

Abstract
Background
Kiwifruit (Actinidia spp.) is a dioecious plant with fruits containing abundant vitamin C and minerals. A handful of kiwifruit species have been domesticated, among which Actinidiaeriantha is increasingly favored in breeding owing to its superior commercial traits. Recently, elite cultivars from A. eriantha have been successfully selected and further studies on their biology and breeding potential require genomic information, which is currently unavailable.
Findings
We assembled a chromosome-scale genome sequence of A. eriantha cultivar White using single-molecular sequencing and chromatin interaction map–based scaffolding. The assembly has a total size of 690.6 megabases and an N50 of 21.7 megabases. Approximately 99% of the assembly were in 29 pseudomolecules corresponding to the 29 kiwifruit chromosomes. Forty-three percent of the A. eriantha genome are repetitive sequences, and the non-repetitive part encodes 42,988 protein-coding genes, of which 39,075 have homologues from other plant species or protein domains. The divergence time between A. eriantha and its close relative Actinidia chinensis is estimated to be 3.3 million years, and after diversification, 1,727 and 1,506 gene families are expanded and contracted in A. eriantha, respectively.
Conclusions
We provide a high-quality reference genome for kiwifruit A. eriantha. This chromosome-scale genome assembly is substantially better than 2 published kiwifruit assemblies from A. chinensis in terms of genome contiguity and completeness. The availability of the A. eriantha genome provides a valuable resource for facilitating kiwifruit breeding and studies of kiwifruit biology.
Keywords: kiwifruit, Actinidia eriantha, Genome assembly, single molecular sequencing, high-throughput chromosome conformation capture
Introduction
Kiwifruit is often referred to as the king of fruits owing to its remarkably high vitamin C content and abundant minerals [1, 2]. Native to China, kiwifruit belongs to the genus Actinidia, which contains 54 species and 75 taxa [3]. All species in this genus are perennial, deciduous, and dioecious plants with a climbing or scrambling growth habit, and they also have many common morphological features including the characteristic radiating arrangement of styles of the female flower and the structure of the fruit [4]. Despite rich germplasm resources in kiwifruit, only a few Actinidia species have been domesticated, such as Actinidia chinensis var. chinensis, A. chinensis var. deliciosa, and Actinidia eriantha, whose fruit size are close to commercial standard [5–7].
Owing to its strong resistance to Pseudomonas syringae pathovarActinidiae, long shelf-life, enriched ascorbic acid, and peelable skin [7–11], A. eriantha (2n = 58) has been favored in kiwifruit breeding. Recently, new cultivars have been selected either from the wild germplasm of A. eriantha such as “White” (Fig. 1) or from the interspecific hybridization between A. eriantha (male) and A. chinensis (female) such as “Jinyan” [7, 12]. White has particularly large fruits (mean, 96 g) with green flesh and favorable flavor and has been widely cultivated in China [7].
Figure 1:

Tree and fruits of A. eriantha cv. White.
Actinidia eriantha (Actinidia eriantha, NCBI:txid165200) has also been used for genetic and genomic studies thanks to its high efficiency in genetic transformation and relatively short phase of juvenility [13]. The flowering and fruiting of A. eriantha can be accomplished within 2 years in greenhouse conditions with a low requirement for winter chilling [13]. In addition, the roots of A. eriantha, which contain many bioactive compounds such as triterpenes and polysaccharides, are used as a traditional Chinese medicine for the treatment of gastric carcinoma, nasopharyngeal carcinoma, breast carcinoma, and hepatitis [12, 14].
Previously, 2 kiwifruit genomes were published and both were varieties of A. chinensis (“Hongyang” and “Red5”) [15, 16]. These short-read–based assemblies are very fragmented, possibly due to the high complexity and heterozygosity of the kiwifruit genomes, as well as technical limitations. Here, we used single-molecular sequencing combined with high-throughput chromosome conformation capture (Hi-C) technology to assemble the genome of the elite kiwifruit cultivar “White” of A. eriantha. The availability of this high-quality chromosome-scale genome sequence not only provides fundamental knowledge regarding kiwifruit biology but also presents a valuable resource for kiwifruit breeding programs.
Sample collection and genome sequencing
Fresh young leaves were collected from a female individual of A. eriantha cv. White. High molecular weight genomic DNA was extracted using the CTAB (cetyl trimethylammonium bromide) method as described in the protocol [17]. To construct genomic libraries (SMRTbell libraries) for Pacific Biosciences (PacBio) long-read sequencing, high molecular weight genomic DNA was sheared into fragments of ∼20 kilobases (kb) using a Covaris g-Tube (KBiosciences part No. 520079), enzymatically repaired, and converted to SMRTbell template following the manufacturer's instructions (DNA Template Prep Kit 1.0, PacBio part No. 100-259-100). The templates were size-selected using a BluePippin (Sage Science, Inc., Beverly, MA, USA) to enrich large DNA fragments (>10 kb) and then sequenced on a PacBio Sequel system. A total of 9 single-molecule real-time (SMRT) cells were sequenced, yielding 3,889,480 million reads with a mean and median length of 10,065 and 15,661 base pairs (bp), respectively, and a total of 39.1 gigabase (Gb) sequences, ∼52.5× coverage of the kiwifruit genome with an estimated size of 745.3 megabases (Mb) based on the flow cytometry analysis (Fig. S1; Table S1).
Three paired-end Illumina libraries with insert sizes of 180, 220, and 500 bp and 7 mate-pair libraries with insert sizes of 3, 4, 5, 8, 10, 15, 17 kb were prepared using Illumina's Genomic DNA Sample Preparation kit and the Nextera Mate Pair Sample Preparation kit (Illumina, San Diego, CA), respectively. All libraries were sequenced on an Illumina HiSeq 2500 system, which yielded ∼80.1 and ∼97.3 Gb of raw sequence data for paired-end and mate-pair libraries, respectively (Table S1). The raw Illumina paired-end reads were processed to remove duplications, adaptors, and low-quality bases using Super-Deduper [18] and Trimmomatic (Trimmomatic, RRID:SCR_011848) [19] (v0.35), and the mate-pair reads were cleaned using NextClip (NextClip, RRID:SCR_005465) [20] (v1.3.1) with default parameters. Finally, we obtained 76.6 and 46.2 Gb high-quality cleaned sequences for paired-end and mate-pair libraries, respectively (Table S1).
To construct the Hi-C library, White plants were grown in a greenhouse, and ∼4–6 g young leaves were then harvested and subsequently fixed in formaldehyde (1% volume/volume [v/v]) for 10 min at room temperature. The fixation was terminated by adding glycine to a final concentration of 0.125 M. The fixed samples were ground into powder in liquid nitrogen and then lysed with the addition of Triton X-100 to a concentration of 1% (v/v). The nuclei were isolated and prepared for Hi-C library construction according to a previously published protocol [21].
Transcriptome sequencing
To improve gene prediction, we generated transcriptome sequences from a pool of mixed tissues of White including root, stem, leaf, flower, and fruits at 7, 30, 60, 90, and 120 days after anthesis. Total RNA was extracted from these tissues using an RNA extraction kit (BIOFIT, Chengdu, Sichuan, China), treated with DNase I and further purified with RNA clean kit (Promega, Madison, WI, USA). RNA sequencing (RNA-Seq) libraries were constructed with the NEBNext Ultra RNA Library Prep Kit (Illumina, USA), and sequenced on an Illumina HiSeq 2500 system using paired-end mode. A total of ∼19.5 million raw read pairs were obtained, which were processed with Trimmomatic to remove adaptors. The cleaned reads were assembled de novo with Trinity (Trinity, RRID:SCR_013048) [22] (v2.4.0). Additionally, we also generated genome-guided assemblies with both Trinity and StringTie (StringTie, RRID:SCR_016323) [23]. Different transcriptome assemblies were eventually integrated by PASA (PASA, RRID:SCR_014656) [24] (v2.3.3) and used as transcript evidence during gene prediction process. Mapping of RNA-Seq reads to the genome assembly was performed with STAR (STAR, RRID:SCR_015899) [25] (v02,0201), and read counting on the coding regions was performed with HTSeq (HTSeq, RRID:SCR_005514) [26] (v0.6.0).
Chromosome-scale assembly of the A. eriantha genome
Actinidia eriantha is a diecious plant with a heterozygous diploid genome. We estimated the heterozygosity level through the k-mer spectrum analysis with GenomeScope [27] using sequences from the paired-end library with an insert size of 180 bp. The depth distribution of the derived 17-mers clearly showed 2 separate peaks, based on which we estimated the heterozygosity level of the A. eriantha cv. White genome to be 1.21% (Fig. S1).
We then estimated the genome size of A. eriantha cv. White using flow cytometry analysis, with tomato (Solanum lycopersicum cv. Ailsa Craig) used as the reference. We also performed flow cytometry analysis on A. chinensis cv. Hongyang. Approximately 1 g of young leaves were washed twice in distilled water and then chopped in ice-cold lysis buffer A (10 mmol/L MgSO4, 50 mmol/L KCl, 3.5 mmol/L HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] pH 7.5, 0.3% [v/v] Triton x-100, 2% polyvinylpyrrolidone 30 weight by volume). After 5 min, the crude lysate was passed through a 75-μm pore size nylon mesh to remove large cellular debris. The filtrate (1 mL) was transferred to a 1.5-mL plastic tube and centrifuged at 1000 rpm for 5 min. The supernatant was discarded, and the nuclei were then resuspended with lysis buffer B (10 mmol/L MgSO4, 50 mmol/L KCl, 3.5 mmol/L HEPES pH 7.5, 0.3% [v/v] Triton x-100, 0.4 mg/mL propidium iodide, 0.04 mg/mL RNase). After 15 min, samples were analyzed using a FACS Vantage SE flow cytometer (Becton-Dickinson, San José, USA). Four biological replicates were performed. Based on the 950-Mb genome of tomato, the genome size of White was estimated to be 745.3 ± 7.9 Mb, similar to the genome size of A. chinensis (Fig. S1) and consistent with that in a previous report (758 Mb; [28]).
We used a strategy that took into account the unique advantage of different assemblers to construct the White genome using PacBio long reads. First, PacBio long reads were corrected and assembled using the Canu program (Canu, RRID:SCR_015880) [29] (v1.7), which is a modularized pipeline consisting of 3 primary stages—read correction, trimming, and assembly. The Canu-corrected reads were also assembled independently with the wtdbg program [30], a fast assembler for long noisy reads. Subsequently, the 2 independent assemblies (one with Canu and another with wtdbg) were merged by Quickmerge [31] (v0.2) to improve the contiguity. The merged assembly was further processed to correct errors using Pilon (Pilon, RRID:SCR_014731) [32] (v1.22) with high-quality cleaned Illumina reads from all paired-end and mate-pair libraries, representing a total genome coverage of 171× (Table S1). This yielded 2,818,370 nucleotides, 2,495,388 insertions, and 1,691,495 deletions being corrected. The resulting final assembled A. eriantha cv. White genome contained 4,076 contigs with an N50 length of 539,246 bp and a cumulative size of 690,376,929 bp (Table 1). The contiguity and completeness of this assembly far exceeds that of 2 published kiwifruit A. chinensis genomes (Table 1).
Table 1:
Assembly statistics
| Parameter | A. eriantha | A. chinensis | |
|---|---|---|---|
| White | Hongyang | Red5 | |
| Contigs | |||
| Total contig No. | 4,076 | 26,721 | 39,868 |
| Total contig length (Mb) | 690.4 | 604.2 | |
| Contig N50 (kb) | 539.2 | 58.9 | |
| Contig N90 (kb) | 50.7 | 11.6 | |
| Longest contig length (kb) | 3,260.20 | 423.5 | |
| Scaffolds | |||
| Total scaffold No. | 1,735 | 7,698 | 3,887 |
| Total scaffold length (Mb) | 690.6 | 616.1 | 550.5 |
| Scaffold N50 (kb) | 23,583.9 | 646.8 | 623.8 |
| Scaffold N90 (kb) | 20,112.1 | 122.7 | 140.7 |
| Longest scaffold length (Mb) | 28.6 | 3.4 | 4.43 |
| Anchored to chromosome (Mb/%) | 682.4/98.84 | 452.4/73.4 | 547.9/98.9 |
| Anchored with order and orientation (Mb/%) | 634.4/91.90 | 333.6/54.1 | |
To scaffold the contigs on the basis of chromatin interaction maps inferred from the Hi-C data, we first used HiC-Pro [33] to evaluate and filter the cleaned Hi-C reads. The Hi-C data usually contain a considerable part of invalid interaction read pairs, which are non-informative and need to be filtered out beforehand. Among the 51 million read pairs that were uniquely aligned to the A. eriantha assembly, 33 million (64.1%) were valid interaction pairs and their insertion size spanned predominantly from dozens to hundreds of kilobases, therefore providing efficient information for scaffolding. As a part of error correction of the assembly, we used valid Hi-C reads to identify misassembled contigs. In principle, a genuine contig should display a continuous Hi-C interaction map whereas the discrete distribution of an interaction map likely indicates a misassembly. We examined the interaction map for each contig and broke 51 that were possibly misassembled. Subsequently, the corrected PacBio assembly was used for scaffolding with the LACHESIS program [34] and parameters “CLUSTER_MIN_RE_SITES = 48, CLUSTER_MAX_LINK_DENSITY = 2, CLUSTER_NONINFORMATIVE_RATIO = 2, ORDER_MIN_N_RES_IN_TRUN = 14, ORDER_MIN_N_RES_IN_SHREDS = 15.” LACHESIS assigned 3,666 contigs with a total size of 682,355,494 bp (98.84% of the assembly) into 29 groups corresponding to the 29 kiwifruit chromosomes (Figs. 2 and 3a), among which 634,430,648 bp (91.90%) had defined order and orientation (Table 1 and Table S2). The final chromosome-scale assembly had a total length of 690,781,529 bp and an N50 of 23,583,865 bp.
Figure 2:

Chromatin interaction map of A. eriantha derived from Hi-C data. Each group represents an individual chromosome.
Figure 3:
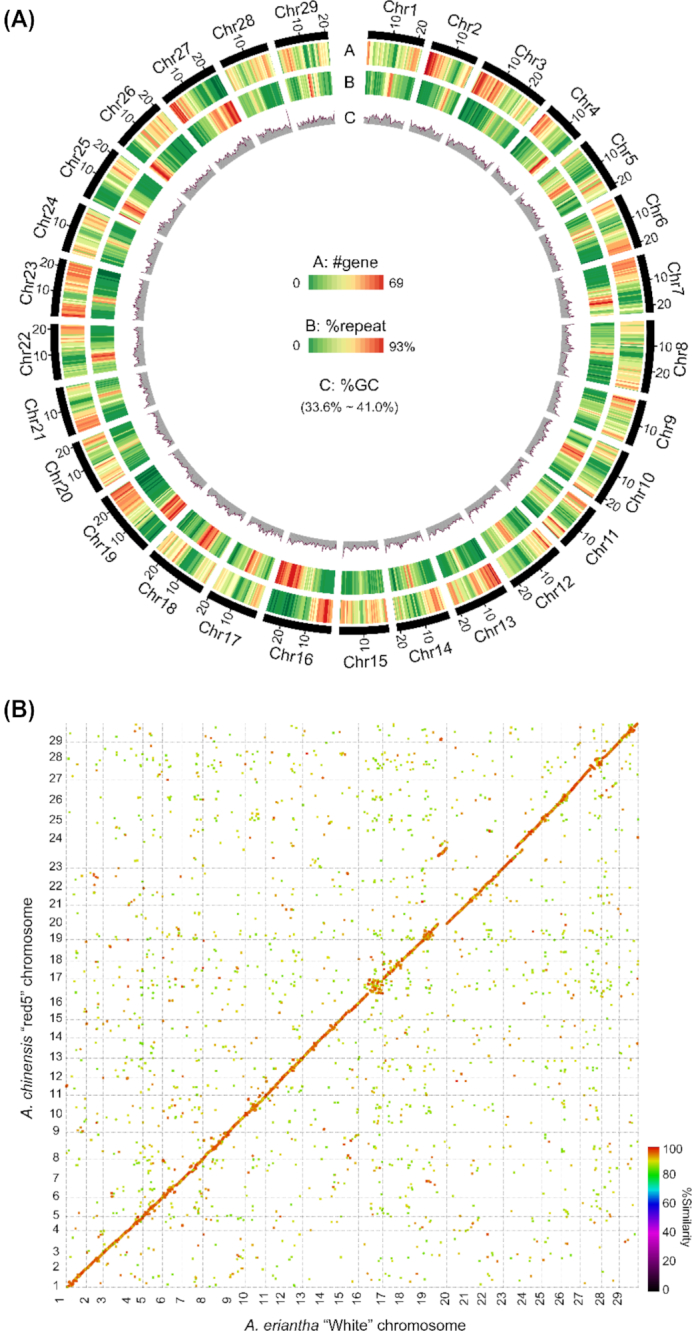
Genome of A. eriantha and synteny between the 2 kiwifruit species. (a) Genome landscape of A. eriantha cv. White. Track A: gene density, Track B: repeat density, Track C: guanine or cytosine (GC) content; all were calculated in a 500-kb window. (b) Genome synteny between A. eriantha cv. White and A. chinensis cv. Red5.
Evaluation of the genome assembly
We first evaluated the quality of the assembled A. eriantha cv. White genome by mapping Illumina genomic and RNA-Seq reads to the assembly. Reads from the paired-end genomic library (with insert size of 180 bp) had a very high mapping rate (98.7%), and the properly paired read mapping rate was 92.0%. For the RNA-Seq reads, 91.7% could be mapped to the genome and 87.1% were uniquely mapped. The high mapping ratio of both genomic and RNA-Seq reads suggests a high quality of the A. eriantha cv. White assembly.
We then identified synteny between the A. eriantha cv. White assembly and the assembly of A. chinensis cv. Red5 using MUMMER [35] (v4.0.0beta2). In general, the 2 assemblies showed a high macro-collinearity, with only a few inconsistencies (Fig. 3b). A detailed check of the major inconsistent regions using genetic maps [36] and mate-pair read alignments confirmed the high quality of the A. eriantha cv. White genome assembly and particularly enabled us to discover that in the Red5 genome a ∼8-Mb region was possibly misassembled into chromosome 23 (Fig. S2).
Repeat annotation
Repeats were annotated following a protocol described in Campbell et al. [37]. The customized repeat library was built to include both known and novel repeat families. We first searched the assembly for miniature inverted transposable elements (MITEs) using MITE-Hunter [38] with default parameters. The long terminal repeat (LTR) retrotransposons were then identified from the A. eriantha cv. White genome using LTRharvest and LTRdigest wrapped in the GenomeTools package [39]. The LTR identification pipeline was run iteratively to collect both recent (sequence similarity ≥99%) and old (sequence similarity ≥85%) LTR retrotransposons. Candidates from each run were filtered on the basis of the elements typically encoded by LTR retrotransposons. The default parameters (–minlenltr 100 –maxlenltr 6000 –mindistltr 1500 –maxdistltr 25,000 –mintsd 5 –maxtsd 5 –motif tgca) were used in LTR calling according to Campbell et al. [37]. An initial repeat masking of A. eriantha cv. White genome was performed with the repeat library derived by combining the identified MITEs and LTR transposons. The repeat-masked genome was fed to RepeatModeler (RepeatModeler, RRID:SCR_015027) [40] to identify novel repeat families. Finally, all identified repeat sequences were combined and searched against a plant protein database from which transposon encoding proteins were excluded. Elements with significant similarity to plant genes were removed. The final repeat library contained 1,670 families, and 526 of them were potentially novel repeat families. We used this species-specific repeat library to mask the A. eriantha cv. White genome. Approximately 43.3% of the A. eriantha cv. White genome was masked, and the largest family of repeats was LTR transposons (Table S3). Repeat content identified in A. eriantha cv. White was much higher than that in A. chinensis (e.g., 36% in Hongyang [15]), and this difference may be largely due to the improvement of the repeat region assembly with PacBio long reads. In addition, divergence between the 2 kiwifruit species could also contribute to this difference.
Prediction and functional annotation of protein-coding genes
Protein-coding genes were predicted from the repeat-masked A. eriantha cv. White genome with the MAKER-P program [37] (v2.31.10), which integrates evidence from protein homology, transcripts, and ab initio predictions. The homology-based evidence was derived by aligning proteomes from 20 plant species to the White genome assembly with Exonerate (Exonerate, RRID:SCR_016088) (v2.26.1) [41]. SNAP [42], AUGUSTUS (Augustus, RRID:SCR_008417) [43] (v3.3), and GeneMark-ES (GeneMark, RRID:SCR_011930) [44] (v4.35) were used for ab initio gene predictions. RNA-Seq data generated in this study were assembled and the assembled contigs were aligned to the White genome assembly to provide transcript evidence. Predictions supported by the 3 different sources of evidence were finally integrated by MAKER-P (MAKER, RRID:SCR_005309), which resulted in a total of 52,514 primitive gene models. We then filtered and polished these gene models by 2 steps. First, we combined our RNA-Seq data with others collected from a previous study [45], and mapped the reads to the White genome using the STAR program [25], and a total of 266 million read pairs were mapped. Based on the mapping, a raw count for each predicted gene model was derived and then normalized to CPM (counts per million mapped read pairs). Gene models with ultra-low expression (CPM < 0.1) were less likely to be real genes. Furthermore, we found that these genes with ultra-low expression had relatively high annotation edit distance score, an indication of low confidence as defined by the MAKER-P program. Therefore, for gene models with CPM < 0.1, we only kept those containing both pfam domains and homologous sequences in the NCBI non-redundant protein database. After this filtering process 42,751 gene models were kept. Second, the predicted protein-coding genes of kiwifruit A. chinensis cv. Red5 have been manually curated [16], and therefore these gene models should have relatively higher accuracy and could be used to modify A. eriantha cv. White gene models whose predictions were not consistently supported by the different types of evidence. To this end, we performed another 2 ab initio predictions using BRAKER [46] and GeMoMa [47] (v1.5.2) with Red5 proteome as the sole evidence. These 2 predictions were compared with the gene models predicted by MAKER-P. Consequently, a total of 237 gene models not predicted by MAKER-P were added and another 415 gene models that had better predictions by BRAKER2 or GeMoMa were used to replace the corresponding gene models predicted by MAKER-P. Finally, we obtained a total of 42,988 protein-coding genes in the A. eriantha cv. White genome, with a mean coding sequence size of 1,004 bp and containing a mean of 5 exons.
The predicted genes were functionally annotated by blasting their protein sequences against TAIR (TAIR, RRID:SCR_004618) [48], Swiss-Prot [49], and TrEMBL [50] databases with an E-value cutoff of 1E–5. Functional descriptions of the protein hits were assembled with the AHRD program [51] and transferred to A. eriantha genes. Protein domains were identified using InterProScan (InterProScan, RRID:SCR_005829) [52] (v5.29–68.0) by searching the protein sequences against domain databases including PANTHER (PANTHER, RRID:SCR_004869) [53], Pfam (Pfam, RRID:SCR_004726) [54], SMART (SMART, RRID:SCR_005026) [55], and PROSITE (PROSITE, RRID:SCR_003457) [56]. The gene ontology terms were assigned to the A. eriantha cv. White predicted genes using the Blast2GO program (Blast2GO, RRID:SCR_005828) [57] with entries from the NCBI protein database and InterProScan. Collectively, 90.9% (n = 39,075) of the predicted genes contain ≥1 annotation from the above databases (Table S4).
Evolutionary and comparative genomic analysis
To infer the divergence time between A. eriantha and A. chinensis, we identified gene orthology between the 2 species using MCScanX [58] and calculated the synonymous substitution rate (Ks) between each orthologous pair. Three additional species, cultivated tomato (Solanum lycopersicum), wild tomato (Solanum pennellii), and potato (Solanum tuberosum), were also included in the analysis. The Ks distribution (Fig. 4a) suggested that the divergence between the 2 kiwifruit species was earlier than that between the 2 tomato species. We dated the divergence by assuming a strict molecular clock [59], and the time when A. eriantha and A. chinensis separated was estimated to be ∼3.3 million years ago (Mya), compared to ∼1.9 Mya between S. lycopersicum and S. pennellii and ∼6.0 Mya between S. lycopersicum and S. tuberosum. Gene family evolution was analyzed by comparing genomes of A. eriantha, A. chinensis, S. lycopersicum, S. tuberosum, Vitis vinifera, Arabidopsis thaliana, and Oryza sativa. A total of 17,593 orthogroups were defined by OrthoFinder [60] (v2.2.6), among which 1,246 were single-copy gene families (Fig. 4b). The single-copy family genes were aligned and concatenated to build a species phylogenetic tree using IQ-TREE [61] (v1.5.5) with a best-fitting model (Fig. 4c). Gene family expansion/contraction along the branches of the phylogenic tree was analyzed by CAFÉ [62] (v4.1). Finally, a total of 1,727 and 1,506 gene families were found apparently expanded and contracted, respectively, in A. eriantha (Fig. 4c).
Figure 4:

Evolutionary and comparative genomic analyses. (a) Distribution of synonymous substitution rate (Ks) between A. eriantha and A. chinensis, S. lycopersicum and S. pennellii, and S. lycopersicum and S. tuberosum. (b) Orthogroups shared by selected species. (c) Species phylogenetic tree and gene family evolution. Numbers on the branch indicate counts of gene families that are under either expansion (red) or contraction (green).
Conclusion
Herein, we report a high-quality reference genome of kiwifruit A. eriantha cv. White. The assembly from single-molecular sequencing combined with Hi-C scaffolding yielded a more highly continuous and complete genome than the 2 previously published kiwifruit genomes. This genome will provide a valuable source for exploration of the genetic basis of unique traits in kiwifruit and also facilitate studies of sexual determination loci in the dioecious plants.
Availability of supporting data and materials
This Whole Genome Shotgun project has been deposited at DBJ/ENA/GenBank under the accession No. QOVS00000000. The version described in this paper is version QOVS01000000. Raw sequencing reads have been deposited in the Sequence Read Archive database under the accession No. SRP155011. The Actinidia eriantha cv. White genome sequence and the annotation are also available via the GigaScience database, GigaDB [63]. Detailed protocols of computational analyses have been deposited in protocols.io [64].
Additional files
Figure S1. Genome characteristics of A. eriantha and A. chinensis. (a) Flow cytometry analyses of A. eriantha cv. White and A. chinensis cv. Hongyang. The main peak (I) indicates G0/G1 cells and the secondary peak (II) represents G2/M cells. (b) Flow cytometry analyses of A. eriantha cv. White and Solanum lycopersicum cv. Ailsa Craig. Peaks a and b represent the G0/G1 cells of “White” and “Ailsa Craig”, respectively. The genome size of “White” was estimated to be 745.3 ± 7.9 Mb using “Ailsa Craig” as the reference. (c) 17-mer distribution of “White” genomic reads (180bp paired-end library).
Figure S2. Examination of assembly inconsistencies between A. eriantha cv. White and A. chinensis cv. Red5. (a) Validation of genome assembly of “White” using genetic maps. Horizontal lines within “White” chromosomes indicate gapped regions and lines between chromosomes of 2 assemblies indicate syntenic regions. (b) A chromosomal segment assembled into the Chr23 in A. chinensis cv. Red5 is syntenic to the region located at the terminus of Chr19 in A. eriantha cv. White. (c) Snapshots of Illumina mate-pair reads mapped to the junctions of the break point as well as nearby regions supporting the assembly of “White.”
Supp_Tables.xlsx
Abbreviations
BLAST: Basic Local Alignment Search Tool; bp: base pair; CPM: counts per million mapped read pairs; CTAB: cetyl trimethylammonium bromide; cv.: cultivar; Gb: gigabase; HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; Hi-C: high-throughput chromosome conformation capture; LTR: long terminal repeat; Mb: megabase; MITE: miniature inverted transposable element; Mya: million years ago; NCBI: National Center for Biotechnology Information; PacBio: Pacific Biosciences; RNA-Seq: RNA sequencing; SMRT: single-molecule real-time; var.: variety; v/v: volume/volume.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31471157 and 31700266), National Foundation for Germplasm Repository of Special Horticultural Crops in Central Mountain Areas of China (NJF2017-69), National Science Fund for Distinguished Young Scholars (30825030), Key Project from the Government of Sichuan Province (2013NZ0014, 2016NZ0105), Key Project from the Government of Anhui Province (2012AKKG0739; 1808085MC57), and the US National Science Foundation (IOS-1339287 and IOS-1539831).
Authors’ contributions
W.T., J.Y., X.T., Y.Y., X.N., M.M., D.Z., S.H., W.S., C.F., and M.L. collected plant samples, extracted DNA/RNA, and performed transcriptome sequencing and gene expression analyses; W.T., X.S., J.Y., X.T., C.J., Z.F., and Y.L. performed DNA sequencing, genome assembly, gene annotation, evolution and comparative genomic analyses, and website construction; X.S., W.T., Z.F., and Y.L. wrote and revised the manuscript; Y.L. and Z.F. conceived strategies, designed experiments, and managed projects. All authors read and approved the manuscript.
Supplementary Material
9/25/2018 Reviewed
12/16/2018 Reviewed
2/1/2019 Reviewed
10/16/2018 Reviewed
References
- 1. Ferguson AR, Ferguson LR. Are kiwifruit really good for you?. Acta Hort. 2013;610:131–8. [Google Scholar]
- 2. Richardson DP, Ansell J, Drummond LN. The nutritional and health attributes of kiwifruit: a review. Eur J Nutr. 2018;57(8):2659–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li JQ, Li XW, Soejarto DD. Actinidiaceae. In: Wu ZY, Raven PH, Hong DY, eds. Flora of China, vol. 12 Beijing, St. Louis: Science Press, Missouri Plant Garden Press; 2007:334–362. [Google Scholar]
- 4. Ferguson AR, Huang H. Genetic resources of kiwifruit: domestication and breeding. Hortic Rev. 2007;33:1–121. [Google Scholar]
- 5. Testolin R. Kiwifruit (Actinidia spp.) in Italy: the history of the industry, international scientific cooperation and recent advances in genetics and breeding. Acta Hortic. 2015; 1096:47–61. [Google Scholar]
- 6. Jo YS, Cho HS, Park MY, et al.. Selection of a sweet Actinidia eriantha ‘bidan’. Acta Hortic. 2017;753:253–8. [Google Scholar]
- 7. Wu Y, Xie M, Zhang Q, et al.. Characteristics of ‘White’: a new easy-peel cultivar of Actinidiaeriantha. N Z J Crop Hortic Sci. 2009;37(4):369–73. [Google Scholar]
- 8. Atkinson RG, Sharma NN, Hallett IC, et al.. Actinidia eriantha: a parental species for breeding kiwifruit with novel peelability and health attributes. N Z J For Sci. 2009;39:207–16. [Google Scholar]
- 9. Guo R, Landis JB, Moore MJ, et al.. Development and application of transcriptome-derived microsatellites in Actinidiaeriantha (Actinidiaceae). Front Plant Sci. 2017;8:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prakash R, Hallett IC, Wong SF, et al.. Cell separation in kiwifruit without development of a specialised detachment zone. BMC Plant Biol. 2017;17(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi ZJ, Zhang HQ, Hui Q, et al.. The resistance evaluation of different kiwifruit varieties to canker. Acta Agric Zhejiang. 2014;26(3):752–9. [Google Scholar]
- 12. Zhang D, Gao C, Li R, et al.. TEOA, a triterpenoid from Actinidiaeriantha, induces autophagy in SW620 cells via endoplasmic reticulum stress and ROS-dependent mitophagy. Arch Pharm Res. 2017;40:(5):579–91. [DOI] [PubMed] [Google Scholar]
- 13. Wang T, Ran Y, Atkinson RG, et al.. Transformation of Actinidiaeriantha: a potential species for functional genomics studies in Actinidia. Plant Cell Rep. 2006;25(5):425–31. [DOI] [PubMed] [Google Scholar]
- 14. Wu JG, Ma L, Lin SH, et al.. Anticancer and anti-angiogenic activities of extract from Actinidiaeriantha Benth root. J Ethnopharmacol. 2017;203:1–10. [DOI] [PubMed] [Google Scholar]
- 15. Huang S, Ding J, Deng D, et al.. Draft genome of the kiwifruit Actinidiachinensis. Nat Commun. 2013;4:2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pilkington SM, Crowhurst R, Hilario E, et al.. A manually annotated Actinidiachinensis var. chinensis (kiwifruit) genome highlights the challenges associated with draft genomes and gene prediction in plants. BMC Genomics. 2018;19(1):257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Preparing Arabidopsis genomic DNA for size-selected ~20 kb SMRTbell™ libraries Pacific Biosciences; https://www.pacb.com/wp-content/uploads/2015/09/Shared-Protocol-Preparing-Arabidopsis-DNA-for-20-kb-SMRTbell-Libraries.pdf.Accessed 15 March 2019. [Google Scholar]
- 18. Petersen KR, Streett DA, Gerritsen AT, et al.. Super deduper, fast PCR duplicate detection in fastq files. In: Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics. New York: ACM; 2015:491–2. [Google Scholar]
- 19. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leggett RM, Clavijo BJ, Clissold L, et al.. NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics. 2013;30(4):566–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rao SS, Huntley MH, Durand NC, et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haas BJ, Papanicolaou A, Yassour M, et al.. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pertea M, Pertea GM, Antonescu CM et al.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haas BJ, Delcher AL, Mount SM, et al.. Improving the Arabidopsisgenome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31(19):5654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dobin A, Davis CA, Schlesinger F, et al.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vurture GW, Sedlazeck FJ, Nattestad M, et al.. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33(14):2202–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hopping ME. Flow cytometric analysis of Actinidia species. N Z J Bot. 1994;32(1):85–93. [Google Scholar]
- 29. Koren S, Walenz BP, Berlin K et al.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. A fuzzy Bruijn graph approach to long noisy reads assembly. https://github.com/ruanjue/wtdbg. Accessed 15 March 2019.
- 31. Chakraborty M, Baldwin-Brown JG, Long AD, et al.. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016;44(19):e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walker BJ, Abeel T, Shea T, et al.. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One. 2014;9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Servant N, Varoquaux N, Lajoie BR, et al.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16(1):259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burton JN, Adey A, Patwardhan RP, et al.. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol. 2013;31(12):1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kurtz S, Phillippy A, Delcher AL, et al.. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Q, Liu C, Liu Y, et al.. High-density interspecific genetic maps of kiwifruit and the identification of sex-specific markers. DNA Res. 2015;22(5):367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campbell M, Law M, Holt C et al.. MAKER-P: a tool-kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 2013;164(2):513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Han Y, Wessler SR. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 2010;38(22):e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gremme G, Steinbiss S, Kurtz S. GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinform. 2013;10(3):645–56. [DOI] [PubMed] [Google Scholar]
- 40. Smit A, Hubley R. RepeatModeler - 1.0.11 Institute for Systems Biology; http://www.repeatmasker.org/RepeatModeler/. Accessed 15 March 2019. [Google Scholar]
- 41. Exonerate. https://www.ebi.ac.uk/about/vertebrate-genomics/software/exonerate. Accessed 15 March 2019.
- 42. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stanke M, Keller O, Gunduz I, et al.. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, et al.. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33(20):6494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Z, Liu Y, Li D, et al.. Identification of circular RNAs in kiwifruit and their species-specific response to bacterial canker pathogen invasion. Front Plant Sci. 2017;8:413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoff KJ, Lange S, Lomsadze A, et al.. BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics. 2016;32(5):767–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Keilwagen J, Wenk M, Erickson JL, et al.. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016;44(9):e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rhee SY, Beavis W, Berardini TZ, et al.. The Arabidopsis Information Resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 2003;31(1):224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bairoch A, Boeckmann B. The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 1991;19(Suppl):2247–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL. Nucleic Acids Res. 1997;25(1):31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. High throughput protein function annotation with Human Readable Description (HRDs) and Gene Ontology (GO) Terms. https://github.com/groupschoof/AHRD. Accessed 15 March 2019.
- 52. Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–8. [DOI] [PubMed] [Google Scholar]
- 53. Mi H, Lazareva-Ulitsky B, Loo R, et al.. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33(Suppl):D284–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Finn RD, Bateman A, Clements J, et al.. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schultz J, Copley RR, Doerks T, et al.. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28(1):231–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bairoch A. PROSITE: a dictionary of sites and patterns in proteins. Nucleic Acids Res. 1991;19(Suppl):2241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Conesa A, Götz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang Y, Tang H, DeBarry JD, et al.. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ossowski S, Schneeberger K, Lucas-Lledó JI, et al.. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327(5961):92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nguyen LT, Schmidt HA, von Haeseler A, et al.. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2014;32(1):268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. De Bie T, Cristianini N, Demuth JP, et al.. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–71. [DOI] [PubMed] [Google Scholar]
- 63. Tang W, Sun X, Yue J, et al.. Supporting data for “Chromosome-scale genome assembly of kiwifruit Actinidia eriantha with single-molecule sequencing and chromatin interaction mapping.”. GigaScience Database. 2019. 10.5524/100568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tang W, Sun X, Yue J, et al.. Protocols for “Chromosome-scale genome assembly of kiwifruit Actinidiaeriantha with single-molecule sequencing and chromatin interaction mapping.”. protocols.io2019. 10.17504/protocols.io.vgse3we [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
9/25/2018 Reviewed
12/16/2018 Reviewed
2/1/2019 Reviewed
10/16/2018 Reviewed