

Abstract
Clostridioides (formerly Clostridium) difficile produces two major toxins, TcdA and TcdB, upon entry into stationary phase. Transcription of tcdA and tcdB requires the specialized sigma factor, σTcdR, which also directs RNA Polymerase to transcribe tcdR itself. We fused a gene for a red fluorescent protein to the tcdA promoter to study toxin gene expression at the level of individual C. difficile cells. Surprisingly, only a subset of cells became red fluorescent upon entry into stationary phase. Breaking the positive feedback loop that controls σTcdR production by engineering cells to express tcdR from a tetracycline-inducible promoter resulted in uniform fluorescence across the population. Experiments with two regulators of tcdR expression, σD and CodY, revealed neither is required for bimodal toxin gene expression. However, σD biased cells towards the Toxin-ON state, while CodY biased cells towards the Toxin-OFF state. Finally, toxin gene expression was observed in sporulating cells. We conclude that (i) toxin production is regulated by a bistable switch governed by σTcdR, which only accumulates to high enough levels to trigger toxin gene expression in a subset of cells, and (ii) toxin production and sporulation are not mutually exclusive developmental programs.
Keywords: sigma factor, signal transduction, toxin regulation, PaLoc, anaerobic RFP, bistability, phase variation
Graphical Abstract
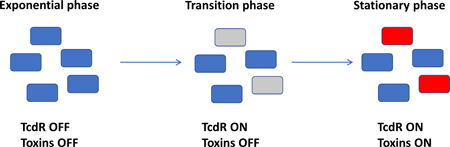
Abbreviated Summary
Clostridioides difficile, the leading cause of antibiotic-associated diarrhea, produces toxins that inactivate host Rho GTPases. We used a fluorescent reporter to visualize toxin gene expression in C. difficile within individual cells. Our findings imply toxin production is an example of bistability governed by cell-to-cell variation in the levels of the sigma factor TcdR, which is directly required for transcription of the toxin genes. TcdR levels are in turn controlled by several metabolic and genetic inputs.
INTRODUCTION
Clostridioides (formerly Clostridium) difficile is a Gram-positive, spore-forming, anaerobic bacterium and an opportunistic pathogen (Lawson et al., 2016). C. difficile infections can cause antibiotic-associated diarrhea and progress to life-threatening conditions, including pseudomembranous colitis and toxic megacolon. Disease is mediated primarily through two exotoxins known as TcdA and TcdB (Lyerly et al., 1982; Lyerly et al., 1985; Triadafilopoulos et al., 1987; Voth and Ballard, 2005). Both toxins are glucosyltransferases that glucosylate host proteins, particularly the Rho family of GTPases (Schirmer and Aktories, 2004; Gerhard et al., 2008; Darkoh et al., 2011; Carter et al., 2012; Shen, 2012; Chandrasekaran and Lacy, 2017). This leads to collapse of the actin cytoskeleton and loss of tight junctions resulting in gastrointestinal distress (Moore et al., 1990; Stubbe et al., 2000; Feltis et al., 2000; Nusrat et al., 2001; Gerhard et al., 2008; Shen, 2012).
TcdA and TcdB are encoded on a 19.6 kb pathogenicity locus (PaLoc), along with three additional toxin-related genes: tcdR, tcdC, and tcdE (Fig. 1) (Cohen et al., 2000). TcdR (σTcdR) is an alternative sigma factor that recruits RNA polymerase to the promoters for tcdA and tcdB, and is required for tcdA and tcdB expression (Mani and Dupuy, 2001). There is also a σTcdR-dependent promoter upstream of tcdR, resulting in a positive-feedback loop whereby σTcdR increases its own production (Mani et al., 2002). TcdC is proposed to function as an anti-sigma for σTcdR and thus negatively regulates toxin production (Matamouros et al., 2007; Dupuy et al., 2008; Carter et al., 2011), but this finding has been challenged (Murray et al., 2009; Cartman et al., 2012). TcdE is a predicted membrane protein with some similarity to the holins that create pores for bacteriophage to escape the cytoplasm (Govind and Dupuy, 2012; Govind et al., 2015). Whereas two studies found TcdE is required for toxin secretion (Kai Soo Tan et al., 2001; Govind and Dupuy, 2012), a third study found TcdE to be completely dispensable (Olling et al., 2012).
Figure 1. Model for regulation of toxin gene expression.
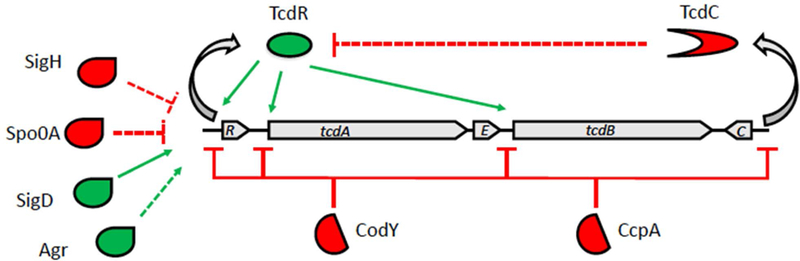
Positive and negative regulators are shown in green and red, respectively. Dashed lines indicate factors that may act indirectly and/or whose contributions are not clearly established. Note that σTcdR activates expression of its own gene (tcdR) and both toxin genes tcdA and tcdB, while σD activates expression of tcdR. The repressor CodY binds to the promoter-regulatory regions at tcdR, tcdA, and tcdB.
Regulation of toxin gene expression is one of the best-studied aspects of C. difficile biology. When C. difficile is cultured in a laboratory in rich media, toxin is produced upon entry into stationary phase (Hundsberger et al., 1997), suggesting expression of tcdA and tcdB responds to nutrient limitation. Consistent with this view, readily metabolized carbon sources like glucose and a variety of amino acids reduce toxin production (Dupuy and Sonenshein, 1998; Karlsson et al., 1999; Karlsson, S., A. Lindberg, L. G. Burman, E. Norin et al., 2000; Karlsson et al., 2003). These effects are thought to be mediated in part by the alternative sigma factor σTcdR, since expression of tcdR is influenced by nutrient availability and temperature (Karlsson et al., 2003).
Toxin gene expression in C. difficile is also influenced by several global regulators (summarized in Fig. 1 and reviewed in (Bouillaut et al., 2015)). CodY is widely distributed in Firmicutes and functions as a repressor when both GTP and branched chain amino acids are abundant in the cell (Guédon et al., 2001; Ratnayake-Lecamwasam et al., 2001). In C. difficile, CodY represses 146 genes, including all five genes in the PaLoc (Dineen et al., 2007; Dineen et al., 2010). In vitro, CodY binds to the promoters for tcdA, tcdB, tcdC, and tcdR, but because its affinity is about 10-fold higher for the tcdR promoter, this is likely to be the most important target for CodY regulation of toxin production (Dineen et al., 2007). CcpA, or carbon catabolite protein A, is a global regulator that responds to readily catabolizable carbohydrates like glucose (Deutscher, 2008). In C. difficile, CcpA regulates ~140 genes (Antunes et al., 2012) and binds directly to the promoter regions of tcdA, tcdB, tcdC, and tcdR (Antunes et al., 2011). Because CcpA has ~10-fold higher affinity for the tcdR promoter than for the other promoters, it is thought to work primarily through controlling expression of tcdR (Antunes et al., 2011). Another important regulator is σD, which is encoded by sigD as part of the flgB operon that contains early stage flagellar genes (Aubry et al., 2012; El Meouche et al., 2013; McKee et al., 2013). σD positively regulates tcdA and tcdB expression by increasing expression of tcdR (McKee et al., 2013). A number of other transcriptional regulators have been reported to impact toxin gene expression: PrdR (Bouillaut et al., 2013), Agr (Martin et al., 2013), Spo0A (Mackin et al., 2013), SigH (Saujet et al., 2011), and RstA (Edwards et al., 2016). These are less well understood and some seem to be restricted to certain C. difficile strains.
Previous studies of toxin production have focused on populations of cells, and thus reflect the “average” behavior of cells in culture. Our development of RFP as a reporter for gene expression in C. difficile enables analysis of tcdA and tcdB expression at the level of individual cells. Here, we used RFP to study toxin regulation in C. difficile. Remarkably, we found toxin gene expression is bimodal; in stationary phase, the population bifurcates into a group of cells that is “TcdA-ON” and a group that is “TcdA-OFF.” In the epidemic strain R20291, about 30% of the cells are in the TcdA-ON state, and the mean fluorescence intensity of these cells is about 50-fold higher than the TcdA-OFF cells. Additional experiments indicate expression of tcdR is the genetic switch that determines whether a cell produces toxin.
RESULTS
tcdA expression is bimodal in C. difficile
Expression of the toxin genes is induced during stationary phase (Ketley et al., 1984; Osgood et al., 1993; Dupuy and Sonenshein, 1998), and several studies have identified global regulators of tcdA expression. However, to date these studies have focused on the bulk population of cells rather than individual cells. We sought to visualize toxin gene expression in single cells by introducing a PtcdA::rfp reporter plasmid into R20291 ribotype 027. As expected, in log phase overall fluorescence of the culture as measured with a plate reader was low and fluorescence microscopy revealed the vast majority of the cells were dark (e.g., sample #1 in Figure 2A, B, C, D). Upon entry into stationary phase overall fluorescence increased ~5 fold and microscopy revealed a striking mixture of bright and dark cells (e.g., sample #5 in Figure 2A, B, C, D). Flow cytometry confirmed that the distribution of fluorescence intensities was bimodal, indicative of two distinct subpopulations, which we will refer to as TcdA-ON and TcdA-OFF. In the experiment shown the TcdA-ON fraction reached a maximum of ~19% of the cells in sample #8 (Fig. 2).
Figure 2. Bistable expression of PtcdA::rfp in C. difficile R20291.
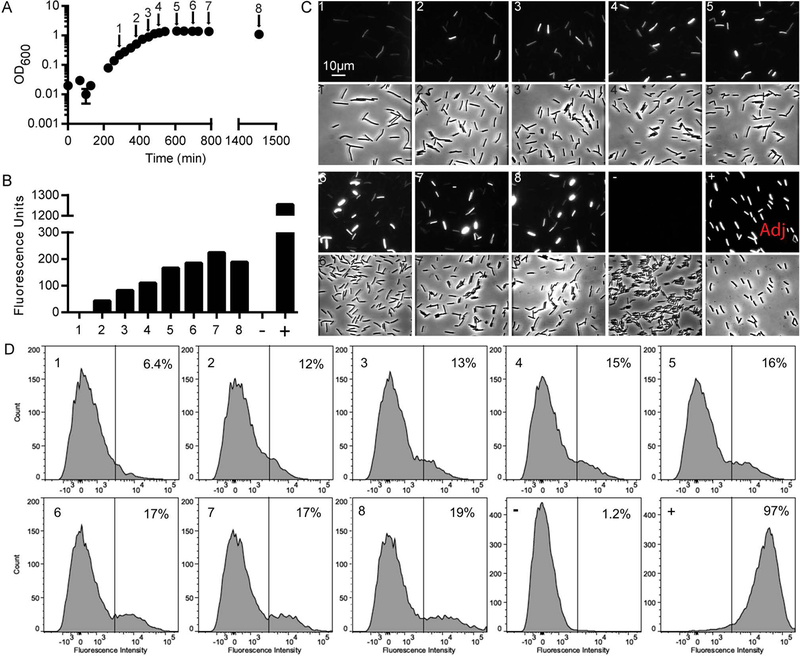
(A) Growth curve of PtcdA::rfp strain. At the times indicated by the arrows, samples of the culture were fixed with paraformaldehyde and exposed to air to allow red fluorescence to develop. (B) Specific fluorescence of PtcdA::rfp population as determined using a plate reader. Numbers on the x-axis refer to samples fixed at the time points indicated in (A). Also shown is a negative control strain containing a Ptet::gus reporter plasmid (−) and a positive control strain carrying a PpdaV::rfp reporter plasmid and induced with lysozyme for 30 min (+). (C) Expression of PtcdA::rfp as assessed by microscopy. Micrographs are paired: fluorescence (above) and phase micrographs (below). All micrographs were captured and processed identically except for one marked “Adj” which indicates the brightness was adjusted down. (D) Expression of PtcdA::rfp as assessed by flow cytometry. The x-axis label is Fluorescence Intensity in arbitrary units. Vertical gate divides TcdA-ON from TcdA-OFF subpopulations, and numbers in the upper right corner of each panel refers to the fraction of cells scored as TcdA-ON.
Multiple lines of evidence rule out the potential artifacts of plasmid segregation and viability issues. First, the plasmid is reported to be very stable (Heap et al., 2009; Ransom et al., 2015). Second, an essentially identical plasmid with a lysozyme-induced PpdaV::rfp (Ransom et al., 2015) fusion provided uniform red fluorescence across the population after exposure of the cells to lysozyme [See (+), for positive control in Figure 2B, C, D]. As a negative control, a plasmid lacking rfp (pRPF185) failed to produce any red fluorescent signal [See (N) in Figure 2B, C, and D]. Third, we sampled cells at various points during the growth curve and plated on TY or TY with thiamphenicol to select for the plasmid. We did not observe a significant drop in cell viability or issues with loss of the reporter plasmid (Fig. S1B–C). Finally, we performed live/dead staining on cells to determine viability. We found that in log phase 100% were viable and after overnight growth 94% remained viable (Fig. S1C–D).
Expression of tcdA is bimodal in multiple C. difficile ribotypes
As noted, the results shown in Figure 2 were obtained with strain R20291 (ribotype 027), but the tcdA promoter region is highly conserved across different C. difficile isolates (Fig. S2), suggesting bimodal expression of tcdA might be a general property of C. difficile. To test this notion, we introduced the PtcdA::rfp reporter plasmid into five additional strains: CD630Δerm and JIR8094, which are independent erythromycin-sensitive derivatives of CD630 (ribotype 012); CD196 (ribotype 027); NAP07 (ribotype 078); NAP08 (ribotype 078); and VPI10463 (ribotype 087). The CD196, NAP07, and NAP08 strains are representative clinical isolates corresponding to the most commonly isolated ribotypes of C. difficile (Wilcox et al., 2012; Walker et al., 2013), while VPI10463 is commonly known as a high toxin producing strain (Akerlund et al., 2008). As expected, expression of PtcdA::rfp was bimodal in all strains, including the three most clinically relevant isolates (Fig. 3). Interestingly, however, two closely related strains, CD630Δerm and JIR8094, exhibited very different fractions of TcdA-ON versus TcdA-OFF subpopulations. In CD630Δerm ~80% of the cells were red fluorescent, as compared to only ~20% in JIR8094 (Fig. 3). As will be explained in more detail below, this difference likely reflects much higher levels of σD production in CD630Δerm (Anjuwon-Foster and Tamayo, 2017; Anjuwon-Foster et al., 2018). σD increases toxin production by activating tcdR transcription (McKee et al., 2013; Anjuwon-Foster and Tamayo, 2017).
Figure 3. Expression of PtcdA::rfp is bistable in multiple C. difficile strains.
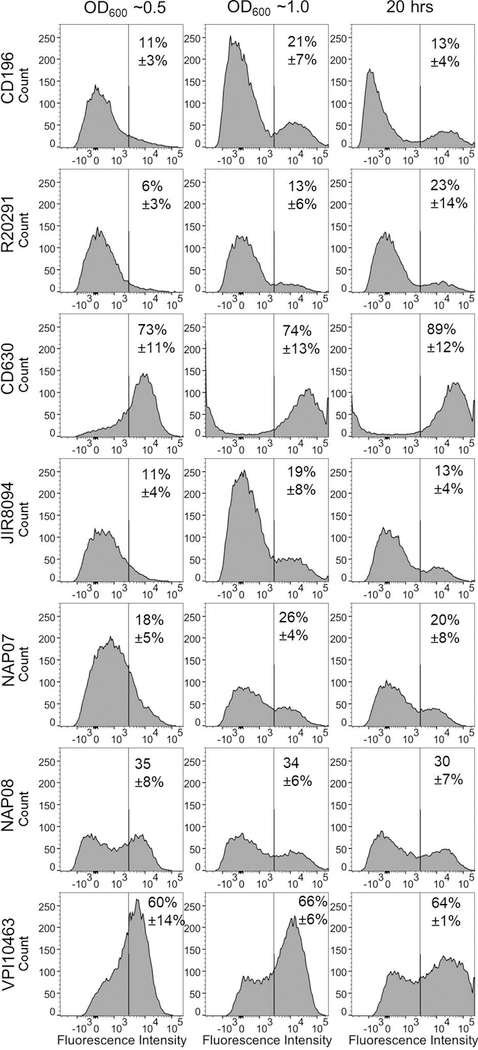
The indicated strains of C. difficile harboring the PtcdA::rfp reporter plasmid were fixed at (A) OD600 ~0.5, (B) OD600 ~1.0 and (C) after 20 hours of growth. Percentages refer to the fraction of cells that were TcdA-ON (mean ± st. dev., n = at least 3 independent experiments). The strains shown represent the following ribotypes: CD196 (ribotype 027), R20291 (ribotype 027), CD630Δerm (ribotype 012), JIR8094 (ribotype 012), NAP07 (ribotype 078), NAP08 (ribotype 078) and VPI10463 (ribotype 087).
Bimodal tcdA expression is dependent upon σTcdR
Bimodal patterns of gene expression can arise from phase variation or from cell-to-cell differences in the levels of a transcriptional activator protein that is part of a positive feedback loop, a mechanism commonly referred to as bistability (Dubnau and Losick, 2006; Dubnau, 2015). Considering the various factors implicated in control of toxin production in C. difficile, the alternative sigma factor σTcdR is a promising candidate for the control point for bistable toxin gene expression. σTcdR increases its own expression (Mani et al., 2002), so higher levels of σTcdR will be self-reinforcing. Moreover, basal expression of tcdR is very low (Mani and Dupuy, 2001), creating a situation in which random fluctuations in σTcdR levels could push a subset of cells over a threshold that locks them into an ON state.
To ask whether tcdR might be the genetic switch that determines whether a given C. difficile cell is TcdA-ON or TcdA-OFF, we sought to break the positive feedback arising from auto regulation of σTcdR production. The first step was to construct a tcdR::erm null mutant in JIR8094 and introduce the PtcdA::rfp reporter plasmid. We did not observe any red fluorescence in the tcdR null mutant strain (Fig. 4A). The absence of TcdA-ON cells was expected because it is well-established that σTcdR is essential for transcription of tcdA (Mani and Dupuy, 2001; Mani et al., 2002; Karlsson et al., 2003). We then added back a tcdR gene under control of a tetracycline-inducible promoter, Ptet, which allowed us to control σTcdR levels by adding increasing amounts of anhydrotetracycline (ATc) to the growth medium. Importantly for our purposes, ATc-induction of Ptet is dose-dependent and uniform across a population of cells in C. difficile (Fagan and Fairweather, 2011; Ransom et al., 2016). When the tcdR::erm/Ptet::tcdR PtcdA::rfp reporter strain was grown in TY broth containing increasing amounts of ATc, we observed a dose-dependent increase in red fluorescence of the cultures (Fig. 4B). Strikingly, however, flow cytometry revealed that red fluorescence was always uniform across the population; there was no concentration of ATc at which C. difficile cultures bifurcated into TcdA-ON and TcdA-OFF subpopulations (Fig. 4C). Thus, breaking the positive-feedback loop that controls tcdR expression breaks the bimodal expression of tcdA, consistent with the hypothesis that toxin gene expression is subject to bistability, and σTcdR is the master regulator.
Figure 4. TcdR mediates bistable expression of tcdA.

(A) TcdR is required for tcdA expression. Wild-type JIR8094/PtcdA::rfp and tcdR::erm/PtcdA::rfp were grown to stationary phase (24 hrs). Samples of each culture were fixed and analyzed for red fluorescence by flow cytometry. (B, C) Breaking the positive feedback loop that controls tcdR expression prevents development of bistability. The tcdR::erm mutant harboring a plasmid with both Ptet::tcdR and PtcdA::rfp was grown to mid-log phase (OD600 = 0.3), at which time ATc was added as indicated to induce expression of tcdR. After 1 hr, samples were fixed and analyzed using a plate reader (B) or by flow cytometry (C). Data shown are from one experiment that is representative of three trials.
σD influences but is not required for bimodal tcdA expression
An alternative underlying cause of bimodal patterns of gene expression is phase variation. In this mechanism, a clonal population of cells becomes genetically heterogenous owing to the generation of (reversible) genetic variants that arise spontaneously. Interestingly, one of the promoters driving production of σTcdR is recognized by σD, and σD expression is subject to phase variation (Anjuwon-Foster and Tamayo, 2017). The gene for σD is located in the flgB operon, which contains flagellar genes and is required for motility (El Meouche et al., 2013; McKee et al., 2013; Anjuwon-Foster and Tamayo, 2017). Expression of the flgB operon is regulated by a 154 bp invertible element flanked by 21 bp inverted repeats located between the promoter and the first gene of the operon. It was recently demonstrated that when the invertible element is in the ON orientation σD is produced, leading to increased expression of tcdR, which in turn drives expression of the toxin genes tcdA and tcdB (Anjuwon-Foster and Tamayo, 2017). Conversely, when the element flips to the OFF orientation, little or no σD is produced and expression of toxin genes is attenuated (Anjuwon-Foster and Tamayo, 2017).
To explore the role of phase variable production of σD in generating a bimodal distribution of toxin gene expression, we first asked whether σD is required for expression of our PtcdA::rfp reporter. To this end, we introduced the reporter plasmid into an R20291 sigD::erm mutant and its isogenic wild-type parent (Anjuwon-Foster and Tamayo, 2017). Eliminating σD reduced the fraction of red fluorescent (TcdA-ON) cells from ~40% to ~5% of the population, consistent with previous reports that assayed toxin gene expression in a bulk population of cell (Fig. 5A). Nevertheless, for our purposes, the drop in toxin gene expression is less important than the fact that a substantial number of cells express PtcdA::rfp despite the complete absence of σD. This finding means bimodal tcdA expression is not simply a consequence of flagellar inversion at flgB and its impact on σD levels.
Figure 5. σD impacts but is not required for bistable expression of tcdA.
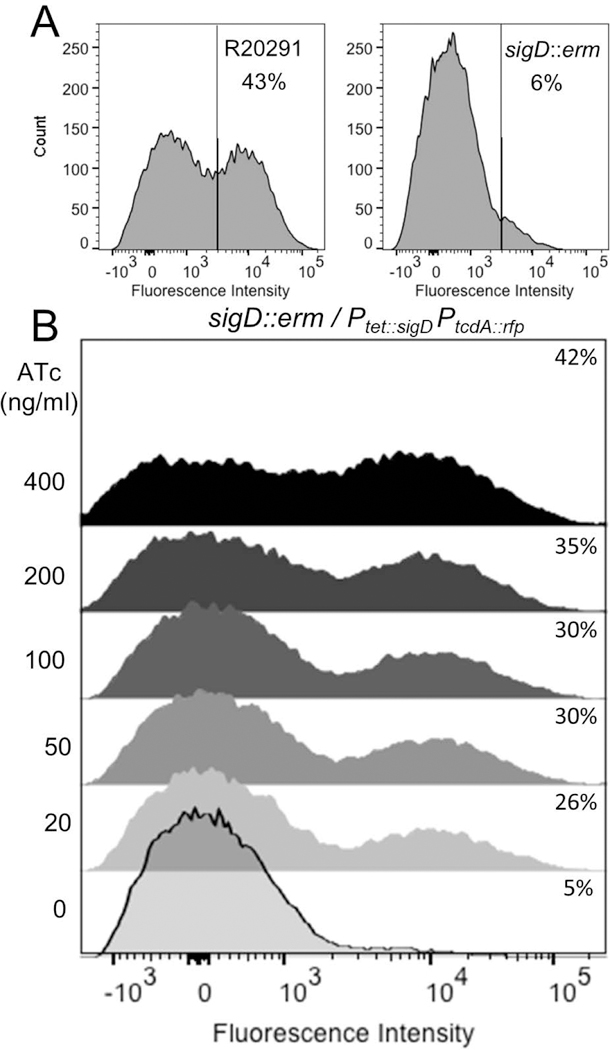
(A) σD is not essential for tcdA expression. Wild-type R20291/PtcdA::rfp and sigD::erm/PtcdA::rfp were grown to stationary phase (24 hrs). Samples of each culture were fixed and analyzed for red fluorescence by flow cytometry. (B) The sigD::erm mutant harboring a plasmid with both Ptet::sigD and PtcdA::rfp was grown to mid-log phase (OD600 = 0.3), at which time ATc was added as indicated to induce expression of sigD. After 2 hrs, samples were fixed and analyzed by flow cytometry. Data shown are from one experiment that is representative of three trials.
Further evidence that flgB phase variation is not required for bifurcation of C. difficile into TcdA-ON and TcdA-OFF subpopulations comes from comparison of CD630Δerm and JIR8094. In these strains the invertible element at flgB is locked in the ON and OFF orientations, respectively, owing to a mutation in the 21 bp inverted repeats flanking the 154 bp invertible sequence (Anjuwon-Foster et al., 2018). If phase variation at flgB were the master regulator of bimodal toxin gene expression, CD630Δerm would be 100% TcdA-ON while JIR8094 would be 100% TcdA-OFF. In reality, however, both strains were bimodal with respect to expression of the PtcdA::rfp reporter, with ~80% TcdA-ON in CD630Δerm and ~20% TcdA-ON in JIR8094 (Fig. 3). Our interpretation of these data is that the flgB ON orientation biases C. difficile towards the TcdA-ON state by driving elevated expression of sigD, but other factors impinge upon, and in some cases override, the contribution of phase variation at flgB. This renders some cells TcdA-OFF even when σD levels are high and some cells TcdA-ON when σD is lacking.
We extended these findings by using a Ptet::sigD construct to assess the effect of modulating sigD transcription on expression of the PtcdA::rfp reporter. Cultures of a sigD::erm mutant harboring a Ptet::sigD/PtcdA::rfp plasmid were grown in TY with increasing amounts of ATc and then assayed for red fluorescence. Although overall fluorescence of the cultures increased with increasing ATc (i.e., increasing σD), examination at the level of individual cells revealed bifurcation into TcdA-ON and TcdA-OFF subpopulations across the entire range of inducer concentrations (Fig. 5B). Even at the highest concentration (400 ng/ml) only ~42% of the cells were TcdA-ON (Fig. 5B). Thus, in contrast to a similar experiment performed with Ptet::tcdR, we were unable to break bimodality by artificially expressing sigD. This result provides further evidence that cell-to-cell differences in σD abundance are not sufficient to explain the bimodal gene expression of tcdA.
The role of environmental signals and global regulators in bimodal tcdA expression
Toxin production is influenced by the state of cellular metabolism [reviewed in (Bouillaut et al., 2015)]. For example, exogenous glucose and cysteine reduce toxin production during entry into stationary phase (Karlsson, S., A. Lindberg, L. G. Burman, E. Norin et al., 2000; Antunes et al., 2012). On the other hand, exogenous butyric acid has been reported to increase toxin production (Karlsson et al., 1999; Karlsson, S., A. Lindberg, L. G. Burman, E. Norin et al., 2000). In principle, different levels of toxin production could reflect changes in the fraction of cells that are TcdA-ON, changes in the level of induction of tcdA in the TcdA-ON subpopulation, or some combination of the two. We used our PtcdA::rfp reporter to examine the effect of glucose, cysteine, and a combination of the two on toxin production in cells grown in TY. As expected, glucose and cysteine reduced toxin production (Fig. S3). Flow cytometry revealed that glucose and cysteine reduced the fraction of TcdA-ON cells (Fig. S3).
Many global regulators have been reported to influence tcdR and thus tcdA and tcdB gene expression in response to these changes in cellular metabolism [reviewed in (Bouillaut et al., 2015)]. To investigate the role of global regulators on cell-to-cell variation in tcdA expression, we introduced the PtcdA::rfp reporter plasmid into C. difficile JIR8094 TargeTron insertion mutants of four global regulators: ccpA, codY, agrB and sigH. Inactivation of ccpA, agrB, and sigH had almost no effect on bimodal PtcdA::rfp expression (Fig. S4). In contrast we observed a 50-fold increase in overall fluorescence in the codY null mutant (Fig. S5), which compares favorably with a previous study showing that inactivating codY increases tcdA mRNA about 50-fold (Dineen et al., 2010). Increased expression of the PtcdA::rfp reporter reflected increases in both the number of cells that were TcdA-ON (~3 fold; Fig. 6A and B) and the mean fluorescence intensity of the TcdA-ON cells (~10 fold; Fig. S5). In summary, CodY biases C. difficile towards the TcdA-OFF state, but it is not required for bimodality per se as toxin expression remains bimodal in the absence of CodY.
Figure 6. CodY impacts but is not essential for bimodal tcdA expression.
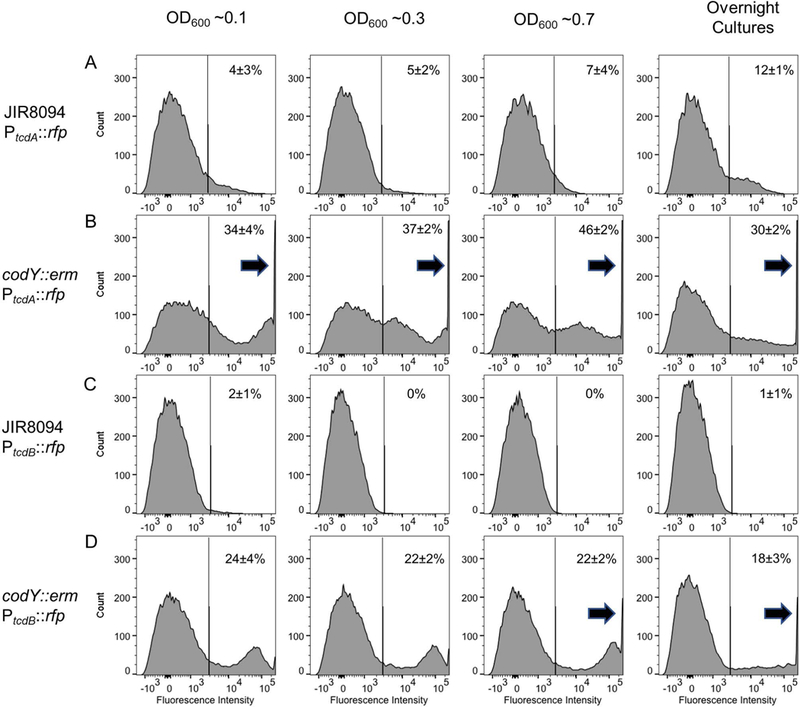
The indicated strains wild-type JIR8094/PtcdA::rfp, codY::erm/PtcdA::rfp, JIR8094/PtcdA::rfp and codY::erm/PtcdA::rfp were grown in TY and samples were harvested at the OD600 noted. Samples were fixed and analyzed by flow cytometry. Note the large number of cells in the codY mutant strains that saturated the fluorescence detector during flow cytometry (arrows). Percentages refer to fraction of TcdA-ON or TcdB-ON cells (mean ± st. dev., n = 3 experiments).
Evidence for bimodal expression of tcdB
Because tcdA expression is bimodal and σTcdR levels appear to be critical for establishing bimodality, we hypothesized that expression of toxin B (TcdB) and the master regulator (σTcdR) would also be bimodal. Unfortunately, wild-type cells carrying a PtcdB::rfp reporter plasmid were not fluorescent even though the reporter is on a plasmid present at ~6 copies per cell (Ransom et al., 2015) (Fig. 6C). Apparently, expression of tcdB was below our detection limit. The expression of tcdB is reported to be 10 to 100 fold lower than tcdA (Merrigan et al., 2010; Vohra and Poxton, 2011; Bakker et al., 2012). However, in a codY::erm mutant background, expression of the PtcdB::rfp reporter was readily detected and bimodal (Fig. 6D). We were unable to assess whether production of σTcdR is bimodal because we could not detect any fluorescence from a PtcdR::rfp reporter plasmid, even in a codY::erm mutant background. Similar reporter constructs incorporating different amounts of DNA from the tcdR promoter region were also non-fluorescent. This finding is not too surprising because expression of tcdR is known to be lower than that of tcdA and tcdB (Dupuy and Sonenshein, 1998).
Sporulation and toxin gene expression are not mutually exclusive
The finding that tcdA expression is bimodal raises the question: How does differential tcdA expression benefit C. difficile? It has been proposed that toxin production and sporulation may be mutually (or temporally) exclusive processes in C. difficile (Saujet et al., 2011; Bouillaut et al., 2015). Only oxygen-tolerant spores can survive outside the host long enough to be ingested by a new host, but spores are metabolically inert and thus not capable of producing the toxins that cause diarrhea. One clever solution to this conundrum would be for stationary phase cultures of C. difficile to differentiate into toxin-producing cells that provoke diarrhea and oxygen-tolerant spores that can survive the journey to the next host. In support of this idea, the master regulator of sporulation, Spo0A, negatively regulates toxin gene expression in some C. difficile strain backgrounds (Mackin et al., 2013). In addition, another positive regulator of sporulation, Spo0H, is said to inhibit toxin production (Saujet et al., 2011). However the relationship between sporulation and toxin production is murky because other studies have come to conflicting conclusions (reviewed in (Martin-Verstraete et al., 2016)).
We used our PtcdA::rfp reporter plasmid in conjunction with microscopy to ask whether toxin production and sporulation can occur in the same cell. Cultures of R20291/PtcdA::rfp and 630∆erm/PtcdA::rfp were sporulated on TY agar containing thiamphenicol to match conditions of our toxin studies. Plates were incubated for 44–76 hrs before samples were harvested and fixed as described previously (Ransom et al., 2016). We were able to identify toxin-producing cells by their red fluorescence and sporulating cells by the fact that spores are phase-bright. Interestingly, TcdA-ON (red) mother cells with spores were readily observed in both strain backgrounds (Fig. 7), and the percentage of cells that were TcdA-ON was similar for vegetative cells and mother cells containing obvious forespores or spores (Table 1). For instance, in R20291, ~15% of cells lacking a spore and ~23% of cells containing a spore were red fluorescent (Table 1). In 630∆erm, which has a much higher percentage of TcdA-ON cells because flgB is locked in the ON orientation (Fig. 3; (Anjuwon-Foster et al., 2018)), the corresponding numbers were 83% and 84% TcdA-ON for cells lacking or containing spores, respectively (Table 1). We also observed a small number of free spores that were red fluorescent. These results demonstrate that sporulation and toxin gene expression can occur within the same cell, although they do not rule out the possibility that these events are sequential, i.e., the red fluorescence observed in mother cells containing spores might be residual RFP protein produced prior to entry into the spore developmental program.
Figure 7. Sporulation and toxin expression are independent processes.
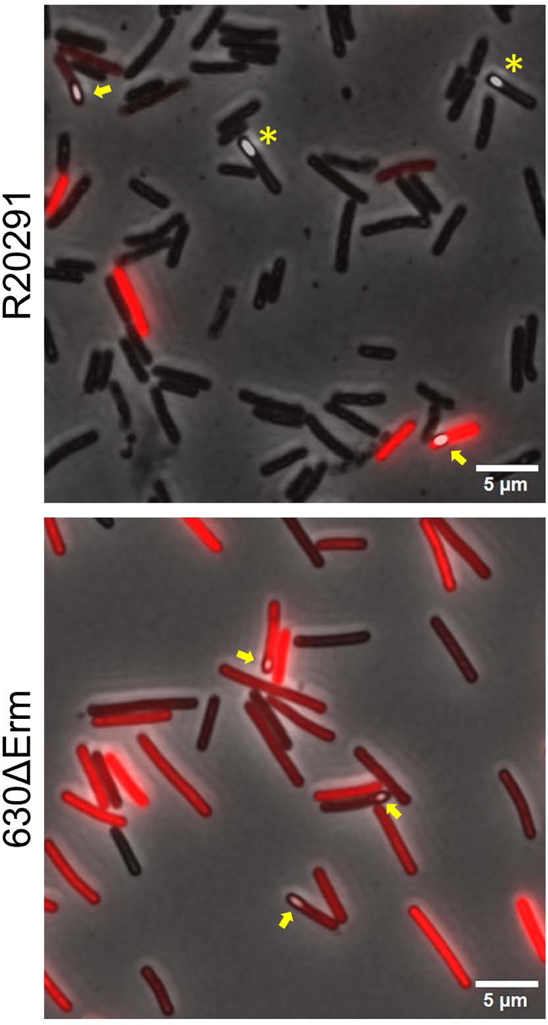
C. difficile strains R20291 and CD630Δerm harboring pRAN737 (PtcdA::rfp) were sporulated on TY agar with thiamphenicol. Cells and spores recovered from plates were fixed, removed from the anaerobic chamber to allow RFP to mature, and then imaged by phase contrast and fluorescence microscopy. Shown are overlays of the phase-contrast images and fluorescence images. Yellow arrows indicate examples of toxin-expressing cells that contain a developing spore. Yellow asterisks indicate sporulating cells that are not expressing the tcdA reporter.
Table 1.
Spore formation and toxin expression
| % expressing PtcdA-rfp |
Total cells counted** |
|
|---|---|---|
| R20291 | ||
| Vegetative | 15.1% | 4580 |
| Forespores* | 22.5% | 129 |
| Endospores* | 19.6% | 46 |
| 630∆erm | ||
| Vegetative | 83% | 2391 |
| Forespores* | 84% | 88 |
| Endospores* | 33% | 3 |
Forespores refers to mother cells containing a phase-bright spore while endospores refers to free spores.
Data pooled from 3 experiments that yielded similar results.
DISCUSSION
Toxin gene expression is bimodal in C. difficile
C. difficile pathogenesis is mediated primarily by two large exotoxins encoded in the PaLoc, TcdA and TcdB. There has been a lot of effort expended to understand how production of these toxins is controlled. Early studies found that the toxins are produced upon entry into stationary phase (Moncrief and Barroso, 1997; Dupuy and Sonenshein, 1998). This response is mediated by a dedicated sigma factor (σTcdR) and by a host of global regulatory proteins, most of which sense various aspects of metabolism [reviewed in (Voth and Ballard, 2005; Bouillaut et al., 2015; Martin-Verstraete et al., 2016)]. All these studies have relied on methods that reflect the average behavior of the cells in the population under the (unstated) assumption that toxin production is relatively uniform across the population. Here we have used a fluorescent protein reporter, RFP, to visualize expression of tcdA in individual cells. Our results indicate that during entry into stationary phase only a subset of C. difficile cells expresses the toxin genes. Expression of the second toxin gene, tcdB, was also bimodal, but visualizing this required working in a codY mutant background to elevate expression sufficiently to detect it using a fluorescent protein reporter.
Bimodal expression of tcdA is probably an example of bistability rather than phase variation, and σTcdR is the genetic switch
Bimodal distributions of gene expression can arise from phase variation or bistability. Our findings point towards the latter, with the toxin-specific sigma factor σTcdR being the master regulator that governs the decision between toxin-ON and toxin-OFF. Studies of bistability in other bacteria have revealed two characteristics that make a regulatory protein well-suited for controlling a bistable switch (Dubnau and Losick, 2006). One is low-level basal expression so that stochastic variation can lead to excursions that tip the balance between an ON and an OFF state. The other is a positive feedback loop that reinforces transient increases in cellular abundance of the activator. In the case of toxin gene regulation in C. difficile, σTcdR fulfills both criteria (Dupuy and Sonenshein, 1998; Mani and Dupuy, 2001; Mani et al., 2002). In support of this notion, we found that graded expression of tcdR using a tetracycline-inducible promoter prevents development of bistability. Instead, as more inducer is added to the culture, toxin production increases uniformly across the cells in the population. These findings imply that in a wild-type background tcdR expression is itself bistable. Unfortunately, efforts to test this idea using an RFP reporter were not successful, owing to the very low level of tcdR expression that resulted in levels of RFP below our detection limit.
Multiple global regulators bias cells towards the toxin-ON or toxin-OFF states
A plethora of global regulators have been implicated in control of toxin production in C. difficile (Dupuy and Sonenshein, 1998; Dineen et al., 2007; Dineen et al., 2010; Antunes et al., 2011; Saujet et al., 2011; Mackin et al., 2013; El Meouche et al., 2013; McKee et al., 2013). We sought to determine which of these global regulators impacted bistable expression of tcdA. Of the major regulators tested, CodY and σD had the most significant impact on the fraction of TcdA-ON cells; however, neither is required for bistability. In the absence of CodY the fraction of cells expressing tcdA increased ~3 fold while the level of toxin production in those cells increased more dramatically by ~10 fold. Nevertheless, populations of the codY mutant still bifurcated into TcdA-ON and TcdA-OFF subpopulations, indicating CodY is not responsible for bistability of toxin gene expression per se. A priori, the motility sigma factor σD was a prime candidate for controlling the decision between TcdA-ON and TcdA-OFF because expression of sigD is regulated by phase variation (Anjuwon-Foster and Tamayo, 2017). Nevertheless, neither a sigD::erm null mutation nor eliminating phase-variable expression of σD broke bistable expression of the PtcdA::rfp reporter. Collectively, our findings indicate σD biases C. difficile towards TcdA-ON, while CodY biases towards TcdA-OFF, but σTcdR is the master regulator that governs the choice between TcdA-ON and TcdA-OFF.
More generally, tcdR expression is affected by multiple physiological inputs and regulatory proteins (Bouillaut et al., 2015; Martin-Verstraete et al., 2016). The sum of these positive and negative inputs poises basal expression of tcdR at a given level, be that relatively high or relatively low, which in turn affects the probability that random fluctuations in σTcdR synthesis and turnover will push cells across a threshold that locks them into the TcdR-ON state that leads to toxin production. For example, when the invertible element at flgB is in the ON orientation, expression of sigD poises σTcdR relatively close to the tipping point and conversion to TcdA-ON is more common. Conversely, in rapidly growing cells replete with energy and amino acids, repression by CodY acting at multiple promoters in the pathogenicity locus renders conversion to TcdA-ON a very rare event.
What are the potential benefits of bistable toxin gene expression?
Bistability has been described as a bet-hedging strategy for dealing with an uncertain and perilous future (Dubnau and Losick, 2006; Davidson and Surette, 2008; Tiwari et al., 2011; García-Pastor et al., 2018). From the perspective of the bacterium, toxin production can be viewed as a means for obtaining food or escaping to a better host. In either case, conditions in the host might change suddenly. If they improve, the large investment in producing the toxins, which are large proteins of over 3000 amino acids that must somehow be transported out of the cell, would be a waste of resources. Conversely, delaying toxin production when conditions are deteriorating comes with its own set of risks. A related possibility is that some of the toxin might remain associated with the cell surface, rendering toxin-producing cells targets for the host immune response. Dilemmas such as these could select for regulatory circuits that incorporate an element of chance into the decision to produce toxins.
An alternative rationale behind bistability invokes division of labor between different cell types needed to achieve a common goal. For example, as noted above, bistable toxin production might be part of a strategy for transmission to a new host whereby some cells produce toxin to provoke diarrhea, while others differentiate into oxygen-tolerant spores that can persist in the environment (Saujet et al., 2011; Bouillaut et al., 2015). However, this explanation seems unlikely as we found that toxin production and spore formation can occur within the same cell.
A third potential explanation for why only a subset of C. difficile cells produce toxins is related to how the toxins are released from the cell. The mechanism of toxin release is not yet known. Some studies have implicated a holin-like protein named TcdE in this process, but that finding has been disputed (Govind and Dupuy, 2012; Govind et al., 2015). In the absence of an obvious export apparatus, it has been suggested toxins might be released by cell lysis, with bystanders reaping the benefits. Obviously not all of the cells in a population can afford to lyse. Our data do not support the notion that toxins are released by lysis because we did not observe a large decrease in cell viability when toxin gene expression is artificially induced by either exogenous expression of tcdR or deletion of codY. Nor did we observe massive lysis of strain CD630Δerm even though 80% of those cells were TcdA-ON. Nevertheless, it remains possible that these mechanisms of driving most cells into the TcdA-ON state do not activate the (putative) lysis mechanisms that might be involved in toxin release. Related to this idea is the potential for toxins to be released when mother cells lyse during spore development. However, this hypothesis implies toxin production would be a step on the pathway towards spore formation, which is inconsistent with our observation that spore development and toxin gene expression appear to be independent phenomena.
Open questions
Using a fluorescent reporter to study toxin gene expression at the level of individual C. difficile cells revealed a bistable switch governed by the toxin-specific sigma factor, σTcdR. This finding makes it a high priority to better understand how σTcdR levels are determined. Another key question that remains to be answered is: What benefit does C. difficile derive from having only a subset of cells produce toxins? Finally, it bears emphasis that all of the studies presented here were performed in laboratory media. In view of the fact that toxin production responds to metabolic inputs and that growth conditions in the host are very different from those in the lab, it will be interesting to visualize toxin-producing cells in the context of an infection model. Among the important unknowns are what fraction of C. difficile cells produce toxins in a host and whether toxin production is restricted to specific regions of the intestines.
MATERIALS AND METHODS
Strains, media, and growth conditions
Bacterial strains used in this study are listed in Table 2. This study included six wild-type C. difficile strains: JIR8094, 630Δerm, R20291, CD196, NAP07, and NAP08. C. difficile mutants were derived from the erythromycin-sensitive isolate JIR8094, a derivative of the sequenced strain CD630 (Sebaihia et al., 2006; O’Connor et al., 2006). C. difficile was routinely grown in Tryptone Yeast (TY) media, supplemented as needed with thiamphenicol at 10 μg/ml, erythromycin at 5 μg/ml, kanamycin at 50 μg/ml, or cefoxitin at 16 μg/ml. TY consisted of 3% tryptone, 2% yeast extract, and 2% agar (for solid media). TY included 0.1% L-cysteine during routine maintenance of C. difficile cultures, but cysteine was generally omitted when assaying toxin production. C. difficile strains were maintained at 37°C in an anaerobic chamber (Coy Laboratory products) in an atmosphere of 10% H2, 5% CO2, and 85% N2.
Table 2.
Strains
| Strain | Genotype and Description | Reference* |
|---|---|---|
| E. coli | ||
| OmniMAX – 2 T1R | F´ [proAB+ lacIq lacZΔM15 Tn10(TetR) Δ(ccdAB)] mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 Δ(lacZYA-argF) U169 endA1 recA1 supE44 thi-1 gyrA96 relA1 tonA panD. | Invitrogen |
| XL1-Blue | endA1 gyrA96(nalR) thi-1 recA1 relA1 lac glnV44 [F’ proAB+ lacIq Δ(lacZ)M15] hsdR17(rK- mK+) Tn10(TetR)] | |
| HB101/pRK24 | F− mcrB mrr hsdS20(rB− mB−) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20 | (Trieu-Cuot et al., 1987) |
| EC3272 | XL1-Blue / pDSW1728 (Ptet::rfp cat) | (Ransom et al., 2015) |
| RAN473 | OmniMAX / pRAN473 (Ptet::rfp –MCS cat) | (Ransom et al., 2015) |
| C. difficile | ||
| JIR8094 | Spontaneous erythromycin-sensitive derivative of strain 630 (Ribotype 012) | (O’Connor et al., 2006) |
| CD630Δerm | Spontaneous erythromycin-sensitive derivative of strain 630 (Ribotype 012) | (Hussain et al., 2005) |
| CD196 | Wild-type C. difficile strain from France (ribotype 027) | |
| NAP07 | Wild-type C. difficile strain ribotype 078 | |
| NAP08 | Wild-type C. difficile strain ribotype 078 | |
| R20291 | Wild-type C. difficile strain from UK outbreak (ribotype 027) | |
| RAN820 | NAP08 / pRAN737 (PtcdA::rfp cat) | |
| RAN828 | JIR8094 / pRAN737 (PtcdA::rfp cat) | |
| RAN829 | JIR8094 / pRAN737 (PpdaV::rfp cat) | (Ransom et al., 2015) |
| RAN912 | JIR8094 / pRAN841 (PtcdB::rfp cat) | |
| RAN913 | JIR8094 / pRAN842 (PtcdR::rfp cat) | |
| RAN925 | R20291 / pRAN737 (PtcdA::rfp cat) | |
| RAN934 | CD196 / pRAN737 (PtcdA::rfp cat) | |
| RAN1101 | NAP07 / pRAN737 (PtcdA::rfp cat) | |
| GMK134 | CD630Δerm / pRAN737 (PtcdA::rfp cat) | |
| RAN1116 | JIR8094 codY330::ltrB::ermB | |
| RAN1123 | JIR8094 tcdR142::ltrB::ermB | |
| RAN1121 | JIR8094 sigH123::ltrB::ermB | |
| RAN1120 | JIR8094 ccpA133::ltrB::ermB | |
| CDE1774 | JIR8094 agrB188::ltrB::ermB | |
| RAN1124 | JIR8094 codY330::ltrB::ermB / pRAN737 (PtcdA::rfp cat) | |
| RAN1127 | JIR8094 tcdR142::ltrB::ermB / pRAN737 (PtcdA::rfp cat) | |
| RAN1126 | JIR8094 sigH123::ltrB::ermB / pRAN737 | |
| RAN1125 | JIR8094 ccpA133::ltrB::ermB / pRAN737 | |
| CDE2770 | JIR8094 agrB188::ltrB::ermB / pRAN737 | |
| TCD20 | JIR8094 csfV63::ltrB::ermB | (Ho et al., 2014) |
| RAN1129 | JIR8094 tcdR142::ltrB::ermB / pRAN1032 | |
| RT1566 | R20291 sigD228::erm | (Anjuwon-Foster and Tamayo, 2017) |
| GMK129 | R20291 sigD228::erm / pRAN737 | |
| GMK130 | R20291 sigD228::erm / pGK110 |
This study unless otherwise noted.
Escherichia coli strains were grown in LB medium at 37°C with chloramphenicol at 20 μg/ml and ampicillin at 100 μg/ml as needed. LB contained 1% tryptone, 0.5% yeast extract, 1% NaCl and, for plates, 1.5% agar.
Plasmid and strain construction
All plasmids are listed in Table 3. Regions of plasmids constructed using PCR were verified by DNA sequencing. The oligonucleotide primers used in this work were synthesized by Integrated DNA Technologies (Coralville, IA). Primers are listed in Table S1. All plasmids were constructed using OmniMax-2 T1R as the cloning host, transformed into HB101/pRK24, and then introduced into C. difficile strains by conjugation (Trieu-Cuot et al., 1987).
Table 3.
Plasmids
| Plasmid | Relevant Features | Reference* |
|---|---|---|
| pRPF185 | E. coli-C. difficile shuttle vector with tetracycline-inducible promoter. Ptet::gusA cat CD6ori RP4oriT-traJ pMB1ori | (Fagan and Fairweather, 2011) |
| pBL100 | E. coli-C. difficile shuttle vector for creating C. difficile mutants using TargeTron mutagenesis. ltrB::ermB::RAM ltrA cat bla CD6ori RP4oriT pMBlori | (Bouillaut et al., 2013) |
| pDSW1728 | Ptet::rfp cat | (Ransom et al., 2015) |
| pCE536 | pBL100 targeted to codY330 | |
| pRAN737 | pDSW1728 derivative with PtcdA::rfp | |
| pRAN841 | pDSW1728 derivative with PtcdB::rfp | |
| pRAN842 | pDSW1728 derivative with PtcdR::rfp | |
| pRAN1018 | PtcdA::rfp / PpdaV::tcdR | |
| pRAN1032 | PtcdA::rfp / Ptet::tcdR | |
| pTHE583 | pBL100 targeted to agrB188 | |
| pCE540 | pBL100 targeted to sigH123 | |
| pCE541 | pBL100 targeted to ccpA133 | |
| pRAN1034 | pBL100 targeted to tcdR142 | |
| pGK110 | PtcdA::rfp / Ptet::sigD |
This study unless otherwise noted.
The C. difficile null mutant of tcdR142 was constructed using modified TargeTron procedures (Sigma-Aldrich) to insert a group II intron conferring Erm resistance (Heap et al., 2007; Heap et al., 2010; Ho and Ellermeier, 2011). Primers for retargeting the group II intron were designed using the ClosTron algorithm (Heap et al., 2010). To retarget the intron to insert after nucleotide 142 of tcdR, the intron template was amplified by PCR as outlined in the TargeTron user manual (Sigma-Aldrich) using an EBS universal primer designated CDE914 in combination with primers RP398, RP399, and RP400. The resulting PCR product and the vector pBL100 (Bouillaut et al., 2013) were digested with HindIII and BsrGI, and then ligated to create plasmid pRAN1034. pRAN1034 was transferred to C. difficile JIR8094 via conjugation and isolates in which the intron had moved to the tcdR142 locus were obtained by selection for Erm-resistance as described previously (Heap et al., 2010; Ho and Ellermeier, 2011). Intron insertion into tcdR142 was confirmed by PCR. Finally, loss of the TargeTron plasmid was confirmed by thiamphenicol sensitivity. To construct additional TargeTron mutants the following primer combinations were used to generate vectors for mutagenesis: codY330, RP323-RP325, pCE536; ccpA133, RP326-RP328, pCE541; agrB188 CDEP1807-CDEP1809, pTHE538; and sigH123, RP335-RP337, pCE540. Mutagenesis was carried out as described above.
For expression studies plasmids were constructed with promoters from tcdA, tcdB, and tcdR. The plasmids are all derivatives of pDSW1728, which has a tetracycline-inducible promoter and codon optimized Red Fluorescent Protein mCherryOpt (Ptet::rfp) (Ransom et al., 2015; Ransom et al., 2016). Promoters were amplified using the following primer sets: PtcdA (RP304 and RP305), PtcdB (RP345 and RP346), and PtcdR (RP347 and RP348). The PCR products were digested with NheI and SacI, then ligated into pDSW1728 digested with the same enzymes to cut out the Ptet promoter. The resulting plasmids were designated pRAN737 (PtcdA), pRAN841 (PtcdB), and pRAN842 (PtcdR).
To regulate tcdR expression in C. difficile, we built two constructs that had tcdR under an inducible promoter: Ptet (Fagan and Fairweather, 2011) or PpdaV (Ho and Ellermeier, 2011; Ho et al., 2014; Ransom et al., 2015). To build PpdaV::tcdR, a synthetic DNA fragment (gBlock) containing both the promoter and gene was synthesized by Integrated DNA Technologies, and the fragment was amplified using RP374 and RP375. This DNA was digested with XmaI and inserted into the XmaI site of pRAN737. The resulting plasmid was designated pRAN1018 (PtcdA::rfp; PpdaV::tcdR). To build Ptet::tcdR, the Ptet promoter was amplified by PCR using primers RP393 and RP394, with pRPF185 as the template (Fagan and Fairweather, 2011). The Ptet promoter was then swapped with the PpdaV promoter in pRAN1018 using a KpnI and SphI digest and ligation. The resulting plasmid was named pRAN1032 (PtcdA::rfp; Ptet::tcdR).
To build Ptet::sigD, the sigD gene was amplified from R20291 chromosomal DNA using primers CDEP3531 and CDEP3532. pRAN1032 was digested using SphI and AscI to remove tcdR. The sigD PCR product was inserted into the cut vector using isothermal assembly resulting in plasmid pGK110.
Fixation protocol
Cells were fixed as previously described (Ransom et al., 2014; Ransom et al., 2016). Briefly, a 500-μl aliquot of cells in growth medium was added directly to a microcentrifuge tube containing 120 μl of a 5X fixation cocktail: 100 μl of 16% (wt/vol) paraformaldehyde aqueous solution (methanol-free; catalog no. AA433689M; Alfa Aesar, Ward Hill, MA) and 20 μl of 1 M NaPO4 buffer (pH 7.4). The sample was mixed, allowed to sit for 15 min, removed from the Coy chamber, and incubated on ice for 45 min. The fixed cells were washed three times with PBS, resuspended in 30 μl of PBS, and left in the dark at 4°C to allow for chromophore maturation.
Microscopy
Microscopy was performed as described previously (Ransom et al., 2016). Cells were immobilized using thin agarose pads (1%). Phase-contrast and fluorescence micrographs were recorded on an Olympus BX60 microscope equipped with a ×100 UPlanApo objective (numerical aperture, 1.35). For RFP the filter set (catalog no. 41004) comprised a 538- to 582-nm excitation filter, a 595-nm dichroic mirror (long pass), and a 582- to 682-nm emission filter. This filter set was from Chroma Technology Corp. (Brattleboro, VT). Micrographs were captured with a Spot 2 CCD camera as described (Ransom et al., 2014; Ransom et al., 2015; Ransom et al., 2016) or with a Hamamatsu ORCA Flash 4.0 V2+ CMOS camera. Typical exposure times for RFP were 3 seconds for the Spot camera and 250 milliseconds for the Flash 4.0 camera. To ensure comparability of fluorescence micrographs, the display range option was adjusted identically for all images. Micrographs were cropped, and figures were assembled in Adobe Illustrator (Adobe Systems, Inc., San Jose, CA) or Olympus cellSens Dimension software.
Flow cytometry
Cells were analyzed at the Flow Cytometry Facility at the University of Iowa. The equipment used in this study includes the Becton Dickinson LSR II with a 561nm laser, 610/20 bandpass filter, and 600 LP dichroic filter, and the Becton Dickinson Aria II. Data was analyzed using BD FACSDiva Software.
Fluorescence measurements with a plate reader
The plate reader was used to measure bulk samples from cultures as described previously (Ransom et al., 2016). Briefly, fluorescence and absorbance (OD600) were measured with an Infinite M200 Pro plate reader (Tecan, Research Triangle Park, NC). Samples were prepared by adding 20 μl of fixed cells in PBS and 180 μl of PBS to the well of a flat-bottom 96-well microtiter plate (AS Plate-PS-96-F-C; AG Advangene, IL). Fluorescence was recorded as follows: excitation, 554 nm; emission, 610 nm; gain setting, 100. The cell density (OD600) was also recorded and used to normalize the fluorescence reading.
Spore Preparation
C. difficile spores were obtained following standard procedures as previously described (Edwards and McBride, 2016). Briefly, C. difficile strains were grown overnight in TY broth with 10 μg/ml thiamphenicol. 200 µl of overnight culture was plated on TY agar with 10 μg/ml thiamphenicol. Plates were incubated at 37°C for 44–76 hours. Cells were scraped from plates and suspended in 500 μl TY broth. Samples were then fixed and visualized as described above.
Supplementary Material
Figure S1. Bimodal tcdA expression is not simply due to loss of cell viability. The R20291 strain containing PtcdA::rfp grown in TY with thiamphenicol was sampled in log phase (OD600 0.5), stationary phase (OD600 1.2), or after overnight growth (20 hrs). (A) To determine the fraction of TcdA-ON cells, samples were fixed and analyzed by flow cytometry. (B) To assess viability and plasmid loss, samples were plated on TY or TY with thiamphenicol and the number of CFUs was determined. (C) To further assess viability, samples were scored after treatment with live/dead stain. (D) Representative micrographs from a live/dead staining experiment, showing top to bottom: phase-contrast, green fluorescence (live), and red fluorescence (dead). Samples were collected without fixation and suspended in PBS and mixed with an equal volume of LIVE/DEAD BacLight staining reagent (Molecular Probes, Invitrogen) which contains a mixture of propidium iodide (30 mM final concentration) and Syto9 (6 mM final concentration) in the anaerobic chamber. The stained cells were removed from the chamber and imaged immediately by microscopy. For Log, Stat, and 20 hrs, the numbers of cells counted were respectively 318, 412, and 959. The red fluorescence from RFP is not detected in these assays because samples were not exposed to air prior to imaging which is required for RFP to become fluorescent (Ransom et al., 2015). There were <1% dead cells in early stationary phase and ~5% dead cells after 24 hrs. Debris that did not stain with either dye was not scored.
Figure S2. Alignment of tcdA promoters from different C. difficile strains. The upstream region of tcdA from different strains was aligned using Clustal Omega (Sievers et al., 2011) (Sievers et al., 2011). The C. difficile strains are CD196, R20291, CD630, NAP07, and NAP08.
Figure S3. Effect of glucose and cysteine on tcdA expression. The R20291 strain containing PtcdA::rfp was grown in TY with thiamphenicol amended with 0.5% glucose or 0.1% cysteine as indicated. (A) Growth was monitored by OD600. At the points indicated, samples were fixed and specific fluorescence (which is total fluorescence normalized to OD) was determined using a plate reader. (B) Cultures in stationary phase (20 hrs after inoculation) were fixed and analyzed by flow cytometry.
Figure S4. Effect of global regulators on bimodal tcdA expression. The indicated strains containing PtcdA::rfp were grown in TY to stationary phase (20 hrs). Samples were fixed and analyzed by flow cytometry.
Figure S5. Expression of PaLoc promoters in a codY null mutant. The indicated strains wild-type JIR8094/PtcdA::rfp, codY::erm/PtcdA::rfp, JIR8094/PtcdB::rfp, and codY::erm/PtcdB::rfp were grown in TY for 20 hrs. Samples were fixed and analyzed by flow cytometry. The mean fluorescence intensity was determined using FlowJo Fluorescence Intensity function for both the entire population of cells and the TcdA-ON subpopulation.
Acknowledgments
We thank R. Tamayo (University of North Carolina) and B. Dupuy (Institut Pasteur) for strains. This work was supported by Public Health Service grant R21 AI121576 to CDE and DSW from the National Institutes for Allergy and Infectious Disease. Some data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine / Holden Comprehensive Cancer Center core research facility at the University of Iowa. The Facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center, and Iowa City Veteran’s Administration Medical Center.
REFERENCES
- Akerlund T, Persson I, Unemo M, Noren T, Svenungsson B, Wullt M, and Burman LG (2008) Increased Sporulation Rate of Epidemic Clostridium difficile Type 027/NAP1. J Clin Microbiol 46: 1530–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjuwon-Foster BR, Maldonado-Vazquez N, and Tamayo R (2018) Characterization of Flagellum and Toxin Phase Variation in Clostridioides difficile Ribotype 012 Isolates. J Bacteriol 200: e00056–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjuwon-Foster BR, and Tamayo R (2017) A genetic switch controls the production of flagella and toxins in Clostridium difficile. PLOS Genet 13: e1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes A, Camiade E, Monot M, Courtois E, Barbut F, Sernova NV, et al. (2012) Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Res 40: 10701–10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes A, Martin-Verstraete I, and Dupuy B (2011) CcpA-mediated repression of Clostridium difficile toxin gene expression. Mol Microbiol 79: 882–99. [DOI] [PubMed] [Google Scholar]
- Aubry A, Hussack G, Chen W, KuoLee R, Twine SM, Fulton KM, et al. (2012) Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect Immun 80: 3521–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker D, Smits WK, Kuijper EJ, and Corver J (2012) TcdC does not significantly repress toxin expression in Clostridium difficile 630ΔErm. PLoS One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillaut L, Dubois T, Sonenshein AL, and Dupuy B (2015) Integration of metabolism and virulence in Clostridium difficile. Res Microbiol 166: 375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillaut L, Self WT, and Sonenshein AL (2013) Proline-dependent regulation of Clostridium difficile Stickland metabolism. J Bacteriol 195: 844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GP, Douce GR, Govind R, Howarth PM, Mackin KE, Spencer J, et al. (2011) The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog 7: e1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GP, Rood JI, and Lyras D (2012) The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol 20: 21–9. [DOI] [PubMed] [Google Scholar]
- Cartman ST, Kelly ML, Heeg D, Heap JT, and Minton NP (2012) Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol 78: 4683–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran R, and Lacy DB (2017) The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev 41: 723–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SH, Tang YJ, and Silva J Jr. (2000) Analysis of the Pathogenicity Locus in Clostridium difficile Strains. J Infect Dis 181: 659–663. [DOI] [PubMed] [Google Scholar]
- Darkoh C, Kaplan HB, and Dupont HL (2011) Harnessing the glucosyltransferase activities of Clostridium difficile for functional studies of toxins A and B. J Clin Microbiol 49: 2933–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson CJ, and Surette MG (2008) Individuality in Bacteria. Annu Rev Genet 42: 253–268. [DOI] [PubMed] [Google Scholar]
- Deutscher J (2008) The mechanisms of carbon catabolite repression in bacteria. Curr Opin Microbiol 11: 87–93. [DOI] [PubMed] [Google Scholar]
- Dineen SS, McBride SM, and Sonenshein AL (2010) Integration of metabolism and virulence by Clostridium difficile CodY. J Bacteriol 192: 5350–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineen SS, Villapakkam AC, Nordman JT, and Sonenshein AL (2007) Repression of Clostridium difficile toxin gene expression by CodY. Mol Microbiol 66: 206–19. [DOI] [PubMed] [Google Scholar]
- Dubnau D (2015) Regulation by the Modulation of Gene Expression Variability. J Bacteriol 197: 1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau D, and Losick R (2006) Bistability in bacteria. Mol Microbiol 61: 564–72. [DOI] [PubMed] [Google Scholar]
- Dupuy B, Govind R, Antunes A, and Matamouros S (2008) Clostridium difficile toxin synthesis is negatively regulated by TcdC. J Med Microbiol 57: 685–9. [DOI] [PubMed] [Google Scholar]
- Dupuy B, and Sonenshein a L. (1998) Regulated transcription of Clostridium difficile toxin genes. Mol Microbiol 27: 107–120. [DOI] [PubMed] [Google Scholar]
- Edwards AN, and McBride SM (2016) Isolating and Purifying Clostridium difficile Spores. In Methods in Molecular Biology Roberts AP, and Mullany P (eds). Springer New York, New York, NY: pp. 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AN, Tamayo R, and McBride SM (2016) A novel regulator controls C lostridium difficile sporulation, motility and toxin production. Mol Microbiol 100: 954–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan RP, and Fairweather NF (2011) Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem 286: 27483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltis BA, Wiesner SM, Kim AS, Erlandsen SL, Lyerly DL, Wilkins TD, and Wells CL (2000) Clostridium difficile toxins A and B can alter epithelial permeability and promote bacterial paracellular migration through HT-29 enterocytes. Shock 14: 629–34. [DOI] [PubMed] [Google Scholar]
- García-Pastor L, Puerta-Fernández E, and Casadesús J (2018) Bistability and phase variation in Salmonella enterica. Biochim Biophys Acta - Gene Regul Mech 0–1. [DOI] [PubMed]
- Gerhard R, Nottrott S, Schoentaube J, Tatge H, Olling A, and Just I (2008) Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J Med Microbiol 57: 765–70. [DOI] [PubMed] [Google Scholar]
- Govind R, and Dupuy B (2012) Secretion of Clostridium difficile toxins A and B requires the holin-like protein TcdE. PLoS Pathog 8: e1002727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind R, Fitzwater L, and Nichols R (2015) Observations on the role of TcdE isoforms in Clostridium difficile toxin secretion. J Bacteriol 197: 2600–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guédon E, Serror P, Ehrlich SD, Renault P, and Delorme C (2001) Pleiotropic transcriptional repressor CodY senses the intracellular pool of branched-chain amino acids in Lactococcus lactis. Mol Microbiol 40: 1227–1239. [DOI] [PubMed] [Google Scholar]
- Heap JT, Kuehne S.a, Ehsaan M, Cartman ST, Cooksley CM, Scott JC, and Minton NP (2010) The ClosTron: Mutagenesis in Clostridium refined and streamlined. J Microbiol Methods 80: 49–55. [DOI] [PubMed] [Google Scholar]
- Heap JT, Pennington OJ, Cartman ST, Carter GP, and Minton NP (2007) The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods 70: 452–64. [DOI] [PubMed] [Google Scholar]
- Heap JT, Pennington OJ, Cartman ST, and Minton NP (2009) A modular system for Clostridium shuttle plasmids. J Microbiol Methods 78: 79–85. [DOI] [PubMed] [Google Scholar]
- Ho TD, and Ellermeier CD (2011) PrsW is required for colonization, resistance to antimicrobial peptides, and expression of extracytoplasmic function σ factors in Clostridium difficile. Infect Immun 79: 3229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TD, Williams KB, Chen Y, Helm RF, Popham DL, and Ellermeier CD (2014) Clostridium difficile extracytoplasmic function σ factor σV regulates lysozyme resistance and is necessary for pathogenesis in the hamster model of infection. Infect Immun 82: 2345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundsberger T, Braun V, Weidmann M, Leukel P, Sauerborn M, and Eichel-Streiber C. von (1997) Transcription analysis of the genes tcdA-E of the pathogenicity locus of Clostridium difficile. Eur J Biochem 244: 735–742. [DOI] [PubMed] [Google Scholar]
- Hussain HA, Roberts AP, and Mullany P (2005) Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Δerm) and demonstration that the conjugative transposon Tn916ΔE enters the genome of this strain at multiple sites. J Med Microbiol 54: 137–141. [DOI] [PubMed] [Google Scholar]
- Tan Kai Soo, Wee Boon Yu, and Song Keang Peng (2001) Evidence for holin function of tcdE gene in the pathogenicity of Clostridium difficile. J Med Microbiol 50: 613–619. [DOI] [PubMed] [Google Scholar]
- Karlsson S, Lindberg A, Burman LG, Norin E, and T.Å., Karlsson S, Lindberg a., Norin E, Burman LG, and Akerlund T (2000) Toxins, butyric acid and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect Immun 68: 5881–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S, Burman LG, and Akerlund T (1999) Suppression of toxin production in Clostridium difficile VPI 10463 by amino acids. Microbiology 1683–1693. [DOI] [PubMed]
- Karlsson S, Dupuy B, Mukherjee K, Norin E, Burman LG, and Åkerlund T (2003) Expression of Clostridium difficile toxins A and B and their sigma factor TcdD is controlled by temperature. Infect Immun 71: 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketley JM, Haslam SC, Mitchell TJ, Stephen J, Candy D.C. a, and Burdon D (1984) Production and Release of Toxins A and B by Clostridium difficile. J Med Microbiol 18: 385–391. [DOI] [PubMed] [Google Scholar]
- Lawson PA, Citron DM, Tyrrell KL, and Finegold SM (2016) Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prévot 1938. Anaerobe 40: 95–99. [DOI] [PubMed] [Google Scholar]
- Lyerly D, Lockwood D, Richardson S, and Wilkins TD (1982) Biological Activities of Toxins A and B of Clostridium difficile. Infect Immun 35: 1032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyerly DM, Saum KE, MacDonald DK, and Wilkins TD (1985) Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun 47: 349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackin KE, Carter GP, Howarth P, Rood JI, and Lyras D (2013) Spo0A differentially regulates toxin production in evolutionarily diverse strains of Clostridium difficile. PLoS One 8: e79666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani N, and Dupuy B (2001) Regulation of toxin synthesis in Clostridium difficile by an alternative RNA polymerase sigma factor. Proc Natl Acad Sci U S A 98: 5844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani N, Lyras D, Barroso L, Howarth P, Wilkins T, Rood JI, et al. (2002) Environmental Response and Autoregulation of Clostridium difficile TxeR, a Sigma Factor for Toxin Gene Expression. J Bacteriol 184: 5971–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Verstraete I, Peltier J, and Dupuy B (2016) The regulatory networks that control Clostridium difficile toxin synthesis. Toxins (Basel) 8: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MJ, Clare S, Goulding D, Faulds-Pain A, Barquist L, Browne HP, et al. (2013) The agr locus regulates virulence and colonization genes in Clostridium difficile 027. J Bacteriol 195: 3672–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matamouros S, England P, and Dupuy B (2007) Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol Microbiol 64: 1274–88. [DOI] [PubMed] [Google Scholar]
- McKee RW, Mangalea MR, Purcell EB, Borchardt EK, and Tamayo R (2013) The Second Messenger Cyclic Di-GMP Regulates Clostridium difficile Toxin Production by Controlling Expression of sigD. J Bacteriol 195: 5174–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meouche El I., Peltier J, Monot M, Soutourina O, Pestel-Caron M, Dupuy B, and Pons J-LL (2013) Characterization of the SigD regulon of Clostridium difficile and its positive control of toxin production through the regulation of tcdR. PLoS One 8: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrigan M, Venugopal A, Mallozzi M, Roxas B, Viswanathan VK, Johnson S, et al. (2010) Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol 192: 4904–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncrief JS, and Barroso L a (1997) Positive regulation of Clostridium difficile toxins. Infect Immun 65: 1105–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R, Pothoulakis C, LaMont JT, Carlson S, and Madara JL (1990) Clostridium difficile toxin A increases intestinal permeability and induces Cl- secretion. Am J Physiol 259: G165–G172. [DOI] [PubMed] [Google Scholar]
- Murray R, Boyd D, Levett PN, Mulvey MR, and Alfa MJ (2009) Truncation in the tcdC region of the Clostridium difficile PathLoc of clinical isolates does not predict increased biological activity of Toxin B or Toxin A. BMC Infect Dis 9: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusrat A, Turner JR, Verkade P, Madara L, and Parkos CA (2001) Clostridium difficile Toxins Disrupt Epithelial Barrier Function by Altering Membrane Microdomain Localization of Tight Junction Proteins Clostridium difficile Toxins Disrupt Epithelial Barrier Function by Altering Membrane Microdomain Localization of Tig. Infect Immun 69: 1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JR, Lyras D, Farrow KA, Adams V, Powell DR, Hinds J, et al. (2006) Construction and analysis of chromosomal Clostridium difficile mutants. Mol Microbiol 61: 1335–51. [DOI] [PubMed] [Google Scholar]
- Olling A, Seehase S, Minton NP, Tatge H, Schröter S, Kohlscheen S, et al. (2012) Release of TcdA and TcdB from Clostridium difficile cdi 630 is not affected by functional inactivation of the tcdE gene. Microb Pathog 52: 92–100. [DOI] [PubMed] [Google Scholar]
- Osgood DP, Wood NP, and Sperry JF (1993) Nutritional aspects of cytotoxin production by Clostridium difficile. Appl Environ Microbiol 59: 3985–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom EM, Ellermeier CD, and Weiss DS (2015) Use of mCherry Red Fluorescent Protein for Studies of Protein Localization and Gene Expression in Clostridium difficile. Appl Environ Microbiol 81: 1652–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom EM, Weiss DS, and Ellermeier CD (2016) Use of mCherryOpt Fluorescent Protein in Clostridium difficile. In Methods in Molecular Biology pp. 53–67. [DOI] [PubMed]
- Ransom EM, Williams KB, Weiss DS, and Ellermeier CD (2014) Identification and characterization of a gene cluster required for proper rod shape, cell division, and pathogenesis in Clostridium difficile. J Bacteriol 196: 2290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnayake-Lecamwasam M, Serror P, Wong KW, and Sonenshein AL (2001) Bacillus subtilis CodY represses early-stationary-phase genes by sensing GTP levels. Genes Dev 15: 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saujet L, Monot M, Dupuy B, Soutourina O, and Martin-Verstraete I (2011) The Key Sigma Factor of Transition Phase, SigH, Controls Sporulation, Metabolism, and Virulence Factor Expression in Clostridium difficile. J Bacteriol 193: 3186–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer J, and Aktories K (2004) Large clostridial cytotoxins: Cellular biology of Rho/Ras-glucosylating toxins. Biochim Biophys Acta - Gen Subj 1673: 66–74. [DOI] [PubMed] [Google Scholar]
- Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, et al. (2006) The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38: 779–86. [DOI] [PubMed] [Google Scholar]
- Shen A (2012) Clostridium difficile toxins: mediators of inflammation. J Innate Immun 4: 149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe H, Berdoz J, Kraehenbuhl J-P, and Corthesy B (2000) Polymeric IgA Is Superior to Monomeric IgA and IgG Carrying the Same Variable Domain in Preventing Clostridium difficile Toxin A Damaging of T84 Monolayers. J Immunol 164: 1952–1960. [DOI] [PubMed] [Google Scholar]
- Tiwari A, Ray JCJ, Narula J, and Igoshin OA (2011) Bistable responses in bacterial genetic networks: Designs and dynamical consequences. Math Biosci 231: 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triadafilopoulos G, Pothoulakis C, O’Brien MJ, and LaMont JT (1987) Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 93: 273–9. [DOI] [PubMed] [Google Scholar]
- Trieu-Cuot P, Arthur M, and Courvalin P (1987) Origin, evolution and dissemination of antibiotic resistance genes. Microbiol Sci 4: 263–6. [PubMed] [Google Scholar]
- Vohra P, and Poxton IR (2011) Comparison of toxin and spore production in clinically relevant strains of Clostridium difficile. Microbiology 157: 1343–1353. [DOI] [PubMed] [Google Scholar]
- Voth DE, and Ballard JD (2005) Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18: 247–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AS, Eyre DW, Wyllie DH, Dingle KE, Griffiths D, Shine B, et al. (2013) Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis 56: 1589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox MH, Shetty N, Fawley WN, Shemko M, Coen P, Birtles A, et al. (2012) Changing epidemiology of Clostridium difficile infection following the introduction of a national ribotyping-based surveillance scheme in England. Clin Infect Dis 55: 1056–1063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Bimodal tcdA expression is not simply due to loss of cell viability. The R20291 strain containing PtcdA::rfp grown in TY with thiamphenicol was sampled in log phase (OD600 0.5), stationary phase (OD600 1.2), or after overnight growth (20 hrs). (A) To determine the fraction of TcdA-ON cells, samples were fixed and analyzed by flow cytometry. (B) To assess viability and plasmid loss, samples were plated on TY or TY with thiamphenicol and the number of CFUs was determined. (C) To further assess viability, samples were scored after treatment with live/dead stain. (D) Representative micrographs from a live/dead staining experiment, showing top to bottom: phase-contrast, green fluorescence (live), and red fluorescence (dead). Samples were collected without fixation and suspended in PBS and mixed with an equal volume of LIVE/DEAD BacLight staining reagent (Molecular Probes, Invitrogen) which contains a mixture of propidium iodide (30 mM final concentration) and Syto9 (6 mM final concentration) in the anaerobic chamber. The stained cells were removed from the chamber and imaged immediately by microscopy. For Log, Stat, and 20 hrs, the numbers of cells counted were respectively 318, 412, and 959. The red fluorescence from RFP is not detected in these assays because samples were not exposed to air prior to imaging which is required for RFP to become fluorescent (Ransom et al., 2015). There were <1% dead cells in early stationary phase and ~5% dead cells after 24 hrs. Debris that did not stain with either dye was not scored.
Figure S2. Alignment of tcdA promoters from different C. difficile strains. The upstream region of tcdA from different strains was aligned using Clustal Omega (Sievers et al., 2011) (Sievers et al., 2011). The C. difficile strains are CD196, R20291, CD630, NAP07, and NAP08.
Figure S3. Effect of glucose and cysteine on tcdA expression. The R20291 strain containing PtcdA::rfp was grown in TY with thiamphenicol amended with 0.5% glucose or 0.1% cysteine as indicated. (A) Growth was monitored by OD600. At the points indicated, samples were fixed and specific fluorescence (which is total fluorescence normalized to OD) was determined using a plate reader. (B) Cultures in stationary phase (20 hrs after inoculation) were fixed and analyzed by flow cytometry.
Figure S4. Effect of global regulators on bimodal tcdA expression. The indicated strains containing PtcdA::rfp were grown in TY to stationary phase (20 hrs). Samples were fixed and analyzed by flow cytometry.
Figure S5. Expression of PaLoc promoters in a codY null mutant. The indicated strains wild-type JIR8094/PtcdA::rfp, codY::erm/PtcdA::rfp, JIR8094/PtcdB::rfp, and codY::erm/PtcdB::rfp were grown in TY for 20 hrs. Samples were fixed and analyzed by flow cytometry. The mean fluorescence intensity was determined using FlowJo Fluorescence Intensity function for both the entire population of cells and the TcdA-ON subpopulation.