

Abstract
Lipopolysaccharides (LPSs) are a major component of Gram-negative bacterial cell wall and play an important role in promoting intestinal inflammatory responses. Recent studies have shown that physiologically relevant concentrations of LPS (0 to 2000 pg/mL) cause an increase in intestinal epithelial tight junction (TJ) permeability without causing cell death. However, the intracellular pathways and the mechanisms that mediate LPS-induced increase in intestinal TJ permeability remain unclear. The aim was to delineate the intracellular pathways that mediate the LPS-induced increase in intestinal permeability using in vitro and in vivo intestinal epithelial models. LPS-induced increase in intestinal epithelial TJ permeability was preceded by an activation of transforming growth factor-β–activating kinase-1 (TAK-1) and canonical NF-κB (p50/p65) pathways. The siRNA silencing of TAK-1 inhibited the activation of NF-κB p50/p65. The siRNA silencing of TAK-1 and p65/p50 subunit inhibited the LPS-induced increase in intestinal TJ permeability and the increase in myosin light chain kinase (MLCK) expression, confirming the regulatory role of TAK-1 and NF-κB p65/p50 in up-regulating MLCK expression and the subsequent increase in TJ permeability. The data also showed that toll-like receptor (TLR)-4/myeloid differentiation primary response (MyD)88 pathway was crucial upstream regulator of TAK-1 and NF-κB p50/p65 activation. In conclusion, activation of TAK-1 by the TLR-4/MyD88 signal transduction pathway and MLCK by NF-κB p65/p50 regulates the LPS-induced increase in intestinal epithelial TJ permeability.
Intestinal epithelial tight junction (TJ) barrier dysfunction contributes to the development of inflammatory bowel disease (IBD) and necrotizing enterocolitis (NEC) by permitting increased paracellular permeation of luminal antigens that elicit and promote inflammatory response.1, 2, 3 Several clinical and animal studies have highlighted the involvement of defective intestinal TJ barrier in the development and the prolongation of intestinal inflammation in IBD and NEC.1, 3, 4, 5 Patients with IBD and NEC have a defective intestinal TJ barrier as shown by an increase in intestinal permeability,5, 6 and animal studies have shown that protecting the intestinal TJ barrier prevents the development of intestinal inflammation in animal models of IBD and NEC.7, 8
Lipopolysaccharides (LPSs) are complex amphiphilic molecules that have a hydrophobic (consisting of lipid A) and a hydrophilic (consisting of carbohydrate core and polysaccharide O-antigen) component and are released from the bacterial cell wall by shedding or through bacterial lysis. In pathologic conditions, the intestinal tissue and circulating LPS levels are markedly elevated and play an important role in mediating inflammatory response. Normally, LPS concentrations are maximum in the gut lumen where the gut bacteria reside and low or undetectable in the circulating plasma (<200 pg/mL) because the intestinal epithelial layer creates an effective barrier against LPS penetration across the healthy intestinal epithelium.6, 9 However, defective intestinal TJ barrier in IBD and NEC leads to paracellular permeation of LPS and other water-soluble luminal antigens, resulting in an increase in the intestinal tissue and plasma concentration of LPS.10, 11 Studies have shown LPS to be an important contributing factor of intestinal inflammation, and removal of circulating LPS accelerates the clinical improvement of IBD and NEC. Clinically achievable concentrations of LPS during active and quiescent IBD and NEC ranges between 200 and 2000 pg/mL. Animal models of NEC and IBD also have increased levels of LPS in the intestinal tissue and in the serum.11, 12, 13 The intestinal tissue expression of toll-like receptor-4 (TLR-4), the pattern recognition receptor that binds the LPS, is also markedly up-regulated in IBD and NEC.14, 15 The TLR-4 polymorphism is associated with an increased risk of IBD and a more extensive colonic involvement in ulcerative colitis.16 Intestinal TJ barrier is primarily regulated by myosin light chain kinase (MLCK).13, 17, 18, 19, 20, 21, 22 MLCK causes the contraction of the peri-junctional actomyosin filaments, leading to a mechanical tension-generated opening of the TJ barrier.17, 18, 22, 23 The intestinal TJ permeability can be increased by physiological concentrations of LPS (0 to 1000 pg/mL) via activation of TLR-4 signal transduction pathway and an increase in MLCK expression and activity.19, 24, 25
NF-κB is a nuclear transcription factor that plays an essential regulatory role in promoting intestinal inflammation. In quiescent cells, NF-κB heterodimers p50/p65 are sequestered in cytosol by a protein inhibitor inhibitory κ B (IκB). The IκB kinase (IKK) complex consists of two catalytic subunits, IKK-β and IKK-α, and a regulatory protein, IKK-γ (alias NF-κB essential modulator). Two distinct NF-κB pathways have been described, the canonical (or classic) and the noncanonical (or alternative) pathways that are involved in NF-κB activation.26, 27, 28, 29, 30, 31 The activation of the canonical pathway leads to the phosphorylation and degradation of inhibitory IκB-α and activation of NF-κB dimer p50/p65,28, 32 whereas the activation of the noncanonical pathway results in the phosphorylation of the p100 subunit, leading to the generation and activation of RelB/p52 dimer.28, 33, 34 NF-κB activation can be triggered by a wide range of proinflammatory mediators, including tumor necrosis factor-α, IL-1β, interferon-γ, and LPS. LPS binds to TLR-4 complex at the cell surface, stimulating a cascade of intracellular signaling events that culminate in the phosphorylation of IκB-α. The LPS activation of both canonical and noncanonical NF-κB pathways has been shown in various models with LPS.35, 36 An important concern with the previous studies is that the LPS concentration used in those studies were at a pharmacologic concentration of 50 μg/mL, which is nonphysiologic and not achievable clinically.35, 36 At a concentration of 50 μg/mL, LPS causes rapid cell apoptosis and cell death.21, 37, 38 In contrast, clinically achievable concentrations of LPS (0 to 2000 pg/mL) do not cause cell death.10, 11, 24 Transforming growth factor-β–activated kinase-1 (TAK-1) has been shown to be a pivotal factor for NF-κB activation in response to the activation of various toll-like receptors, including TLR-2, TLR-4, and TLR-5.39 The phosphorylation of TAK-1 is a common signal for the activation of downstream targets, including IKK, Jun kinase, and p38 mitogen-activated protein kinase.40, 41 Previously, it was shown that the LPS-induced increase in intestinal TJ permeability depended on activation of a TLR-4 signal transduction pathway.24 However, the intracellular signaling processes that mediate the TLR-4 signal transduction pathway modulation of MLCK expression and TJ permeability remain unclear. Here, the involvement of TAK-1 and NF-κB pathways was examined in the LPS-induced increase in intestinal TJ permeability with the use of intestinal epithelial model systems that consisted of filter-grown Caco-2 monolayers and live mice.
Materials and Methods
Reagents
Dulbecco's Modified Eagle Medium, trypsin, fetal bovine serum, penicillin, streptomycin, and phosphate-buffered saline (PBS) were purchased from Gibco BRL (Grand Island, NY). LPS (O111:B4) and TAK-1 inhibitor, 5Z-7-oxozeaenol, NF-κB inhibitors, ammonium pyrrolidinedithiocarbamate (PDTC) and Bay-11, 5Z-7-oxozeaenol were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies [IκB-α, p65, phospho- (p)65, p50, p52, p100, IKK-α, IKK-β, TAK-1, pTAK-1, IL-1 receptor-associated kinase 4, phospho–mitogen-activated kinase kinase (pMEKK)-1, MEKK-1, phospho NF-κB–inducing kinase (pNIK), NIK, MLCK, pMLC, TLR-4, myeloid differentiation primary response (MyD)88, and anti–β-actin] were obtained from Abcam (Cambridge, MA). Phospho–IKK-α/β antibodies were also obtained from Abcam and Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase–conjugated secondary antibodies for Western blot analysis were purchased from Invitrogen (San Francisco, CA). All other chemicals were purchased from Sigma-Aldrich, VWR (West Chester, PA), or Fisher Scientific (Pittsburgh, PA). Curcumin, PDTC, and Bay-11 were purchased from Sigma-Aldrich, VWR (Aurora, CO), or Fisher Scientific. siRNA for p65, p50, p100, p52, IKK-α, IKK-β TAK-1, NIK, MEKK-1, MLCK, TLR-4, and MyD88 were purchased from Dharmacon (Lafayette, CO).
Cell Culture
Caco-2 cells (passage 20) were purchased from the ATCC (Manassas, VA) and maintained at 37°C in Dulbecco's Modified Eagle Medium composed of 4.5 mg/mL glucose, 50 U/mL penicillin, 50 U/mL streptomycin, 4 mmol/L glutamine, 25 mmol/L HEPES, and 10% fetal bovine serum as previously described.19 Caco-2 cells were used between passages 22 and 30 in this study. The cells were kept at 37°C in a 5% CO2 environment. For filter-grown cells, high-density cells (1 × 105 cells) were plated on Transwell filters with 0.4-μm pore (Corning Incorporated, Corning, NY) and monitored regularly by visualization with an inverted microscope (Eclipse TS100/100-F; Nikon, Melville, NY) and by epithelial resistance measurements.
Assessment of Protein Expression by Western Blot Analysis
Protein expression from Caco-2 cells and mouse tissue (intestinal scrapings that contained mostly enterocytes) was assessed by Western blot analysis as previously described.19 Cells and mouse intestinal scrapings were lyzed with lysis buffer [50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 500 μmol/L NaF, 2 mmol/L EDTA, 100 μmol/L vanadate, 100 μmol/L phenylmethylsulfonyl fluoride, 1 μg/mL leupeptin, 1 μg/mL pepstatin A, 40 mmol/L paranitrophenyl phosphate, 1 μg/mL aprotinin, and 1% Triton X-100] on ice for 30 minutes. For cytosolic and nuclear fractionation the NE-PER kit from Thermo Scientific (Pittsburgh, PA) was used according to the manufacturer's instructions. The lysates were centrifuged at 10,000 × g for 10 minutes in an Eppendorf Centrifuge (5417R; Hauppauge, NY) to obtain a clear lysate. The supernatant was collected, and protein concentration was determined with the Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). Laemmli buffer (Bio-Rad Laboratories) was added to the lysate that contained 20 to 40 μg of protein and boiled at 100°C for 7 minutes, after which proteins were separated on an SDS-PAGE gel. Proteins from the gel were transferred to the membrane (Trans-Blot Transfer Medium, Nitrocellulose Membrane; Bio-Rad Laboratories) overnight. The membrane was incubated for 2 hours in blocking solution (5% dry milk or 5% bovine serum albumin in tris-buffered saline–Tween 20 buffer). The membrane was then incubated with antibody in blocking solution. After a wash in tris-buffered saline–1% Tween buffer, the membrane was incubated in secondary antibody and developed with the use of the Santa Cruz Western Blotting Luminal Reagents (Santa Cruz Biotechnology) on the Kodak Bio-Max MS film (Fisher Scientific, Pittsburgh, PA). ImageJ version 1.50d (NIH, Bethesda, MD; https://imagej.nih.gov/ij) was used for densitometry analysis for the quantification of the Western blot analyses.
Confocal Immunofluorescence Microscopy
The filter-grown cells were fixed with absolute methanol and stored at −80°C until used. The cells were thawed, rinsed in PBS, permeabilized with 0.1% Triton X-100, blocked with normal serum, and incubated overnight at 4°C in primary antibody. The cells were washed thoroughly and incubated in secondary antibodies conjugated with fluorescent dyes Alexa Fluor 488 or cyanine 3. After washings in PBS, the cells were mounted in Prolong Gold antifade reagent (Invitrogen) that contained DAPI as a nuclear stain and examined with a Zeiss LSM 510 microscope (Zeiss, Jena, Germany) equipped with a digital camera (Fluorescence Microscopy Shared Resource, Health Sciences Center, University of New Mexico). For mice intestines, cryosections were fixed in methanol before immunofluorescence staining. Images were processed with ZEN LSM software version 1.0 en 04/2018 (Zeiss).
Determination of Epithelial Monolayer Resistance and Paracellular Permeability
Caco-2 transepithelial electrical resistance (TER) was measured by using an epithelial voltmeter (EVOM; World Precision Instruments, Sarasota, FL) as previously reported.19 Both apical and basolateral sides of the epithelium were bathed with buffer solution. Electrical resistance was measured until similar values were recorded on three consecutive measurements. Caco-2 paracellular permeability was determined by using an established paracellular marker inulin.24 Known concentrations of permeability marker (10 μmol/L) and its radioactive tracer were added to the apical solution.42 Low concentrations of permeability markers were used to ensure that negligible osmotic or concentration gradient was introduced.
siRNA Transfection
Caco-2 monolayers were transiently transfected with siRNA using Dharmafect transfection reagent (Dharmacon) as previously described.19, 24, 43, 44, 45 Briefly, cells (1 × 105/filter) were seeded into a Transwell plate and grown to 70% confluency. Caco-2 monolayers were then washed with PBS twice, and 0.5 mL of Accell medium (Thermo Scientific) was added to the apical compartment of each filter, and 1.5 mL was added to the basolateral compartment of each filter. siRNA (5 ng) of interest and Dharmafect reagent (2 μL) were preincubated in Accell medium. After 5 minutes of incubation, the two solutions were mixed, and the mixture was added to the apical compartment of each filter. For LPS experiments, Caco-2 cells were transfected with siRNA for 24 hours before the LPS treatment. The Caco-2 TJ barrier assessments were performed at day 3 or 3.5 and at the end of LPS treatment.
Flow Cytometry
To determine the activation of the NF-κB pathway, filter-grown Caco-2 monolayers were treated with or without LPS (300 pg/mL) for 3 to 4 days. The treated and untreated monolayers were detached by incubation with 0.25% EDTA in calcium- and magnesium-free PBS (pH 7.2). Cells were immediately fixed and prepped for intranuclear permeabilization with the use of ice-cold BD Phosflow Perm Buffer III (BD Biosciences Inc., San Jose, CA).46, 47 The resulting permeabilized cells were stained with phospho-Iκβ alpha (S32/S36) (RILYB3R) eFluor 660 ebioscience or concentration-matched Mouse IgG2b kappa Isotype control eFluor 660 ebioscience (Thermo Fisher Scientific Inc.) or phospho–NF-kB p65 (ser536) (clone93H1) rabbit monoclonal antibody (phosphatidylethanolamine conjugate) or concentration-matched Rabbit (DA1E) monoclonal antibody IgG XP Isotype Control (phosphatidylethanolamine Conjugate) (Cell Signaling Technology, Inc.) in cold 0.1% bovine serum albumin/PBS for 30 minutes. All samples were collected on a LSR Fortessa (Becton Dickinson, Franklin Lakes, NJ), and data were analyzed with FlowJo software version 10 (TreeStar Inc., Ashland, OR).
In Vivo Determination of Mouse Intestinal Permeability
The Laboratory Animal Care and Use Committee at the University of New Mexico approved all experimental protocols. C57BL/6 mice, TLR-4, and MyD88 knockout mice (9 to 10 weeks of age) were purchased from The Jackson Laboratories (Bar Harbor, ME). The mice were kept two per cage in a temperature-controlled room at 25°C with a 12:12 hour light/dark cycle. Diet and drinking water were provided ad libitum.
LPS effect on intestinal permeability in an in vivo mouse model system was determined with the use of a re-cycling intestinal perfusion method as previously described.19, 24, 25 Briefly, male or female mice were injected with LPS intraperitoneally (0.1 mg/kg body weight) every 24 hours for up to 5 days of experimental period. LPS caused similar increase in intestinal permeability in male and female mice. For experimental NF-κB inhibition, mice were injected with PDTC (10 mg/kg body weight) or Bay-11 (5 mg/kg body weight) dissolved in dimethyl sulfoxide or without LPS for 5 days. Similarly, for experimental TAK-1 inhibition, mice were also injected with 5Z-7-oxozeaenol (5 mg/kg body weight) with or without LPS for 5 days. A 6-cm segment of mouse small intestine was isolated and cannulated with a small-diameter plastic tube (in an anesthetized mouse maintained in 1% isoflurane in oxygen) and continuously perfused with 5 mL of Krebs-PBS for a 2-hour perfusion period. An external recirculating pump (Econo Pump; Bio-Rad Laboratories) was used to recirculate the perfusate at a constant flow rate (0.75 mL/minute). The body temperature of mice was maintained at 37°C with a temperature-controlled warming blanket. The intestinal permeability was assessed by measuring flux rate of paracellular probe, Texas Red-labeled dextran (molecular weight = 10,000 g/mol). Water absorption was determined by using a nonabsorbable marker sodium ferrocyanide or by measuring the difference between initial and final volume of the perfusate.
Statistical Analysis
Data shown represent the means ± SEM. Statistical analyses were performed by using GraphPad Prism software version 7 (GraphPad Software, La Jolla, CA). Differences between groups were determined by U-test or one-way analysis of variance or by two-way analysis of variance with Bonferroni or Tukey posttest. A P value < 0.05 was considered statistically significant.
Results
LPS-Induced Increase in Caco-2 TJ Permeability Is Mediated by an Activation of NF-κB Pathway
LPS caused a time- and concentration-dependent increase in Caco-2 TJ permeability (measured by mucosal-to-serosal flux of paracellular marker inulin) (Table 1) starting between 3 and 4 days after LPS exposure.19 However, the signaling pathways and mechanisms that mediated LPS-induced increase in MLCK gene expression and TJ permeability remained unclear. Because LPS was known to activate NF-κB (both canonical and noncanonical pathway), it was hypothesized that the LPS-induced increase in intestinal TJ permeability was mediated in part by TLR4/MyD88 signal transduction pathway activation of NF-κB. To test this hypothesis, the effects of pharmacologic NF-κB inhibitor, PDTC, were first examined on LPS-induced increase in Caco-2 TJ permeability. PDTC prevented the LPS-induced drop in Caco-2 TER (Figure 1A) and increase in mucosal-to-serosal inulin flux (Figure 1B). These data suggested that the activation of NF-κB was required for the LPS-induced increase in Caco-2 TJ permeability.
Table 1.
Time-Course Effect of LPS (0.3 ng/mL) on Mucosal-to-Serosal Flux of Paracellular Probe Inulin in Filter-Grown Caco-2 Monolayers
| Time, days | Relative inulin flux/cm2 per minute |
|---|---|
| 0 | 1.00 ± 0.021 |
| 1 | 0.9574 ± 0.0524 |
| 2 | 0.8033 ± 0.0628 |
| 3 | 1.5423 ± 0.1.256 |
| 4 | 4.123 ± 0.6597∗ |
| 5 | 5.627 ± 1.752∗ |
Adapted from Nighot et al19 with permission from Elsevier. Inulin was measured over a 5-day experimental period of basolateral exposure of Caco-2 monolayers to lipopolysaccharide (LPS).19, 24, 25 LPS caused a time-dependent increase in Caco-2 tight junction permeability, starting at days 3 to 4 after LPS exposure (n = 6). Data are expressed as means ± SEM.
P < 0.01 versus control untreated.
Figure 1.

Effect of pharmacologic inhibition of NF-κB on lipopolysaccharide (LPS)-induced increase in intestinal epithelial tight junction permeability. A: Filter-grown Caco-2 monolayers were treated with 300 pg/mL LPS for a 5-day experimental period. Pharmacologic inhibition of NF-κB by 10 μm ammonium pyrrolidinedithiocarbamate (PDTC) inhibited the LPS (physiological dose of 300 pg/mL)-induced drop in Caco-2 transepithelial electrical resistance. B: Pharmacologic inhibition of NF-κB by PDTC prevented the LPS-induced increase in inulin flux. NF-κB inhibitor PDTC was added 1 hour before LPS treatment. All experimental treatments were renewed every 24 hours for the 5-day experimental period. Data are expressed as means ± SEM. n = 4 independent experiments. ∗∗∗P < 0.001 versus control; †††P < 0.001 versus LPS.
Canonical but Not Noncanonical NF-κB Pathway Mediates the LPS-Induced Increase in Caco-2 TJ Permeability
In the following studies, the possible involvement of canonical (NF-κB p50/p65) or noncanonical (NF-κB p52/p100) pathway in the LPS modulation of Caco-2 TJ permeability was examined. The effect of LPS (300 pg/mL) on activation of canonical or noncanonical pathway was determined by immunoblot analysis and immunostaining. LPS caused an increase in nuclear expression of p65 but not p52 (Figure 2, A and B). LPS also caused an increase in cytoplasmic-to-nuclear translocation of NF-κB p65 but not NF-κB p52 (Figure 2, C and D). In separate experiments, the activation of canonical pathway was also determined by degradation of inhibitory protein IκB-α. LPS caused a degradation of IκB-α in Caco-2 cells by 3 to 3.5 days (Figure 2, E and F). To further confirm the LPS-induced activation of canonical NF-κB pathway, the phosphorylation of IκB-α and p65 was determined by flow cytometry. LPS caused an increase in phospho IκB-α (mean fluorescence intensity: control, 175 ± 12.12, versus LPS, 292 ± 25.63) and phospho-p65 (mean fluorescence intensity: Control, 3307 ± 154.7, versus LPS, 4329.3 ± 169.4) in Caco-2 cells by 3 to 3.5 days of LPS exposure (Figure 2, G and H). Together these data demonstrated that LPS induced the degradation of IκB-α and activation and nuclear translocation of NF-κB p50/p65. In the following studies, the role of IKKs, IKK-α or IKK-β, in mediating the LPS-induced increase in intestinal TJ permeability was examined. In these studies, the effect of LPS on IKK-α or IKK-β activation was examined by immunoblot. LPS (300 pg/mL) treatment caused phosphorylation of both IKK-α and IKK-β by day 3 to 3.5 (Figure 3A). To further evaluate the requirement of IKK-α or IKK-β on LPS-induced increase in Caco-2 TJ permeability, IKK-α or IKK-β was silenced by transfection with IKK-α or IKK-β siRNA (Figure 3, B and C). The LPS-induced increase in inulin flux was completely inhibited by IKK-β siRNA and transfection, mostly prevented by IKK-α siRNA transfection, but to a lesser degree than IKK-β transfection (Figure 3, D and E).
Figure 2.

Time course effect of lipopolysaccharide (LPS) on Caco-2 NF-κB pathway (p65 and p52) activation. A: LPS at the concentration of 300 pg/mL caused significant increase in p65 protein expression in Caco-2 cells on day 3 to 3.5. The expression of p52 and p100 did not change with LPS treatment. β-Actin was used as an internal control for protein loading. B: Relative densitometry analysis for p65 protein levels. C: Confocal immunofluorescence of Caco-2 cells treated with 300 pg/mL LPS (3 to 3.5 days) indicated p65 (red) translocation to the nucleus (blue) (arrowheads). D: P52 (green) did not change after LPS treatment (arowheads). E: LPS caused degradation of inhibitory κ B (IκB)-α expression (3 to 3.5 days), as assessed by Western blot analysis. β-Actin was used as an internal control for protein loading. F: Densitometry analysis of LPS treatment showed significant decrease in IκB-α level on day 3 to 3.5 compared with control untreated cells. G and H: LPS-treated Caco-2 cells (3 to 3.5 days) analyzed by flow cytometry for phospho-IκB-α and phospho-p65, respectively, showed increased expression compared with control untreated cells. Gray indicates isotope control (Cont); blue, control untreated; red, LPS treated day 3. [Mean fluorescence intensity (MFI): pIκB-α, 175 ± 12.12, versus 292 ± 25.63; pp65, 3307 ± 154.7, versus 4329.3 ± 169.4]. Data are expressed as means ± SEM. n = 3 independent experiments. ∗∗∗P < 0.001 versus control; †††P < 0.01 versus LPS (day 4); ‡‡‡P < 0.01 versus LPS (day 3.5). Scale bars = 5 μm (C and D).
Figure 3.

Effect of lipopolysaccharide (LPS) on inhibitory κ B kinases (IKKs) and intestinal epithelial tight junction permeability. A: LPS at 300 pg/mL in Caco-2 cells caused significant increase in activation of both IKK-α and IKK-β on day 3 to 3.5 after LPS treatment. β-Actin was used as an internal control (C; Con; Cont) for protein loading. Densitometry analysis of LPS treatment showed significant increase in IKK-α and IKK-β, respectively, on day 3 and 3.5 compared with control untreated cells. B and C: siRNA transfection of IKK-α and IKK-β in Caco-2 cells inhibited the protein expression of IKK-α and IKK-β. D: siRNA transfection of IKK-α in Caco-2 cells partially prevented the LPS-induced increase in inulin flux. E: siRNA transfection of IKK-β in Caco-2 cells inhibited the LPS-induced increase in inulin flux. Data are expressed as means ± SEM. n = 6 experiments (B, C, and E). ∗∗∗P < 0.001 versus control untreated cells; †††P < 0.001 versus control (siNT); ‡‡‡P < 0.001 versus nontarget (NT) siRNA; §§§P < 0.001 versus LPS.
Next, the requirement of NF-κB p50/p65 or NF-κB p52/p100 on LPS-induced increase in Caco-2 TJ permeability was determined by siRNA-induced silencing of NF-κB p50/p65 or NF-κB 52/p100. The siRNA transfections of p65, p50, p52, or p100 caused a marked depletion of respective target protein (Figure 4, A–D). The siRNA-induced silencing of p50 or p65 subunit resulted in an inhibition of LPS-induced drop in TER and an increase in inulin flux in Caco-2 monolayers (Figure 4, E and F). In contrast, siRNA-induced knockdown of p52 or p100 did not have significant effect on the LPS-induced drop in TER or increase in inulin flux (Figure 4, G and H). Collectively, these data demonstrated that LPS caused an activation of the canonical NF-κB pathway and that NF-κB p50/p65 activation was required for the LPS-induced increase in Caco-2 TJ permeability.
Figure 4.

Effects of genetic knockdown of p65/p50 and p52/p100 on lipopolysaccharide (LPS)-induced increase in intestinal epithelial tight junction permeability. A: p65 siRNA transfection resulted in a near-complete depletion of p65. Relative densitometry analysis of p65 protein expression levels. B: The siRNA of p50 transfection resulted in a near-complete depletion of p50 protein expression. Relative densitometry of p50 protein expression levels. C: The p52 siRNA transfection resulted in a near-complete depletion of p52 as shown in densitometry analysis. D: p100 siRNA transfection resulted in a near-complete depletion of p100, as shown in densitometry analysis. E: p65 and p50 siRNA transfection in Caco-2 monolayers inhibited the LPS-induced drop in Caco-2 cell transepithelial electrical resistance (TER). F: p65 and p50 siRNA transfection inhibited the LPS-induced increase in inulin flux. G: p52 and p100 siRNA transfection in Caco-2 monolayers did not prevent the LPS-induced drop in TER. H: p52 and p100 siRNA transfection did not prevent the LPS-induced increase in inulin flux. The Western blot analysis was performed 72 hours after all siRNA transfections. n = 4 experiments (A–D and F); n = 3 experiments (H). ∗∗∗P < 0.001 versus control; †††P < 0.001 versus LPS. Con, control; NT, nontarget.
TAK-1 Mediates the LPS-Induced Activation of Canonical NF-κB Pathway and Increase in Caco-2 TJ Permeability
TAK-1 has been implicated to play an important regulatory role in toll-like receptor activation of NF-κB,34, 48, 49, 50, 51, 52 and recent studies have also shown both NIK and MEKK-1 to be an important regulator of NF-κB p50/p65 activation.34, 53 To determine the upstream protein kinases responsible for the LPS activation of NF-κB p50/p65, the possible involvement of TAK-1, NIK, and MEKK-1 was examined. LPS (300 pg/mL) caused an increase in TAK-1 phosphorylation by day 3 in Caco-2 monolayers (Figure 5A). Immunostaining studies also showed that LPS caused an increase in phosphorylated TAK-1 in the cytoplasm by day 3 of LPS treatment (Figure 5D). TAK-1, NIK, and MEKK-1 siRNA transfection resulted in a marked depletion of TAK-1, NIK, and MEKK-1 proteins, respectively (Figure 5E). The siRNA knockdown of TAK-1 inhibited the LPS-induced drop in Caco-2 cell TER and increase in inulin flux (Figure 5, F and G). Next, the effect of LPS on MEKK-1 or NIK activation was determined by assessing MEK-1 and NIK phosphorylation. LPS did not cause an increase in NIK or MEKK-1 phosphorylation (Figure 5, B and C). The siRNA silencing of NIK or MEKK-1 (Figure 5H) did not inhibit the LPS-induced drop in TER (Figure 5H). Next, the effect of TAK-1 knockdown on LPS-induced activation of NF-κB p50/p65 was determined. The siRNA-induced silencing of TAK-1 inhibited the LPS-induced degradation of IκB-α and the cytoplasmic-to-nuclear translocation of NF-κB p50/p65 (Figure 6, A and C).In addition, TAK-1 inhibition prevented activation of LPS-induced activation of IKK-α and IKK-β phosphorylation (Figure 6E). Together, these data suggested that TAK-1 activation was required for the LPS-induced activation of canonical NF-κB pathway and increase in Caco-2 TJ permeability.
Figure 5.

Role of transforming growth factor-β–activating kinase (TAK)-1 in the lipopolysaccharide (LPS)-induced activation of canonical NF-κB pathway and Caco-2 tight junction permeability. A: LPS treatment at the concentration of 300 pg/mL caused activation of phospho-TAK-1. B and C: LPS treatment did not induce phosphorylation of NF-κB–inducing kinase (NIK) or mitogen-activated kinase kinase (MEKK)-1 at day 3 after LPS exposure compared with untreated Caco-2 cells. D: Confocal immunofluorescence of Caco-2 cells treated with 300 pg/mL LPS for 5 days indicated increase in pTAK-1 (green) on day 3 and 3.5 after LPS treatment. Nucleus, blue. E: TAK-1, NIK, and MEKK-1 siRNA transfections in Caco-2 cells significantly reduced TAK-1, NIK, and MEKK-1 protein expression, as analyzed by Western blot analysis and relative densitometry, respectively. F: TAK-1 siRNA transfection prevented the LPS-induced drop in Caco-2 cell transepithelial electrical resistance (TER). G: TAK-1 siRNA transfection inhibited the LPS-induced increase in Caco-2 inulin flux. H: NIK or MEKK-1 siRNA in transfection in Caco-2 cells did not prevent the LPS-induced drop in Caco-2 TER. Data are expressed as means ± SEM. n = 4 experiments (A). ∗∗∗P < 0.001 versus control; ††P < 0.01 versus LPS day 3; ‡‡P < 0.01 versus LPS day 3.5; §§P < 0.01 versus nontarget (NT) siRNA. Scale bars = 5 μm. C, control.
Figure 6.

Effect of toll-like receptor (TLR)-4, myeloid differentiation primary response (MyD)88, and transforming growth factor-β–activating kinase (TAK)-1 siRNA on lipopolysaccharide (LPS)-induced activation of NF-κB canonical pathway in Caco-2 cells. A: TAK-1 siRNA transfection in Caco-2 cells prevented LPS-induced degradation of inhibitory κ B (IκB)-α protein expression compared with nontarget (NT) siRNA-transfected LPS-treated cells. Relative densitometry analysis of IκB-α protein levels. B: TLR-4 siRNA and MyD88 siRNA transfection in Caco-2 cells prevented LPS-induced increase in TAK-1 phosphorylation. Relative densitometry analysis of pTAK-1 protein levels. C: Confocal immunofluorescence showed that TLR-4, MyD88, and TAK-1 siRNA transfection of Caco-2 monolayers prevented the LPS-induced p65 (red) translocation to the nucleus (blue) (arrowhead) at 3 day after LPS exposure D: TLR-4 siRNA and MyD88 siRNA transfection in Caco-2 cells prevented LPS-induced degradation of IκB-α protein expression. Densitometry of IκB-α protein levels. E: TAK-1 siRNA transfection in Caco-2 cells prevented LPS-induced activation of IκB kinase (IKK)-α and IKK-β compared with nontarget (NT) siRNA-transfected LPS-treated cells. Relative densitometry analysis of IKK-α and IKK-β is also shown. n = 4 experiments. ∗∗P < 0.01, ∗∗∗P < 0.001 versus control; ††P < 0.01, †††P < 0.001 versus LPS. Scale bars: 5 μm (C). C, control.
LPS-Induced Activation of TAK-1 and NF-κB p50/p65 Pathway Is Regulated by TLR-4/MyD88 Signal Transduction Axis
In the following studies, the regulatory role of TLR-4 receptor complex on TAK-1 and canonical NF-κB pathway activation was examined. In these studies, the effect of siRNA silencing of TLR-4 or MyD88 on TAK-1 and NF-κB p50/p65 was determined. The siRNA silencing of TLR-4 or MyD88 inhibited the LPS-induced increase in TAK-1 phosphorylation (Figure 6B). The TLR-4 or MyD88 siRNA transfection of Caco-2 monolayers also inhibited the LPS-induced IκB-α degradation (Figure 6D) and the cytoplasmic-to-nuclear translocation of NF-κB p50/p65 dimer (Figure 6C). These findings suggested that the LPS-induced activation of TAK-1 and NF-κB p50/p65 was mediated by the TLR-4/MyD88 signal-transduction pathway.
LPS-Induced Increase in MLCK Expression Is Mediated by TAK-1–Dependent Activation of NF-κB p50/p65
Previous studies from our laboratory showed that MLCK was an effector protein responsible for the LPS-induced increase in Caco-2 TJ permeability.19 In the following studies, the involvement of TAK-1 and NF-κB p50/p65 in LPS-induced increase in MLCK gene and protein expression was examined. The TAK-1 siRNA transfection inhibited the LPS-induced increase in MLCK mRNA and phosphorylated MLC and MLCK protein expression (Figure 7, A and B). The NF-κB p50 or p65 siRNA transfection also inhibited the LPS-induced increase in MLCK mRNA and protein expression in Caco-2 monolayers (Figure 7, C and D). These findings suggested that the LPS-induced increase in Caco-2 MLCK mRNA and protein expression in Caco-2 monolayers was mediated by TAK-1–dependent activation of NF-κB p50/p65.
Figure 7.

Effect of genetic knockdown of (p65/p50) canonical pathway and transforming growth factor-β–activating kinase (TAK)-1 on lipopolysaccharide (LPS)-induced activation of myosin light chain kinase (MLCK) protein expression. A: TAK-1 siRNA transfection in Caco-2 cells prevented LPS-induced increase in MLCK mRNA. B: Similarly, TAK-1 siRNA transfection in Caco-2 cells also inhibited LPS-induced increase in MLCK and phosphoMLC protein expression by Western blot analysis and was shown by relative densitometry of MLCK protein. C: p65 siRNA transfection in Caco-2 cells prevented LPS-induced increase in MLCK mRNA. D: p50 and p65 siRNA transfection in Caco-2 cells caused marked inhibition of LPS-induced increase in MLCK protein expression, as analyzed by Western blot analysis and relative analysis of MLCK protein levels by densitometry. n = 3 experiments (A); n = 4 experiments (B and D). ∗∗P < 0.01 versus nontarget siRNA control; ††P < 0.01 versus LPS. C, control; NT, nontarget.
LPS-Induced Increase in Mouse Intestinal Permeability in Vivo Is Mediated by an Enterocyte NF-κB p50/p65 Activation
Previous studies from our laboratory showed that the i.p. injection of LPS (0.1 mg/kg body weight) in mice caused a serum steady state LPS concentrations of 0.405 ng/mL and peak level of 2.35 ng/mL.19, 24, 25 In the following studies, the in vivo effect of LPS on mouse enterocyte NF-κB p50/p65 activation was determined by assessing IκB-α degradation and cytoplasmic-to-nuclear NF-κB p65 translocation. The i.p. LPS administration (0.1 mg/kg body weight) resulted in an activation of mouse enterocyte NF-κB p50/p65 as evidenced by marked decrease in intestinal tissue IκB-α expression and an increase in cytoplasmic-to nuclear translocation of NF-κB p50/p65 in mouse enterocytes (Figure 8, A–C). Next, to determine the requirement of NF-κB p50/p65 activation in LPS-induced increase in mouse intestinal permeability, the effects of pharmacologic NF-κB inhibitor PDTC and Bay-11 on LPS-induced increase in mouse intestinal permeability were examined. The i.p. injection of PDTC inhibited the LPS-induced increase in mouse intestinal permeability to 10K dextran (10kd) (Figure 8D). Similarly, Bay-11 (5 mg/kg body weight), which inhibited IκB-degradation,54, 55, 56, 57 also inhibited the LPS-induced increase in mouse intestinal permeability (Figure 8D), confirming that the LPS-induced increase in intestinal permeability required the activation of NF-κB p50/p65.
Figure 8.

Activation of p65/p50 canonical pathway by lipopolysaccharide (LPS) in mice enterocytes and effect of NF-κB inhibitors on LPS-induced increase in mouse intestinal epithelial tight junction permeability. A: LPS i.p. injections (0.1 mg/kg body weight) in mice caused activation of canonical p65/p50 pathway, as assessed by degradation of inhibitory κ B (IκB)-α protein expression on day 3 in mice enterocytes. Densitometry of IκB-α protein levels. B: The immunoblot analysis from LPS-treated mice enterocytes revealed significant increase in nuclear p65 protein expression on day 3 compared with untreated mice enterocytes. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Lamin B were used as loading controls for cytoplasmic (cyto) and nuclear (nuc) fractions, respectively C: Confocal immunofluorescence of mouse intestines treated with LPS (0.1 mg/kg body weight) on day 3 indicated p65 (red) (arrowheads) translocation to the nucleus (blue) compared with control (C) mouse enterocytes. D: NF-κB inhibitor, ammonium pyrrolidinedithiocarbamate (PDTC; 10 mg/kg body weight), and Bay-11 (5 mg/kg body weight) pretreatment prevented the LPS-induced increase in 10K dextran flux. PDTC and Bay-11 were dissolved in dimethyl sulfoxide and injected 1 hour before LPS treatment. Data are expressed as means ± SEM. n = 4 experiments (A and C); n = 3 experiments (B). ∗∗P < 0.01 versus control vehicle; ††P < 0.01 versus LPS. Scale bars = 5 μm.
TAK-1 Mediates the LPS-Induced Activation of Mouse Enterocyte NF-κB p50/p65 in Vivo
In the following studies, the in vivo effect of LPS on TAK-1 activation in mouse enterocyte was determined. LPS i.p. injection (0.1 mg/kg body weight) resulted in an increase in TAK-1 phosphorylation in mouse intestinal enterocytes by day 3 (Figure 9, A and B). Next, the effect of TAK-1 inhibition was examined on LPS-induced increase in mouse intestinal permeability. An i.p. administration of pharmacologic inhibitor of TAK-1, oxozeaenol58 (5 mg/kg body weight), inhibited the LPS-induced increase in mouse intestinal permeability (Figure 9C), confirming that TAK-1 activation was also required for the LPS-induced increase in intestinal permeability.
Figure 9.

Lipopolysaccharide (LPS) activated transforming growth factor-β–activating kinase (TAK)-1 in vivo, and TAK-1 inhibition prevented the LPS-induced increase in mouse intestinal epithelial tight junction permeability. A: LPS i.p. injections (0.1 mg/kg body weight) in mice caused activation of TAK-1 (phopshoTAK-1) expression by day 3. Densitometry of phosphoTAK-1 protein levels. B: Confocal immunofluorescence showed increase in phopshoTAK-1 expression (green) (arrowheads) (nucleus, blue) in the intestines (enterocytes) of mice treated with LPS (0.1 mg/kg body weight). C: Pretreatment with TAK-1 inhibitor (Inh), oxozeaenol (5 mg/kg body weight), prevented the LPS-induced increase in 10K dextran flux. Oxozeaenol was dissolved in dimethyl sulfoxide and injected intraperitoneally, 1 hour before LPS injection. n = 3 experiments (A and B). ∗∗P < 0.01, ∗∗∗P < 0.001 versus control; †††P < 0.001 versus LPS. Scale bars = 5 μm. C, control.
TLR-4/MyD88 Signal Transduction Pathway and TAK-1 Mediate the LPS-Induced Activation of NF-κB p50/p65 in Mouse Intestine in Vivo
To study the role of TLR4 and MyD88 pathway in LPS-induced activation of NF-κB p50/p65 in mouse intestinal enterocytes, the effects of LPS on IκB-α degradation were examined in TLR-4– and MyD88-deficient mice enterocytes. The LPS-induced increase in IκB-α degradation in mice enterocytes was inhibited in TLR-4−/− and MyD88−/− mice, supporting the regulatory involvement of TLR-4/MyD88 axis in NF-κB p50/p65 pathway activation in mice (Figure 10A). In separate studies, the effect of TAK-1 inhibition by oxozeaenol on LPS-induced IκB-α degradation in mouse enterocytes was also examined. LPS treatment with TAK-1 inhibitor (oxozeaenol, 5 mg/kg body weight) also inhibited the enterocytes IκB-α degradation (Figure 10B). These data further supported the regulatory role of TLR-4/My D88 transduction pathways and TAK-1 in the activation of NF-κB p50/p65 in in vivo.
Figure 10.
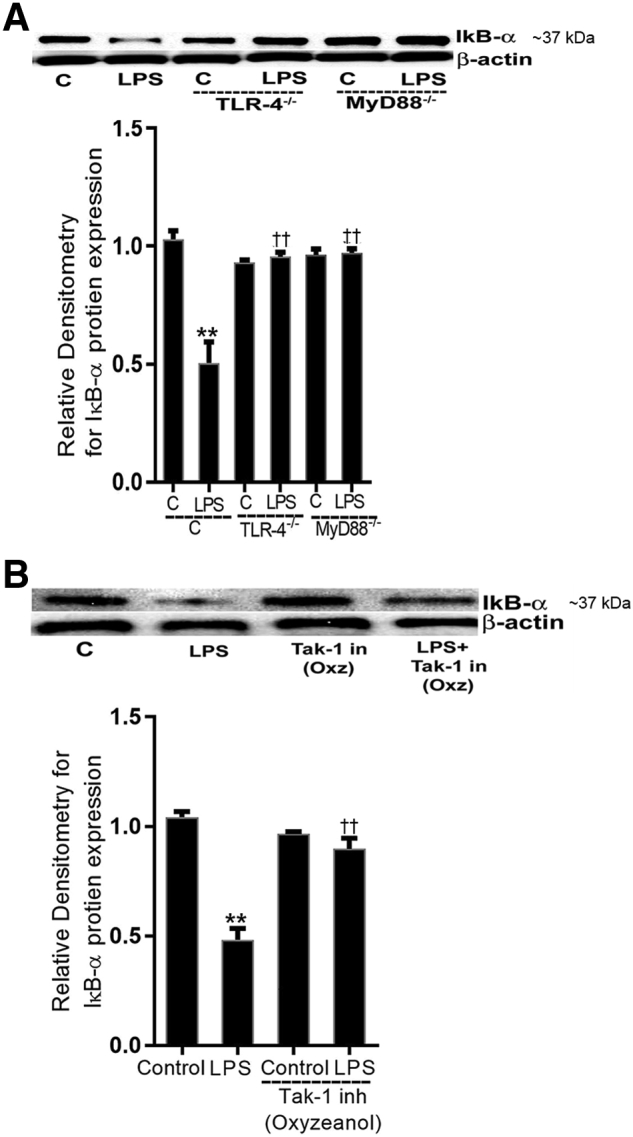
Lipopolysaccharide (LPS)-induced activation of NF-κB canonical pathway was inhibited in toll-like receptor (TLR)-4−/− and myeloid differentiation primary response (MyD)88−/− mice and with transforming growth factor-β–activating kinase (TAK)-1 inhibition. A: LPS i.p. injections (0.1 mg/kg body weight) in TLR-4−/− and MyD88−/− mice did not induce inhibitory κ B (IκB)-α degradation (day 3) compared with TLR4+/+ and MyD88+/+ control (C) mice, respectively. Densitometry of IκB-α protein. B: Pretreatment with TAK-1 inhibitor (in; inh), oxyzeanol (Oxz; 5 mg/kg body weight), prevented the LPS-induced degradation of IκB-α expression (day 3) compared with vehicle- or LPS-treated mice. Densitometry of IκB-α protein levels. n = 3 experiments. ∗∗P < 0.01 versus control; ††P < 0.01 versus LPS.
TAK-1 and NF-κB Pathway Mediates the LPS-Induced Increase in MLCK Expression in Mouse Intestine in Vivo
In the following studies, the involvement of TAK-1 and NF-κB p50/p65 on LPS-induced increase in MLCK protein in mouse intestinal tissue (enterocytes) was examined. Mice were injected with PDTC (10 mg/kg body weight) and Bay-11 (5 mg/kg body weight) or TAK-1 inhibitor, oxozeaenol (5 mg/kg body weight) in dimethyl sulfoxide with or without LPS (0.1 mg/kg body weight by i.p. injection) daily for 5 days. The mouse intestinal tissue (enterocytes) was prepared for immunoblotting to study MLCK protein expression. PDTC treatment inhibited the LPS-induced increase in MLCK protein expression in mouse enterocytes (Figure 11A). Similarly, Bay-11 also inhibited the LPS-induced increase in MLCK protein expression (Figure 11B). The TAK-1 inhibitor oxyzeonol also prevented the LPS-induced increase in MLCK protein expression (Figure 11C).
Figure 11.

Effect of inhibition of NF-κB pathway and transforming growth factor-β–activating kinase (TAK)-1 on lipopolysaccharide (LPS)-induced activation of myosin light chain kinase (MLCK) expression in mouse enterocytes. A: NF-κB inhibitor ammonium pyrrolidinedithiocarbamate (PDTC) prevented LPS-induced increase in MLCK protein expression (5 day) compared with vehicle- or LPS-treated mice. Densitometry of MLCK protein levels. B: NF-κB inhibitor Bay-11 also prevented LPS-induced increase in MLCK protein expression (5 day) compared with vehicle- or /LPS-treated mice. Densitometry of MLCK protein levels. C: Pretreatment with TAK-1 inhibitor (in; inh), oxyzeanol (Oxz; 5 mg/kg body weight), prevented the LPS-induced increase in MLCK protein expression (5 day) compared with vehicle- or LPS-treated mice. Densitometry of MLCK protein levels. n = 3 experiments. ∗∗P < 0.01 versus control; ††P < 0.01 versus LPS. C, control.
Discussion
Defective intestinal epithelial TJ barrier is an important pathogenic factor that contributes to the development of intestinal inflammation and systemic inflammatory responses by allowing increased intestinal permeation and systemic circulation of gut-derived bacterial antigens.1, 13 Plasma LPS levels are markedly elevated in IBD and NEC and play an important pathogenic role in the inflammatory process by activating and prolonging the immune response. In previous studies, pharmacologic concentrations of LPS, ranging from 1 to 100 μg/mL, induced rapid intestinal epithelial cell death within minutes of LPS exposure, leading to large gaps and openings in the epithelial layer and a rapid increase in paracellular epithelial permeability in both cell culture systems and live animals.19, 24 The most commonly used pharmacologic concentrations of LPS (50 μg/mL) exceed the clinically achievable serum concentrations of LPS (100 to 1000 pg/mL) by approximately 50,000 to 500,000 times.21, 59 However, LPS at lower clinically achievable concentrations, ranging from 0 to 2000 pg/mL, does not cause cell death but causes a selective time-dependent increase in intestinal TJ permeability.19, 24, 25 At clinically achievable concentrations, LPS-induced increase in intestinal TJ permeability is mediated by an increase in the expression of TJ effector protein MLCK.19 MLCK induces the opening of the TJ barrier by activating MLC phosphorylation and Mg++/myosin ATPase-dependent contraction of perijunctional actin and myosin filaments, which in turn generates mechanical tension and centripetal pulling apart of the TJ barrier.13, 17, 18, 20, 60 MLCK plays a central mechanistic role in inducing an increase in intestinal TJ permeability in a number of inflammatory conditions.19, 24, 25 In healthy individuals, serum LPS levels usually range from undetectable levels up to approximately 200 pg/mL. In patients with IBD and other inflammatory conditions such as necrotic enterocolitis and alcoholic liver disease, the serum LPS levels can range as high as 2000 pg/mL, but levels >3000 pg/mL are extremely rare and difficult to achieve.11, 61 LPS concentrations up to 2000 pg/mL do not cause intestinal cell death.10, 11, 12, 24 Clinically relevant concentration of LPS (300 pg/mL) were used in this study to decipher the intracellular processes that mediate the LPS-induced increase in intestinal TJ permeability.
In the studies described herein, the intracellular signaling pathway that mediates the LPS-induced increase in intestinal epithelial TJ permeability was studied. LPS-induced increase in intestinal TJ permeability is regulated in part by its binding and activation of TLR-4 receptor complex, and NF-κB is an important downstream target of the TLR-4 signal transduction pathway. This study examined the possible involvement of NF-κB in LPS modulation of the intestinal TJ barrier. Previous studies have suggested that both canonical and noncanonical NF-κB pathways play a regulatory role in intestinal TJ barrier regulation.24, 34 Our data showed that LPS causes an activation of canonical NF-κB pathway (p50/p65) but not the noncanonical pathway (p52/p100). The siRNA knockdown of p50/p65 but not p52/p100 inhibited the increase in Caco-2 TJ permeability. Thus, our studies show that the activation of NF-κB canonical pathway was required for the LPS-induced increase in Caco-2 TJ permeability.
To delineate the up-stream signaling kinases involved in NF-κB activation, the possible involvement of TAK-1, MEKK-1, and NIK was examined. Both MEKK-1 and NIK have been shown to regulate the IKK activity and TJ barrier opening.24, 34, 53 Interestingly, neither MEKK-1 nor NIK was activated by LPS, and their knockdown did not affect the NF-κB p50/p65 activation or the increase in Caco-2 TJ permeability. However, LPS caused an activation of TAK-1, and TAK-1 knockdown inhibited the LPS-induced activation of IKK-α, IKK-β, NF-κB p50/p65 and the increase in the Caco-2 TJ permeability. Our results herein describe a new distinct kinase pathway regulation of LPS-induced increase in intestinal TJ permeability that involves TAK-1. In contrast, the prior studies showed that proinflammatory mediators tumor necrosis factor-α and IL-1β24, 53 cause an increase in intestinal TJ permeability by MEKK-1– and NIK-dependent activation of NF-κB p50/p65. The present results show that TAK-1 mediates the LPS-induced NF-κB p50/p65 activation and increase in intestinal TJ permeability, independent of MEKK-1 or NIK. TAK-1 has been demonstrated to be involved in the activation of IKK-β in neural cells, and recent studies have shown that polyubiquitination of signaling proteins through lysine (Lys)-63–linked polyubiquitin chains plays an important role in the activation of TAK1 and IKK.32, 52 Our data also showed that TAK-1 knockdown inhibits the LPS-induced activation on IKK-α, IKK-β, IκB-α. Thus, these findings demonstrated the regulatory role of TAK-1 and NF-κB p50/p65 in mediating the LPS-induced increase in TJ permeability.
As for the effector protein involved, the previous studies have shown that MLCK mediates the LPS-induced increase in Caco-2 TJ permeability by inducing MLC phosphorylation and opening of TJ barrier.19 The present studies showed that TAK-1–dependent activation of NF-κB p50/p65 was responsible for the increase in MLCK mRNA and protein expression. In the present studies, the siRNA induced knockdown of TAK-1 or NF-κB p50/p65 inhibited the LPS-induced increase in MLCK activity, expression, and TJ permeability. Thus, these findings show that NF-κB p50/p65 mediates the LPS-induced increase in TJ permeability by targeting the expression of TJ effector protein MLCK.
Interestingly, the LPS-induced increase in intestinal TJ permeability only occurs after 3 to 4 days of LPS exposure. The increase in TJ permeability was mediated by an increase in basolateral membrane localization of protein components of TLR-4 receptor complex, including TLR-4, CD14, and MD-2.19, 24, 25 The results described herein provide a mechanistic explanation for the observed time course of LPS-induced increase in Caco-2 permeability. The time course of LPS-induced activation of TAK-1 and NF-κB p50/p65 occurring at day 3 paralleled the activation of TLR-4 and MyD88.24, 25 The LPS-induced increase in MLCK expression and Caco-2 TJ permeability occurred after the activation of NF-κB p50/p65 and was inhibited by pharmacologic inhibition or siRNA-induced silencing of NF-κB p50/p65. The present studies also showed that the LPS-induced increase in TAK-1 and NF-κB p50/p65 was mediated by TLR-4/MyD88 signal-transduction pathway, and siRNA knockdown of TLR-4 or MyD88 inhibited the activation of TAK-1, NF-κB p50/p65, MLCK expression, and the increase in TJ permeability.19 Thus, in combination with previous studies,19, 24, 25 the present findings suggest that LPS binding to the TLR-4 receptor complex activates the MyD88 signaling pathway, which, in turn, activates TAK-1 and NF-κB p50/65 leading to the increase in MLCK gene activity and expression and an increase in Caco-2 TJ permeability (Figure 12).
Figure 12.

Proposed scheme of the intracellular pathways involved in lipopolysaccharide (LPS)-induced activation of canonical NF-κB pathway. LPS treatment results in activation of the toll-like receptor (TLR)-4 signal transduction and myeloid differentiation primary response (MyD)88-dependent signaling cascade. Activation of TLR-4/MyD88 signal transduction pathway leads to the activation of IL-1 receptor-associated kinase (IRAK)-4 and phosphorylation of transforming growth factor-β–activating kinase (TAK)-1, leading to the activation of canonical NF-κB pathway of both inhibitory κ B (IκB) kinases (IKKs), IKK-α and IKK-β, which further leads to degradation of IκB-α and nuclear translocation of NF-κB p65/p50. The activation of TAK-1 and canonical NF-κB p65/p50 causes up-regulation of myosin light chain kinase (MLCK) gene and protein expression, ultimately increasing tight junction (TJ) permeability in vitro and in vivo.
The IKK complex plays an integral role in the regulation of NF-κB activity by inducing the degradation of IκB-α.31, 62 The IKK complex consists of two catalytic subunits IKK-α and IKK-β and a regulatory subunit IKK-γ/NEMO. IKK-α and IKK-β are protein kinases and have an enzymatic function.53, 63 The activation of IKK complex leads to the phosphorylation of serine residues within the activation loop of IKK catalytic subunits. The activated IKK catalytic subunits phosphorylate IκB proteins at Ser32 and Ser36, leading to a rapid degradation of phosphorylated IκB-α by 26S proteasome and concurrent activation and nuclear translocation of NF-κB p65/p50 dimer.53 The involvement of IKK catalytic subunits in the regulation of TJ barrier function has been previously reported.34, 53, 64 These data show that TAK-1–dependent activation of NF-κB p50/p65 was mediated by IKKs, IKK-α and IKK-β. Although both IKK-α and IKK-β were involved in the activation of NF-κB p50/p65, IKK-β appeared to have a greater effect than IKK-α.
The mouse intestinal perfusion studies were also performed to determine the in vivo involvement of the TAK-1 and NF-κB p50/p65 in LPS-induced increase in mouse intestinal permeability. Similar to the cell culture studies with filter-grown Caco-2 monolayers, the in vivo mouse studies also showed that LPS administration caused an activation of TAK-1 in mouse small intestine. The TAK-1 inhibition by pharmacologic TAK-1 inhibitor (oxyzeonol) inhibited the LPS-induced increase in mouse enterocyte NF-κB p50/p65 activation, MLCK expression, and intestinal permeability in mice intestines. LPS also activated the canonical NF-κB p50/p65 pathway, and administration of NF-κB inhibitors, curcumin, PDTC, and Bay-11 prevented the LPS-induced increase in MLCK expression and mouse intestinal permeability. In addition, the LPS-induced activation of TAK-1 and NF-κB p50/p65 was prevented in TLR-4 and My D88 knockout mice. These data suggested that LPS-induced activation of TAK-1 and canonical NF-κB p50/p65 in mouse enterocytes was required for the increase in intestinal MLCK expression and mouse intestinal permeability. In vivo studies also showed that TAK-1 was a key regulator of LPS-induced IκB-α degradation and NF-κB p50/p65 activation in mouse enterocytes. Thus, in vivo data indicated that TAK-1 also plays an integral role in mediating the LPS-induced activation of the canonical pathway, increase in MLCK gene expression, and increase in mouse intestinal permeability.
Conclusions
These results provide important new insight into the intracellular signaling processes that mediate the LPS-induced increase in intestinal TJ permeability in vitro and in vivo. They show that LPS, at physiologically relevant concentrations, causes a TLR-4/MyD88 signal transduction pathway–dependent phosphorylation and activation of TAK-1. The activation of TAK-1 then in a step-wise manner induces the activation of catalytic components of IKK complex, IKK-α and IKK-β, degradation of IκB-α, and activation of NF-κB p50/p65 pathway. The activation of NF-κB p50/p65 then leads to an increase in MLCK activity, gene and protein expression, and MLCK-induced opening of the intestinal TJ barrier.
Acknowledgments
We thank the Clinical and Translational Science Center Laboratory and the Fluorescence Microscopy Shared Resource, Health Sciences Center (University of New Mexico) for excellent technical assistance and the Penn State College of Medicine Core facility (Hershey Medical Center) for assistance.
M.N. conceived and designed the research; M.N., M.R., R.A.-S., E.F.C., and P.N. performed experiments; M.N. analyzed data, prepared figures, and wrote the manuscript; M.N. and T.Y.M. interpreted results; T.Y.M. edited and revised the manuscript and approved the final version.
Footnotes
Supported by a Veterans Affairs (VA) Merit Review grant from the VA Research Service and National Institute of Diabetes and Digestive and Kidney Diseases grants R0-1-DK-64165–01, R0-1-DK-106072-01 (T.Y.M.), and R0-1-DK114024 (P.N.).
Disclosures: None declared.
References
- 1.Turner J.R. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 2.Turner J.R. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169:1901–1909. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark J.A., Doelle S.M., Halpern M.D., Saunders T.A., Holubec H., Dvorak K., Boitano S.A., Dvorak B. Intestinal barrier failure during experimental necrotizing enterocolitis: protective effect of EGF treatment. Am J Physiol Gastrointest Liver Physiol. 2006;291:G938–G949. doi: 10.1152/ajpgi.00090.2006. [DOI] [PubMed] [Google Scholar]
- 4.Oriishi T., Sata M., Toyonaga A., Sasaki E., Tanikawa K. Evaluation of intestinal permeability in patients with inflammatory bowel disease using lactulose and measuring antibodies to lipid A. Gut. 1995;36:891–896. doi: 10.1136/gut.36.6.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arrieta M.C., Madsen K., Doyle J., Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benoit R., Rowe S., Watkins S.C., Boyle P., Garrett M., Alber S., Wiener J., Rowe M.I., Ford H.R. Pure endotoxin does not pass across the intestinal epithelium in vitro. Shock. 1998;10:43–48. doi: 10.1097/00024382-199807000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Mennigen R., Nolte K., Rijcken E., Utech M., Loeffler B., Senninger N., Bruewer M. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1140–G1149. doi: 10.1152/ajpgi.90534.2008. [DOI] [PubMed] [Google Scholar]
- 8.Ge Y., Ezzell R.M., Warren H.S. Localization of endotoxin in the rat intestinal epithelium. J Infect Dis. 2000;182:873–881. doi: 10.1086/315784. [DOI] [PubMed] [Google Scholar]
- 9.Hurley J.C. Endotoxemia: methods of detection and clinical correlates. Clin Microbiol Rev. 1995;8:268–292. doi: 10.1128/cmr.8.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andreasen A.S., Krabbe K.S., Krogh-Madsen R., Taudorf S., Pedersen B.K., Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008;15:1697–1705. doi: 10.2174/092986708784872393. [DOI] [PubMed] [Google Scholar]
- 11.Wellmann W., Fink P.C., Benner F., Schmidt F.W. Endotoxaemia in active Crohn's disease. Treatment with whole gut irrigation and 5-aminosalicylic acid. Gut. 1986;27:814–820. doi: 10.1136/gut.27.7.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma R., Tepas J.J., III, Hudak M.L., Mollitt D.L., Wludyka P.S., Teng R.J., Premachandra B.R. Neonatal gut barrier and multiple organ failure: role of endotoxin and proinflammatory cytokines in sepsis and necrotizing enterocolitis. J Pediatr Surg. 2007;42:454–461. doi: 10.1016/j.jpedsurg.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 13.Ma T.Y., Anderson J.M. Physiology of the Gastrointestinal Tract. ed 4. Elsevier Academic Press; Burlington, MA: 2006. Tight junctions and the intestinal barrier. [Google Scholar]
- 14.Leal R.F., Milanski M., Ayrizono Mde L., Coope A., Rodrigues V.S., Portovedo M., Oliveira L.M., Fagundes J.J., Coy C.S., Velloso L.A. Toll-like receptor 4, F4/80 and pro-inflammatory cytokines in intestinal and mesenteric fat tissue of Crohn's disease. Int J Clin Exp Med. 2013;6:98–104. [PMC free article] [PubMed] [Google Scholar]
- 15.Le Mandat Schultz A., Bonnard A., Barreau F., Aigrain Y., Pierre-Louis C., Berrebi D., Peuchmaur M. Expression of TLR-2, TLR-4, NOD2 and pNF-kappaB in a neonatal rat model of necrotizing enterocolitis. PLoS One. 2007;2:e1102. doi: 10.1371/journal.pone.0001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Jager P.L., Franchimont D., Waliszewska A., Bitton A., Cohen A., Langelier D., Belaiche J., Vermeire S., Farwell L., Goris A., Libioulle C., Jani N., Dassopoulos T., Bromfield G.P., Dubois B., Cho J.H., Brant S.R., Duerr R.H., Yang H., Rotter J.I., Silverberg M.S., Steinhart A.H., Daly M.J., Podolsky D.K., Louis E., Hafler D.A., Rioux J.D., Quebec IBD Genetics Consortium. NIDDK IBD Genetics Consortium The role of the Toll receptor pathway in susceptibility to inflammatory bowel diseases. Genes Immun. 2007;8:387–397. doi: 10.1038/sj.gene.6364398. [DOI] [PubMed] [Google Scholar]
- 17.Ma T.Y., Tran D., Hoa N., Nguyen D., Merryfield M., Tarnawski A. Mechanism of extracellular calcium regulation of intestinal epithelial tight junction permeability: role of cytoskeletal involvement. Microsc Res Tech. 2000;51:156–168. doi: 10.1002/1097-0029(20001015)51:2<156::AID-JEMT7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18.Ma T.Y., Hoa N.T., Tran D.D., Bui V., Pedram A., Mills S., Merryfield M. Cytochalasin B modulation of Caco-2 tight junction barrier: role of myosin light chain kinase. Am J Physiol Gastrointest Liver Physiol. 2000;279:G875–G885. doi: 10.1152/ajpgi.2000.279.5.G875. [DOI] [PubMed] [Google Scholar]
- 19.Nighot M., Al-Sadi R., Guo S.H., Rawat M., Nighot P., Watterson M.D., Ma T.Y. Lipopolysaccharide-induced increase in intestinal epithelial tight permeability is mediated by Toll-like receptor 4/myeloid differentiation primary response 88 (MyD88) activation of myosin light chain kinase expression. Am J Pathol. 2017;187:2698–2710. doi: 10.1016/j.ajpath.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma T.Y., Boivin M.A., Ye D., Pedram A., Said H.M. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422–G430. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 21.Yu L.C., Flynn A.N., Turner J.R., Buret A.G. SGLT-1-mediated glucose uptake protects intestinal epithelial cells against LPS-induced apoptosis and barrier defects: a novel cellular rescue mechanism? FASEB J. 2005;19:1822–1835. doi: 10.1096/fj.05-4226com. [DOI] [PubMed] [Google Scholar]
- 22.Turner J.R., Rill B.K., Carlson S.L., Carnes D., Kerner R., Mrsny R.J., Madara J.L. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 23.Turner J.R., Buschmann M.M., Romero-Calvo I., Sailer A., Shen L. The role of molecular remodeling in differential regulation of tight junction permeability. Semin Cell Dev Biol. 2014;36:204–212. doi: 10.1016/j.semcdb.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo S., Al-Sadi R., Said H.M., Ma T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. 2013;182:375–387. doi: 10.1016/j.ajpath.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo S., Nighot M., Al-Sadi R., Alhmoud T., Nighot P., Ma T.Y. Lipopolysaccharide regulation of intestinal tight junction permeability is mediated by TLR4 signal transduction pathway activation of FAK and MyD88. J Immunol. 2015;195:4999–5010. doi: 10.4049/jimmunol.1402598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J.-Y., Morgan M., Kim D.-G., Lee J.-Y., Bai L., Lin Y., Liu Z.-g., Kim Y.-S. TNFα-induced noncanonical NF-κB activation is attenuated by RIP1 through stabilization of TRAF2. J Cell Sci. 2011;124:647–656. doi: 10.1242/jcs.075770. [DOI] [PubMed] [Google Scholar]
- 27.Zarnegar B., Yamazaki S., He J.Q., Cheng G. Control of canonical NF-kappaB activation through the NIK–IKK complex pathway. Proc Natl Acad Sci U S A. 2008;105:3503–3508. doi: 10.1073/pnas.0707959105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramakrishnan P., Wang W., Wallach D. Receptor-specific signaling for both the alternative and the canonical NF-kappaB activation pathways by NF-kappaB-inducing kinase. Immunity. 2004;21:477–489. doi: 10.1016/j.immuni.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 29.Baldwin A.S., Jr. The NF-kappaB and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh S., May M.J., Kopp E.B. NF-kappa B and REL proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 31.Pomerantz J.L., Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10:693–695. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 32.Widera D., Mikenberg I., Elvers M., Kaltschmidt C., Kaltschmidt B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006;7:64. doi: 10.1186/1471-2202-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun S.-C. The non-canonical NF-[kappa]B pathway in immunity and inflammation. Nat Rev Immunol. 2017;17:545–558. doi: 10.1038/nri.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al-Sadi R., Guo S., Ye D., Rawat M., Ma T.Y. TNF-alpha modulation of intestinal tight junction permeability is mediated by NIK/IKK-alpha axis activation of the canonical NF-kappaB pathway. Am J Pathol. 2016;186:1151–1165. doi: 10.1016/j.ajpath.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guha M., Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 36.Gay N.J., Gangloff M. Structure and function of Toll receptors and their ligands. Annu Rev Biochem. 2007;76:141–165. doi: 10.1146/annurev.biochem.76.060305.151318. [DOI] [PubMed] [Google Scholar]
- 37.Forsythe R.M., Xu D.Z., Lu Q., Deitch E.A. Lipopolysaccharide-induced enterocyte-derived nitric oxide induces intestinal monolayer permeability in an autocrine fashion. Shock. 2002;17:180–184. doi: 10.1097/00024382-200203000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Liu C., Li A., Weng Y.B., Duan M.L., Wang B.E., Zhang S.W. Changes in intestinal mucosal immune barrier in rats with endotoxemia. World J Gastroenterol. 2009;15:5843–5850. doi: 10.3748/wjg.15.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato S., Sanjo H., Takeda K., Ninomiya-Tsuji J., Yamamoto M., Kawai T., Matsumoto K., Takeuchi O., Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 40.Cuevas B.D., Abell A.N., Johnson G.L. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26:3159–3171. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- 41.Chen Z.J., Bhoj V., Seth R.B. Ubiquitin, TAK1 and IKK: is there a connection? Cell Death Differ. 2006;13:687–692. doi: 10.1038/sj.cdd.4401869. [DOI] [PubMed] [Google Scholar]
- 42.Ye D., Guo S., Al-Sadi R., Ma T.Y. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141:1323–1333. doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Sadi R., Khatib K., Guo S., Ye D., Youssef M., Ma T. Occludin regulates macromolecule flux across the intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2011;300:G1054–G1064. doi: 10.1152/ajpgi.00055.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Sadi R., Guo S., Ye D., Ma T.Y. TNF-alpha modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am J Pathol. 2013;183:1871–1884. doi: 10.1016/j.ajpath.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Al-Sadi R., Guo S., Ye D., Dokladny K., Alhmoud T., Ereifej L., Said H.M., Ma T.Y. Mechanism of IL-1beta modulation of intestinal epithelial barrier involves p38 kinase and activating transcription factor-2 activation. J Immunol. 2013;190:6596–6606. doi: 10.4049/jimmunol.1201876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krutzik P.O., Trejo A., Schulz K.R., Nolan G.P. Phospho flow cytometry methods for the analysis of kinase signaling in cell lines and primary human blood samples. Methods Mol Biol. 2011;699:179–202. doi: 10.1007/978-1-61737-950-5_9. [DOI] [PubMed] [Google Scholar]
- 47.Castillo E.F., Acero L.F., Stonier S.W., Zhou D., Schluns K.S. Thymic and peripheral microenvironments differentially mediate development and maturation of iNKT cells by IL-15 transpresentation. Blood. 2010;116:2494–2503. doi: 10.1182/blood-2010-03-277103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cubillos-Zapata C., Hernández-Jiménez E., Toledano V., Esteban-Burgos L., Fernández-Ruíz I., Gómez-Piña V., del Fresno C., Siliceo M., Prieto-Chinchiña P., Pérez de Diego R., Boscá L., Fresno M., Arnalich F., López-Collazo E. NFκB2/p100 is a key factor for endotoxin tolerance in human monocytes: a demonstration using primary human monocytes from patients with sepsis. J Immunol. 2014;193:4195–4202. doi: 10.4049/jimmunol.1400721. [DOI] [PubMed] [Google Scholar]
- 49.Bozinovski S., Jones J.E., Vlahos R., Hamilton J.A., Anderson G.P. Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappaB and AP-1 in vivo. J Biol Chem. 2002;277:42808–42814. doi: 10.1074/jbc.M207840200. [DOI] [PubMed] [Google Scholar]
- 50.Hayes J.B., Sircy L.M., Heusinkveld L.E., Ding W., Leander R.N., McClelland E.E., Nelson D.E. Modulation of macrophage inflammatory nuclear factor κB (NF-κB) signaling by intracellular Cryptococcus neoformans. J Biol Chem. 2016;291:15614–15627. doi: 10.1074/jbc.M116.738187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takaesu G., Surabhi R.M., Park K.-J., Ninomiya-Tsuji J., Matsumoto K., Gaynor R.B. TAK1 is Critical for IkappaB kinase-mediated activation of the NF-kappaB Pathway. J Mol Biol. 2003;326:105–115. doi: 10.1016/s0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 52.Adhikari A., Xu M., Chen Z.J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene. 2007;26:3214–3226. doi: 10.1038/sj.onc.1210413. [DOI] [PubMed] [Google Scholar]
- 53.Al-Sadi R., Ye D., Said H.M., Ma T.Y. IL-1beta-induced increase in intestinal epithelial tight junction permeability is mediated by MEKK-1 activation of canonical NF-kappaB pathway. Am J Pathol. 2010;177:2310–2322. doi: 10.2353/ajpath.2010.100371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White D.E., Burchill S.A. BAY 11-7082 induces cell death through NF-kappaB-independent mechanisms in the Ewing's sarcoma family of tumours. Cancer Lett. 2008;268:212–224. doi: 10.1016/j.canlet.2008.03.045. [DOI] [PubMed] [Google Scholar]
- 55.Strickson S., Campbell D.G., Emmerich C.H., Knebel A., Plater L., Ritorto M.S., Shpiro N., Cohen P. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem J. 2013;451:427–437. doi: 10.1042/BJ20121651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miyamoto R., Ito T., Nomura S., Amakawa R., Amuro H., Katashiba Y., Ogata M., Murakami N., Shimamoto K., Yamazaki C., Hoshino K., Kaisho T., Fukuhara S. Inhibitor of IkappaB kinase activity, BAY 11-7082, interferes with interferon regulatory factor 7 nuclear translocation and type I interferon production by plasmacytoid dendritic cells. Arthritis Res Ther. 2010;12:R87. doi: 10.1186/ar3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keller S.A., Hernandez-Hopkins D., Vider J., Ponomarev V., Hyjek E., Schattner E.J., Cesarman E. NF-kappaB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood. 2006;107:3295–3302. doi: 10.1182/blood-2005-07-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ivshina M., Alexandrov I.M., Vertii A., Doxsey S., Richter J.D. CPEB regulation of TAK1 synthesis mediates cytokine production and the inflammatory immune response. Mol Cell Biol. 2015;35:610–618. doi: 10.1128/MCB.00800-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sukhotnik I., Mogilner J., Krausz M.M., Lurie M., Hirsh M., Coran A.G., Shiloni E. Oral arginine reduces gut mucosal injury caused by lipopolysaccharide endotoxemia in rat. J Surg Res. 2004;122:256–262. doi: 10.1016/j.jss.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 60.Madara J.L., Marcial M.A. Structural correlates of intestinal tight-junction permeability. Kroc Found Ser. 1984;17:77–100. [PubMed] [Google Scholar]
- 61.Hinzpeter A., Lipecka J., Brouillard F., Baudoin-Legros M., Dadlez M., Edelman A., Fritsch J. Association between Hsp90 and the ClC-2 chloride channel upregulates channel function. Am J Physiol Cell Physiol. 2006;290:C45–C56. doi: 10.1152/ajpcell.00209.2005. [DOI] [PubMed] [Google Scholar]
- 62.Karin M., Delhase M. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 63.Sun Z., Wang X., Lasson A., Borjesson A., Leveau P., Haraldsen P., Andersson R. Roles of platelet-activating factor, interleukin-1beta and interleukin-6 in intestinal barrier dysfunction induced by mesenteric arterial ischemia and reperfusion. J Surg Res. 1999;87:90–100. doi: 10.1006/jsre.1999.5746. [DOI] [PubMed] [Google Scholar]
- 64.Al-Sadi R., Boivin M., Ma T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci (Landmark Ed) 2009;14:2765–2778. doi: 10.2741/3413. [DOI] [PMC free article] [PubMed] [Google Scholar]