

Abstract
Ubiquitin domain‐containing protein 1 (UBTD1) is highly evolutionary conserved and has been described to interact with E2 enzymes of the ubiquitin–proteasome system. However, its biological role and the functional significance of this interaction remain largely unknown. Here, we demonstrate that depletion of UBTD1 drastically affects the mechanical properties of epithelial cancer cells via RhoA activation and strongly promotes their aggressiveness. On a stiff matrix, UBTD1 expression is regulated by cell–cell contacts, and the protein is associated with β‐catenin at cell junctions. Yes‐associated protein (YAP) is a major cell mechano‐transducer, and we show that UBTD1 is associated with components of the YAP degradation complex. Interestingly, UBTD1 promotes the interaction of YAP with its E3 ubiquitin ligase β‐TrCP. Consequently, in cancer cells, UBTD1 depletion decreases YAP ubiquitylation and triggers robust ROCK2‐dependent YAP activation and downstream signaling. Data from lung and prostate cancer patients further corroborate the in cellulo results, confirming that low levels of UBTD1 are associated with poor patient survival, suggesting that biological functions of UBTD1 could be beneficial in limiting cancer progression.
Keywords: matrix stiffness, ROCK2, UBTD1, YAP, β‐TrCP
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
Cells and tissues sense their environment not only via chemical signals but also through mechanical inputs. Mechanical cues are detected and translated into biochemical signals and transcriptional programs that allow the cell to adapt to environmental changes. This ability is essential for numerous biological processes ranging from tissue development and regeneration to pathological states like muscle dystrophies or cancers 1, 2. The Yes‐associated protein (YAP) and PDZ‐binding motif (TAZ) transcriptional co‐activators are major mechano‐transducers mediating various cell responses, mainly cell proliferation, migration, and invasion or suppression of apoptosis through TEA domain family member (TEAD) transcription factor family responsive genes 3.
YAP/TAZ activity is tightly controlled by the Hippo signaling pathway, which is composed of the core components, large tumor suppressor kinases (LATS1/2) and Ste20‐like kinases 1/2 (MST1/2) 4. Upon activation, like cell contact inhibition, MST1/2 phosphorylate LATS1/2, which in turn phosphorylate YAP/TAZ and prevent their nuclear translocation by promoting their interaction with 14.3.3 5, 6. Phosphorylation by Hippo on serine 127 and 381 (S127 and S381) primes YAP for subsequent phosphorylation by creatine kinase 1 (CK1) and induces its proteasomal degradation by SCFβ‐TRCP E3 ubiquitin ligase (β‐TRCP) 7. Interestingly, the β‐TRCP degradation complex also constitutes a critical checkpoint for the Wnt signaling pathway by degrading β‐catenin, suggesting a crosstalk between Wnt and YAP/TAZ signaling. This hypothesis is supported by the presence of YAP and β‐catenin in the β‐TRCP destruction complex and by the coordination of these pathways in response to Hippo inactivation 2, 8. Beside Hippo proteins, several other upstream regulators of YAP/TAZ have been identified 3. Notably, even if the mechanism and signaling intermediates are not yet elucidated, Rho‐associated protein kinase (ROCK)‐dependent actomyosin contraction driven by matrix stiffness induces robust YAP/TAZ activation, confirming the versatility of the YAP/TAZ pathway to be activated by different mechanical constraints.
Ubiquitin domain‐containing protein 1 (UBTD1) is an ubiquitin‐like protein. This protein is evolutionarily conserved among species and interacts with some E3 (ubiquitin ligase) and E2 (ubiquitin‐conjugating) enzymes of the ubiquitin–proteasome system (UPS) both in vitro and in vivo 9. Even if its biological functions remain largely unknown, UBTD1 expression is regulated by P53 and induces cellular senescence by stabilizing P53 through degradation of murine double minute 2 (MDM2). UBTD1 is down‐regulated in gastric cancer cells and tissues, and its expression correlates with patient survival suggesting that it could act as a tumor suppressor 10. Moreover, UBTD1 is highly expressed in cells/tissues that are subjected to constant or intermittent mechanical constraints, such as cardiac or smooth muscle cells and basal cells of the epithelium (skin, intestine, and lung) or glands (prostate and thyroid) 11, 12. Considering the specific expression profile of UBTD1 in cells experiencing mechanical stress and its potential role in cancer progression, we sought to elucidate the biological function of UBTD1 in mechano‐sensing and cancer.
Results
UBTD1 regulates traction forces and is controlled by mechanical cues
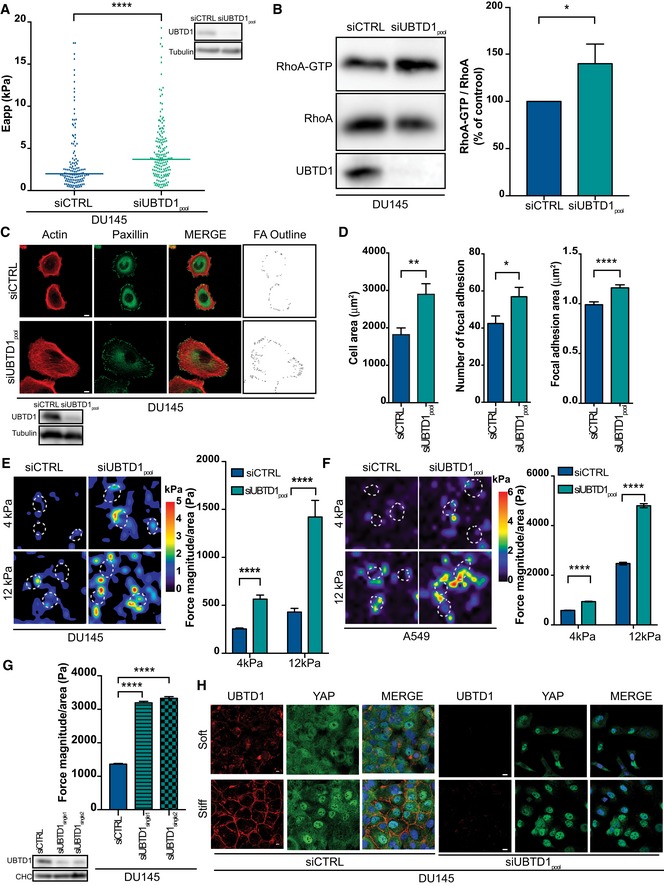
Within epithelium, the basal cell layer is constantly challenged by physical forces and cancer frequently arises from these cells. To gain insight into the role of UBTD1 in cell mechanical properties, we studied two representative basal epithelial‐derived cancer cell lines expressing high levels of UBTD1: A549 (lung) and DU145 (prostate) 13. To test our hypothesis, we performed atomic force microscopy (AFM) to measure cell elasticity. In UBTD1‐depleted cells, we observed a significant increase in the elastic modulus showing a lower deformability of the cell (Fig 1A). Modifications of cell elasticity mainly reflect changes in the cellular cortex induced by cytoskeletal network rearrangements, especially actomyosin contractility 14, 15, 16. The small GTPase, RhoA, is an important mediator of actomyosin dynamics; thus, we performed a RhoA‐GTP pull‐down assay to monitor RhoA activity. In UBTD1‐depleted cells, we observed a robust activation of RhoA, confirming the effect of UBTD1 on cytoskeleton‐driven cell mechanical properties (Fig 1B). Additionally, we analyzed cell adhesion parameters in A549 and DU145 cell lines (Figs 1C and EV1A). In cells that were UBTD1‐depleted and plated on fibronectin, we observed a striking increase in the cell area together with an increase in the number and area of focal adhesions (Figs 1D and EV1B). Subsequently, we investigated the role of UBTD1 on the cell traction forces applied by cells on the extracellular matrix (ECM). Thus, sub‐confluent DU145 and A549 cells depleted for UBTD1 were seeded on ECMs of two different stiffnesses (4 and 12 kPa) and substrate deformation was measured by traction force microscopy (Figs 1E and F, and EV1C). Traction forces were increased in isolated UBTD1‐depleted cells, both at low and high matrix rigidity. Similarly, in confluent cells, on a stiff matrix (12 kPa), UBTD1 depletion induced a threefold increase in force magnitude, demonstrating a role for UBTD1 in force generation independently of cell density (Figs 1G and EV1D). Higher substrate stiffness resulted in increased focal adhesion assembly, a process that is known to drive increased actomyosin contractility 17. Interestingly, the effect of UBTD1 depletion on traction forces was noticeably higher in stiff compared to soft ECM, confirming the active involvement of UBTD1 in the control of cell contractility (Fig 1E and F).
Figure 1. UBTD1 regulates cell mechanical properties via RhoA activation.
-
AThe scatter plot shows the apparent Young's moduli obtained by AFM analysis. Bars represent the median values of the distributions. The immunoblot shows UBTD1 siRNA knock‐down efficiency.
-
BImmunoblots (left) and quantification (right) show levels of GST‐ROCK‐associated GTP‐bound RhoA (RhoA‐GTP) and total RhoA in lysates (RhoA) in confluent cells. Immunoblot of UBTD1 shows the level of siRNA depletion.
-
CRepresentative confocal images of focal adhesion (paxillin) and actin cytoskeleton (phalloidin) and outlines of focal adhesions using threshold‐based image. Immunoblot of UBTD1 shows the level of siRNA‐mediated depletion.
-
DCell area, number, and area of focal adhesion were quantified (ImageJ) using the analysis as shown (C).
-
E, FSub‐confluent DU145 (E) or A549 (F) cells were seeded on two matrices of different stiffnesses (4 and 12 kPa). Representative heat map (left) and quantification (right) showing contractile forces generated by cells plated on 4 kPa or 12 kPa hydrogel.
-
GConfluent DU145 cells were seeded on 12 kPa matrix. Representative heat map (left) and quantification (right) showing contractile forces generated by cells plated on 12 kPa hydrogel.
-
HRepresentative confocal images of YAP and UBTD1 localization on two matrices of different stiffness. DU145 cells were grown on collagen/acrylamide‐coated glass coverslips (soft) or directly on glass coverslips (stiff). Nuclei were stained with DAPI (blue) on the MERGE image.
Figure EV1. UBTD1 regulates cell mechanical properties via RhoA activation.

-
A–EA549 cells were transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool or siUBTD1single1 or single2) for 24h and plated on fibronectin‐coated glass coverslips. (A) Representative confocal images of focal adhesion (paxillin) and actin cytoskeleton (phalloidin) and outlines of focal adhesions using threshold‐based image. The immunoblot shows UBTD1 siRNA knock‐down efficiency. (B) Cell area, number, and area of focal adhesions were quantified (ImageJ) using the analysis as shown in (A). (C) Sub‐confluent DU145 or A549 cells were seeded on 12 kPa. Quantification showing contractile forces generated by cells plated on 12 kPa hydrogel. Substrate deformation was measured by traction force microscopy and analyzed. (D) Confluent A549 cells were seeded on 12 kPa. Quantification showing contractile forces generated by cells plated on 12 kPa hydrogel. Substrate deformation was measured by traction force microscopy and analyzed. (E) Representative confocal images of YAP and UBTD1 localization in two matrices of different stiffnesses. A549 cells were grown on collagen/acrylamide‐coated glass coverslips (soft) or directly on glass coverslips (stiff).
-
FRepresentative confocal images of YAP and UBTD1 localization in two different cell densities of DU145 and A549 cells.
-
GRepresentative confocal images of UBTD1 localization at steady state or after 30 min of uniaxial stretch in confluent A549 cells.
-
HRepresentative confocal images of YAP and UBTD1 localization depending on the actin cytoskeleton using cytochalasin D (Cyto D) treatment in confluent DU145 and A549 cells. YAP is used as a control.
-
IRepresentative confocal images of UBTD1 localization depending on the RhoA pathway using ROCK inhibitor (Y‐27632) in confluent DU145 cells.
Matrix compliance modifies cell force generation and is a well‐defined experimental model to monitor cell mechano‐responses 18, 19. To evaluate the effect of ECM stiffness on UBTD1 subcellular localization, DU145 and A549 cells were grown on collagen‐coated acrylamide hydrogels with varying levels of rigidity. As shown in Figs 1H and EV1E and extensively described elsewhere 19, YAP plays a central role in mechano‐sensing and localizes to the nucleus on stiff ECM but becomes predominantly cytoplasmic on softer ECM. Under the same experimental conditions, UBTD1 accumulates at cell junctions on stiff ECM whereas it remains mostly diffuse in the cytoplasm on soft ECM, indicating that UBTD1 localization is drastically modified by the mechanical forces exerted by the ECM (Figs 1H and EV1E). Likewise, increased cell density or mechanical stretching performed on soft supports had a similar effect on UBTD1 re‐localization near the membrane (Fig EV1F and G). Furthermore, inhibition of actomyosin contraction by a ROCK inhibitor (Y‐27632), or actin depolymerization using cytochalasin D (Cyto D), drastically changed the UBTD1 subcellular distribution by promoting its delocalization from the membrane into the cytosol (Fig EV1H and I). Hence, UBTD1 localization requires an intact actin network and is dependent on actomyosin contractility, demonstrating that UBTD1 is functionally linked to the cellular contractile machinery. Moreover, we also found that UBTD1 is involved in the regulation of the mechanical properties of the cell and activates RhoA.
UBTD1 is associated with β‐catenin at cell–cell adhesion sites
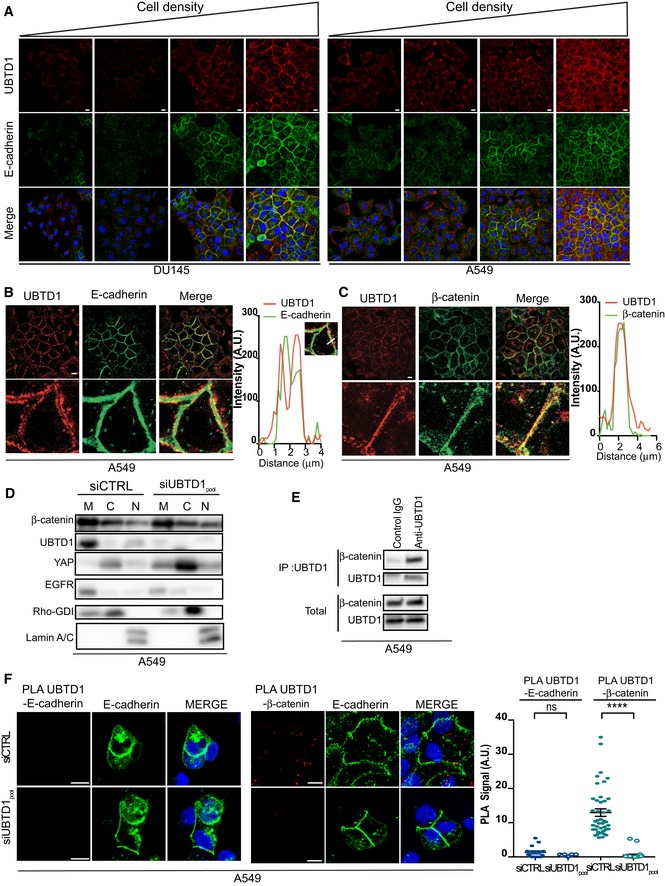
The role of UBTD1 in cell mechanical forces and its ECM stiffness‐dependent accumulation at cell junctions led us to postulate that UBTD1 could be linked to cell adhesion. The core components of the cell adhesion complex include β‐catenin, which associates with the cytoplasmic tail of E‐cadherin and functionally links E‐cadherin with the actin cytoskeleton through α‐catenin 20. We therefore monitored UBTD1 levels across increasing cell densities in A549 and DU145 cultures (Figs 2A and EV2A). As expected, markers for the cell adhesion complex (E‐cadherin and β‐catenin) increased proportionally with cell density, i.e., with the number of cell contacts 20, 21. Similarly, the expression of UBTD1 increased with cell density suggesting a close relationship between adhesion complex formation and UBTD1 expression or stability (Figs 2A and EV2A). Co‐localization experiments revealed that, in confluent cells, UBTD1 localized close to the cell membrane, in the vicinity of cell–cell contact sites (Figs 2B and C, and EV2B and C). Accurate image analysis demonstrated that UBTD1 is juxtaposed with E‐cadherin, suggesting that UBTD1 is associated with the adhesion complex but not with E‐cadherin per se, as confirmed by proximity ligation assay (PLA) experiments (Figs 2D and EV2D). As shown in Figs 2C and EV2C, UBTD1 consistently co‐localized with β‐catenin suggesting an association at the adhesion site. To challenge this hypothesis, we performed cellular fractionation experiments in confluent cells (Figs 2E and EV2E). In accordance with previous data, both β‐catenin and UBTD1 were highly enriched in the membrane fraction. Additionally, UBTD1 depletion did not modify either β‐catenin levels or cellular distribution. We then performed co‐immunoprecipitation experiments and showed that, in both DU145 and A549 cell lines, UBTD1 is associated with β‐catenin (Figs 2F and EV2F). The close proximity of UBTD1 and β‐catenin was further validated by PLA in vivo (Figs 2D and EV2D). PLA fluorescent signals between UBTD1 and β‐catenin were observed in both the cytoplasm and closed to the membrane as reflected by E‐cadherin staining. Specificity of the PLA association was further confirmed by UBTD1 knock‐down. We subsequently performed a “calcium switch assay” to cause rapid disassembly of the adhesion complex as monitored by E‐cadherin staining (Appendix Fig S1A). Upon calcium chelation with EGTA, we observed that UBTD1 was displaced from the cell membrane to the cytoplasm and that the addition of calcium (recovery; Rec) restored the localization of UBTD1 at the cell membrane (Fig 2G and Appendix Fig S1B). Concordantly, β‐catenin moves back to the membrane when the adhesion complexes are re‐assembled. Of note, during the recovery period, the return of UBTD1 to the cell membrane exhibited the same kinetics as β‐catenin, reinforcing our initial hypothesis that UBTD1 could be associated with cell–cell adhesion. We next performed a more physiological assay to displace the adhesion complex by treating cells with hepatocyte growth factor (HGF), a growth factor that is well known to induce the dispersion of clustered cells into single cells (scattering) in various epithelial cell types, including prostate cancer cells 22. In HGF or Cyto D‐treated cells, UBTD1 was no longer localized at the cell membrane, but displayed a cytoplasmic distribution instead (Fig 2H and Appendix Fig S1C and D). In concordance with these findings, β‐catenin also re‐localized from cell–cell adhesion junctions to the cytoplasm. Additionally, we showed that, on a soft–stiff matrix or in sparse‐confluent cell culture conditions, UBTD1 depletion did not modify β‐catenin localization (Appendix Fig S1E and F). These experiments clearly demonstrate that UBTD1 is dynamically recruited to cell–cell adhesion sites and is found in the adhesion complex, where it associates with β‐catenin.
Figure 2. UBTD1 is associated with β‐catenin at the cell–cell adhesion site.

-
AImmunoblot of DU145 and A549 cells at various cell densities ranging from 20 to 100% confluence showing the increased expression of UBTD1 and β‐catenin with cell confluency. E‐cadherin was used as a positive control. Tubulin was used as a loading control.
-
B, CLeft, immunofluorescent labeling of UBTD1 and E‐cadherin (B) or β‐catenin (C) at the cell–cell contact in confluent DU145 cells and analyzed by confocal microscopy. Right, representative histogram of a co‐localization profile prepared with RGB profiler plugin of ImageJ.
-
DProximal ligation assay monitoring (left) and quantification (right) of UBTD1 association with E‐cadherin or β‐catenin in DU145 cells transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). Nuclei were stained with DAPI (blue) on the MERGE image. n ≥ 3 independent experiments.
-
EImmunoblots of UBTD1, β‐catenin, and YAP after subcellular fractionation of confluent DU145 cells transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). EGFR, RhoGDI, and Lamin A/C were used as controls for the membrane (M), cytosol (C), and nucleus (N) compartments, respectively.
-
FCo‐immunoprecipitation between endogenous β‐catenin and UBTD1 was performed in confluent DU145 cells. IgG isotype was used as a negative control.
-
GRepresentative confocal images of β‐catenin and UBTD1 in DU145 under resting (Ctrl), after calcium chelator treatment (EGTA) and recovery (Rec) conditions. Nuclei were stained with DAPI (blue) on the MERGE image.
-
HRepresentative confocal images of β‐catenin and UBTD1 in DU145 after cell scattering induced by HGF treatment (24 h). Nuclei were stained with DAPI (blue) on the MERGE image.
Figure EV2. UBTD1 is associated with β‐catenin at cell–cell adhesion sites.
- A
-
B, CLeft, immunofluorescent labeling of UBTD1 and E‐cadherin (B) or β‐catenin (C) at cell–cell contacts in A549 cells and analyzed by confocal microscopy. Right, representative histogram of co‐localization profile prepared with RGB profiler plugin of ImageJ.
-
DImmunoblots of UBTD1, β‐catenin, and YAP after subcellular fractionation of confluent A549 cells transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). EGFR, RhoGDI, and Lamin A/C were used as controls for the membrane (M), cytosol (C), and nucleus (N) compartments, respectively.
-
ECo‐immunoprecipitation between endogenous β‐catenin and UBTD1 was performed in confluent A549 cells. IgG isotype was used as a negative control. (D) Proximal ligation assay monitoring (left) and quantification (right) of UBTD1 interaction with E‐cadherin or β‐catenin in A549 cells.
-
FProximal ligation assay monitoring (left) and quantification (right) of UBTD1 association with E‐cadherin or β‐catenin in A549 cells transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool).
UBTD1 promotes YAP degradation by the UPS independently of the Hippo signaling pathway
Yes‐associated protein signaling is a central node in mechano‐sensing, and the Hippo pathway is an important regulator of YAP signaling in the detection of cell–cell contacts 2, 23. Upon activation by cell–cell contact, the Hippo kinases LATS1/2 and MST1/2 are activated and phosphorylate YAP (S127), which inhibits its nuclear translocation and subsequent transcriptional program. Our findings that UBTD1 modifies cellular mechanical properties and localizes at the adhesion complex in confluent cells led us to hypothesize that UBTD1 could modify the YAP signaling pathway. As expected, upon increased cell density, YAP phosphorylation (S127) (p‐YAP) by the Hippo signaling cascade increased drastically (Figs 3A and EV2A). In UBTD1‐depleted cells, we noticed that the YAP level increased significantly (x2) either in low or in high cell density, resulting in a sharp decrease (25%) in the level of YAP phosphorylation (p‐(S127)‐YAP/total YAP ratio) (Figs 3B and EV3A). Concordantly, the Hippo signaling cascade, monitored by LATS1/2 and MST1/2 phosphorylation, was not inhibited in confluent UBTD1‐depleted cells, suggesting that UBTD1 affects YAP abundance independently of the canonical Hippo pathway (Figs 3C and EV3B). This result prompted us to consider how UBTD1 could mechanistically act on the YAP protein level. UBTD1 is structurally considered to be a ubiquitin‐like protein, which interacts stoichiometrically with the UbcH5c ubiquitin‐conjugating enzyme, both in vitro and in vivo 9. The YAP ubiquitylation complex encompasses UbcH5c, β‐TrCP, and β‐catenin 7, 8. It is thus tempting to speculate that UBTD1 participates in YAP degradation via its ubiquitylation complex. To explore this hypothesis, we first looked at the effect of UBTD1 depletion on YAP protein stability in confluent cells (Fig 3D and E). In control cells, treatment with a global translation inhibitor (cycloheximide, CHX) induced a rapid decrease in YAP protein levels, whereas in UBTD1‐depleted cells, the YAP level remained broadly unchanged. At low cell density, YAP protein levels were stable and UBTD1 depletion modified YAP stability (Appendix Fig S2A). To verify that UBTD1 is involved in the proteasome‐dependent degradation of YAP, confluent cells were treated with the proteasome inhibitor (MG132) for 6 h. As depicted in Fig 3F, inhibition of proteasomal degradation induces an increase in YAP protein levels in control cells. However, in UBTD1‐depleted cells, we did not observe a YAP accumulation upon MG132 treatment, demonstrating that the proteasome‐dependent degradation of YAP was already impaired by UBTD1 depletion.
Figure 3. UBTD1 promotes YAP degradation by the UPS independently of the Hippo signaling pathway.

-
AImmunoblot of DU145 and A549 cells at various cell densities ranging from 20 to 100% confluence showing the increased expression of UBTD1, p‐YAP (ser127), and YAP levels with the cell confluency. Tubulin was used as a loading control.
-
B–EDU145 cells or (F) A549 cells were transfected for 48h with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). (B) Immunoblots of p‐YAP (ser127), YAP, and UBTD1 (left) and quantification of YAP and p‐YAP/YAP levels (right) in control or UBTD1‐depleted DU145 at low and high cell density. Immunoblot of UBTD1 shows the level of siRNA‐mediated depletion. Actin was used as a loading control. (C) Immunoblots of the Hippo signaling pathway using phosphorylated (p‐ser909LATS and p‐thr183MST1/p‐thr180MST2) and total LATS, and MST at low and high DU145 cells density. Immunoblot of UBTD1 shows the level of siRNA‐mediated depletion. Actin and RhoGDI were used as loading controls. In (B) and (C), immunoblots for the loading control (actin) and UBTD1 knock‐down efficiency (UBTD1) are duplicated because they belong to the same experiment shown in both panels. (D, E) Immunoblots (up) and quantification (down) of YAP levels in confluent (D) DU145 or (E) A549 UBTD1‐depleted cells treated or not with cycloheximide (CHX) for 2 h. Immunoblot of UBTD1 shows the level of siRNA‐mediated depletion. Actin was used as a loading control.
-
FImmunoblots showing YAP levels in UBTD1‐depleted confluent DU145 (left) or A549 (right) cells treated or not with MG132 for 6h. Immunoblot of UBTD1 shows the level of siRNA‐mediated depletion. Actin was used as a loading control. Numbers at the bottom of the Western blots represent the ratio between 6h treatment/0h for each condition.
Figure EV3. UBTD1 promotes YAP degradation by the UPS by modulating its association with β‐TrCP .

-
A, B(A) Immunoblots of p‐YAP (ser127), YAP, and UBTD1 (left) and quantification of YAP and p‐YAP/YAP levels (right) in control or UBTD1‐depleted A549 at low and high cell density. Immunoblot of UBTD1 shows the level of siRNA depletion. Actin was used as a loading control. (B) Immunoblots of the Hippo signaling pathway using phosphorylated (p‐ser909LATS and p‐thr183MST1/p‐thr180MST2) and total LATS, and MST at low and high A549 cells density. Immunoblot of UBTD1 shows the level of siRNA depletion. Actin and RhoGDI were used as loading controls. In (A and B), immunoblots for the loading control (actin) and UBTD1 knock‐down efficiency (UBTD1) are duplicated because they belong to the same experiment shown in both panels.
-
CCo‐immunoprecipitation in confluent A549 cells between endogenous β‐TRCP, UbcH5c, and UBTD1. UBTD1 was used as bait. The IgG isotype was used as a negative control.
-
DProximal ligation assay monitoring and quantification of β‐TrCP associated with UbcH5, YAP, or β‐catenin of A549 cells transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). Nuclei were stained with DAPI (blue) on the MERGE image. Scale bar = 10 μm.
UBTD1 regulates YAP ubiquitylation by modulating its association with β‐TrCP
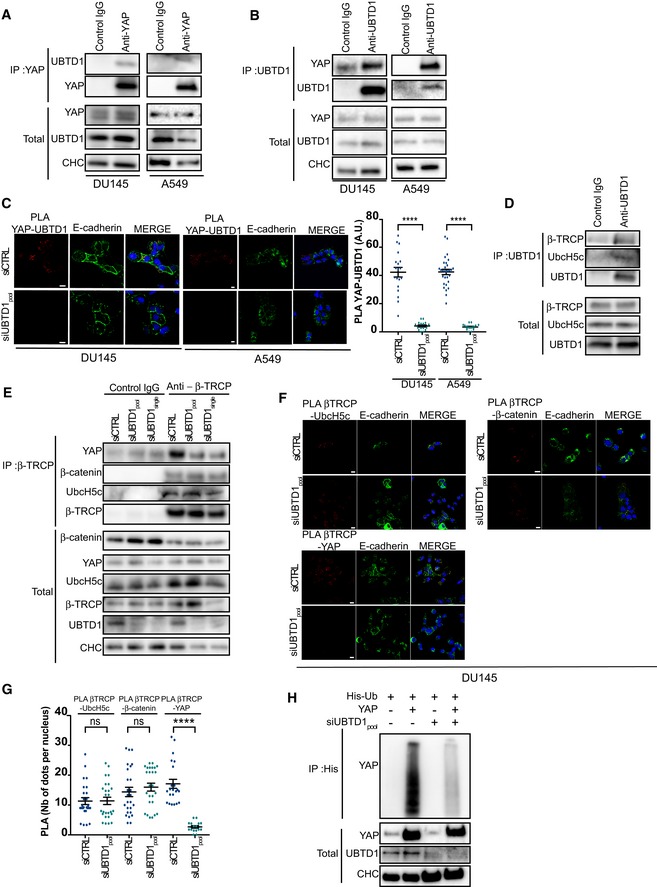
To decipher more precisely how UBTD1 acts on YAP degradation, we first demonstrated, by co‐immunoprecipitation experiments, that UBTD1 is associated with YAP (Fig 4A and B). We confirmed this association by performing a PLA experiment between UBTD1 and YAP in both DU145 and A549 cells (Fig 4C). Hence, in control cells, we observed PLA association dots, which were almost completely lost in UBTD1‐depleted cells. Furthermore, the PLA experiments showed that interactions between YAP and UBTD1 occurred mainly in the cytoplasm, in agreement with previous studies demonstrating that YAP degradation takes place in the cytoplasmic compartment 7. Unexpectedly, we observed less PLA signals when UBTD1 was localized at the cell–cell contact sites instead of the cytoplasm. Furthermore, we were able to further confirm that the YAP‐UBTD1 association relied on the actin cytoskeleton network since actin depolymerization induced by Cyto D treatment completely abolished this association (Appendix Fig S2B).
Figure 4. UBTD1 regulates YAP ubiquitylation by modulating its association with β‐TrCP .
-
ACo‐immunoprecipitation in DU145 and A549 cells between endogenous YAP and UBTD1. YAP was used as bait. The IgG isotype was used as a negative control.
-
BCo‐immunoprecipitation in DU145 and A549 cells between endogenous YAP and UBTD1. UBTD1 was used as bait. The IgG isotype was used as a negative control.
-
CProximal ligation assay monitoring (left) and quantification (right) of UBTD1 associated with YAP of DU145 and A549 cells transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). Nuclei were stained with DAPI (blue) on the MERGE image.
-
DCo‐immunoprecipitation in confluent DU145 cells between endogenous β‐TrCP, UbcH5c, and UBTD1. UBTD1 was used as bait. IgG isotype was used as a negative control.
-
ECo‐immunoprecipitation in confluent DU145 cells between endogenous β‐TrCP, UbcH5c, YAP, and β‐catenin. DU145 cells were transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool or siUBTD1single). β‐TrCP was used as bait. IgG isotype was used as a negative control. Immunoblot of UBTD1 shows the level of siRNA‐mediated depletion.
-
F, GProximal ligation assay monitoring (F) and quantification (G) of β‐TrCP associated with UbcH5, YAP, or β‐catenin in DU145 cells transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). Nuclei were stained with DAPI (blue) on the MERGE image.
-
HImmunoblots show YAP ubiquitylation in HEK cells in different experimental conditions. Cells were transfected, as indicated, with expression vectors of histidine‐tagged ubiquitin (His‐Ub) together with control siRNA or UBTD1 siRNA. His‐Ub crosslinked forms of YAP were purified (IP: His), and the immunoblot of YAP showed YAP ubiquitylation. The immunoblot of YAP (lower panel) was performed in parallel to verify the amounts of YAP protein engaged in the His‐Ub purifications. The immunoblot of UBTD1 shows the level of siRNA depletion.
We next performed co‐immunoprecipitation experiments between UBTD1 and components of the YAP degradation complex, UbcH5c and β‐TrCP (Figs 4D and EV3C). In A549 and DU145 cell lines, UBTD1 was associated with both UbcH5c and β‐TrCP, the E3 ligase that is responsible for targeting YAP for degradation 7, 8. In addition, we showed that UBTD1 is not associated with ubiquitin (Appendix Fig S2C), demonstrating that UBTD1 is not implicated in ubiquitin transfer from UbcH5c to YAP, but more likely in the formation or in the functionality of the ubiquitylation complex 24. To define the role of UBTD1 in the YAP degradation complex, we then immunoprecipitated β‐TrCP (Fig 4E). UBTD1 depletion did not alter the level of the core components (UbcH5c, β‐TrCP, and β‐catenin) of the degradation complex. However, the co‐immunoprecipitation between YAP and β‐TrCP was drastically reduced in UBTD1‐depleted conditions, demonstrating that UBTD1 is important to promote the association between YAP and β‐TrCP. To confirm this result, we performed PLA experiments between components of the degradation complex (Figs 4F and G, and EV3D). UBTD1 depletion did not modify either UbcH5c‐β‐TrCP or β‐TrCP‐β‐catenin associations, as estimated by PLA dot number. Nevertheless, confirming the co‐immunoprecipitation result, we observed a sharp drop in the number of interacted dots between β‐TrCP and YAP. To functionally validate these data, we performed a YAP ubiquitylation assay (Fig 4H and Appendix Fig S2D). We observed that the YAP polyubiquitylation level was drastically decreased in UBTD1‐depleted cells, demonstrating that UBTD1 is required for the efficient ubiquitylation of YAP. Collectively, our results show that UBTD1 promotes association between YAP and β‐TrCP to induce YAP ubiquitylation.
UBTD1 depletion stimulates YAP signaling via ROCK2 and independently of Hippo signaling pathway
We next explored the effect of UBTD1 modulation on YAP signaling. Thus, we measured the YAP nuclear/cytoplasmic index and found that UBTD1 depletion promoted YAP nuclear accumulation (Figs 5A and EV4A). In line with these data, we observed that the mRNA levels of YAP target genes (BIRC5, CYR61, and CTGF1) increased by up to fourfold to sixfold, confirming that UBTD1 depletion resulted in an increase in both YAP accumulation in the nucleus and transcriptional activity (Figs 5B and EV4B). We next performed a rescue experiment by expressing a GFP‐fused UBTD1 recombinant protein (GFP‐UBTD1) in either control (siCTRL) or UBTD1‐depleted cells (Figs 5C and D, and EV4C and D). It should be noted that we performed these experiments by using a siRNA targeting a UBTD1 mRNA untranslated sequence (siUBTD1 single2). As expected, UBTD1 depletion increased YAP level, and transient over‐expression of GFP‐UBTD1 in UBTD1‐depleted cells significantly decreased YAP to the basal level (Figs 5C and EV4C). Notably, over‐expression of GFP‐UBTD1 in control cells (siCTRL) induced a slight decrease in YAP levels. Then, we quantified YAP nuclear translocation in the same experimental settings (Figs 5D and EV4D). GFP‐UBTD1 expression induces a slight decrease in the YAP nuclear content, whereas UBTD1 depletion increases YAP nuclear translocation. In accordance with our previous result, in UBTD1‐depleted cells, GFP‐UBTD1 expression decreases significantly YAP nuclear content. Hence, this set of experiments establishes that UBTD1 depletion is sufficient to induce a major upregulation of YAP signaling.
Figure 5. Loss of UBTD1 increases YAP nuclear translocation and its activity via ROCK2.

-
A, BDU145 cells were transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). (A) Representative wide‐field immunofluorescence images (left) and quantification (right) showing endogenous YAP nuclear translocation in DU145 cells after UBTD1 depletion. Nuclei were stained with DAPI (blue) on the MERGE image. (B) Quantification of CTGF1, CYR61, and BIRC5 mRNA levels.
-
CImmunoblots of YAP in DU145 cells depleted (siUBTD1single2) or not (siCTRL) for UBTD1 and transfected or not with GFP‐UBTD1 (+). Immunoblot of UBTD1 shows the level of siRNA‐mediated depletion. Immunoblot of GFP shows the level of UBTD1 transfection. CHC was used as a loading control.
-
DRepresentative confocal immunofluorescence images (left) and quantification (right) showing endogenous YAP nuclear translocation in DU145 cells depleted (siUBTD1single2) or not (siCTRL) for UBTD1 and transfected or not with GFP‐UBTD1. Nuclei were stained with DAPI (blue) on the MERGE image.
-
EImmunoblots of p‐YAP and YAP in DU145 control cells (siCTRL) or depleted for LATS1 and LATS2 (siLATS1 + siLATS2) and/or UBTD1 (siUBTD1pool). Immunoblot of p‐YAP reflects the activity of the Hippo pathway. Immunoblot of UBTD1 and LATS shows siRNA knock‐down efficiency. Actin was used as a loading control.
-
F, GRepresentative confocal immunofluorescence images (F) and quantification (G) showing endogenous YAP nuclear translocation in DU145 control cells (siCTRL) or depleted for LATS1 and LATS2 (siLATS1 + siLATS2) and/or UBTD1 (siUBTD1pool). Nuclei were stained with DAPI (blue) on the MERGE image.
-
HImmunoblots of YAP in DU145 control (siCTRL) or depleted for ROCK1 (siROCK1) or ROCK2 (siROCK2) and/or UBTD1 (siUBTD1pool). Immunoblots of UBTD1, ROCK1, and ROCK2 show the level of siRNA knock‐down efficiency. Actin was used as a loading control.
-
I, JRepresentative confocal immunofluorescence images (I) and quantification (J) showing endogenous YAP localization in DU145 control cells (siCTRL) or depleted for ROCK1 (siROCK1) or ROCK2 (siROCK2) and/or UBTD1 (siUBTD1pool). Nuclei were stained with DAPI (blue) on the MERGE image.
-
KQuantification of CTGF1, Cyr61, UBTD1, and BIRC5 mRNA in DU145 control cells (siCTRL) or depleted for ROCK1 (siROCK1) or ROCK2 (siROCK2) and/or UBTD1 (siUBTD1pool).
Figure EV4. Loss of UBTD1 increases YAP nuclear translocation and its activity via ROCK2.

-
A, BA549 cells were transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool). (A) Representative wide‐field immunofluorescence images (left) and quantification (right) showing endogenous YAP nuclear translocation in A549 cells after UBTD1 depletion. (B) Quantification of CTGF1, CYR61, and BIRC5 mRNA levels.
-
CImmunoblots of YAP in confluent A549 cells depleted (siUBTD1single2) or not (siCTRL) for UBTD1 and transfected or not with GFP‐UBTD1. Immunoblot of UBTD1 shows the level of siRNA depletion. Immunoblot of GFP shows the level of UBTD1 transfection. CHC was used as a loading control.
-
DRepresentative confocal immunofluorescence images (left) and quantification (right) showing endogenous YAP nuclear translocation in A549 cells depleted (siUBTD1single2) or not (siCTRL) for UBTD1 and transfected or not with GFP‐UBTD1.
-
EImmunoblots of YAP in confluent A549 control (siCTRL) or depleted for ROCK1 (siROCK1) or ROCK2 (siROCK2) and/or UBTD1 (siUBTD1pool). Immunoblots of UBTD1, ROCK1, and ROCK2 show the level of siRNA knock‐down efficiency. Actin was used as a loading control.
-
F, GRepresentative confocal immunofluorescence images (F) and quantification (G) showing endogenous YAP localization in A549 cells control (siCTRL) or depleted for ROCK1 (siROCK1) or ROCK2 (siROCK2) and/or UBTD1 (siUBTD1pool). For microscopy imaging panels (A, D, F), nuclei were stained with DAPI (blue) on the MERGE image.
-
HQuantification of CTGF1, CYR61, UBTD1, and BIRC5 mRNA in A549 control cells (siCTRL) or depleted for ROCK1 (siROCK1) or ROCK2 (siROCK2) and/or UBTD1 (siUBTD1pool).
We next determined whether YAP signaling induced by UBTD1 depletion was dependent on the Hippo pathway. Depletion of LATS1/2 alone or with siUBTD1 in confluent DU145 and A549 cells decreased YAP phosphorylation (siLATS1 + siLATS2), showing that the Hippo pathway is inhibited (Fig 5E and Appendix Fig S3A). In these conditions, we quantified the YAP nuclear/cytoplasmic ratio (Fig 5F and G, and Appendix Fig S3B, C). In Hippo‐inactivated confluent cells (siLATS1 + siLATS2), the level of nuclear YAP was slightly increased. In UBTD1‐depleted cells, this level was significantly higher than in Hippo‐inactivated cells. Interestingly, UBTD1 depletion in Hippo‐inactivated cells (siUBTD1pool + siLATS1 + siLATS2) caused a significant increase in YAP nuclear content suggesting an additive effect of UBTD1 depletion and Hippo inactivation on YAP nuclear translocation. Based on these data, we propose that UBTD1 acts on YAP localization mostly in a Hippo‐independent way.
Besides Hippo, other modulators of YAP/TAZ localization have been described 3, 25. Interestingly, YAP/TAZ activity is regulated by matrix stiffness relayed by tension of the F‐actin cytoskeleton and ROCK activity 19. As shown in Fig 1, UBTD1 depletion induces RhoA activation and robustly increases cell traction force magnitude on the matrix. Therefore, we speculate that UBTD1 acts on YAP signaling through a RhoA‐dependent pathway. Indeed, besides its central role in mechano‐sensing, the Rho/ROCK axis has been shown to control YAP nuclear shuttling 18, 19, 26, 27. To test our hypothesis, we separately knocked down the two ROCK isoforms, ROCK1 and ROCK2 (Figs 5H and EV4E) 27. Neither ROCK1 nor ROCK2 modified the increased YAP level induced by UBTD1 depletion. Using the same experimental conditions, we then measured YAP nuclear translocation (Figs 5I and J, and EV4F and G). ROCK2 depletion induced a sharp decrease in YAP nuclear translocation, whereas ROCK1 depletion did not modulate YAP localization. Remarkably, in UBTD1‐depleted cells, while ROCK1 knock‐down did not affect YAP nuclear content, ROCK2 drastically abrogated the increase in YAP translocation suggesting that UBTD1 acts on YAP localization in a ROCK2‐specific manner. Finally, we measured the mRNA levels of YAP target genes (Figs 5K and EV4H). ROCK2 depletion led to a significant decrease in YAP target genes expression, whereas ROCK1 depletion had no effect. Thus, the increase in YAP transcriptional activity induced by UBTD1 depletion is mediated by ROCK2. Collectively, these data demonstrate that UBTD1 depletion modulates YAP nuclear translocation and transcriptional activity via ROCK2.
UBTD1 represses cancer aggressiveness, and high expression of UBTD1 enhances cancer patient survival
We next investigated the pathophysiological relevance of our observations. It has been recently demonstrated that YAP/TAZ signaling could drive epithelial‐to‐mesenchymal transition (EMT) 28, 29, 30. EMT is a key step in cancer dissemination and metastasis 31, by allowing a normal polarized epithelial cell, which normally interacts with the basement membrane, to undergo multiple events that confer a mesenchymal cell phenotype, including enhanced migratory capacity, invasiveness, and elevated resistance to apoptosis 31. Thus, we analyzed some EMT markers at both the transcriptional and protein level in UBTD1‐depleted cells (Figs 6A and B, and EV5A–C). We observed a significant increase in the mRNA levels of EMT‐related transcription factors (snail and slug) and mesenchymal markers, such as vimentin, fibronectin, or α‐SMA associated with a drop in E‐cadherin (Figs 6A and EV5A). All these effects are YAP/TAZ‐dependent since YAP/TAZ depletion abrogates this transcriptional response in both A549 and DU145 cell lines. Likewise, the EMT transcriptional signature was confirmed at the protein level (Figs 6B and EV5B and C). To confirm the involvement of YAP/TAZ signaling in this EMT signature, we knocked down YAP and TAZ in UBTD1‐depleted cells and analyzed the expression of EMT markers: E‐cadherin and vimentin (Fig EV5C). In addition, we also confirmed in both cell lines that the increase in YAP target gene expression (CYR61, BIRC5, and CTGF1) induced by UBTD1 depletion was specific to YAP/TAZ signaling (Appendix Fig S4A and B). We next monitored the invasive and migratory properties of both A549 and DU145 cancer cells. UBTD1 depletion promoted both migration (×2.5) and invasion (×2), indicating that UBTD1 inhibits cancer cell aggressiveness in vitro (Figs 6C and D, and EV5D and E). Additionally, we showed that the acquisition of these migratory and invasive properties is under the control YAP/TAZ since knocking down YAP/TAZ fully reverses the UBTD1 depletion phenotype. To reinforce these data, we took advantage of human organoid technology to evaluate the effect of UBTD1 on 3D cancer cell invasion generated from human prostate tumor biopsies 32, 33, 34. As observed in Fig 6E, UBTD1 depletion greatly increased the 3D invasiveness of human tumoroids and increased the expression of AMACR, a molecular marker of prostate carcinoma (Appendix Fig S4C). Collectively, our data suggest that UBTD1 controls via YAP/TAZ inhibition, the migration, and invasion properties of cancer cells that are hallmarks of the metastatic phenotype. More importantly, in a 3D invasion assay of human prostate tumoroids, UBTD1 depletion greatly enhanced invasion, suggesting that UBTD1 acts as a tumor suppressor. Based on these observations, we investigated whether the level of UBTD1 expression could impact cancer progression and survival in patients. Interestingly, we observed that UBTD1 mRNA levels were significantly decreased in prostate tumoral tissues compared to adjacent normal tissues (Fig 6F). Concordantly, immunohistochemistry analysis of 8 prostate tumor samples showed a complete disappearance of UBTD1 staining corresponding to the basal cell layer and a marked decrease in cancerous cells compared to normal luminal cells (Fig 6G). Immunohistochemistry analysis of 6 lung tumor samples showed that UBTD1 staining was very high in the non‐tumoral pulmonary alveolar wall thickening, whereas in lung adenocarcinoma, UBTD1 expression was restricted to the residual pulmonary alveolar wall and totally negative in tumoral cells (Fig 6H). We next used a bioinformatics analysis on patient sample databases to extend our results to a larger cohort of prostate cancer and lung adenocarcinoma patients. Based on prostate cancer patient survival and disease progression, we observed that high UBTD1 expression was associated with slower disease progression and better patient survival in prostate cancer (grade 3), but, because of the limited size of the sample, this association remains below statistical significance (Figs 6I and EV5F). Moreover, in a larger lung carcinoma cohort, UBTD1 expression was highly correlated with both disease progression (P = 0.0011) and patient survival (P = 0.0011; Figs 6J and EV5G) and absent in lung adenocarcinoma metastasis (Fig EV5H). Finally, in both lung and prostate patient cohorts, the expression of UBTD1 was inversely correlated with the mRNA level of Rho‐associated kinase (Rho‐kinase/ROCK/ROK), an effector of RhoA implicated in tumor promotion 35, and BIRC5, a YAP target gene, suggesting an important role for UBTD1 in the control of cell mechanical properties and cancer progression (Fig EV5I and J).
Figure 6. Loss of UBTD1 is associated with increased prostate cancer cell aggressiveness and poor prognosis for patients.

-
A–EDU145 cells were transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool or siUBTD1single1; UBTD1 + YAP + TAZ, siUBTD1single1 + siYAPsingle1 + siTAZsingle1 or siUBTD1single1 + siYAPsingle2 + siTAZsingle2). (A) Quantification of EMT marker genes (vimentin, slug, snail, fibronectin, α‐SMA, and E‐cadherin) and UBTD1. (B) Immunoblots of EMT markers (vimentin, E‐cadherin, and slug). Immunoblot of UBTD1 shows siRNA knock‐down efficiency. Hsp90 and tubulin were used as loading controls. (C) Quantification of DU145 cell migration in Transwell chamber inserts. (D) Quantification of DU145 cell invasion in Transwell chamber inserts. (E) Invasive properties of human prostate tumoroids depleted for UBTD1.
-
FQuantification of UBTD1 mRNA levels in prostate tumor (n = 70) and adjacent normal tissues (n = 47).
-
GRepresentative UBTD1 immunohistochemistry staining on sections of human prostate tumor and normal tissues (n = 8).
-
HRepresentative UBTD1 immunohistochemistry staining on sections of human lung tumor and normal tissues (n = 6).
-
IThe levels of UBTD1 mRNA in grade 3 prostate adenocarcinoma patients (56 patients) are correlated with overall survival (OS). OS was calculated from patient subgroups with mRNA levels that were less or greater than the first quartile value.
-
JThe levels of UBTD1 mRNA in lung adenocarcinoma patients (522 patients) are correlated with OS.
Figure EV5. Loss of UBTD1 is associated with increased lung cancer cell aggressiveness and poor prognosis for patients.

-
A–EA549 or DU145 cells were transfected with the indicated siRNA (control, siCTRL; UBTD1, siUBTD1pool or siUBTD1single1; UBTD1 + YAP + TAZ, siUBTD1single1 + siYAPsingle1 + siTAZsingle1 or siUBTD1single1 + siYAPsingle2 + siTAZsingle2). (A) Quantification of EMT marker genes (vimentin, slug, snail, fibronectin, α‐SMA, and E‐cadherin) and UBTD1. (B, C) Immunoblots of EMT markers (vimentin and E‐cadherin). Immunoblots of UBTD1 and YAP show siRNA knock‐down efficiency. Hsp90 and tubulin were used as loading controls. (D) Quantification of A549 cell migration in Transwell chamber inserts. (E) Quantification of A549 cell invasion in Transwell chamber inserts.
-
FThe levels of UBTD1 mRNA in grade 3 prostate adenocarcinoma patients (56 patients) are correlated with disease‐free survival (DFS). DFS was calculated from patient subgroups with mRNA levels that were less or greater than the first quartile value.
-
GThe levels of UBTD1 mRNA in lung adenocarcinoma patients (522 patients) are correlated with DFS. DFS was calculated from patient subgroups with mRNA levels that were less or greater than the median value.
-
HUBTD1 immunohistochemistry staining on a section of metastasized human lung adenocarcinoma. Scale bar = 100 μm.
-
I, JmRNA levels of BIRC5 (I) and ROCK2 (J) are stratified in two groups expressing low or high levels of UBTD1 from prostate or lung adenocarcinoma patients.
Discussion
Cell contractility represents an important issue for numerous biological or pathophysiological processes 15. In cancer cells, it is necessary, inter alia, for cell rear retraction during mesenchymal migration or cell adhesion. We here identified UBTD1, a previously uncharacterized ubiquitin‐like protein, as an important player controlling some mechanical properties of the cell. Indeed, UBTD1 depletion drastically modified traction force magnitudes and activated the RhoA pathway in cancer cells. This phenotypic alteration probably reflects some major modification in the actomyosin contractile machinery. The actin network could be radically rearranged by the classical RhoA/ROCK pathway through actin polymerization and actomyosin contraction 27. Therefore, UBTD1 can sequentially activate RhoA to modify actomyosin contractility. Another explanation would be that UBTD1 acts on the regulatory proteins of the contractile system. The RhoA/ROCK signaling pathway induces non‐muscle myosin contraction via activation of myosin light chain kinase (MLCK) and a concomitant inhibition of myosin light chain phosphatase (MLCP) 15. Therefore, we could not exclude that UBTD1 modifies the MLCK/MLCP balance downstream of RhoA/ROCK to straightforwardly change cell contractility and, thereby, activate RhoA. Considering the central role of cell contractility in several aspects of cancer progression, this point needs further investigation. In addition, we showed that UBTD1 localization could be modified by mechanical forces including ECM stiffness suggesting a complex interplay between UBTD1 and the RhoA/ROCK pathway in the control of force generation by the cell.
Crucially, we noticed that UBTD1 protein expression was regulated by cell density in our epithelial cancer cell models. Moreover, its localization was not restricted to the cytoplasm, but, in confluent cell culture, UBTD1 partially re‐localized closed to the cell membrane, suggesting a possible involvement in the cell adhesion complex. We thus tested whether UBTD1 participates in the E‐cadherin/catenin system. Based on our data, we demonstrated that UBTD1 is associated with β‐catenin in the adhesion complex. Since the initial discovery of the YAP/TAZ signaling pathway as a key controller of organ size in Drosophila, mechano‐signaling is under intense scientific debate and represents a rapidly growing field in many aspects of biology 36. In cancer biology, the crucial role of this pathway has been widely emphasized at many stages of cancer progression, e.g., proliferation, migration, and invasion or stroma remodeling, and deregulation of YAP/TAZ signaling is oncogenic in humans 3. Historically, the Hippo pathway was considered as the only controller of YAP/TAZ. However, the YAP/TAZ pathway has been reported to be much more versatile upon mechanical stimulation than initially thought. Indeed, this pathway can be activated by various mechanical stresses but also through metabolic cues 37. As described by S. Piccolo's group, YAP/TAZ can be activated by cytoskeleton remodeling and ROCK signaling in response to matrix rigidity 19. This aspect is particularly important for cancer progression, since ECM stiffness becomes more rigid during tumorigenesis 38, 39. Moreover, the Rho/ROCK axis is considered as an important signaling platform for cancer progression. Based on the role of UBTD1 in cell contractility and its association with β‐catenin at cell adhesion sites, we logically wondered whether some phenotypic features induced by UBTD1 were linked to the Hippo pathway and more precisely to YAP/TAZ signaling. To address this issue, we first showed that UBTD1 depletion induces a robust activation of the YAP/TAZ signaling mostly independently of the canonical Hippo pathway. Consistent with our data showing that UBTD1 depletion increases cell contractility and activates RhoA, we demonstrated that YAP signaling was ROCK2‐dependent. These data illustrate the initial work of S. Piccolo's group and should be seen in light of a recently published article demonstrating that the RhoA/ROCK2 pathway triggers YAP nuclear shuttling to coordinate focal adhesion assembly 19, 26. Concomitantly, we noticed that the level of YAP was strikingly higher upon UBTD1 depletion suggesting a possible role of UBTD1 in YAP protein stability. It has been previously described that following phosphorylation, YAP could be retained in the cytoplasm by 14.3.3 and/or degraded by the UPS 6, 7. The YAP degradation complex includes the E3 ligase β‐TRCP, the E2 ubiquitin‐conjugating enzyme UbcH5, and β‐catenin 8. We previously described a stoichiometric interaction between UBTD1 and UbcH5, and others described some interactions with some E3 ligases suggesting a role of UBTD1 in the UPS 9, 40, 41. Our results provide converging evidence indicating that UBTD1 is associated with the YAP degradation complex and participates in its ubiquitylation by β‐TRCP. Contrary to what has been described for MDM2, it seems that UBTD1 acts mainly on β‐TRCP functionality rather than E3 ligase stability 10. Thus, we can extrapolate that MDM2 and β‐TRCP are probably not the only E3 ligases regulated by UBTD1. However, the way that UBTD1 controls protein degradation by the UPS is certainly complex because we have not been able to demonstrate that UBTD1 acts similarly to YAP for β‐catenin in our cell models. Clearly, further work is required to decipher the role of UBTD1 in the UPS.
Importantly, we also demonstrated that the level of UBTD1 is decreased in human cancer tissues and is inversely correlated with the expression of both ROCK2 and the YAP target gene, BIRC5. Hence, UBTD1 expression is highly correlated with both disease progression and patient survival in lung carcinoma. This observation is corroborated by in cellulo experiments demonstrating that UBTD1 depletion controls a YAP‐dependent EMT program which dramatically increases invasion and migration of cancer cells.
Collectively, we here characterized UBTD1 as a new mechano‐regulator linked to the cellular contractile machinery (Fig 7). At the same time, and probably by an intricate manner involving ROCK2, UBTD1 controls YAP signaling, the major cell mechano‐transducer. Finally, UBTD1 also participates in YAP degradation by modulating its ubiquitylation, emphasizing a possible tumor suppressor function.
Figure 7. A proposed model for UBTD1 controlling cancer cell aggressiveness through YAP signaling and ubiquitylation.
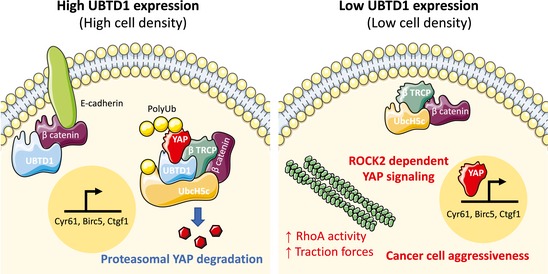
At high cell density, UBTD1 is highly expressed and is associated with β‐catenin at the cell–cell adhesion complex. Cytoplasmic UBTD1 is associated with the YAP degradation complex composed of UbcH5, β‐TrCP, and β‐catenin and promotes YAP ubiquitylation for proteasomal degradation (Left). At low cell density, or during cancer progression, UBTD1 is weakly expressed. Low UBTD1 expression induces RhoA activation, increases cell traction forces, and activates YAP signaling via ROCK2 fostering cancer cell aggressiveness (Right).
Materials and Methods
Reagents and antibodies
Hepatocyte growth factor (40 ng/ml) was purchased from PeproTech (100‐39H). Cytochalasin D (10 μM), EGTA (4 mM), anti‐UBTD1 (HPA034825), and anti‐α‐tubulin (T6199) were purchased from Sigma‐Aldrich. Anti‐YAP (sc‐101199), anti‐HSP90 (sc‐13119), anti‐RhoGDI (sc‐360), anti‐RhoA (sc‐418), anti‐Ubiquitin (sc‐8017), anti‐Lamin A/C (sc‐7292), anti‐ROCK1 (sc‐17794), and anti‐ERK2 (sc‐1647) were purchased from Santa Cruz Biotechnology. Anti‐β‐TRCP (4394), anti‐p‐YAP (4911), anti‐vimentin (3932), anti‐Slug (9585), anti‐MST1 (3682), anti‐p‐MST1/MST2 (3681), anti‐LATS1 (9153), anti‐p‐LATS1 (9157), and anti‐EGF receptor (2232) were purchased from Cell Signaling. Anti‐UbcH5 (Ab58251) was purchased from Abcam. Anti‐E‐cadherin (610181), anti‐Clathrin Heavy Chain (610499), anti‐β‐catenin (610153), and anti‐E‐cadherin (610181) were purchased from BD Biosciences. Anti‐ROCK2 (07‐443) was purchased from EMD Millipore. Anti‐GFP (11814460001) was purchased from Roche. Anti‐Histidine (34660) was purchased from Qiagen. Anti‐paxillin (clone 5H11) was purchased from Thermo Fisher. Texas Red phalloidin (T7471) was purchased from Invitrogen. HRP‐conjugated donkey anti‐mouse IgG (715‐035‐150) and HRP‐conjugated anti‐mouse IgG (711‐035‐152) were purchased from Jackson ImmunoResearch Laboratories.
Cell culture
DU145 and A549 cells were purchased from the American Type Culture Collection (ATCC). All cells used in this study were within 20 passages after thawing. A549 and DU145 cells were cultured (37°C, 5% CO2) in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (Gibco) and Penicillin/Streptomycin (1%, Gibco). For the studies dependent on matrix stiffness, collagen‐coated hydrogel pre‐plated in culture wells (Matrigen) was generated from a mix of acrylamide and bis‐acrylamide coated with collagen. Cells were cultured, passaged, and harvested while on top of the hydrogel, using standard cell culture techniques as previously described 42.
Human prostate organoids
Human prostate organoid cultures were generated and maintained as described previously 32. Briefly, human prostate tumor tissue was dissected and enzymatically digested with collagenase type II (Gibco) and TrypLE (Gibco). Cells were embedded in growth factor reduced Matrigel (Corning) at a 1:5 ratio and overlaid with ADMEM/F12 medium supplemented with B27 (Life Technologies), 10 mM HEPES (Life technologies), Glutamax (Life technologies), Penicillin/Streptomycin, Primocin (InvivoGen), and the following growth factors: EGF (PeproTech); FGF2 (PeproTech); FGF10 (PeproTech); Prostaglandin E2 (Tocris); R‐spondin 1 and Noggin conditioned medium and the TGF‐β/ALK inhibitor A83‐01 500 nM (Santa Cruz); Rock Inhibitor Y27632 (Selleck Chemical) 10 μM; n‐acetylcysteine (N‐AC) (Sigma‐Aldrich) 1.25 mM; and Dihydrotestosterone (DHT) (Sigma‐Aldrich) 1 nM. Organoids were isolated and transfected with siRNAmax according to the manufacturer (Invitrogen). Next, organoids were added to in a collagen I (Corning) matrix and images of invasion were obtained 48 h later.
siRNA and plasmid transfection
Transfections were performed with Lipofectamine RNAiMAX according to the manufacturer's instructions (Invitrogen) using siRNA SMARTpool ON‐TARGETplus Human UBTD1 (80019), ON‐TARGETplus Non‐Targeting Control siRNAs, or single ON‐TARGETplus siRNAs for YAP (J‐012200‐07 and J‐012200‐05) and TAZ (WWTR1; J‐016083‐05 and J‐016083‐06) (GE Healthcare). Sequences of the other single siRNA are provided in the Appendix Table S1.
Lipofectamine 2000 was used for plasmid transfection according to the manufacturer's instructions (11668, Invitrogen). Histidine‐tagged ubiquitin plasmid (pCI‐His‐hUbi, #31815) and the YAP1 (18881) coding sequence were purchased from Addgene. The YAP1 (18881) coding sequence was sub‐cloned in the pCDH‐CMV‐MCS‐EF1‐copGFP vector (System Biosciences) using EcoRI and NotI restriction sites. The lentiviral parent vector expressing GFP was used as a control. Codon‐optimized (DNA 2.0) DNA constructs corresponding to the mature form of human UBTD1 were cloned into the pEGFP‐N1 vector (Novagen).
Immunofluorescence
Cells were fixed with PBS/PFA 4% for 10 min and permeabilized with PBS/Triton X‐100 0.2% for 5 min. After blocking with PBS/BSA 0.2% for 1 h, the cells were then incubated with primary antibodies (1/100) at room temperature for 1 h. Secondary antibodies coupled with Alexa‐594 and/or Alexa‐488 (A‐11012, A‐11001, Life Technologies) were used at 1/500 for 1 h. Nuclei were counterstained with DAPI (Sigma‐Aldrich). Images were obtained using a LSM510 confocal microscope (Zeiss).
Proximity ligation assay
The PLA kit was purchased from Sigma‐Aldrich, and the assay was performed according to the manufacturer's protocol. Cells were fixed in 4% PFA for 10 min at room temperature, quenched in 50 mM NH4Cl for 10 min, permeabilized with 0.2% Triton X‐100 (w/v) for 10 min, and blocked in PBS/BSA. Cells were incubated with primary antibodies for 45 min in PBS/BSA. Coverslips were mounted in Fluoromount with DAPI to stain nuclei. PLA signals were visible as fluorescent dots and imaged using an inverted epifluorescence Leica DM6000B microscope equipped with a HCX PL Apo 63× NA 1.32 oil immersion objective and an EMCCD camera (Photometrics CoolSNAP HQ). Fluorescent dots were quantified using ImageJ. Cells and nuclei were delineated to create masks. After a max entropy threshold, the PLA dots were quantified in both masks with the ImageJ Analyze Particles plugin. All counts were divided by the number of cells.
Nuclear, cytosolic, and membrane extraction
Nuclear–cytoplasmic–membrane fractionation was performed using the Subcellular Protein Fractionation Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. The cytoplasmic fraction contains soluble cytoplasmic contents; the membrane fraction contains plasma, mitochondria, and ER/Golgi membranes; and the nuclear fraction contains the soluble nuclear extract and chromatin‐bound nuclear proteins. Lysates were analyzed by SDS–PAGE and immunoblotted for lamin A/C as a marker of the nuclear fraction, for RhoGDI as a marker of the cytoplasmic fraction, and for EGF receptor (EGFR) as a marker of the membrane fraction.
Western blot assays
Cells were lysed in RIPA buffer (Pierce) or directly in Laemmli's buffer. After denaturation, protein lysates were resolved by SDS–PAGE and transferred onto a PVDF membrane (Millipore). Membranes were blocked with 2% BSA in TBS Tween‐20 0.1% and incubated in the presence of the primary and then secondary antibodies. After washing, immunoreactive bands were visualized with ECL (Millipore) and analyzed on PXi (Syngene).
Ubiquitylation assays
HEK293 cells (5 × 106) were transfected with 10 μg of His‐tagged ubiquitin WT expression vectors together with YAP and siRNA targeting UBTD1. Ubiquitylated proteins were recovered by His‐tag affinity purification on cobalt resin in urea denaturing conditions, as described 43.
Pull‐down and immunoprecipitation
Activated RhoA pull‐down using GST‐ROCK is described in Doye et al 44. For endogenous immunoprecipitation, cells were harvested and lysed in RIPA buffer containing a protease inhibitor cocktail (Thermo Fisher). The lysates (16,000 g, 10 min, 4°C) were incubated for 2 h with the indicated antibody (IP) at 4°C. Then, 10 μl Dynabeads protein A (Invitrogen) was added to each aliquot for 45 min at 4°C. Beads were washed 3–5 times with RIPA and eluted by boiling in 2× sample buffer at 95°C for 10 min. The eluted fractions were analyzed by Western blot.
RNA isolation and RT–PCR from cell lines
Total RNA was extracted by TRIzol reagent according to the manufacturer's instructions (Invitrogen). RNA quantity and quality were determined using NanoDrop™ One Spectrophotometer (Thermo Scientific). One microgram of total RNA was reverse‐transcribed into cDNA (A3500, Promega). Real‐time quantitative PCR was performed using Fast SYBR Green Master Mix (Applied Biosystems) on a StepOnePlus System (Applied Biosystems). The gene‐specific primer sets were used at a final concentration of 1 μM in a 10 μl final volume. RPLP0 mRNA levels were used as an endogenous control to normalize relative expression values of each target gene. The relative expression was calculated by the comparative C t method. All real‐time RT–PCR assays were performed in triplicate with three independent experiments. Primers are provided in Appendix Table S2. Primers were published in http://pga.mgh.harvard.edu/primerbank/.
RNA isolation and RT–PCR from paraffin‐embedded prostate tumor tissues
Total RNA was extracted with PureLink™ FFPE RNA Isolation Kit (K156002) according to the manufacturer's instructions (Invitrogen). For each RNA purification, six pieces of 10‐μm‐thick sections of paraffin‐embedded prostate tissues, with a tissue surface area of 0.5–1 cm2, were used. For each patient, a sample of both normal and tumor tissue was collected. One microgram of total RNA was reverse‐transcribed into first‐strand cDNA using Maxima First Strand cDNA Synthesis Kit (K1671) according to the manufacturer's instructions (Thermo Scientific). Real‐time quantitative PCR was performed with TaqMan Fast Universal Master Mix (Applied Biosystems) using the ABI PRISM 7500/Step‐One Fast Real Time PCR System following the manufacturer's protocols (Applied Biosystems). UBTD1 (Hs00227913_m1) TaqMan gene expression assays were purchased from Applied Biosystems. Gene expression values were normalized to the value of the housekeeping genes 18S (Hs99999901_s1) and calculated based on the comparative cycle threshold Ct method (ΔΔC t).
Boyden chamber assay
Migration and invasion assays were performed using cell culture inserts with 8.0‐μm pore transparent PET membrane (353097, Falcon) coated with 10 μg/ml fibronectin (Sigma). For invasion experiments, the inserts were coated with 25 μg/μl of Matrigel (354234, Corning) and incubated for 3 h at 37°C in a CO2 incubator for migration assays; transwells were left uncoated. Briefly, overnight serum‐starved cells (12×104 cells) were seeded into the top chamber in medium without FBS, while medium with 10% FBS was present in the bottom chamber. The cells were incubated for 3–6 h or 24 h for migration and invasion, respectively. The media and remaining cells were removed from the top chamber with a cotton swab and washed two times with PBS. The bottom chamber was aspirated and washed two times with PBS. The inserts were fixed with 4% PFA and incubated with a DAPI solution (2 mg/ml) to label cell nuclei. Cell migration and invasion were determined by counting all cells on the total areas at the lower surface of the membrane with a Nikon A1R Confocal Laser Microscope System. Nuclei were counted and analyzed using ImageJ software and normalized to area.
Calcium switch assay
Cells were allowed to grow on glass slides. At 80% confluence, cell–cell junctions were disrupted by incubation with EGTA (4 mM)‐containing medium. Thirty minutes later, cell–cell junction formation was stimulated by putting back normal medium (recovery). Cells were fixed and immunostained at various times during the recovery period and imaged.
Atomic force microscopy
Cells, grown on a 50‐mm WillCo Glass Bottom Dish, were first washed twice with 3 ml of Leibovitz's medium (Life Technologies) supplemented with heat‐inactivated 10% fetal bovine serum (FBS), and then covered with 3 ml of the same medium. The mechanical properties of samples were studied using a BioScope Catalyst atomic force microscope (Bruker Nano Surfaces, Santa Barbara, CA, USA) equipped with a NanoScope V controller and coupled with an optical microscope (Leica DMI6000B, Leica Microsystems Ltd., UK). All the experiments were performed at 37°C. For each sample, at least 30 cells were analyzed using the “Point and Shoot” method, collecting at least 200 force–distance curves at just as many discrete points (on average five points for each cell in the perinuclear area). The experiments were performed using a probe with a borosilicate glass spherical tip (5 μm diameter) and a cantilever with a nominal spring constant of 0.06 N/m (Novascan Technologies, Ames, IA USA). After determining both the deflection sensitivity of the system in the Leibovitz's medium using a clean WillCo Glass Bottom Dish and the spring constant of the cantilever by means of the thermal tune method, force–distance curves were collected on samples using a velocity of 2 μm/s, in relative trigger mode and by setting the trigger threshold to 2 nN. The apparent Young's modulus was calculated using the NanoScope Analysis 1.80 software (Bruker Nano Surfaces, Santa Barbara, CA, USA) applying to the force curves, after the baseline correction, the Hertz spherical indentation model using a Poisson's ratio of 0.5. For each force curve, the force fit boundaries to perform the fit were chosen between 5% and 25%. To avoid any tilt effect due to the base correction step, only the force curves having their maximum value at 2 nN were taken into account for performing the fit and only the apparent Young's modulus values corresponding to a fit with R 2 > 0.90 were accepted.
Uniaxial mechanical stretch
Cells were cultured in 4‐cm2 silicone chambers (STB‐CH‐04; Strex, Osaka, Japan) coated with fibronectin (Santa Cruz). A uniaxial stretch was applied to the chamber using a stretching apparatus for 30 min. 20% strain was applied to the chamber.
Traction force microscopy
Contractile forces exerted by cells on different stiffness gels were assessed by traction force microscopy essentially as described 45. Briefly, polyacrylamide substrates with shear moduli of 4, or 12 kPa conjugated with fluorescent bead latex microspheres (0.5 μm, 505/515 nm ex/em), were purchased from Matrigen. After transfection, cells were plated on fluorescent bead‐conjugated discrete stiffness gels and grown for 24 h. Images of gel surface‐conjugated fluorescent beads were acquired for each cell before and after cell removal using an Axiovert 200M motorized microscope stand (Zeiss) and a ×32 magnification objective. Tractions exerted by cells were estimated by measuring bead displacement fields, computing corresponding traction fields using Fourier transform traction microscopy, and calculating root‐mean‐square traction using the PIV (particle image velocimetry) and TFM (traction force microscopy) package on ImageJ. To measure baseline noise, the same procedure was performed on a cell‐free region.
Immunohistochemistry
UBTD1 IHC was performed on 2‐μm‐thick sections. Immunolabeling and detection were performed using a Dako Autostainer AutoMate, as per the manufacturer's recommendations. The detection was performed using the Envision Flex Kit (Dako), with 3–3′ diaminobenzidine as a chromogen.
Gene expression microarray analysis
Normalized RNA sequencing (RNA‐Seq) data produced by The Cancer Genome Atlas (TCGA) were downloaded from cBioPortal (www.cbioportal.org, TCGA Provisional; RNA‐Seq V2). Data were available for 522 lung adenocarcinoma tumor samples and 56 grade 3 prostate adenocarcinoma tumor samples TCGA subjected to mRNA expression profiling. The results published here are in whole or in part based upon data generated by the TCGA Research Network 46, 47. PFS was defined as the time between diagnosis and progression, or death from any cause, censoring live patients and progression free at last follow‐up. OS was defined as the time between surgery and the date of death from any cause, censoring those alive at last follow‐up. The Kaplan–Meier method was used to produce survival curves, and analyses of censored data were performed using Cox models.
Statistical analysis
All analyses were performed using Prism 6.0 software (GraphPad Inc.). A two‐tailed t‐test was used if comparing only two conditions. For comparing more than two conditions, one‐way ANOVA was used with the following: Bonferroni's multiple comparison test or Dunnett's multiple comparison test (if comparing all conditions to the control condition). Significance of mean comparison is marked on the graphs by asterisks. Error bars denote SEM.
Author contributions
ST, FB, and SC conceived, designed, and performed the study. FRR performed organoid experiments, SP performed AFM experiments, and CG designed and supervised the AFM experiments. MD performed bioinformatic analysis of lung and prostate cancer patients, TB performed traction force experiments, KL and LK performed RNA experiments on patient tissues, and DA and JFM performed immunohistochemistry experiments. MG performed image processing and analysis. All authors contributed to the data collection and analysis. SC, ST, and FB wrote the paper. CL, EL, JPU, and AM participated actively to data interpretation and critical revision of the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank the GIS‐IBISA multi‐sites platform “Microscopie Imagerie Côte d'Azur” (MICA), and particularly the imaging site of C3M and IRCAN (PICMI), which are supported by the “Conseil General 06” and “Conseil Départemental 06”. The PICMI AFM was supported by the Association pour la Recherche sur le Cancer (ARC) and by the “Conseil General 06 de la Région Provence Alpes‐Côte”. This work has been supported by the French government, through the UCA‐JEDI Investments in the Future project managed by the National Research Agency (ANR) with the reference number ANR‐15‐IDEX‐01. S.T. is a recipient of a post‐doctoral fellowship from “La Fondation de France”. L.K. is a recipient of a doctoral fellowship from the French ministry of research. F.B. is a CNRS researcher.
EMBO Reports (2019) 20: e46570
Contributor Information
Frédéric Bost, Email: bost@unice.fr.
Stéphan Clavel, Email: stephan.clavel@unice.fr.
References
- 1. Jaalouk DE, Lammerding J (2009) Mechanotransduction gone awry. Nat Rev Mol Cell Biol 10: 63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Panciera T, Azzolin L, Cordenonsi M, Piccolo S (2017) Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol 18: 758–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Totaro A, Panciera T, Piccolo S (2018) YAP/TAZ upstream signals and downstream responses. Nat Cell Biol 20: 888–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meng Z, Moroishi T, Guan KL (2016) Mechanisms of Hippo pathway regulation. Genes Dev 30: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D (2007) Elucidation of a universal size‐control mechanism in Drosophila and mammals. Cell 130: 1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L et al (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta‐TRCP). Genes Dev 24: 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V et al (2014) YAP/TAZ incorporation in the beta‐catenin destruction complex orchestrates the Wnt response. Cell 158: 157–170 [DOI] [PubMed] [Google Scholar]
- 9. Uhler JP, Spahr H, Farge G, Clavel S, Larsson NG, Falkenberg M, Samuelsson T, Gustafsson CM (2014) The UbL protein UBTD1 stably interacts with the UBE2D family of E2 ubiquitin conjugating enzymes. Biochem Biophys Res Commun 443: 7–12 [DOI] [PubMed] [Google Scholar]
- 10. Zhang XW, Wang XF, Ni SJ, Qin W, Zhao LQ, Hua RX, Lu YW, Li J, Dimri GP, Guo WJ (2015) UBTD1 induces cellular senescence through an UBTD1‐Mdm2/p53 positive feedback loop. J Pathol 235: 656–667 [DOI] [PubMed] [Google Scholar]
- 11. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- 12. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S et al (2010) Towards a knowledge‐based human protein atlas. Nat Biotechnol 28: 1248–1250 [DOI] [PubMed] [Google Scholar]
- 13. Zhang D, Park D, Zhong Y, Lu Y, Rycaj K, Gong S, Chen X, Liu X, Chao HP, Whitney P et al (2016) Stem cell and neurogenic gene‐expression profiles link prostate basal cells to aggressive prostate cancer. Nat Commun 7: 10798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schillers H, Walte M, Urbanova K, Oberleithner H (2010) Real‐time monitoring of cell elasticity reveals oscillating myosin activity. Biophys J 99: 3639–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vicente‐Manzanares M, Ma X, Adelstein RS, Horwitz AR (2009) Non‐muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10: 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark AG, Wartlick O, Salbreux G, Paluch EK (2014) Stresses at the cell surface during animal cell morphogenesis. Curr Biol 24: R484–R494 [DOI] [PubMed] [Google Scholar]
- 17. Wei WC, Lin HH, Shen MR, Tang MJ (2008) Mechanosensing machinery for cells under low substratum rigidity. Am J Physiol Cell Physiol 295: C1579–C1589 [DOI] [PubMed] [Google Scholar]
- 18. Halder G, Dupont S, Piccolo S (2012) Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol 13: 591–600 [DOI] [PubMed] [Google Scholar]
- 19. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183 [DOI] [PubMed] [Google Scholar]
- 20. Nelson WJ (2008) Regulation of cell‐cell adhesion by the cadherin‐catenin complex. Biochem Soc Trans 36: 149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gumbiner BM (2005) Regulation of cadherin‐mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol 6: 622–634 [DOI] [PubMed] [Google Scholar]
- 22. Miura H, Nishimura K, Tsujimura A, Matsumiya K, Matsumoto K, Nakamura T, Okuyama A (2001) Effects of hepatocyte growth factor on E‐cadherin‐mediated cell‐cell adhesion in DU145 prostate cancer cells. Urology 58: 1064–1069 [DOI] [PubMed] [Google Scholar]
- 23. Yu FX, Zhang Y, Park HW, Jewell JL, Chen Q, Deng Y, Pan D, Taylor SS, Lai ZC, Guan KL (2013) Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev 27: 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z, Liu P, Inuzuka H, Wei W (2014) Roles of F‐box proteins in cancer. Nat Rev Cancer 14: 233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S (2013) A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin‐processing factors. Cell 154: 1047–1059 [DOI] [PubMed] [Google Scholar]
- 26. Nardone G, Oliver‐De La Cruz J, Vrbsky J, Martini C, Pribyl J, Skladal P, Pesl M, Caluori G, Pagliari S, Martino F et al (2017) YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun 8: 15321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Julian L, Olson MF (2014) Rho‐associated coiled‐coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases 5: e29846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, Meng Z, Lin KC, Yu FX, Alexander CM et al (2015) Alternative Wnt signaling activates YAP/TAZ. Cell 162: 780–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu FX, Zhao B, Guan KL (2015) Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163: 811–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zanconato F, Cordenonsi M, Piccolo S (2016) YAP/TAZ at the roots of cancer. Cancer Cell 29: 783–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kalluri R, Weinberg RA (2009) The basics of epithelial‐mesenchymal transition. J Clin Invest 119: 1420–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, Clevers H (2016) Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc 11: 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, Bergren SK, Badani KK, McKiernan JM, Benson MC, Hibshoosh H et al (2014) Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol 16: 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK et al (2014) Organoid cultures derived from patients with advanced prostate cancer. Cell 159: 176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, Munro J, Schroder E, Zhou J, Brunton VG et al (2011) Actomyosin‐mediated cellular tension drives increased tissue stiffness and beta‐catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 19: 776–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pan D (2007) Hippo signaling in organ size control. Genes Dev 21: 886–897 [DOI] [PubMed] [Google Scholar]
- 37. Koo JH, Guan KL (2018) Interplay between YAP/TAZ and Metabolism. Cell Metab 28: 196–206 [DOI] [PubMed] [Google Scholar]
- 38. DuFort CC, Paszek MJ, Weaver VM (2011) Balancing forces: architectural control of mechanotransduction. Nat Rev Mol Cell Biol 12: 308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu H, Mouw JK, Weaver VM (2011) Forcing form and function: biomechanical regulation of tumor evolution. Trends Cell Biol 21: 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K et al (2015) The BioPlex network: a systematic exploration of the human interactome. Cell 162: 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R et al (2014) A proteome‐scale map of the human interactome network. Cell 159: 1212–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang YY et al (2016) Vascular stiffness mechanoactivates YAP/TAZ‐dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 126: 3313–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Torrino S, Visvikis O, Doye A, Boyer L, Stefani C, Munro P, Bertoglio J, Gacon G, Mettouchi A, Lemichez E (2011) The E3 ubiquitin‐ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev Cell 21: 959–965 [DOI] [PubMed] [Google Scholar]
- 44. Doye A, Boyer L, Mettouchi A, Lemichez E (2006) Ubiquitin‐mediated proteasomal degradation of Rho proteins by the CNF1 toxin. Methods Enzymol 406: 447–456 [DOI] [PubMed] [Google Scholar]
- 45. Liu K, Yuan Y, Huang J, Wei Q, Pang M, Xiong C, Fang J (2013) Improved‐throughput traction microscopy based on fluorescence micropattern for manual microscopy. PLoS One 8: e70122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6