substrate storage, metabolic dysfunction, and obesity. Involuntary weight loss and an energy‐deprived metabolic state on the other hand are also coupled to severe metabolic dysfunction, including insulin resistance, fatty liver, and dyslipidemia 1. Chronic wasting conditions such as cancer‐induced cachexia, sepsis, and burn injuries share many of these metabolic phenotypes, suggesting that distinct etiologies may trigger common downstream cellular events eventually responsible for systemic body wasting. These common biochemical wasting cycles may include the classical Cori Cycle 2, enhanced lipid turnover 3, and creatine kinase‐dependent futile cycling 4 but in general remain still largely unexplored. In cancer cachexia, tumor metabolism may also be at least partially responsible for the wasting, since the aerobic glycolysis typical for tumor cells is energetically highly inefficient (Warburg effect).
The existence of tumors with identical origins and of equal size, one of which induces cachexia while the other does not, suggests that only a very limited number of genetic or gene expression events, e.g., in metabolic genes 5, may underlie the tumor‐associated cachexia phenotype. This may also be extrapolated to non‐tumorous wasting disorders as well as the peripheral target organs.
Interestingly, anorexia (loss of appetite, leading to reduced food intake) is a common hallmark of cancer, cancer therapies, and other wasting disorders. Multiple factors lead to anorexia, including secretion of proinflammatory cytokines from tumor and host, hormonal and neuroendocrine changes, and psychological changes such as depression or pain 6. While these factors are fundamentally involved in cachexia development, they form only one part of the equation summing up to the disease‐related weight loss. Indeed, body weight loss in cachexia cannot be reversed by treatment of anorexia, and conventional nutritional support neither improves muscle or adipose tissue loss nor functional impairment 7. Thus, decreased energy intake and its central control alone cannot be the sole reason for the wasting, suggesting that peripheral energy metabolism must be altered in the cachectic state. By tracking the body's main peripheral organ systems, we here discuss our current knowledge on tissue‐specific wasting mechanisms that impact systemic energy homeostasis and may thus represent key sites in the pathogenesis of severe human wasting disorders (Fig 1).
Figure 1. Overview of energy consuming processes in cachexia.
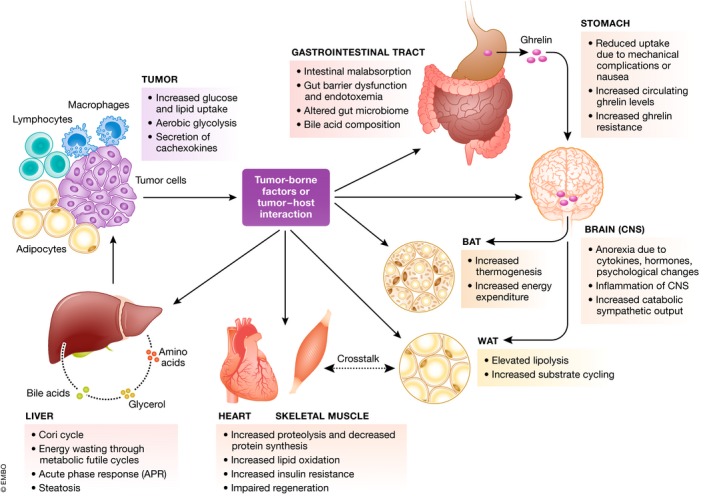
Tumor‐secreted factors or tumor/host interactions reduce energy uptake and activate energy‐wasting processes in different organ systems, acting on brain/CNS, adipose tissues, gastrointestinal system, liver, and muscles.
Absorption/malabsorption of energy substrates
Intestinal malabsorption can lead to reduced metabolic efficiency
Efficient recovery and absorption of nutrients by stomach and intestine are the first steps in whole‐body energy metabolism. In patients with gastrointestinal cancer, mechanical limitations such as difficulties in swallowing, as well as feelings of satiety or nausea, restrict energy uptake and recovery. Hence, weight loss occurs in up to 83% of patients with upper gastrointestinal malignancy 8. Further, nutrient malabsorption by the intestine can lead to reduced metabolic efficiency and, with equal energy intake, to wasting not only in patients with gastrointestinal cancer. Indeed, rodents with cancer‐induced cachexia show a marked decrease in intestinal lipid and carbohydrate uptake 9, 10. Reduced lipid uptake is also observed in patients with cachexia induced by chronic congestive heart failure 11. Protein or amino acid uptake, on the other hand, is either unaltered or slightly enhanced 10, 12. Malabsorption in cachexia due to chronic heart failure is thought to result from mesenteric ischemia and disturbed microcirculation of the intestine 13, while in cancer‐associated cachexia, chemotherapies are likely to contribute to the development of intestinal absorptive dysfunction 14. The molecular mechanisms contributing to intestinal malabsorption in cachexia are not clear yet. However, alterations in expression or localization of the major intestinal glucose (SGLT1, GLUT5, GLUT2) and lipid (CD36, NPC1L1, SRB1) transporters, as seen upon fasting and refeeding 15, are likely. Altered intestinal gluconeogenesis, a recently identified regulator of the central control of glucose and energy homeostasis, may further contribute to the energetic imbalance observed in cachexia: Tumor‐bearing rats show ameliorated wasting when fed an l‐glutamine‐rich diet, activating intestinal gluconeogenesis and thereby improving glycemia 16.
Microbiota and inflammation by gut leakage
The intestine has yet another role to play in wasting diseases. Gut barrier dysfunction and subsequent endotoxemia occur when the intestinal mucosal barrier leaks or breaks down, causing gut microbiota or bacterial cell wall components to enter the circulation and cause an inflammatory response. Measuring gut permeability to the neutral hydrophilic polymer FITC‐dextran, Puppa et al 17 have reported a marked reduction in intestinal gut barrier integrity in cachectic Apc(Min/+) mice with colorectal cancer. This is accompanied by significantly elevated plasma endotoxin concentrations. It may be speculated that the multiple intestinal lesions in this model contribute to gut dysfunction. Arguing against this and supporting a systemic effect also in gastrointestinal cancer, increased gut leakage is also apparent in an additional mouse model where colon cancer cells are injected subcutaneously, excluding a direct mechanical effect of the primary tumor. In the latter study, gut integrity is restored by treatment with an anti‐IL‐6 antibody, demonstrating the involvement of the inflammatory system in intestinal dysfunction in cachexia 18. Inflammation therefore seems to be both cause and consequence of gut dysfunction, at least in rodent models. Causality is less clear in humans, but as shown in patients with gastric cancer, increased bacterial translocation through the intestinal mucosa is associated with increased cytokine production, altered levels and activation of T cells, and worse prognosis 19. As discussed in Box 1, inflammatory mediators play a pivotal role in energy wasting in cachexia by multiple mechanisms.
Box 1: Inflammation and energy expenditure in brief.
An inflammatory status is an established stimulator of resting energy expenditure in cancer 7, originating both from cytokine secretion by the tumor and from the immune response of the host. Inflammation, mainly high levels of IL‐6 and TNFα, in cachexia triggers both high energy expenditure and muscle loss 7. In fact, injection of TNFα‐expressing tumor cells causes cachexia, while this is not the case for cells that do not express the cytokine 129. TNFα inhibits both adipocyte and myocyte differentiation, stimulates lipolysis, impairs insulin signaling, influences food intake, and directly causes muscle atrophy 7, 79, 130. It has also been shown to activate an intracellular futile substrate cycle between fructose 6‐phosphate and fructose 1,6‐bisphosphate, creating an energy sink 71. Intracerebroventricular administration of TNFα induces body weight loss and brown AT stimulation 131. In a recent retrospective cohort study with weight‐losing patients, a SNP in the TNF gene was newly associated with weight loss and a low skeletal muscle index in cachexia 132. SNPs in the genes influencing levels of other cytokines, such as IL‐1β, IL‐6, and IL‐10, are associated with cachexia in pancreatic and gastric cancer 133. Circulating IL‐6 levels correlate with weight loss and survival in patients with cancer 7, and IL‐6 has been shown to stimulate acute phase response in the liver 134. In summary, inflammation influences energy expenditure on the level of most organs and cells and is therefore of central importance for cachexia development. In fact, hypermetabolism is commonly associated with inflammation in all forms of cachexia, be it cachexia associated with chronic kidney disease 135, AIDS 136, burn injury 137, rheumatism 138, or cancer 7, 139.
Healthy energy metabolism is further modulated by the amount and quality (species) of gut microbiota. Balanced gut microbiota influence nutrition of the host as they determine nutrient metabolic efficiency, and microbiota‐derived metabolites alter inflammation, gut barrier function, and energy expenditure. For example, colonocytes usually metabolize microbiota‐derived SCFA such as butyrate but switch from oxidizing butyrate to fermenting glucose into lactate when butyrate is absent, resulting in an energetic defect of the host cell as insufficient ATP is produced 20. The gut microbiota produces SCFA by fermentation of non‐digestible carbohydrates, which may contribute to up to 10% of the daily energy requirements in humans. Dietary supplementation of the SCFA butyrate increases energy expenditure and mitochondrial function in mice 21.
The contribution of gut microbiota to metabolic health has been thoroughly studied in obesity/diabetes, in particular with regard to inflammation, SCFA action, and bile acids and cholesterol metabolism 22. Gut microbiota and their metabolites have also been proposed as potential targets for the prevention and treatment of other metabolic diseases such as cardiovascular disease, heart failure, and chronic kidney disease. The role of gut microbiota for energy metabolism in cachexia is less clear, but a common microbial signature for cachectic mice has recently been demonstrated 23, 24. For example, the selective modulation of Lactobacillus spp. affects muscle atrophy, inflammatory marker levels, and cachexia in a mouse leukemia model 23, suggesting a gut‐muscle signaling axis contributing to wasting. If tumor cells directly influence the gut microbiota through secreted factors or inflammatory mediators, or if the altered food and nutrient uptake are responsible for the observed changes is unclear so far. Likewise, the molecular mechanisms of this phenomenon are yet to be determined, but it is conceivable that microbiota‐derived metabolites impact energy metabolism of muscle, liver, or adipose tissue. The gut microbiota also influences the regulation of bile acids and cholesterol metabolism 22, with impact on fat resorption and energy expenditure. Importantly, restoring the lactobacilli levels in a strain‐ and/or species‐specific manner restores the molecular signs of muscle atrophy in cachectic mice 23. In rodents, synbiotics restoring the gut microbial signature have since been used to successfully treat muscle atrophy and cachexia‐associated body weight loss, independent of the site of the original tumor 24. Transfer of gut microbiota from healthy patients has been shown to improve insulin sensitivity in obese patients with metabolic syndrome, as shown by a landmark study from Vrieze et al 25. Thus, synbiotics or microbiota transplants may be tempting approaches for future cachexia therapy.
Ghrelin effects on wasting metabolism are closely linked to anorexia
Gut hormones have gained substantial interest with regard to body weight and metabolic regulation in recent years. In particular, the stomach‐derived ghrelin 26, the so‐called “hunger‐hormone”, has been extensively studied in the context of cachexia. Ghrelin was first found to improve cardiac cachexia in a rodent model of chronic heart failure (a syndrome also often associated with cachexia in patients) 27 and has since been explored for the treatment of other forms of cachexia and anorexia. Ghrelin's function in regulation of energy homeostasis has initially been described in the context of obesity 28, and ghrelin has since been shown to be involved in the regulation of appetite, gut motility and gastric acid secretion, white and brown adipose tissue function, and glucose metabolism, all of which influence energy homeostasis 29. Ghrelin levels are elevated upon fasting 28 and in multiple forms of cancer cachexia 30, 31, although this is not always the case 32. This increase may be closely linked to anorexia: A study comparing anorectic, cachectic, and anorectic–cachectic patients with non‐small‐cell lung cancer clearly demonstrated anorexia as determinant of circulating ghrelin levels 33. However, opposite to fasting, the increased ghrelin levels in cachexia fail to induce appetite and energy storage, potentially hinting to ghrelin resistance in these patients 31. Resistance to ghrelin action has been confirmed in cachectic rats 34.
Nonetheless, ghrelin has proven effective against both tumor‐ and chemotherapy‐induced cachexia in mice, acting directly on muscle cells to prevent atrophy by reducing inflammation, p38/CEBP‐β/myostatin, and activating Akt, myogenin, and myoD 35, 36. Ghrelin increases the energy intake in cancer patients with impaired appetite 37, and a large body of evidence from clinical trials now highlights the positive effect of ghrelin on patients with cachexia, in particular with regard to growth hormone plasma levels, weight gain, increases in lean mass, and reductions in loss of adipose tissue 38. Anamorelin, a ghrelin receptor agonist with anabolic and appetite‐enhancing activities, has successfully been tested in phase 3 studies in the treatment of non‐small‐cell lung cancer, where it ameliorates body weight and symptom burden 39. It should be noted however that handgrip strength was unaltered in these studies.
Other gut hormones, GLP‐1 and GIP, have less well‐defined roles in cachexia, despite their profound influence on metabolism. GLP‐1 promotes insulin secretion and has been extensively explored for the treatment of metabolic diseases such as obesity and type 2 diabetes. Altered glucose homeostasis and insulin sensitivity/insulin secretion have also been shown in cachexia 40, so a role for GLP‐1 is likely. Indeed, the GLP‐1 agonist and insulin sensitizer exendin‐4 partially prevents cachexia in tumor‐bearing rats 41. Recent evidence further suggests that brainstem GLP‐1 signaling influences body weight loss and lean/fat mass in cachectic rodents 42. As for GIP, its plasma levels decrease in cachexia in rheumatoid arthritis patients 43. Thus, gut hormones may be further explored for cachexia therapy in the future.
Bile acids as metabolic regulators
Bile acids (BA) are not only important for digestion and fat absorption but are also involved in the regulation of appetite and food intake, and plasma BA levels positively correlate with BMI, at least in obese patients 44. BA administration has been shown to suppress appetite in patients with a high BMI, yet plasma BA levels are negatively correlated with cognitive restraint of eating in obese patients 44. The high plasma BA levels in obesity may thus represent a compensatory mechanism to prevent further overeating. Treatment of healthy volunteers with the BA taurocholic acid dose dependently stimulates the secretion of GLP‐1 and PYY and increases the feeling of fullness 45. In mice, the administration of BA increases energy expenditure in brown adipose tissue and prevents obesity and insulin resistance upon high fat diet feeding 46. Mechanistically, BA induce energy expenditure by promoting intracellular thyroid hormone activation via D2, which converts inactive T4 into active T3. In the study by Watanabe et al, brown adipose tissue D2 levels and activity are induced by BA treatment. The BA effects on D2 and subsequent increase of oxygen consumption are mediated by the G protein‐coupled receptor TGR5. The BA‐D2‐TGR5 axis and positive effect on energy expenditure are also apparent in human primary myoblasts 46.
Opposite to what one might expect from these studies, plasma BA levels seem to be decreased rather than elevated in cachexia, at least in mice 47. The fat malabsorption seen in patients with cardiac cachexia further hints toward lower BA levels in wasting 11. Accordingly, the BA ursodeoxycholic acid has been successfully used to improve tissue loss in animals with progressive weight loss in the Yoshida hepatoma model of cancer cachexia, albeit to a low extent 48. Thus, the role of BA in regulating energy expenditure and food intake in the wasting pathology is not fully elucidated yet. It is conceivable that the composition of BA rather than total levels may be important for regulation of appetite and energy expenditure. This is further supported by a study by Roberts et al showing the correlation between GLP‐1 and ghrelin levels with a range of different BA. In response to a test meal in healthy patients, the levels of the BA glycochenodeoxycholic acid and glycodeoxycholic acid correlate positively with plasma levels of the anorexigenic GLP‐1, while the orexigenic ghrelin correlates negatively with taurochenodeoxycholic acid and taurodeoxycholic acid 49. In the context of cachexia, BA have shown limited potential under the tested circumstances so far. Thus, further studies are needed to fully understand the contribution of bile acids to energy metabolism in wasting diseases.
Dysfunctional peripheral handling of energy substrates
Energy dissipation through futile cycles in the liver
The liver as a highly metabolic organ contributes to energy wasting through metabolic futile cycles (Fig 2). This is true under both physiological and pathological conditions, such as in glycogen storage diseases, end‐stage liver diseases, and cachexia. Futile cycles are substrate cycles that dissipate energy without any anabolic or catabolic function. Futile cycles affecting the liver can be found in glycolysis/gluconeogenesis, where both pathways operate even under high glucose concentrations 50. Glycolytic products such as lactate remain constant not through pausing of glycolysis but rather through equal rates of lactate production and removal. The use of anaerobic muscle derived lactate by the liver through gluconeogenesis for glucose production is part of a large substrate cycle known as the Cori cycle. This is particularly evident in cancer patients, where lactate derived from tumors is reconverted to glucose via hepatic gluconeogenesis in a very energy consuming futile cycle that accounts for a big portion of energy loss 51. Hepatic gluconeogenesis can also be fueled by amino acids derived from muscle protein degradation 52, 53. It has further been suggested that energy is dissipated in a futile cycle initiated by mismatched amino acid composition originating from the liver acute phase response 54, which is activated in cachexia 55.
Figure 2. Overview of inter‐ and intra‐organ futile energy‐wasting cycles.
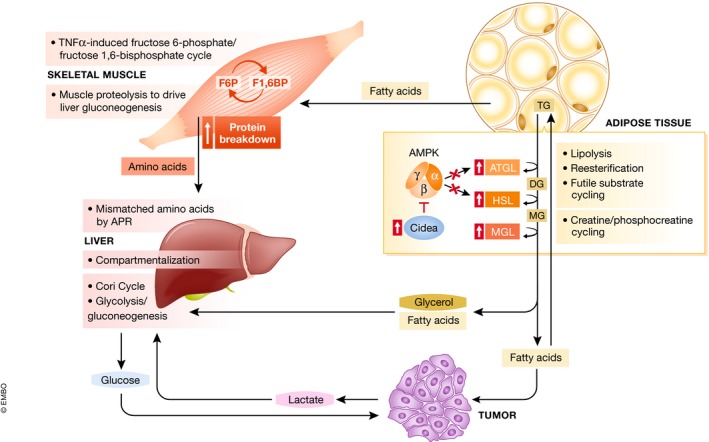
Futile cycles are mechanisms to regulate and fine‐tune metabolic processes under physiologic conditions. In muscle, adipose and liver, futile cycling is increased in cachectic conditions, and released metabolites are involved in inter‐organ cycles.
Interestingly, mathematical modeling of a genome‐wide human metabolic network revealed high amounts of futile cycles in humans, which highlight the importance of compartmentalization present in eukaryotes 56. These cellular compartments include cytosol, ER, Golgi apparatus, lysosomes, mitochondria, nucleus, and peroxisomes. Substrate cycles that employ multiple compartments are known to participate in energy dissipation through transport reactions between the different cellular organelles. An example in carbon metabolism is the hexokinase/glucose‐6‐phosphatase reaction, where in the liver hexokinase is replaced by GK, which is regulated by GKRP. Under fasting, GKRP inactivates and sequesters GK in the nucleus to prevent futile cycles of glucose phosphorylation during gluconeogenesis 57. Liver hexokinase activity is markedly increased in cachexia 52. However, it is not completely clear whether the amount of futile cycles changes between healthy individuals and patients with metabolic disorders, since the mathematical model is a combination of multi‐omics data without distinguishing the origin. It would be very interesting to construct two different mathematical models for healthy and disease conditions to compare the different metabolic fluxes and futile cycles in the two states.
Energy wasting in the liver under cachectic conditions leads to a reduction in the oxidative phosphorylation capacity of mitochondria due to an increase in mitochondrial cardiolipin content 58. This is accompanied by an increase of TNFα resulting in enhanced expression of phosphatidylglycerolphosphate synthase 59, which mediates cardiolipin expression 60. In addition, decreased usage of hepatic triglyceride stores has been demonstrated in cancer cachexia, due to enhanced expression of the transcriptional cofactors RIP140 61 and TSC22D4 62, leading to hepatic steatosis. Increased liver lipogenesis, as observed in cachectic rats, may further contribute to the hyperlipidemia 9. Hepatic steatosis is a prerequisite of nonalcoholic fatty liver disease and is associated with sarcopenia or muscle loss 63, which also occurs in the majority of patients with chronic liver disease. Liver cirrhosis causes an imbalance in the metabolic rate, leading to increased energy expenditure, insulin resistance, and increased fat turnover, resulting in a hypermetabolic state 64. Apparently, an increase in blood ammonia levels induces skeletal muscle autophagy and up‐regulation of myostatins 65, 66; however, the direct underlying molecular mechanisms are not understood. In summary, there seems to be a strong inter‐organ relationship between liver and muscle under energy‐wasting conditions.
There are still many open questions regarding the role of the liver for cachexia, as exemplified by other forms of energy wasting in the liver, such as glycogen storage diseases, rare human diseases caused by abnormalities in proteins that regulate the synthesis or degradation of glycogen. Liver glycogen storage diseases are mainly characterized by hypoglycemia and, when occurring during infancy, are associated with poor prognosis. The importance of cellular compartments, such as the ER and lysosomes, is evident in glycogen storage disease type 1 (von Gierke disease or G6Pase deficiency) as well as glycogen storage disease type 2 (Pompe disease). G6Pase is an ER resident protein that catalyzes the terminal reaction of glycogenolysis and gluconeogenesis by hydrolyzing G6P to glucose and phosphate 67. Multiple transport systems exist to shuttle the different substrates in and out of the ER. Deficiency of G6Pase or any of the transporters causes an accumulation of glycogen in the cytosol and a swelling of the ER probably due to the accumulation of glucose‐6‐phosphate. Pompe disease is a lysosomal storage disease characterized by mutations in the GAA enzyme, which is responsible for the cleavage of α‐1,4‐ and α‐1,6‐glycosidic bonds of glycogen leading to lysosomal accumulation of glycogen in particular in skeletal and cardiac muscle, but also in liver 68. The relevance of the liver in this storage disease is emphasized by a liver‐specific enzyme replacement therapy approach using AAV8 vectors for GAA transgene expression in mice with Pompe disease. Expression of GAA in liver resulted in GAA secretion from the liver and enhanced GAA activity in plasma and peripheral tissues, leading to the correction of glycogen accumulation not only in liver but also skeletal muscles, emphasizing the importance of tissue crosstalk between liver and muscle in this disease 69. Given its importance for other metabolic diseases, surprisingly, cellular compartmentalization in cachexia has not been studied so far.
Skeletal muscle and cardiac muscle atrophy are hallmarks of cachexia
Skeletal muscle is a highly plastic tissue with the ability to adapt its structure and metabolism in response to different physiological stimuli. Under pathological conditions like cancer cachexia, several cues derived either from the host or tumor compromise muscle homeostasis, resulting in wasting and impairment of function and metabolism of this tissue. Being the main site of protein storage in the body, muscle both balances the metabolic demands of other organs and serves as a protein reserve for energy production, for example, during starvation. It is well established that inflammatory mediators including IL‐1b, IL‐6, or TNFα play a crucial role in muscle wasting, and recent studies have identified additional candidates like soluble proteins, exosomes, and metabolites that are associated with cachexia and potentially induce muscle atrophy 70. TNFα has been shown to activate a futile cycle between fructose 6‐phosphate and fructose 1,6‐bisphosphate which acts as ATP sink for elevated glycolytic activity, leading to increased energy expenditure, heat production, and tissue wasting in C2C12 myotubes 71. An inter‐organ futile energy‐wasting cycle is generated by increased muscle proteolysis and release of amino acids to sustain tumor protein synthesis and liver gluconeogenesis 52, 53. Efficiency is low when amino acids are used for energy production, and the removal of waste nitrogen building up during this process requires additional energy, profoundly altering the normal homeostatic energy balance and contributing to energy wasting in cachexia 51.
Although increased futile cycling in the muscle occurs and might contribute to wasting, it is probably not a major factor causing atrophy. Instead, muscle atrophy during cancer cachexia is induced by increased protein breakdown mediated mainly by the activation of the UPS 72 and the autophagic–lysosomal system 73. In the UPS, the 26S proteasome complex recognizes the substrates covalently attached to a chain of ubiquitin molecules for breakdown 74. Many classes of enzymes (E1, E2, and E3) are involved in protein ubiquitination, but the E3 ubiquitin ligases are rate‐limiting for the ubiquitination process and are well‐known markers of UPS activity 74. MuRF1 and Atrogin 1 were the first E3‐ligases identified and are considered master markers of muscle atrophy. These E3‐ligases are strongly upregulated in some mouse models of cancer cachexia 75, 76, 77. MuRF1 is involved in the ubiquitination and degradation of muscle structural proteins including beta/slow MHC, MHCIIa, actin, myosin binding protein C and myosin light chains 1 and 2, and troponin I 74. Atrogin 1‐induced atrophy is mediated by degradation of eIF3f, which suppresses S6K1 activation induced by mTOR as well as MyoD breakdown 78, blocking differentiation and inhibiting myotube formation. These data might partially explain the reduced protein synthesis 74 and impaired muscle regeneration in atrophic muscle during cancer cachexia observed in previous studies 79, 80. Importantly, mice lacking Atrogin 1 81 and MuRF1 75 are partially protected from fasting‐ and denervation‐induced muscle wasting, respectively. Current studies have shown the involvement of newly emerged E3‐ligases TRAF6 82 and MUSA1 83 as potential targets of cachexia in rodent models of muscle atrophy. However, whether or not these E3‐ligases spare muscle loss in cachexia still needs to be investigated.
Autophagy is an evolutionarily conserved process involved in the degradation of target cytoplasmic materials in a lysosome‐dependent manner. Autophagy is activated during starvation, where its role is to recycle intracellular components to maintain mitochondrial metabolic function, thereby maintaining energy homeostasis. The recycled material can thus be seen as a cellular nutrient store which is released upon activation of autophagy. Although lysosome‐dependent proteolysis has been shown to be activated in muscle cells during catabolic conditions several years ago 84, 85, 86, only in the last few years enhanced autophagy has been found in muscle 73 and heart 87 of tumor‐bearing animals. As previously observed in atrophic models of muscle denervation or starvation 84, 85, autophagy also plays a role in muscle atrophy in Apc(Min/+), C26 73, and Lewis Lung Carcinoma tumor‐bearing mice 86. Reinforcing the role of autophagy in muscle atrophy, Penna et al 73 have observed that this proteolytic pathway was induced in muscle of three different models of cancer cachexia as well as in glucocorticoid‐treated mice. Further, several autophagy markers such as Atg5, Beclin1, and Gabarap are induced in skeletal muscle of cachectic patients affected by pancreatic cancer, esophageal cancer, and gastric cancer 88, 89. There is conflicting evidence to whether or not cancer cachexia affects fiber type composition 90. As often the case in this field, this can partly be explained by the criteria employed to define cachexia. Several studies have shown a predominant loss of type II fibers under different wasting conditions and in cachectic mouse models 90. Interestingly, type II skeletal muscle fiber atrophy associated with altered autophagy is also observed in atrophic conditions induced by Pompe disease, fasting, and sarcopenia 91. The relative preservation of the slow‐twitch oxidative type I fibers potentially reflects the body's attempt at conserving energy. Taken together, these results demonstrate that autophagy, either excessive or defective, contributes to the complicated network that leads to muscle atrophy during cachexia 73.
Beyond the induction of UPS and autophagy, excessive muscle fatty acid oxidation has been demonstrated as a driver of muscle atrophy in a mouse model of kidney cancer 92. Cachectic kidney cancer cell lines secret a cocktail of inflammatory factors that rapidly lead to high levels of fatty acid oxidation and activation of a p38 MAPK‐response signature in skeletal muscles, which precedes the manifestation of atrophy 92. Interestingly, the in vitro and in vivo pharmacological blockage of fatty acid oxidation rescues the muscle atrophy induced by kidney cancer cells 92, suggesting a possible crosstalk between adipose tissues and skeletal muscle. Aberrant fatty acid metabolism might also be involved in cardiac atrophy induced by C26 tumor‐bearing mice. Importantly, this study by Schäfer et al 93 has identified a signature panel of “cachexokines” including Ataxin‐10 which are sufficient and necessary to trigger the aberrant fatty acid metabolism and cardiac atrophy. Ataxin‐10 serum levels are also elevated in cachectic cancer patients and might be a potential target to treat heart wasting 93. Since cachectic patients in advanced cancer stages often become intolerant to anticancer therapies, and those with severe wasting of skeletal muscles and cardiac muscle often die prematurely due to respiratory and cardiac failure, identification of extracellular triggers or intracellular mediators of muscle wasting might be critical for the treatment of cancer cachexia and prolong the rate of survival.
Disturbed adipose tissue energy handling
White adipose tissue
The major role of the white adipose tissue is to store energy as fat, a storage form approximately sixfold more efficient than carbohydrates due to fat's hydrophobic nature. Complete oxidation of fatty acids yields about 9 kcal/g, while carbohydrates and proteins yield about 4 kcal/g. When needed, as upon exercise or after prolonged fasting, energy is mobilized from the adipose tissue by triglyceride hydrolysis (lipolysis), releasing fatty acids and glycerol into the circulation for oxidation by other organs. Tumor‐bearing animals show increased lipid utilization, and hence, adipose tissue is degraded 3. Lipolysis is a sequential process orchestrated by three lipases, ATGL, HSL, and MGL. Elevated lipolysis is a characteristic of cachexia in both patients and rodent models 94, 95, leading to adipose tissue wasting and elevated circulating free fatty acid levels 96. Altered levels and/or activity of ATGL and HSL in white adipose tissue are causal for this 95, 97. Knockout of ATGL or HSL in mice partly protects against cancer‐induced wasting 95. Signal for the increased energy demand in fasting are catecholamines, and lipolysis can also be activated by cortisol, natriuretic peptides, and proinflammatory cytokines, while insulin inhibits lipolysis. Adipocytes isolated from cachectic patients show stronger catecholamine‐ and natriuretic peptide‐induced lipolysis 98. Weight‐losing patients are also more sensitive to catecholamine signaling 99. Transcriptome profiling of adipose tissue from gastrointestinal cancer patients with cachexia reveals pathways regulating energy turnover are upregulated 100. Further, insulin resistance or reduced insulin secretion often occur in patients with cachexia 101, potentially preventing insulin from exerting its anti‐lipolytic effect. The importance of insulin for adipose tissue integrity is emphasized by the severe lipodystrophy of mice lacking the insulin receptor specifically in adipose tissue 102. In cachexia, lipolysis has been shown to be further activated by tumor‐secreted ZAG, levels of which are elevated in patients with cancer cachexia, contributing to weight loss in patients with cancer 103, 104. Similar to lipolytic hormones, ZAG binds to a β3‐adrenergic receptor and stimulates adenylyl cyclase in a GTP‐dependent process 105. Inflammatory cytokines such as TNFα or IL‐6 contribute to adipose loss both by directly activating lipolysis and by impairing insulin sensitivity 1, 106.
Glycerol released from adipose tissue during lipolysis can serve as a substrate for gluconeogenesis in the liver, leading to an energy‐wasting effect as part of futile substrate cycling. Fatty acids generated during lipolysis are mostly distributed to other organs for energy usage, but some are also re‐esterified to triglycerides during lipogenesis in the adipose tissue, again creating a futile cycle (Fig 2). Adipocyte lipogenesis has indeed been shown to increase in cachexia 97, 107. With both lipolysis and lipogenesis induced upon stimulation by tumor‐secreted factors, enhanced substrate cycling occurs. This is an energy costly process, creating an enhanced energy demand which, in light of the undersupply frequently occurring in cachexia, is seldom met and contributes to adipose tissue wasting. Indeed, intracellular ATP levels are low in cachectic adipocytes 97. The enhanced lipolysis–lipogenesis futile cycling is mediated by impaired signaling through AMPK, activity of which is reduced in cachectic adipose tissue 97. Interestingly, as shown by Mulligan et al, de novo lipogenesis occurs not only in adipose tissue but also in kidney and liver of cachectic mice. This may result in reduced glucose availability and hence loss of utilizable carbohydrate energy, further favoring catabolism by increasing overall energy requirements 107.
Brown and brite adipose tissue
While white adipose tissue serves mainly as energy storing organ, brown adipose tissue uses stored energy for heat production during thermogenesis, a highly energy costly process. Brown adipose tissue is found in cachectic mice and patients with cancer 108, 109. Catecholamine signaling in brown adipose tissue leads to expression of UCP1, causing heat generation by uncoupling oxidative phosphorylation from ATP production. Accordingly, catecholamine levels correlate with brown adipose tissue activity and BMI, also in humans 110. Catecholamine signaling is enhanced in cachectic mice, but increased thermogenesis can be prevented by β‐adrenergic blockade with propranolol 108. Expression profiling of cachectic mice with colorectal cancer reveals alterations in key regulators of lipid accumulation and fatty acid β‐oxidation, including increased UCP1 expression, indicative of active brown adipose tissue 111. Brown fat activity also correlates positively with cancer stage when adjusted to ambient temperature 112. Thus, there is ample evidence that brown adipose tissue is activated in various conditions of cachexia in mice and men.
It is estimated that humans possess up to 60 g brown fat, contributing to 15–25 kcal/day 113. Other studies estimate the contribution of brown AT to total basal metabolic rate to be around 3–5% 114. Considering the small overall amount of brown fat, it is thus questionable if this is sufficient to substantially contribute to increased energy expenditure and wasting. In contrast, skeletal muscle accounts for up to 40% of body weight, accounting for 20–30% of total resting energy expenditure 115.
In addition to classical brown adipose tissue, white adipose tissue can contain varying amounts of brite (“brown in white”) adipocytes that are thermogenically active. Patients with pheochromocytoma (catecholamine‐secreting tumors) display white‐to‐brown fat conversion as indicated by elevated UCP1 staining of the omental adipose tissue 116. Unfortunately, no observations regarding body weight or energy expenditure were made in this study. Increased UCP1 mRNA and protein levels are also described in both patients and mice with cachexia 117, 118, as is increased energy uptake, measured by oxygen consumption rate but also heat generation. IL‐6 was identified as one of the drivers for this browning effect 106, 117.
While multiple lines of evidence suggest that brite fat‐induced thermogenesis contributes to increased energy expenditure in cachexia 106, 117, 118, 119, 120, there is also data questioning the prominent role of browning for adipose and body wasting. Brown or brite adipose tissue thermogenesis contributes to the body's ability to defend against cold temperatures, so more brown/brite AT would suggest elevated body temperature or cold tolerance. However, in temperature preference studies, mice carrying tumors select a higher ambient temperature than mice without tumors, indicating elevated cold stress in tumor‐bearing mice 121. Further, cachectic Apc(Min/+) mice experience a gradual decline in body temperature during development of body wasting 17. Finally, mice housed at thermoneutrality develop cachexia exactly parallel to mice housed at room temperature (a mild cold stress activating brown/brite AT) 97—in contrast, tumor growth and inflammation are aggravated by housing at room temperature 121. While increased AT UCP1 levels are recorded for several murine and human cancers 106, 117, 118, 119, 120, this is not consistently seen, as some reports demonstrate unchanged or even reduced adipose UCP1 levels 97, 122. Mice lacking UCP1 are not protected from cachexia development or adipose tissue loss, questioning the prominent role of browning for wasting 97. Further, 6‐hydroxydopamine‐induced adrenergic blockade does not protect from tumor‐induced AT wasting 97. However, other, UCP1 independent energy dissipation mechanisms have been described in brite fat, albeit not in the context of cachexia so far. These include activation of creatine/phosphocreatine cycling 4, and ATP‐dependent Ca2+ cycling 123, again emphasizing the importance of cellular compartmentalization for energy homeostasis. Thus, to further clarify the role of adipose tissue it will certainly be helpful to gain further insights into futile energy‐wasting cycles, quantify the capacity of brown/brite fat activation to increase whole‐body energy expenditure, and to quantify its contribution to wasting in patients with cachexia.
Future directions
The present review discusses pathways in which energy metabolism is at fault under pathologic wasting conditions, be it through altered energy uptake or energy dissipation for example by inter‐organ or intracellular futile cycles. Many of the presented studies show increased energy expenditure in cachexia. It is thus surprising that a large systematic literature review of domains associated with involuntary weight loss in cancer shows no correlation between energy expenditure and weight loss 124. Indeed, after correction for changes in body composition, no alterations in energy expenditure are observed in patients with pancreatic cancer 125. Likewise, in cachectic mice, even with severe cachexia, increased energy expenditure is not always seen 97, or only after dividing oxygen consumption by body weight, which is arguably lower in cachexia.
This may indicate a compensatory decrease in energy expenditure in response to poor caloric intake. Indeed, in a pilot study with 10 subjects, intensive, biometric parameter‐oriented dietary counseling was sufficient to improve cachexia and prolong life in cachectic cancer patients 126. However, meta‐analyses have largely failed to show an improvement of mortality due to nutritional intervention 127, partially due to low compliance to the therapeutic intervention 128. This overall suggests that even behavioral and/or psychological aspects have to be considered in order to improve metabolic dysfunction as associated with metabolic wasting.
While research over the past years has provided significant progress in our understanding of peripheral wasting mechanisms (Fig 1), the system‐wide integration of site‐specific contributions to organismal energy remains a grand challenge for future research and therapy development. This relates in particular to the identification of common therapeutic targets between distinct wasting disorders as well as between distinct tumor entities.
In addition, the contribution of the brain‐periphery axis to systemic wasting (beyond appetite control) has been largely ignored up to date but will definitely provide critical insights into integrative wasting mechanisms.
Given the remarkable similarity in metabolic phenotypes between obesity and metabolic wasting, addressing the abovementioned neglected areas as well as improving technical approaches to reliably determine energy expenditure particularly in humans will represent important research avenues for the years to come. It can be envisaged that key mechanisms in wasting disorders might eventually be used in anti‐obesity therapies and vice versa, thereby further highlighting the necessity to intensify biomedical research and translation in wasting disorders in the future.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by grants from the Helmholtz Association program AMPro to SH.
EMBO Reports (2019) 20: e47258
See the Glossary for abbreviations used in this article.
References
- 1. Vegiopoulos A, Rohm M, Herzig S (2017) Adipose tissue: between the extremes. EMBO J 36: 1999–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Argilés JM, Fontes‐Oliveira CC, Toledo M, López‐Soriano FJ, Busquets S (2014) Cachexia: a problem of energetic inefficiency. J Cachexia Sarcopenia Muscle 5: 279–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mulligan HD, Beck SA, Tisdale MJ (1992) Lipid metabolism in cancer cachexia. Br J Cancer 66: 57–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kazak L, Chouchani ET, Jedrychowski MP, Erickson BK, Shinoda K, Cohen P, Vetrivelan R, Lu GZ, Laznik‐Bogoslavski D, Hasenfuss SC et al (2015) A creatine‐driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 163: 643–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Monitto CL, Berkowitz D, Lee KM, Pin S, Li D, Breslow M, O'Malley B, Schiller M (2001) Differential gene expression in a murine model of cancer cachexia. Am J Physiol Endocrinol Metab 281: E289–E297 [DOI] [PubMed] [Google Scholar]
- 6. Ezeoke CC, Morley JE (2015) Pathophysiology of anorexia in the cancer cachexia syndrome. J Cachexia Sarcopenia Muscle 6: 287–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fearon KCH, Glass DJ, Guttridge DC (2012) Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166 [DOI] [PubMed] [Google Scholar]
- 8. Deans DAC, Tan BH, Wigmore SJ, Ross JA, de Beaux AC, Paterson‐Brown S, Fearon KCH (2009) The influence of systemic inflammation, dietary intake and stage of disease on rate of weight loss in patients with gastro‐oesophageal cancer. Br J Cancer 100: 63–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. López‐Soriano J, Argilés JM, López‐Soriano FJ (1996) Lipid metabolism in rats bearing the Yoshida AH‐130 ascites hepatoma. Mol Cell Biochem 165: 17–23 [DOI] [PubMed] [Google Scholar]
- 10. Gomes‐Marcondes MC, Honma HN, Areas MA, Cury L (1998) Effect of Walker 256 tumor growth on intestinal absorption of leucine, methionine and glucose in newly weaned and mature rats. Braz J Med Biol Res 31: 1345–1348 [DOI] [PubMed] [Google Scholar]
- 11. King D, Smith ML, Chapman TJ, Stockdale HR, Lye M (1996) Fat malabsorption in elderly patients with cardiac cachexia. Age Ageing 25: 144–149 [DOI] [PubMed] [Google Scholar]
- 12. King D, Smith ML, Lye M (1996) Gastro‐intestinal protein loss in elderly patients with cardiac cachexia. Age Ageing 25: 221–223 [DOI] [PubMed] [Google Scholar]
- 13. Sandek A, Rauchhaus M, Anker SD, von Haehling S (2008) The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care 11: 632–639 [DOI] [PubMed] [Google Scholar]
- 14. Keefe DM, Cummins AG, Dale BM, Kotasek D, Robb TA, Sage RE (1997) Effect of high‐dose chemotherapy on intestinal permeability in humans. Clin Sci (Lond) 92: 385–389 [DOI] [PubMed] [Google Scholar]
- 15. Habold C, Foltzer‐Jourdainne C, Le Maho Y, Lignot J‐H, Oudart H (2005) Intestinal gluconeogenesis and glucose transport according to body fuel availability in rats. J Physiol 566: 575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martins HA, Bazotte RB, Vicentini GE, Lima MM, Guarnier FA, Hermes‐Uliana C, Frez FCV, Bossolani GDP, Fracaro L, Fávaro LDS et al (2017) l‐Glutamine supplementation promotes an improved energetic balance in Walker‐256 tumor‐bearing rats. Tumour Biol 39: 1010428317695960 [DOI] [PubMed] [Google Scholar]
- 17. Puppa MJ, White JP, Sato S, Cairns M, Baynes JW, Carson JA (2011) Gut barrier dysfunction in the ApcMin/+mouse model of colon cancer cachexia. Biochim Biophys Acta Mol Basis Dis 1812: 1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bindels LB, Neyrinck AM, Loumaye A, Catry E, Walgrave H, Cherbuy C, Leclercq S, Van Hul M, Plovier H, Pachikian B et al (2018) Increased gut permeability in cancer cachexia: mechanisms and clinical relevance. Oncotarget 9: 18224–18238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mi L, Lin J, Zheng H, Xu X, Zhang J, Zhang D (2012) Bacterial translocation contributes to cachexia from locally advanced gastric cancer. Hepatogastroenterology 59: 2348–2351 [DOI] [PubMed] [Google Scholar]
- 20. Donohoe DR, Wali A, Brylawski BP, Bultman SJ (2012) Microbial regulation of glucose metabolism and cell‐cycle progression in mammalian colonocytes. PLoS One 7: e46589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J (2009) Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58: 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baothman OA, Zamzami MA, Taher I, Abubaker J, Abu‐Farha M (2016) The role of Gut Microbiota in the development of obesity and Diabetes. Lipids Health Dis 15: 108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bindels LB, Beck R, Schakman O, Martin JC, De Backer F, Sohet FM, Dewulf EM, Pachikian BD, Neyrinck AM, Thissen J‐P et al (2012) Restoring specific lactobacilli levels decreases inflammation and muscle atrophy markers in an acute leukemia mouse model. PLoS One 7: e37971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bindels LB, Neyrinck AM, Claus SP, Le Roy CI, Grangette C, Pot B, Martinez I, Walter J, Cani PD, Delzenne NM (2016) Synbiotic approach restores intestinal homeostasis and prolongs survival in leukaemic mice with cachexia. ISME J 10: 1456–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JFWM, Dallinga‐Thie GM, Ackermans MT, Serlie MJ, Oozeer R et al (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143: 913–916 e7 [DOI] [PubMed] [Google Scholar]
- 26. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999) Ghrelin is a growth‐hormone‐releasing acylated peptide from stomach. Nature 402: 656–660 [DOI] [PubMed] [Google Scholar]
- 27. Nagaya N, Uematsu M, Kojima M, Ikeda Y, Yoshihara F, Shimizu W, Hosoda H, Hirota Y, Ishida H, Mori H et al (2001) Chronic administration of ghrelin improves left ventricular dysfunction and attenuates development of cardiac cachexia in rats with heart failure. Circulation 104: 1430–1435 [DOI] [PubMed] [Google Scholar]
- 28. Tschöp M, Smiley DL, Heiman ML (2000) Ghrelin induces adiposity in rodents. Nature 407: 908–913 [DOI] [PubMed] [Google Scholar]
- 29. Müller TD, Nogueiras R, Andermann ML, Andrews ZB, Anker SD, Argente J, Batterham RL, Benoit SC, Bowers CY, Broglio F et al (2015) Ghrelin. Mol Metab 4: 437–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takahashi M, Terashima M, Takagane A, Oyama K, Fujiwara H, Wakabayashi G (2009) Ghrelin and leptin levels in cachectic patients with cancer of the digestive organs. Int J Clin Oncol 14: 315–320 [DOI] [PubMed] [Google Scholar]
- 31. Shimizu Y, Nagaya N, Isobe T, Imazu M, Okumura H, Hosoda H, Kojima M, Kangawa K, Kohno N (2003) Increased plasma ghrelin level in lung cancer cachexia. Clin Cancer Res 9: 774–778 [PubMed] [Google Scholar]
- 32. Huang Q, Fan YZ, Ge BJ, Zhu Q, Tu ZY (2007) Circulating ghrelin in patients with gastric or colorectal cancer. Dig Dis Sci 52: 803–809 [DOI] [PubMed] [Google Scholar]
- 33. Blauwhoff‐Buskermolen S, Langius JAE, Heijboer AC, Becker A, de van der Schueren MAE, Verheul HMW (2017) Plasma Ghrelin levels are associated with anorexia but not cachexia in patients with NSCLC. Front Physiol 8: 119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Terawaki K, Kashiwase Y, Sawada Y, Hashimoto H, Yoshimura M, Ohbuchi K, Sudo Y, Suzuki M, Miyano K, Shiraishi S et al (2017) Development of ghrelin resistance in a cancer cachexia rat model using human gastric cancer‐derived 85As2 cells and the palliative effects of the Kampo medicine rikkunshito on the model. PLoS One 12: e0173113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen J‐A, Splenser A, Guillory B, Luo J, Mendiratta M, Belinova B, Halder T, Zhang G, Li Y‐P, Garcia JM (2015) Ghrelin prevents tumour‐ and cisplatin‐induced muscle wasting: characterization of multiple mechanisms involved. J Cachexia Sarcopenia Muscle 6: 132–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zeng X, Chen S, Yang Y, Ke Z (2017) Acylated and unacylated ghrelin inhibit atrophy in myotubes co‐cultured with colon carcinoma cells. Oncotarget 8: 72872–72885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neary NM, Small CJ, Wren AM, Lee JL, Druce MR, Palmieri C, Frost GS, Ghatei MA, Coombes RC, Bloom SR (2004) Ghrelin increases energy intake in cancer patients with impaired appetite: acute, randomized, placebo‐controlled trial. J Clin Endocrinol Metab 89: 2832–2836 [DOI] [PubMed] [Google Scholar]
- 38. Mansson JV, Alves FD, Biolo A, Souza GC (2016) Use of ghrelin in cachexia syndrome: a systematic review of clinical trials. Nutr Rev 74: 659–669 [DOI] [PubMed] [Google Scholar]
- 39. Temel JS, Abernethy AP, Currow DC, Friend J, Duus EM, Yan Y, Fearon KC (2016) Anamorelin in patients with non‐small‐cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomised, double‐blind, phase 3 trials. Lancet Oncol 17: 519–531 [DOI] [PubMed] [Google Scholar]
- 40. Holroyde CP, Skutches CL, Boden G, Reichard GA (1984) Glucose metabolism in cachectic patients with colorectal cancer. Cancer Res 44: 5910–5913 [PubMed] [Google Scholar]
- 41. Honors MA, Kinzig KP (2014) Chronic exendin‐4 treatment prevents the development of cancer cachexia symptoms in male rats bearing the Yoshida sarcoma. Horm Cancer 5: 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Borner T, Liberini CG, Lutz TA, Riediger T (2018) Brainstem GLP‐1 signalling contributes to cancer anorexia‐cachexia syndrome in the rat. Neuropharmacology 131: 282–290 [DOI] [PubMed] [Google Scholar]
- 43. Chen C‐Y, Tsai C‐Y, Lee P‐C, Lee S‐D (2013) Long‐term etanercept therapy favors weight gain and ameliorates cachexia in rheumatoid arthritis patients: roles of gut hormones and leptin. Curr Pharm Des 19: 1956–1964 [DOI] [PubMed] [Google Scholar]
- 44. Prinz P, Hofmann T, Ahnis A, Elbelt U, Goebel‐Stengel M, Klapp BF, Rose M, Stengel A (2015) Plasma bile acids show a positive correlation with body mass index and are negatively associated with cognitive restraint of eating in obese patients. Front Neurosci 9: 199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu T, Bound MJ, Standfield SD, Gedulin B, Jones KL, Horowitz M, Rayner CK (2013) Effects of rectal administration of taurocholic acid on glucagon‐like peptide‐1 and peptide YY secretion in healthy humans. Diabetes Obes Metab 15: 474–477 [DOI] [PubMed] [Google Scholar]
- 46. Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T et al (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439: 484–489 [DOI] [PubMed] [Google Scholar]
- 47. Castellani C, Singer G, Kaiser M, Kaiser T, Huang J, Sperl D, Kashofer K, Fauler G, Guertl‐Lackner B, Höfler G et al (2017) Neuroblastoma causes alterations of the intestinal microbiome, gut hormones, inflammatory cytokines, and bile acid composition. Pediatr Blood Cancer 64 [DOI] [PubMed] [Google Scholar]
- 48. Tschirner A, von Haehling S, Palus S, Doehner W, Anker SD, Springer J (2012) Ursodeoxycholic acid treatment in a rat model of cancer cachexia. J Cachexia Sarcopenia Muscle 3: 31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roberts RE, Glicksman C, Alaghband‐Zadeh J, Sherwood RA, Akuji N, le Roux CW (2011) The relationship between postprandial bile acid concentration, GLP‐1, PYY and ghrelin. Clin Endocrinol 74: 67–72 [DOI] [PubMed] [Google Scholar]
- 50. Phillips JW, Clark DG, Henly DC, Berry MN (1995) The contribution of glucose cycling to the maintenance of steady‐state levels of lactate by hepatocytes during glycolysis and gluconeogenesis. Eur J Biochem 227: 352–358 [DOI] [PubMed] [Google Scholar]
- 51. Friesen DE, Baracos VE, Tuszynski JA (2015) Modeling the energetic cost of cancer as a result of altered energy metabolism: implications for cachexia. Theor Biol Med Model 12: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Argilés JM, Campos N, Lopez‐Pedrosa JM, Rueda R, Rodriguez‐Mañas L (2016) Skeletal muscle regulates metabolism via interorgan crosstalk: roles in health and disease. J Am Med Dir Assoc 17: 789–796 [DOI] [PubMed] [Google Scholar]
- 53. Argilés JM, López‐Soriano FJ (1991) The energy state of tumor‐bearing rats. J Biol Chem 266: 2978–2982 [PubMed] [Google Scholar]
- 54. Fearon KC, Barber MD, Falconer JS, McMillan DC, Ross JA, Preston T (1999) Pancreatic cancer as a model: inflammatory mediators, acute‐phase response, and cancer cachexia. World J Surg 23: 584–588 [DOI] [PubMed] [Google Scholar]
- 55. Narsale AA, Enos RT, Puppa MJ, Chatterjee S, Murphy EA, Fayad R, Pena MO, Durstine JL, Carson JA (2015) Liver inflammation and metabolic signaling in ApcMin/+ Mice: the role of cachexia progression. PLoS One 10: e0119888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gebauer J, Schuster S, de Figueiredo LF, Kaleta C (2012) Detecting and investigating substrate cycles in a genome‐scale human metabolic network. FEBS J 279: 3192–3202 [DOI] [PubMed] [Google Scholar]
- 57. Lloyd DJ, St Jean DJ, Kurzeja RJM, Wahl RC, Michelsen K, Cupples R, Chen M, Wu J, Sivits G, Helmering J et al (2013) Antidiabetic effects of glucokinase regulatory protein small‐molecule disruptors. Nature 504: 437–440 [DOI] [PubMed] [Google Scholar]
- 58. Julienne CM, Tardieu M, Chevalier S, Pinault M, Bougnoux P, Labarthe F, Couet C, Servais S, Dumas J‐F (2014) Cardiolipin content is involved in liver mitochondrial energy wasting associated with cancer‐induced cachexia without the involvement of adenine nucleotide translocase. Biochim Biophys Acta 1842: 726–733 [DOI] [PubMed] [Google Scholar]
- 59. Peyta L, Jarnouen K, Pinault M, Coulouarn C, Guimaraes C, Goupille C, de Barros J‐PP, Chevalier S, Dumas J‐F, Maillot F et al (2015) Regulation of hepatic cardiolipin metabolism by TNFα: implication in cancer cachexia. Biochim Biophys Acta 1851: 1490–1500 [DOI] [PubMed] [Google Scholar]
- 60. Morita S, Terada T (2015) Enzymatic measurement of phosphatidylglycerol and cardiolipin in cultured cells and mitochondria. Sci Rep 5: 11737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Berriel Diaz M, Krones‐Herzig A, Metzger D, Ziegler A, Vegiopoulos A, Klingenspor M, Müller‐Decker K, Herzig S (2008) Nuclear receptor cofactor receptor interacting protein 140 controls hepatic triglyceride metabolism during wasting in mice. Hepatology 48: 782–791 [DOI] [PubMed] [Google Scholar]
- 62. Jones A, Friedrich K, Rohm M, Schäfer M, Algire C, Kulozik P, Seibert O, Müller‐Decker K, Sijmonsma T, Strzoda D et al (2013) TSC22D4 is a molecular output of hepatic wasting metabolism. EMBO Mol Med 5: 294–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bhanji RA, Narayanan P, Allen AM, Malhi H, Watt KD (2017) Sarcopenia in hiding: the risk and consequence of underestimating muscle dysfunction in nonalcoholic steatohepatitis. Hepatology 66: 2055–2065 [DOI] [PubMed] [Google Scholar]
- 64. O'Brien A, Williams R (2008) Nutrition in end‐stage liver disease: principles and practice. Gastroenterology 134: 1729–1740 [DOI] [PubMed] [Google Scholar]
- 65. Qiu J, Tsien C, Thapalaya S, Narayanan A, Weihl CC, Ching JK, Eghtesad B, Singh K, Fu X, Dubyak G et al (2012) Hyperammonemia‐mediated autophagy in skeletal muscle contributes to sarcopenia of cirrhosis. Am J Physiol Endocrinol Metab 303: E983–E993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Qiu J, Thapaliya S, Runkana A, Yang Y, Tsien C, Mohan ML, Narayanan A, Eghtesad B, Mozdziak PE, McDonald C et al (2013) Hyperammonemia in cirrhosis induces transcriptional regulation of myostatin by an NF‐κB‐mediated mechanism. Proc Natl Acad Sci USA 110: 18162–18167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nordlie RC, Foster JD, Lange AJ (1999) Regulation of glucose production by the liver. Annu Rev Nutr 19: 379–406 [DOI] [PubMed] [Google Scholar]
- 68. van der Ploeg AT, Reuser AJJ (2008) Pompe's disease. Lancet 372: 1342–1353 [DOI] [PubMed] [Google Scholar]
- 69. Puzzo F, Colella P, Biferi MG, Bali D, Paulk NK, Vidal P, Collaud F, Simon‐Sola M, Charles S, Hardet R et al (2017) Rescue of Pompe disease in mice by AAV‐mediated liver delivery of secretable acid α‐glucosidase. Sci Transl Med 9: eaam6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH (2018) Cancer‐associated cachexia. Nat Rev Dis Primers 4: 17105 [DOI] [PubMed] [Google Scholar]
- 71. Zentella A, Manogue K, Cerami A (1993) Cachectin/TNF‐mediated lactate production in cultured myocytes is linked to activation of a futile substrate cycle. Cytokine 5: 436–447 [DOI] [PubMed] [Google Scholar]
- 72. Zhang L, Tang H, Kou Y, Li R, Zheng Y, Wang Q, Zhou X, Jin L (2013) MG132‐mediated inhibition of the ubiquitin‐proteasome pathway ameliorates cancer cachexia. J Cancer Res Clin Oncol 139: 1105–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, Cavallini G, Bonelli G, Baccino FM, Costelli P (2013) Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 182: 1367–1378 [DOI] [PubMed] [Google Scholar]
- 74. Sandri M (2016) Protein breakdown in cancer cachexia. Semin Cell Dev Biol 54: 11–19 [DOI] [PubMed] [Google Scholar]
- 75. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K et al (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708 [DOI] [PubMed] [Google Scholar]
- 76. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL (2001) Atrogin‐1, a muscle‐specific F‐box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98: 14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51 [DOI] [PubMed] [Google Scholar]
- 78. Tintignac LA, Lagirand J, Batonnet S, Sirri V, Leibovitch MP, Leibovitch SA (2005) Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J Biol Chem 280: 2847–2856 [DOI] [PubMed] [Google Scholar]
- 79. Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin J (2000) NF‐κB‐induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289: 2363–2365 [DOI] [PubMed] [Google Scholar]
- 80. Marchildon F, Lamarche É, Lala‐Tabbert N, St‐Louis C, Wiper‐Bergeron N (2015) Expression of CCAAT/enhancer binding protein beta in muscle satellite cells inhibits myogenesis in cancer cachexia. PLoS One 10: e0145583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cong H, Sun L, Liu C, Tien P (2011) Inhibition of atrogin‐1/MAFbx expression by adenovirus‐delivered small hairpin RNAs attenuates muscle atrophy in fasting mice. Hum Gene Ther 22: 313–324 [DOI] [PubMed] [Google Scholar]
- 82. Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, Kumar A (2010) Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol 191: 1395–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A et al (2013) BMP signaling controls muscle mass. Nat Genet 45: 1309–1318 [DOI] [PubMed] [Google Scholar]
- 84. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M (2009) Autophagy is required to maintain muscle mass. Cell Metab 10: 507–515 [DOI] [PubMed] [Google Scholar]
- 85. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J et al (2007) FoxO3 controls autophagy in skeletal muscle in vivo . Cell Metab 6: 458–471 [DOI] [PubMed] [Google Scholar]
- 86. Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6: 472–483 [DOI] [PubMed] [Google Scholar]
- 87. Cosper PF, Leinwand LA (2011) Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res 71: 1710–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tardif N, Klaude M, Lundell L, Thorell A, Rooyackers O (2013) Autophagic‐lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am J Clin Nutr 98: 1485–1492 [DOI] [PubMed] [Google Scholar]
- 89. Stephens NA, Skipworth RJE, Gallagher IJ, Greig CA, Guttridge DC, Ross JA, Fearon KCH (2015) Evaluating potential biomarkers of cachexia and survival in skeletal muscle of upper gastrointestinal cancer patients. J Cachexia Sarcopenia Muscle 6: 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Johns N, Hatakeyama S, Stephens NA, Degen M, Degen S, Frieauff W, Lambert C, Ross JA, Roubenoff R, Glass DJ et al (2014) Clinical classification of cancer cachexia: phenotypic correlates in human skeletal muscle. PLoS One 9: e83618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Neel BA, Lin Y, Pessin JE (2013) Skeletal muscle autophagy: a new metabolic regulator. Trends Endocrinol Metab 24: 635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fukawa T, Yan‐Jiang BC, Min‐Wen JC, Jun‐Hao ET, Huang D, Qian C‐N, Ong P, Li Z, Chen S, Mak SY et al (2016) Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med 22: 666–671 [DOI] [PubMed] [Google Scholar]
- 93. Schäfer M, Oeing CU, Rohm M, Baysal‐Temel E, Lehmann LH, Bauer R, Volz HC, Boutros M, Sohn D, Sticht C et al (2016) Ataxin‐10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol Metab 5: 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zuijdgeest‐van Leeuwen SD, van den Berg JW, Wattimena JL, van der Gaast A, Swart GR, Wilson JH, Dagnelie PC (2000) Lipolysis and lipid oxidation in weight‐losing cancer patients and healthy subjects. Metabolism 49: 931–936 [DOI] [PubMed] [Google Scholar]
- 95. Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M et al (2011) Adipose triglyceride lipase contributes to cancer‐associated cachexia. Science 333: 233–238 [DOI] [PubMed] [Google Scholar]
- 96. Tisdale MJ (2009) Mechanisms of cancer cachexia. Physiol Rev 89: 381–410 [DOI] [PubMed] [Google Scholar]
- 97. Rohm M, Schäfer M, Laurent V, Üstünel BE, Niopek K, Algire C, Hautzinger O, Sijmonsma TP, Zota A, Medrikova D et al (2016) An AMP‐activated protein kinase‐stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat Med 22: 1120–1130 [DOI] [PubMed] [Google Scholar]
- 98. Agustsson T, Rydén M, Hoffstedt J, van Harmelen V, Dicker A, Laurencikiene J, Isaksson B, Permert J, Arner P (2007) Mechanism of increased lipolysis in cancer cachexia. Cancer Res 67: 5531–5537 [DOI] [PubMed] [Google Scholar]
- 99. Drott C, Persson H, Lundholm K (1989) Cardiovascular and metabolic response to adrenaline infusion in weight‐losing patients with and without cancer. Clin Physiol 9: 427–439 [DOI] [PubMed] [Google Scholar]
- 100. Dahlman I, Mejhert N, Linder K, Agustsson T, Mutch DM, Kulyte A, Isaksson B, Permert J, Petrovic N, Nedergaard J et al (2010) Adipose tissue pathways involved in weight loss of cancer cachexia. Br J Cancer 102: 1541–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rofe AM, Bourgeois CS, Coyle P, Taylor A, Abdi EA (1994) Altered insulin response to glucose in weight‐losing cancer patients. Anticancer Res 14: 647–650 [PubMed] [Google Scholar]
- 102. Qiang G, Whang Kong H, Xu S, Pham HA, Parlee SD, Burr AA, Gil V, Pang J, Hughes A, Gu X et al (2016) Lipodystrophy and severe metabolic dysfunction in mice with adipose tissue‐specific insulin receptor ablation. Mol Metab 5: 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Todorov PT, McDevitt TM, Meyer DJ, Ueyama H, Ohkubo I, Tisdale MJ (1998) Purification and characterization of a tumor lipid‐mobilizing factor. Cancer Res 58: 2353–2358 [PubMed] [Google Scholar]
- 104. Mracek T, Stephens NA, Gao D, Bao Y, Ross JA, Rydén M, Arner P, Trayhurn P, Fearon KCH, Bing C (2011) Enhanced ZAG production by subcutaneous adipose tissue is linked to weight loss in gastrointestinal cancer patients. Br J Cancer 104: 441–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Russell ST, Hirai K, Tisdale MJ (2002) Role of beta3‐adrenergic receptors in the action of a tumour lipid mobilizing factor. Br J Cancer 86: 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Han J, Meng Q, Shen L, Wu G (2018) Interleukin‐6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis 17: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mulligan HD, Tisdale MJ (1991) Lipogenesis in tumour and host tissues in mice bearing colonic adenocarcinomas. Br J Cancer 63: 719–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Brooks SL, Neville AM, Rothwell NJ, Stock MJ, Wilson S (1981) Sympathetic activation of brown‐adipose‐tissue thermogenesis in cachexia. Biosci Rep 1: 509–517 [DOI] [PubMed] [Google Scholar]
- 109. Shellock FG, Riedinger MS, Fishbein MC (1986) Brown adipose tissue in cancer patients: possible cause of cancer‐induced cachexia. J Cancer Res Clin Oncol 111: 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang Q, Zhang M, Ning G, Gu W, Su T, Xu M, Li B, Wang W (2011) Brown adipose tissue in humans is activated by elevated plasma catecholamines levels and is inversely related to central obesity. PLoS One 6: e21006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tsoli M, Moore M, Burg D, Painter A, Taylor R, Lockie SH, Turner N, Warren A, Cooney G, Oldfield B et al (2012) Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res 72: 4372–4382 [DOI] [PubMed] [Google Scholar]
- 112. Huang Y‐C, Chen T‐B, Hsu C‐C, Li S‐H, Wang P‐W, Lee B‐F, Kuo C‐Y, Chiu N‐T (2011) The relationship between brown adipose tissue activity and neoplastic status: an (18)F‐FDG PET/CT study in the tropics. Lipids Health Dis 10: 238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Muzik O, Mangner TJ, Leonard WR, Kumar A, Janisse J, Granneman JG (2013) 15O PET measurement of blood flow and oxygen consumption in cold‐activated human brown fat. J Nucl Med 54: 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Beijer E, Schoenmakers J, Vijgen G, Kessels F, Dingemans A‐M, Schrauwen P, Wouters M, van Marken Lichtenbelt W, Teule J, Brans B (2012) A role of active brown adipose tissue in cancer cachexia? Oncol Rev 6: e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zurlo F, Nemeth PM, Choksi RM, Sesodia S, Ravussin E (1994) Whole‐body energy metabolism and skeletal muscle biochemical characteristics. Metabolism 43: 481–486 [DOI] [PubMed] [Google Scholar]
- 116. Frontini A, Vitali A, Perugini J, Murano I, Romiti C, Ricquier D, Guerrieri M, Cinti S (2013) White‐to‐brown transdifferentiation of omental adipocytes in patients affected by pheochromocytoma. Biochim Biophys Acta 1831: 950–959 [DOI] [PubMed] [Google Scholar]
- 117. Petruzzelli M, Schweiger M, Schreiber R, Campos‐Olivas R, Tsoli M, Allen J, Swarbrick M, Rose‐John S, Rincon M, Robertson G et al (2014) A switch from white to brown fat increases energy expenditure in cancer‐associated cachexia. Cell Metab 20: 433–447 [DOI] [PubMed] [Google Scholar]
- 118. Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, Spiegelman BM (2014) Tumour‐derived PTH‐related protein triggers adipose tissue browning and cancer cachexia. Nature 513: 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Elattar S, Dimri M, Satyanarayana A (2018) The tumor secretory factor ZAG promotes white adipose tissue browning and energy wasting. FASEB J 32: 4727–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hu W, Ru Z, Xiao W, Xiong Z, Wang C, Yuan C, Zhang X, Yang H (2018) Adipose tissue browning in cancer‐associated cachexia can be attenuated by inhibition of exosome generation. Biochem Biophys Res Commun 506: 122–129 [DOI] [PubMed] [Google Scholar]
- 121. Kokolus KM, Capitano ML, Lee C‐T, Eng JW‐L, Waight JD, Hylander BL, Sexton S, Hong C‐C, Gordon CJ, Abrams SI et al (2013) Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci USA 110: 20176–20181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Michaelis KA, Zhu X, Burfeind KG, Krasnow SM, Levasseur PR, Morgan TK, Marks DL (2017) Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle 8: 824–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ikeda K, Kang Q, Yoneshiro T, Camporez JP, Maki H, Homma M, Shinoda K, Chen Y, Lu X, Maretich P et al (2017) UCP1‐independent signaling involving SERCA2b‐mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat Med 23: 1454–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Blum D, Omlin A, Baracos VE, Solheim TS, Tan BHL, Stone P, Kaasa S, Fearon K, Strasser F, European Palliative Care Research Collaborative (2011) Cancer cachexia: a systematic literature review of items and domains associated with involuntary weight loss in cancer. Crit Rev Oncol Hematol 80: 114–144 [DOI] [PubMed] [Google Scholar]
- 125. Vaisman N, Lusthaus M, Niv E, Santo E, Shacham‐Shmueli E, Geva R, Figer A (2012) Effect of tumor load on energy expenditure in patients with pancreatic cancer. Pancreas 41: 230–232 [DOI] [PubMed] [Google Scholar]
- 126. De Waele E, Mattens S, Honoré PM, Spapen H, De Grève J, Pen JJ (2015) Nutrition therapy in cachectic cancer patients. The Tight Caloric Control (TiCaCo) pilot trial. Appetite 91: 298–301 [DOI] [PubMed] [Google Scholar]
- 127. Baldwin C, Spiro A, Ahern R, Emery PW (2012) Oral nutritional interventions in malnourished patients with cancer: a systematic review and meta‐analysis. J Natl Cancer Inst 104: 371–385 [DOI] [PubMed] [Google Scholar]
- 128. Solheim TS, Laird BJA, Balstad TR, Stene GB, Bye A, Johns N, Pettersen CH, Fallon M, Fayers P, Fearon K et al (2017) A randomized phase II feasibility trial of a multimodal intervention for the management of cachexia in lung and pancreatic cancer. J Cachexia Sarcopenia Muscle 8: 778–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Oliff A, Defeo‐Jones D, Boyer M, Martinez D, Kiefer D, Vuocolo G, Wolfe A, Socher SH (1987) Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 50: 555–563 [DOI] [PubMed] [Google Scholar]
- 130. Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF (2002) Tumor necrosis factor‐alpha suppresses adipocyte‐specific genes and activates expression of preadipocyte genes in 3T3‐L1 adipocytes: nuclear factor‐kappaB activation by TNF‐alpha is obligatory. Diabetes 51: 1319–1336 [DOI] [PubMed] [Google Scholar]
- 131. Arruda AP, Milanski M, Romanatto T, Solon C, Coope A, Alberici LC, Festuccia WT, Hirabara SM, Ropelle E, Curi R et al (2010) Hypothalamic actions of tumor necrosis factor alpha provide the thermogenic core for the wastage syndrome in cachexia. Endocrinology 151: 683–694 [DOI] [PubMed] [Google Scholar]
- 132. Johns N, Stretch C, Tan BHL, Solheim TS, Sørhaug S, Stephens NA, Gioulbasanis I, Skipworth RJE, Deans DAC, Vigano A et al (2017) New genetic signatures associated with cancer cachexia as defined by low skeletal muscle index and weight loss. J Cachexia Sarcopenia Muscle 8: 122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Tan BHL, Ross JA, Kaasa S, Skorpen F, Fearon KCH, European Palliative Care Research Collaborative (2011) Identification of possible genetic polymorphisms involved in cancer cachexia: a systematic review. J Genet 90: 165–177 [DOI] [PubMed] [Google Scholar]
- 134. Castell JV, Gómez‐Lechón MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC (1989) Interleukin‐6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett 242: 237–239 [DOI] [PubMed] [Google Scholar]
- 135. Utaka S, Avesani CM, Draibe SA, Kamimura MA, Andreoni S, Cuppari L (2005) Inflammation is associated with increased energy expenditure in patients with chronic kidney disease. Am J Clin Nutr 82: 801–805 [DOI] [PubMed] [Google Scholar]
- 136. García‐Lorda P, Serrano P, Jiménez‐Expósito MJ, Fraile J, Bulló M, Alonso C, Bonada A, Viciana P, Luna PP, Salas‐Salvadó J (2000) Cytokine‐driven inflammatory response is associated with the hypermetabolism of AIDS patients with opportunistic infections. JPEN J Parenter Enteral Nutr 24: 317–322 [DOI] [PubMed] [Google Scholar]
- 137. Abdullahi A, Chen P, Stanojcic M, Sadri A‐R, Coburn N, Jeschke MG (2017) IL‐6 signal from the bone marrow is required for the browning of white adipose tissue post burn injury. Shock 47: 33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Roubenoff R, Roubenoff RA, Cannon JG, Kehayias JJ, Zhuang H, Dawson‐Hughes B, Dinarello CA, Rosenberg IH (1994) Rheumatoid cachexia: cytokine‐driven hypermetabolism accompanying reduced body cell mass in chronic inflammation. J Clin Invest 93: 2379–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Falconer JS, Fearon KC, Plester CE, Ross JA, Carter DC (1994) Cytokines, the acute‐phase response, and resting energy expenditure in cachectic patients with pancreatic cancer. Ann Surg 219: 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]