

Abstract
In order to understand the nature of the relationship between cerebral blood flow (CBF) and primary headaches, we have conducted a literature review with particular emphasis on the role of perivascular neurotransmitters. Primary headaches are in general considered complex polygenic disorders (genetic and environmental influence) with pathophysiological neurovascular alterations. Identified candidate headache genes are associated with neuro- and gliogenesis, vascular development and diseases, and regulation of vascular tone. These findings support a role for the vasculature in primary headache disorders. Moreover, neuronal hyperexcitability and other abnormalities have been observed in primary headaches and related to changes in hemodynamic factors. In particular, this relates to migraine aura and spreading depression. During headache attacks, ganglia such as trigeminal and sphenopalatine (located outside the blood-brain barrier) are variably activated and sensitized which gives rise to vasoactive neurotransmitter release. Sympathetic, parasympathetic and sensory nerves to the cerebral vasculature are activated. During migraine attacks, altered CBF has been observed in brain regions such as the somatosensory cortex, brainstem and thalamus. In regulation of CBF, the individual roles of neurotransmitters are partly known, but much needs to be unraveled with respect to headache disorders.
Keywords: Blood–brain barrier, cerebral blood flow, migraine, parasympathetic nervous system, sympathetic nervous system
Introduction
Migraine is a frequent primary headache disorder, with higher prevalence in females (15.5%) than males (4.5%),1 and associated with clinical symptoms (typical headache, and phonophobia/photophobia/nausea/vomiting that occur over 4–72 h).2 On the contrary, cluster headache is more prevalent in males than females for most age groups.3 The origin of headache attacks is still under discussion, with particular focus on whether they represent a vascular or neurogenic disorder.4,5 During the different phases of the migraine attack (premonitory and during attacks), there are variations in cerebral blood flow (CBF), coinciding with changes in brain activity, observed in the somatosensory cortex, brainstem and thalamus.6,7 In headache-free periods (inter-ictal), elevated CBF in the primary somatosensory cortex has been reported for migraineurs. This effect was positively correlated to headache attack frequency, but also to the number of cutaneous allodynia symptoms.6 During headache attacks, autonomic and sensory ganglia are known to be variably activated and sensitized,8,9 which gives rise to neurotransmitter release, believed to be important in the migraine pathophysiology.10,11 In particular, trigeminal ganglion (TG) stimulation in cats resulted in increased CBF and calcitonin gene-related peptide (CGRP) release.12 Several signaling molecules are involved in both migraine and innervation of the cerebral vasculature.
The cerebral vasculature is known to be innervated by (i) sympathetic nerves storing adenosine triphosphate (ATP), noradrenaline and neuropeptide Y (NPY), (ii) parasympathetic nerves with acetylcholine, vasoactive intestinal peptide (VIP), peptide histidine methionine, pituitary adenylate cyclase-activating peptide (PACAP) and nitric oxide (NO), and (iii) sensory nerves containing CGRP, PACAP, NO, substance P (SP) and neurokinin A (NKA).13,14 The extrinsic innervation ends close to the entry of the vasculature into the brain parenchyma; a transition zone in the Virchow-Robin space. Moreover, the intraparenchymal nerve supply to arterioles and microvessels stems primarily from central nervous system (CNS) neurons.14 Therefore, it is necessary to discuss migraine in the light of extrinsic and intrinsic neural inputs, and their role in regulation of CBF.
The link between headache, CNS symptoms, intracranial vessel diameter and regulation of blood flow has long been pursued. In general, primary headaches are considered complex polygenic disorders (genetic and environmental influence) with pathophysiological neurovascular alterations.15–17 Several primary headache theories have been proposed over time, with the purpose of elucidating the underlying cause of headache attacks and associated characteristics. Early studies on involvement of extracranial vasodilation and a link between intracranial vasospasm and cortical spreading depression (CSD) in migraine with aura led to the vascular theory of migraine, originally forwarded by Wolff in the early 1940s.18,19 Although a pure vascular theory has now lost its role as the leading hypothesis to explain migraine, involvement of extracranial and intracranial vasculature has persisted over time as an etiological or coexisting factor.20,21 Direct experimental evidence, contrary to the vascular theory, came from a magnetic resonance angiography study showing no/minor alterations in the diameter of meningeal and cerebral arteries in genuine migraine attacks.22 Nevertheless, this study is not entirely conclusive, as the intracranial middle meningeal artery (MMA) was not measured due to technical limitations.23
Today CSD remains one of the possible early links between vascular observations and the neurogenic theory. CSD is a self-propagating wave of neuronal and glial cortical depolarization resulting in biophysical and biochemical alterations such as changes in parenchymal blood flow and ionic concentrations.24 The aura, a clinical symptom (mostly visual) seen in about 20–30% migraineurs,25 is supposedly due to CSD.26,27 It has been suggested that CSD directly activates trigeminal sensory nerve terminals in dura mater/meninges followed by trigeminovascular system activation.24,28 For decades, activation and sensitization of the trigeminovascular system have been considered a pathophysiological mechanism/consequence behind primary headaches.29–31 Interestingly, CSD not only affects meningeal arteries, but also cortical cerebral arterioles and CBF.32 An alternative hypothesis involves activation of CNS regions by CSD which in turn, via thalamus and brainstem, causes activation of the trigeminovascular system with dural inflammation being secondary to the CNS activation. Moreover, during the headache phase in genuine attacks, waves of flow reductions slowly spread over the cortex bilaterally. This pattern resembled CSD even though the subject did not have obvious aura symptoms.33 Other studies also report the occurrence of local depression of CBF in migraine.34 More details on this research topic has been discussed by Pietrobon and Moskowitz26,27 and by Ayata and Lauritzen.35
Today, leading scientists advocate that already in the premonitory phase of an acute migraine attack, distinct blood flow alterations within, e.g. hypothalamus-thalamus and the adjacent brainstem (periaqueductal gray) can be observed.36,37 The proposal of a “migraine generator” originates from Weiller et al.38 demonstrating activation of brain regions such as the periaqueductal gray, raphe nuclei, and locus coeruleus in the early phase of a migraine attack. Following injection of sumatriptan for acute treatment of migraine, pain subsided but the areas remained activated. After some hours, the pain returned, probably due to disappearance of the triptan from the involved receptors.38 Thus, evidence suggests that migraine pathophysiology includes dysfunction of subcortical structures such as diencephalic and brainstem nuclei which can modulate the trigeminovascular system and perhaps also participate in CSD generation.39
Complexity of neurovascular coupling and CBF
Brain homeostasis is known to be controlled by a tight regulation between neuronal function, local blood flow and maintenance of neurovascular coupling.40,41 More precisely, the CBF is controlled by (i) autoregulation, maintenance of adequate CBF regardless of blood pressure (BP) fluctuations (within some limits),42,43 (ii) chemical and metabolic factors, e.g. interstitial pH44 and CO2,45 and (iii) neurogenic factors such as perivascular innervation.46,47 Regulatory mechanisms of CBF depend on variations in the rate of local brain metabolism.48
The changes of CBF in primary headaches are obviously not as drastic as observed in ischemic stroke. Weiller et al.38 reported minor changes in cortical blood flow which was significant only if all migraine patients were normalized and grouped (about 5% of resting flow). In migraineurs with aura, a reduction in CBF was observed closely in time to the aura symptoms.49,50 In the interictal period, Loehrer et al.51 reported a higher total and parenchymal CBF in migraineurs compared to controls, suggested to be driven by a higher basilar artery flow. In experimental animals, induction of CSD causes an abrupt rise in CBF within a minute followed by depression of function and flow.52
Relevant for primary headaches, an association between neurovascular dysfunction and CSD has been proposed.52,53 It is important to clarify that the intraparenchymal neurovascular system is distinctly different from that of the large cerebral arteries and arterioles located around the brain and leading into the intraparenchymal system.14,41 The extraparenchymal vasculature (arteries belonging to the circle of Willis, and arteries located on the cortical surface) receives a dense supply of perivascular nerves that originate in cranial ganglia.47 The intraparenchymal neurovascular system is thin and mainly contains pericytes and endothelial cells. Their connections with the local environment come from local neurons and astrocytes, inter alia. They are not supplied by innervation from the cranial ganglia, and47 hence there are two different sites of neuronal regulation of the cerebral circulation.
Extrinsic and intrinsic cerebral vasculature: Neurogenic factors and neurotransmitter release
Extrinsic perivascular nerves in the cerebral circulation and dura mater
Three centuries ago, Thomas Willis noted nerve profiles on major cerebral arteries and speculated on a role in regulation of brain blood flow.54 The role of perivascular nerves took a step forward in 1967 when Nielsen and Owman55 demonstrated sympathetic noradrenaline-containing perivascular nerves. As a result, a new era began with analysis and demonstration of a multitude of associated receptors.56 Originally, it was thought that they were an anomaly based on experiments showing that denervation had no effect on basic regulation of flow-metabolism coupling, chemical control or autoregulation.47,57 However, it is now known that the upper and lower limits of autoregulation are modified by sympathetic nerves arising from the superior cervical ganglia (SCG).58 The parasympathetic fibers originate from the sphenopalatine (SPG) and otic ganglia (OTG), storing acetylcholine and numerous neuropeptides (the first to be found was VIP59). Currently, these ganglia have been linked to facial symptoms in, e.g. cluster headache.60 The third system innervating the cerebral circulation is the sensory system which originates in the TG.13 For long, we were seeking a role of this system, and in 1986, we reported that it was a reflex designated to counterbalance any reductions in local CBF.61 Recent work has focused on understanding of the intimate coupling process. CBF alterations, induced by neuronal responses, have been studied by functional magnetic resonance imaging (fMRI) as an indirect measure of neurovascular coupling.62 Transient photophobia was investigated by fMRI in a healthy human subject and was found to be associated with trigeminal system activation.63
Advances in our understanding of the neuronal innervation of the extraparenchymal cerebral vasculature
In the 1970s, characterization of neurotransmitters in perivascular nerve fibers was demonstrated in more detail due to the availability of novel histochemical methodologies, immunohistochemistry in particular. This increased the theoretical understanding of possible pathophysiological mechanisms involved in primary headaches (e.g. regulatory mechanisms of CBF). The origin of perivascular nerve endings on cerebral vasculature64,65 has been investigated by a nerve tracing and immunohistochemical localization study. Using True Blue applied on the vessel walls, we traced the dye to both uni- and bilateral ganglia, and showed co-localizations with NPY (sympathetic), VIP (parasympathetic) and SP and CGRP (sensory).66–68 The brainstem was subsequently analyzed using transganglionic tracers and the sensory system was found to localize in the trigeminal nucleus caudalis and brainstem C1-3, laminae I-II.69–71 Innervation studies of the trigeminal system showed that CGRP and SP originate in the TG.72 Another study showed that PACAP-containing rat nerve fibers, innervating the extrinsic cerebral vasculature, primarily originate from neurons in the parasympathetic SPG and OTG and to a lesser extent from neurons in the sensory TG.64 Most of PACAP and VIP were expressed in the SPG and OTG while PACAP only showed minor expression in TG,73 also shown in man.74,75 The TG has, by far, retained most focus over time when investigating the pathophysiology of primary headaches. Neuronal sensory innervation of cerebral blood vessels, by vasoactive neuropeptides, has been linked to the TG.64,72 Electrical stimulation of TG resulted in increased CGRP, PACAP, SP and NKA levels,29,76–78 in agreement with immunohistochemical localization studies investigating TG and perivascular nerve fibers.64,79,80 PACAP immunoreactivity was also found in nerve fibers on cerebral arteries in cats.81 Interestingly, PACAP-38 was increased in the plasma and trigeminal nucleus caudalis, but not in the TG itself or in the spinal cord78 indicating stimulated signaling pathways upon TG activation.
Somatic afferents of the trigeminal nerve innervate considerable regions of the face, scalp, and meninges amongst others.82 In coherence, increased ipsilateral facial skin blood flow is induced by experimental activation of TG in rats.83 The trigeminal nerve is linked with the SPG,84,85 which has gained interest, particularly in cluster headache research. Repetitive transnasal blockade of SPG has shown effectiveness in treating both cluster headache86 and chronic migraine.87,88 The structure is located in the pterygopalatine fossa below the maxillary nerve (trigeminal nerve branch, V2) where sympathetic and parasympathetic nerve fibers pass through. The superior salivary nucleus in the pons is connected to the SPG through nerve fibers82 followed by nerve projections to the cranial vasculature from the parasympathetic ganglion.39 Nerve projections originating from the SPG were traced to the internal ethmoidal, internal carotid and cerebral arteries by anterograde labelling of nerve fibers.89 A study observed increased CBF in the ipsilateral and contralateral parietal cortex as a response to electrical SPG stimulation in rats90 possibly as a response to neurotransmitter release.91 Immunohistochemical localization studies revealed expression of PACAP, VIP and NO synthase (NOS) in rat and human SPG85,92 and expression of CGRP nerve fibers likely from the TG.84 SPG extirpation, on the other hand, affects expression of VIP and acetylcholine in cerebrovascular structures.93 Together these results indicate that there is a link between autonomic and sensory ganglia, CBF and migraine.
Nerve projections from the SCG, cervical C2 dorsal root ganglia (DRG) and OTG onto extrinsic cerebral arteries have been studied by retrograde tracing showing that the ganglia contain NPY, CGRP and SP, and VIP immunoreactive neurons, respectively.66 The effects of these ganglia have been investigated less intensively compared to that of the TG and SPG with respect to primary headaches. In general, sympathetic nervous activity has been associated with cerebrovascular constriction and parasympathetic nervous activity with cerebrovascular dilation41 of which parasympathetic activation is known to result in sympathetic hypoactivity and vice versa.94 DRG has been associated with neuropathic pain as a response to pathological changes in the structure.95 Since headache only seems to arise as a response to C1–C3 nerve innervations referred to as cervicogenic headache, the DRG of interest are the ones positioned at the C1–C3 level of the cervical spine.96,97 Autonomic and sensory ganglionic innervation of intracranial vessels by neuronal messenger molecules is illustrated in Figure 1.
Figure 1.

Overview of innervation of intracranial vasculature. (a) Whole-mounted intracranial vessel immunohistochemically processed using anti-transmitter antibodies. Asterisk point at transmitter innervation of a nerve, and also at the region of perpendicular section shown in Figure 3(b). (b) An intracranial vessel (from the inside and out): endothelium (red), lamina elastic interna (green), unstained smooth muscle cells, transmitter immunoreactive nerves (red) and nuclei (blue). Transmitter innervation is displayed by schematic drawings of neurons. Abbreviations: Ach: acetylcholine; ATP: adenosine triphosphate; CGRP: calcitonin gene-related peptide; NA: noradrenaline; NKA: neurokinin A; NO: nitric oxide; NPY: neuropeptide Y; PACAP: pituitary adenylate cyclase-activating polypeptide; SP: substance P; VIP: vasoactive intestinal peptide.
At this stage, it is pertinent to state that we, so far, have discussed innervation of the extraparenchymal system regulating the major cerebral and meningeal circulation. The current view on intraparenchymal regulation is still under analysis; so far, the prevailing view is a balance between local neuron and astrocyte activity, both in terms of their metabolism and direct activation of pericytes, smooth muscle cells and endothelium actions.98,99
Vasoconstrictive agents
In the relation to the development of a headache/migraine, the primary focus has been on vasodilating neurotransmitters. However, vasoconstrictive neurotransmitters are important in regulation of CBF and could also counteract a possible migraine-induced dilation. Therefore, studies have been performed on endothelin-1 (ET-1),100 NPY101 and 5-hydroxytryptamine (5-HT).102,103
High plasma levels of ET-1 have been recorded in the beginning of a migraine attack, which then decrease significantly over time. Increased levels of ET-1 have been proposed to account for part of local vasoconstriction observed early during migraine attacks.104 It might be a counterbalance-mechanism to ganglionic release of vasodilatory neurotransmitters in order to maintain vascular tone, possibly until vasodilation overpowers the vasoconstrictive properties, e.g. of ET-1. There are currently two lines of research carried out regarding ET-1 and its receptors: (i) Uddman et al.105 showed presence of ET receptors in TG neuronal cell bodies, and (ii) the drug industry developed and tested ET receptor blockade in migraine; however, the result was not positive.106 NPY has been detected in nerve fibers on human cerebral arteries, in addition to CGRP, SP and VIP, reporting expressional variations between the neuropeptides (Figure 1).80
Studies of 5-HT (or serotonin) indicate that migraine with aura leads to lower interictal 5-hydroxytryptophan (5-HTP) plasma levels, a serotonin precursor, compared to controls and migraineurs without aura.107 Multiple studies have shown low interictal 5-HT levels with elevations recorded during attacks, however, with conflicting results.108 Intravenous 5-HT and 5-HTP injection lead to symptomatic pain relief in migraineurs, however, with substantially varying outcome, for example with different injection rates.103 The triptans (5-HT analogues) were further developed, and have been a partial successful treatment in migraine. The 5-HT1B/1D agonists are still the preferred treatments for acute migraine headaches.
Vasodilating agents
Numerous studies have examined the effects of vasodilator agents in patients in order to find key candidates that cause specific migraine attacks. Over the years, glyceryl trinitrate (GTN) and other headache-inducing agents have attracted much interest. It has been known for centuries, that GTN causes headache, in particular noted with workers in the dynamite industry. It is well known by clinicians that GTN, used to treat angina pectoris, results in headache at an early point in time. However, in a study in healthy volunteers, Daugaard et al.109 reported that after 20 min of intravenous infusion, the recording of GTN-induced headache and a subsequent reduction in headache, a late phase called migraine-like headache occurred after some hours. Subsequently, >10 other agents have been tested in human pharmacological models resulting, not only in extracranial vessel dilatation, but also in different types of headache.110 Of particular interest, CGRP has attracted vast attention since the 1980s in the search for underlying mechanisms explaining part of primary headache etiologies.111 In cerebral arteries and arterioles (more precisely the middle cerebral arteries (MCA) and pial vessels), studies demonstrate that vasodilation caused by CGRP was more prominent and potent compared to that of SP and VIP.61,112 CGRP was also more potent compared to NKA, NKB, PACAP-38, dynorphine, galanin, peptide histidine methionine/isoleucine and nociceptin. Over time, this trend was observed in multiple studies investigating the effect of CGRP in headache disorders. Lately, this resulted in tests of CGRP receptor antagonists for prophylaxis and acute treatment of migraine.113 Following CGRP, PACAP was isolated from ovine hypothalamic tissues.114 Originally, PACAP was shown to have its main expression in the SPG and OTG, in parallel with VIP.74 While VIP is not present in the trigeminal system, a subpopulation of CGRP positive neurons expresses PACAP-38 in the TG.79 Interestingly, PACAP immunoreactivity was also seen along with CGRP, SP and neuronal NOS in the trigeminal nucleus caudalis and C1-2 laminae I-II, which are the central projection areas from the TG.115 Infusion of PACAP-38 demonstrated vasodilation of extracranial arteries (external carotid artery and superficial temporal artery), but not intracranial arteries (internal carotid artery, basilar artery and MCA) using magnetic resonance angiography.116,117 In humans without a history of headache/migraine, 89% and 100% of the subjects developed headache in the immediate and delayed phase, respectively.116 In functional studies, it was demonstrated that PACAP-38 and PACAP-27 both possess vasodilatory effects on MCA. It was suggested that the neuropeptides were involved in neuronal regulation of CBF reporting increases in CBF, as a response to PACAP-38 injection.81 In addition to direct vasoactive effects, the use of PACAP knockout (−/−) mice has provided insight into the role of PACAP in migraine. PACAP has an essential role in activation and sensitization of the trigeminovascular system but also in occurrence of photophobia and vasodilatory responses.118 Both α-CGRP and PACAP knockout (−/−) mice are viable, which suggest the presence of compensatory mechanisms. This might explain why there are so few adverse effects of the drugs that target CGRP and its receptor in humans.
The effect of exogenous administration of vasoactive peptides
Intravenous CGRP/PACAP-38 administration triggers migraine-like attacks in migraine subjects.117,119 Furthermore, MMA vasodilation has been detected upon α-CGRP and PACAP-38 infusions.116,120,121 The MMA consists of several segments: An extracranial (branch from the external carotid artery), intraosseous and intracranial part122 of which we mainly are interested in the intracranial part due to the focus on CBF in primary headaches. Moreover, the inability of infused neuropeptides to influence intracranial arteries in vivo is proposed to be due to an incapability to pass the blood–brain barrier (BBB) based on intravenous infusion and myograph studies.117,123 Tuor et al.124 investigated intracarotid/intracerebral NPY injection in rats and detected a cortical CBF reduction, although emphasizing that the change is only observable if NPY is administrated on the abluminal side of the BBB. In 2011, a myograph study revisited vasodilation of cerebral arteries, and observed vasodilation induced by PACAP and VIP.125 In vivo, vasodilatory effects of VIP are, however, not as pronounced as the ones for PACAP-38.117 In contrast, PACAP-38 did not produce vasodilation in isolated human intracranial MMA.126 It is worth pointing out that intravenous infusion of VIP (18%) did not result in generation of migraine-like attacks to the same extent as intravenous infusion of CGRP and PACAP-38 (63–73%) in migraineurs without aura, even though vasodilation was recorded.117,119,127
Plasma–protein extravasation
Neurotransmitter release has been associated with plasma–protein extravasation,120,128,129 which might cause local edema.130–132 Extravasation has been described as follows: “proteins accumulate in the extravascular space when the rate of delivery exceeds the rate of dispersion into the surrounding tissue.”133 Electrical TG and SPG stimulation have been associated with plasma–protein extravasation predominantly around small blood vessels in dura mater (which lacks BBB), with SP and NKA believed to be the most influential molecules involved.134,135 In contrast, CGRP did not induce plasma–protein extravasation,136 but only vasodilatation. Perivascular projections from the TG have been detected on meningeal blood vessels in cats by tracing horseradish peroxidase,65 and in rats using True Blue.66 Moreover, induction of CSD, believed to stimulate trigeminal axons, resulted in plasma–protein extravasation in dura mater in rats.130 Intracranial plasma extravasation has also been detected during a migraine attack in a female patient with episodic migraine evaluated by single photon emission computerized tomography.137 Perivascular neurotransmitter release might affect vascular permeability since intravenous SP injection enhanced dural vascular permeability in rats possibly as a response to alterations of endothelial cells. Widening of intercellular junctions, elevated amount of gaps and vesicles and structural modifications of endothelial cells might occur.138 More than a decade ago, neurokinins were considered the key molecules, and neurogenic inflammation the mechanism and target for migraine therapy.139,140 However, after the industry produced a series of specific neurokinin blockers and tested them in migraine (without significant effect), this line of research was abandoned.141 In fact, SP was at this time not considered a major peptide involved in pain.142
Integration of vasoconstrictors and vasodilators
In headache-free periods, research on blood samples revealed a significantly higher neurotransmitter baseline level of CGRP, SP and VIP in migraineurs when compared to controls,143–146 and significantly lower baseline levels of ET-1.147 In chronic migraine, interictal VIP levels were significantly increased, whereas interictal PACAP levels were not significantly different between groups (chronic migraine, episodic migraine and controls).148
CGRP in particular, is released in excess during headache attacks.10,11,149,150 As a response to CGRP depletion in mice, elevated baseline systolic BP and mean arterial pressure have been reported thereby revealing general effects of CGRP on vascular function.151 Even though CGRP has been reported to be the main peptide in primary headaches, PACAP is also considered an important player. A significant change in the neuropeptide plasma levels was seen when comparing the interictal and ictal phase in migraine and cluster headache.150,152,153 In clinical tests, intravenous PACAP-38 infusion resulted in decreased MCA blood flow velocity in the beginning of the ictal phase (experimental).154 Relative release of CGRP, PACAP, SP and VIP in migraine is summarized in Figure 2.
Figure 2.
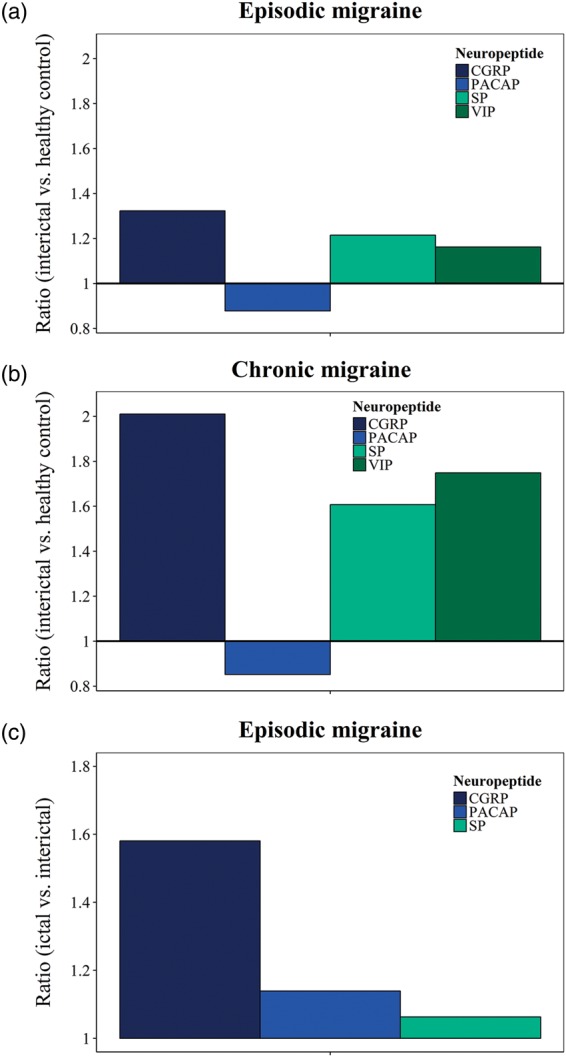
Summary of neurotransmitter release into the bloodstream in episodic and chronic migraine. Neurotransmitter baselines of calcitonin gene-related peptide (CGRP),11,143,145,146,155,258 pituitary adenylate cyclase-activating polypeptide (PACAP),148,150,259 substance P (SP)11,146,258 and vasoactive intestinal peptide (VIP)143,148 in episodic migraine (a) and chronic migraine (b) comparing the interictal headache period with healthy controls (measured as ratio). Neurotransmitter release of CGRP,10,11,149,150 PACAP150 and SP11 in episodic migraine (c) comparing the ictal with the interictal headache period (measured as ratio).
Even though migraine and cluster headache have been associated with autonomic and sensory neurotransmitter release during attacks,10–12,15,29,104,155,156 there still exists some degree of inconsistency between the findings.157 Our view is mainly based on the very short half-life of released neuromodulators in plasma, high levels of peptidases in the blood and lack of proper site of sampling, important factors to consider for the detection of an effect and to obtain consistent results across studies.158,159 Moreover, because of the inability of peptides to pass the BBB, it is likely that neuropeptide release from cranial ganglia emanates from sites outside the BBB. In addition, Hoffmann et al.160 investigated the effect of destroying primary trigeminal afferents on CGRP release into the jugular vein and cerebrospinal fluid, and suggested that stimulus-induced CGRP release mainly come from trigeminal afferents.
Multiple studies report an interconnection between neurotransmitter release in migraine, but also between neurotransmitter release and symptomatology/clinical phenotypes.146,161,162 To summarize, CGRP, PACAP, SP and VIP are considered cerebrovascular dilators, and ET-1, NPY and 5-HT cerebrovascular constrictors. However, potency and site of action vary between neurotransmitters. The observed neurotransmitter fluctuations have been proposed as a failing self-defense response163 and being due to dysfunctional biosynthesis (e.g. de novo synthesis), release and uptake.107
Possible importance of the endothelium
Endothelial dysfunction in migraine has been suggested by some authors.164 Indeed, a considerable number of patients show signs of dysfunctional endothelial cells in cerebral arteries evaluated using a breath holding or reactive hyperaemia index.165,166 Increases in CBF cause flow-mediated dilation induced by production of NO in endothelial cells, as a response to shear forces, which depend on blood flow and velocity, and pressure on the vessel walls. If the regulatory mechanisms of CBF cannot keep up with changes in physiological conditions, cellular stress, morphological abnormalities and altered gene expression patterns might occur and give rise to inflammatory responses.167,168 Parameters such as intima-media thickness, flow-mediated dilation, intracellular adhesion molecules (ICAM) and vascular cell adhesion molecules (VCAM) have all been described as important factors in the evaluation of endothelial function. Elevated serum levels of VCAM and ICAM have been linked to migraine in children and young adults.169 Cellular adhesion molecules are essential for immune cell transmigration across the BBB.170,171 Interestingly, there is a strong correlation between serum levels of ICAM-1 and interleukin 6 (IL-6) (proinflammatory cytokine) during migraine attacks; however, no correlation was observed in the headache-free period172 suggesting shared regulatory ictal mechanisms. In addition, tumor necrosis factor α (TNF-α) (proinflammatory molecule) results in increased VCAM-1 expression in human cerebral endothelial cells.170
Inflammation and BBB integrity and permeability
At present there is only limited evidence that points to an altered BBB permeability in migraineurs.173–175 BBB integrity has been investigated in animal models by means of Evans blue.176 The marker revealed that cranial ganglia are not protected by the BBB including the TG,79,175 while the brain and cerebral vessels have a patent BBB. Figure 3 visualizes the spinal cord and selected ganglia from Sprague-Dawley male rats in relation to the BBB. It shows that TG, C1-C3 DRG, SPG, OTG and SCG are located outside the BBB (blue structures colored primarily by the Evans blue albumin complex, but also other protein complexes,176 which indicates endothelial permeability). SPG stimulation is known to give rise to vasodilation and increased CBF, but these changes do not seem to affect the BBB permeability in rats.91 Experimentally induced dural inflammation, and secondarily ganglionic inflammation, does not seem to alter BBB passage either,175 even though inflammatory pain has been linked to BBB damage.177,178
Figure 3.

Sensory and autonomic ganglia in rats outside the blood–brain barrier (BBB) evaluated by means of in vivo staining with Evans Blue. (a) Trigeminal nerves, encircled by white dashed lines, after removal of the brain (dorsal view). The trigeminal nerves are covered by dura mater,82 colored by Evans Blue and thus outside the BBB. (b) Trigeminal nerve (removal of dura mater) revealing Evans Blue leakage at sites of the trigeminal ganglion (black arrows). (c) Spinal cord, brainstem and cerebellum (dorsal view) protected by the BBB (not colored by Evans Blue). Dura mater, encasing the spinal cord, is stained by Evans Blue (outside the BBB). The cervical nerves C1–C3 are marked and the C3 dorsal root ganglion (DRG) visible. (d,e) C1–C3 DRG, not protected by the BBB (blue), after removal of dura mater. (f,g) Sphenopalatine ganglion (SPG) (black arrows) located below the maxillary nerve (white arrow).82 The SPG is also outside the BBB (colored by Evans Blue). (h) Superior cervical ganglion (black arrow) not protected by the BBB (blue). (I) Otic ganglion (black arrow), outside the BBB (blue), positioned on the medial surface of the mandibular nerve (white arrow). The methodology is described by Eftekhari et al.79
If BBB disruption occurs in an episodic disorder such as migraine, it would for example occur through endothelial tight junctions to result in increased permeability.179 CGRP, SP and interleukin 1β (IL-1β), involved in primary headache pathophysiology, seem to have an effect on the brain endothelial permeability.180
In an ischemic stroke model, results indicate that expression of tight junction proteins gets enhanced when inhibiting matrix metallopeptidase (MMP), which points to BBB restoration.181 Focusing on primary headaches, specific MMP-2 and MMP-9 gene or haplotype polymorphisms have been associated with increased levels of MMP-21,82 and MMP-91,83 in the blood of migraineurs and are believed to be associated with alteration in BBB patency (however only studied in peripheral blood samples). Another study reported that CSD resulted in elevated MMP-9 in the ipsilateral cortex suggesting subsequent disruption of the BBB. Plasma protein leakage and edema were also observed, but not in MMP-9 null mice.184 An ex vivo study revealed that cytokines (e.g. TNF and interleukin subtypes) also might contribute to disruption of the BBB. A NeuroVascular Unit microfluidic two chamber system (vascular and brain chamber) was used to investigate the effect of inflammation on the BBB. TNF and interleukin subtypes were released as a response to lipopolysaccharide exposure, but the release varied in regards to chamber and exposure time when comparing each cytokine separately.185 Brown et al.185 suggested that cytokine release patterns possibly are involved in alteration of membrane permeability over time.185
In a BBB study involving gadolinium-enhanced MRI, cortical opening of the BBB and edema was identified in a familial hemiplegic migraine (FHM) type 2 patient where the attack was classified as severe.132 However, there are no clinical studies to date that link primary headaches and opening of the BBB. This is reviewed by Edvinsson and Tfelt-Hansen.174 In agreement, Schankin et al.173 recently found no change in the BBB (interictally and ictally) in acute GTN-induced migraine attacks in humans. This field requires more studies in order to prove or disprove the effect of primary headaches on BBB integrity and permeability.
Internal and external factors and genetic predisposition in primary headaches
Internal and external factors and genetic predisposition are known to affect the susceptibility to develop primary headaches. Therefore, individual variation in regards to response to triggers, neuronal hyperexcitability and vascular regulation might exist. Figure 4 illustrates a proposed hypothesis of migraine development due to variable predispositions from genetic and environmental factors. It is evident when comparing subject B-D (migraineurs), that there can be individual differences in the factors leading to development of migraine, and hence they will probably respond differently to treatment strategies (e.g. medication). Therefore, creation of individual treatment strategies might be beneficial for treating primary headache disorders.
Figure 4.

Genetic and environmental factors and migraine predisposition. Migraine conditions are primarily considered polygenic complex disorders of which genetic (inherited) and environmental risk factors, as well as interactions, constitute headache predisposition. (a) The bar plot illustrates a proposed hypothesis of individual migraine development. The migraine threshold, termed cutoff, shows the point at which subjects are diagnosed with migraine (ratio ≥ 1). Subject A is a non-migraineur and subject B-D migraineurs. (b) Subject B has developed migraine due to an equal predisposition from genetic (50%) and environmental factors (50%), subject C mainly due to environmental predisposition (92%) and subject D mainly due to genetic predisposition (75%).
Neuronal hyperexcitability and over-activated neuronal circuits are considered functional abnormalities in migraine and believed to be important in triggering headache/migraine attacks.186 More information on multiple internal and external headache triggers (e.g. sleep disturbance, stress, light, odors) can be found in thorough reviews by Ho et al.15 and Borkum.187 Migraine patients have subjectively reported a link between headache attacks and triggers (e.g. alcohol, food, visual triggers, stress/tiredness, environment), categorized by a principal component analysis.188 Migraine triggers have been proposed to lead to fluctuations in neurotransmitter levels and hypersensitivity in specific regions of the brain.12,15 In humans, short exposure to noise resulted in sensitization in subjects without headache; however, prolonged exposure led to desensitization. For headache patients, the trend was not clear.189 The effect of stress in rats included satellite glial cell (SGC) activation and elevated SP expression in the TG. IL-1β and interleukin 1 receptor type 1 (IL-1R1) expression in TG neurons were also elevated.190
Development of migraine and cluster headache also seems to be age- and gender-dependent,3,191,192 e.g. with particular dependence on gender hormones (e.g. rate of estrogen withdrawal in females).193 During the menstrual cycle, fluctuations in steroid hormones seem to be associated with changes in cerebrovascular reactivity of which vasodilation primarily is observed in the right hemisphere.194 Regarding the effect of age and sex on cerebrovascular regulation, inconsistency exists between human studies. One study reported that these two factors did not seem to affect cerebrovascular regulation to any major extent,195 while another study suggested an effect of age on BP and CBF.196 Moreover, neuronal activity and CBF are coupled tightly to brain metabolism.197,198 Increased brain activity causes increased local energy expenditure and is thus correlated with increased local CBF. For long, it has been discussed which physiological parameter comes first, metabolism or flow? Recent studies suggest that they indeed may be closely coupled.62,199 Moreover, Gantenbein et al.200 put forward a hypothesis stating that processing of sensory stimuli in migraine has a higher energy demand for neuronal function compared to that of healthy subjects, possibly linked to the theory of neuronal hyperexcitability.
In terms of genetic migraine predisposition, there are several lines of evidence including rare mutations in known proteins, single nucleotide polymorphisms and microsatellite markers. For the rare condition FHM, there are now at least three subtypes (FHM1-3) reporting gene mutations of calcium voltage-gated channel subunit alpha1 A (CACNA1A), ATPase Na+/K+ transporting subunit alpha 2 (ATP1A2) and sodium voltage-gated channel alpha subunit 1 (SCN1A).201 The subtypes seem to share the potential to modify glutamate signaling in the CNS, making the neurons more susceptible to depolarize (cortical hyperexcitability).202 The first mutation (FHM1) was detected in locus 19p13203 which relates to the P/Q calcium channel in the CNS, and affects calcium-dependent facilitation and synaptic plasticity.204 In a genetic family study, a considerable number of individuals (86%), carrying a mutation in ATP1A2, were diagnosed with FHM,205 which implies a high effect size.
More recently, genome-wide association studies (GWAS) have revolutionized the discovery of susceptibility variants for common traits and diseases (low-moderate effect size of markers), including primary headaches.16,17 The first GWAS paper investigating single nucleotide polymorphisms implicated in primary headaches, and consequently pathological mechanisms, was published in 2010 according to the GWAS catalog.206 Numerous headache-related single nucleotide polymorphisms and suggested migraine candidate genes (e.g. LDL receptor related protein 1 (LRP1), gap junction protein alpha 1 (GJA1), phosphatase and actin regulator 1 (PHACTR1)) have been identified and are involved in molecular mechanisms of ion homeostasis, oxidative stress and regulation of vascular tone.207,208 Although the vascular theory is no longer the primary theory on the origin of migraine, GWAS show that several candidate migraine genes are indeed related to the vasculature, based on data from recent meta-analyses.207,208 Table 1 summarizes selected candidate migraine genes detected by GWAS and their relation to cerebral vasculature in order to reveal potential interconnected biological mechanisms and effects on vascular function. The candidate genes seem to be involved in vascular diseases/disorders (e.g. cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), arteriovenous malformations (AVMs)), hemodynamics, regulation of transport across the BBB, neurogenesis and gliogenesis amongst others (Table 1). Other genome wide scans have been carried out investigating microsatellite markers in a cluster headache cohort. The hypocretin receptor 2 (HCRTR2) was detected as a candidate cluster headache gene,209 also detected by other genetic studies.210,211 HCRTR2 has also been associated with heart failure. The findings included (i) upregulation of HCRTR2 in dilated cardiomyopathy in humans and (ii) dysfunctional diastolic BP in HCRTR2 transcription-disrupted mice.212 In addition, ADCYAP1R1, a PACAP receptor gene believed to be involved in signal transmission in migraine, was detected by GWAS to be associated with the susceptibility to develop cluster headache.17 These findings could again give life to the importance of the vasculature in primary headaches, maybe with focus on hemodynamics and regulation of intracerebral flow and the neurovascular unit.
Table 1.
Selected candidate migraine genes detected by GWAS meta-analyses.
| Candidate genes | Gene description | Potential linkage to cerebral vasculature and primary headaches | References |
|---|---|---|---|
| HTRA1*a | HtrA serine peptidase 1 | ▪ Associated with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), a cerebral small vessel disease. | 222,260–262 |
| ▪ CARASIL: Reported vascular smooth muscle cell (vSMC) loss, change of small vessels in cerebral write matter and basal ganglia, and extracellular matrix breakdown. | |||
| ▪ Migraine with aura has been referred to as a central feature of CADASIL, the autosomal dominant variant. | |||
| GJA1* | Gap junction protein alpha 1 (also known as gap junction protein connexin43) | ▪ Functions in synaptic transmission and homeostasis of the brain/immune system. | 263,264 |
| ▪ Connexin43 (a main astroglial connexin) forming gap junction channels and hemichannels involved in (dys)regulation of BBB transportation in human endothelial cells. | |||
| ▪ Detected, especially, in the end-feetb of astrocytes, interacting with the endothelium, implicated in maintenance of BBB integrity. | |||
| HEY2*a | Hes related family bHLH transcription factor with YRPW motif 2 | ▪ HEY2 is expressed in the cerebral cortex and cranial ganglia, and believed to be involved in neuro- and gliogenesis in the developing brain, but also cognitive impairment. | 265,266 |
| ▪ HEY regulated genes are involved in neurogenesis, apoptosis and vascular development. | |||
| JAG1* | Jagged 1 | ▪ Associated with cerebral arteriovenous malformations (AVMs) and endothelial differentiation. | 267–269 |
| ▪ Lesions in AVMs might be linked to increased blood flow and vessel wall shear stress, hemodynamic factors. These hemodynamic factors might trigger release of angiogenic factors and inflammatory markers. | |||
| LRP1**a | Low density lipoprotein receptor-related protein 1 | ▪ LRP1 is involved in amyloid uptake by vSMCs and possibly cerebral amyloid angiopathy. | 270,271 |
| ▪ Cerebral amyloid angiopathy might be implicated in late onset of aura reporting occipital haemorrhage and subcortex haemosiderin deposition. | |||
| NRP1* | Neuropilin 1 | ▪ Implicated in vascular development (e.g. angiogenesis and vascularization) and formation of arteriovenous connections. | 272,273 |
| ▪ Knockdown of NRP1 in human brain microvascular endothelial cells affected chemokine levels through TNF-α and IFNγ, inflammatory responses. It also showed that NRP1 is involved in BBB function, (de)myelination of neurons and lymphocyte infiltration. | |||
| RNF213* | Ring finger protein 213 | ▪ Link between RNF213 and intracranial vasculopathies. | 274–277 |
| ▪ RNF213 variants affect the susceptibility to develop intracranial atherosclerotic stenosis and moyamoya disease. | |||
| ▪ Migraine-like attacks with aura observed in a female patient showing signs of moyamoya disease evaluated by computed tomography and angiograms. | |||
| SLC24A3* | Solute carrier family 24 member 3 (also known as NCKX3) | ▪ NCKX3 (Na+–Ca2+ exchanger dependent on K+) is believed to be involved in vision and olfaction through its regulatory effects on calcium homeostasis and thus Ca2+ flux. | 278,279 |
| ▪ Gene variants associated with variations in systolic blood pressure possibly through altered vascular tone as a response to changes in intracellular Ca2+ concentrations. This implies that NCKX3 might be involved in the vasoconstriction and vasodilation system. | |||
| TGFBR2** | Transforming growth factor beta receptor 2, | ▪ Implicated in intracerebral haemorrhages, expression of angiogenic factors, BBB integrity and permeability, vessel morphology including vascular branching investigated in conditional TGFBR2 knock-out mice (neural deletion of TGFBR2). This implies potential neurovascular interactions. | 280,281 |
| ▪ Gene variants of TGFBR2 suggested being involved in cerebral cavernous malformation (CCM) type 1 in terms of severity of the disease but also phenotypic variability. |
GWAS: genome-wide association studies.
Note: Results indicate involvement of the selected candidate genes in vascular disease/disorder, smooth muscle contractility or regulation of vascular tone amongst others (*Gormley et al.208; **Anttila et al.207 and Gormley et al.208)
Reported as ARMS2–HTRA1, HEY2–NCOA7 and LRP1-STAT6-SDR9C7 in Gormley et al.,208 respectively.
End-feet defined as “specialized foot-processes of perivascular astrocytes that are closely opposed to the outer surface of brain microvessels, and have specialized functions in inducing and regulating the BBB” by Abbott et al.282
Neuronal hyperexcitability and vascular regulation
Neurophysiological alterations such as altered neural plasticity, hyperexcitability and lack of habituation to sensory stimuli have been associated with primary headaches.213–215 Recent research focuses on the epigenome, which is believed to affect the susceptibility to develop migraine,216 with the field being at an emerging state. For example, a stressful environment during childhood has been linked to stress-induced alterations of homeostasis systems and development of migraine later in life217 thereby including the psychological aspect of primary headaches. One postulation is that epigenetic changes, occurring as a response to chronic stress, results in hyperexcitable neurons,218 thereby linking the epigenome to the CSD and trigeminovascular theory. However, methylation patterns have been found to vary considerably both between tissues and genes reported in a study investigating migraine-related genes.219
Focusing on vascular diseases, hypoperfusion in the white matter has been observed in CADASIL patients,220 who seem to have prolonged and more frequent CSD221 possibly due to parenchymal hyperexcitability.35 Interestingly, a study showed that migraine is present in 75% of CADASIL patients of which many reported aura. The migraine aura seemed to increase the odds of encephalopathy.222 Moreover, a microvascular vasoconstrictor anomaly has been associated with CADASIL in noradrenaline and angiotensin pathways, but not with a vasodilator anomaly.223 There are also some gender differences; brain vessels from males seem to be more sensitive to vasoconstrictors (angiotensin II and endothelin) than from females.224 Overall, these parameters might predispose individuals to enhanced migraine susceptibility or vice versa.
As a response to neuronal hyperexcitability, one would expect increased neurotransmitter release from perivascular ganglionic nerve endings on the cerebral vasculature. Moreover, neurotransmitters might be released directly into the extracellular space surrounding the neuronal cell bodies in the ganglia, triggered by neuronal depolarization evoking SGC activation through neurotransmitter-receptor binding.225,226 CGRP evoked release of NO and inflammatory responses in these glial cells, which was blocked by a CGRP receptor antagonist.226 The exact functional role of SGCs is still unknown; however, they are believed to be involved in control of the neuronal microenvironment.190,227 Neuronal excitability has been associated with SGC activation227 and various pain syndromes seem to be related to diverse activation states of the glial cells.228 The SGCs may thus be an important player in the sensitization process and chronification of migraine, in addition to the phenomena discussed in the previous sections. The SGCs constitute >90% of the total number of cells in the TG, and are linked to neurons via gap junctions (e.g. connexin) implicated in cell-cell signaling.226,229 Connexin, but also pannexin are involved in neuronal excitability and synaptic plasticity in the CNS forming gap junctions/hemichannels facilitating neuronal transmission.230,231 Experimental CSD provoked opening of pannexin1 channels, which was suggested to affect activation of the trigeminovascular system.232 Ashina et al.145 suggested that migraineurs suffer from abnormal neurogenic vascular control enduringly or permanently. Figure 5 gives an overview of possible pathophysiological effects, which occur due to migraine susceptible gene pools and external and internal headache triggers, all resulting in altered CBF.
Figure 5.

Genetic headache predisposition, headache triggers and cerebral blood flow (CBF). Potential effects of and link between genetic headache susceptibility (input: green), extrinsic and intrinsic headache triggers (input: green) and CBF (output: red) through multiple pathophysiological steps (simplified flow diagram).
Primary headaches and sensitization
There are several primary headache disorders; migraine and trigeminal autonomic cephalgias (cluster headache, chronic paroxysmal hemicranias, inter alia) of which all are characterized by pain varying in duration, location and quality with accompanied autonomic and sensory neurological signs/symptoms depending on the diagnosis.2 Cluster headache patients experience autonomic symptoms such as rhinorrhea, lacrimation and abnormal sweating (parasympathetic influence) and miosis and ptosis (sympathetic influence).2,233 Cranial sensory symptoms include experience of allodynia, and abnormalities in sensory perception of visual, olfactory and auditory stimuli.234 This implies involvement of the parasympathetic, sympathetic and sensory nervous systems which form complex functional units implicated in processing of impulses.235
During the headache attack phase, ganglia such as the TG and SPG, located outside the BBB, are activated and sensitized. The discovery of the sensitization process was pioneered by Burstein et al.236 Basically, upon repeated activation/stimulation (heat, cold or pressure stimuli), the subsequent response to the same stimulus was enhanced, being referred to as sensitization. Sensitization has been associated with release of neurotransmitters, including CGRP, and could be a part of the mechanism behind allodynia.236 Despite that the sensory trigeminal system contains several messenger molecules (e.g. CGRP, SP/NKA, PACAP and NO), only CGRP seems to be clearly associated with headache in migraine attacks.156,237,238 So far, release of CGRP/PACAP has only been positively linked to acute attacks of migraine, or in bouts of cluster headache.12,150,152,153,156,157 Moreover, patients with nasal congestion and rhinorrhea show release of VIP,237 which is observed in chronic migraine as well.143 Putatively, other molecules such as NO and cytokines are involved.149,239
Perivascular nerve terminals originating from sensory, parasympathetic and sympathetic ganglia, believed to be involved in primary headaches, have been observed on cerebral arteries and arterioles.47 In addition to the involvement of multiple neurotransmitters in CBF regulation, they are part of: (i) the neurovascular unit in the brain parenchyma, and (ii) larger arteries belonging to the circle of Willis and pial cortex arterioles.
Chronification of pain in primary headaches
Migraine experienced at least 15 days per month for >3 months is considered chronic. Chronic cluster headache, on the other hand, should last for at least a year with or without remission periods (<1 month).2 Chronic headache and headache frequency correlated significantly in adults240 possibly due to the detrimental effects of pathophysiological changes (e.g. trigeminovascular activation and sensitization) arising during headache attacks. In coherence, chronic migraine seems to increase with age.241,242 Trigeminovascular activation has also been observed as a response to inflammation, which could be involved in headache progression.243 At a cellular level, systemic inflammation of L4/5 DRG, involved in transmission of sensory signal, seems to modify the SGCs. Abnormal growth, formation of new gap junctions and increased ATP sensitivity of SGCs could potentially contribute to pain.244 Structural and functional modification of SGCs might be connected to cellular stress and inflammation.190,243,245 Other findings suggest that SGC activation, as a response to inflammation, results in modulated excitability of neurons in TG and DRG.244,246 We hypothesize that neurogenic inflammation might have a reinforcing effect on neuronal hyperexcitability, a feed-forward loop, probably contributing to development of chronic headache.
The chronic pain state of the brain is believed to result in further structural, functional and chemical modifications. Chronic pain might arise due to impairment of pathways in the CNS, but also of the peripheral nervous system, which collectively manifest phenotypically as altered sensation, emotion and cognition or modulated pain response.247 These changes are believed to be involved in disease progression from the episodic to chronic state. Therefore, it appears to be important for the future migraine management strategy to treat primary headaches early to avoid chronification bearing in mind that many primary headaches are considered complex polygenic disorders.
Episodic to chronic headache: Neurogenic inflammation and chronification
It is often argued, based on clinical observations, that a “migraine highway” is present in many patients. In the beginning, occasional attacks occur which, over the years, become frequent episodic migraine with up to 14 migraine headache days per month (according to ICHD-III beta2). This may, in some cases, expand even more, sometimes associated with migraine medication overuse.248,249 The mechanisms involved are still not clear. Vasodilation, release of vasoactive peptides and plasma-protein extravasation constitute factors that are involved in development of inflammatory responses, also referred to as neurogenic inflammation,76,139,250 which can be triggered by electrical stimulation of ganglia.76 Research shows that PACAP-38 is involved in central sensitization,154 neuronal and satellite glial cyclic adenosine monophosphate (cAMP) production in TG,251 and neurogenic inflammation.252 CGRP and VIP are associated with cAMP accumulation occurring simultaneously with cerebral vasodilation.112 Sarchielli et al.10 showed that cAMP and cGMP release occurred after release of CGRP, NKA and nitrites in migraine attacks measured in internal jugular venous blood. In addition, both CGRP and cAMP induced NO and inducible NOS release from TG glial cells in culture.226 However, single nucleotide polymorphisms in the neuronal NOS gene do not seem to increase the risk of migraine.253 Moreover, in migraineurs, release of other inflammatory markers such as interleukin (IL) subtypes (e.g. IL-1β, IL-6, IL-10) and TNF have been observed,239,254 which seems to be most likely in frequent or chronic migraine. A tendency of higher lymphocyte number in the peripheral blood was observed in chronic migraine compared to episodic migraine suggested to be linked to the inflammatory state of the patients.255 In the calvarial periosteum of chronic migraineurs, gene expression of proinflammatory markers was increased (e.g. IL-6, IL-1 receptor type 2 (IL1R2), C-C motif chemokine ligand 2 (CCL2), C-X3-C motif chemokine ligand 1 (CX3CL1)), whereas gene expression of markers involved in suppression of inflammation was decreased.256 Some of these markers might affect nociception. In addition, results show that application of IL-1β to the meninges is involved in activation and sensitization of nociceptors, which was not observed to the same degree for IL-6.257
Summary
Primary headaches are considered cerebral neurovascular disorders of which abnormal brain activity and CBF changes have been observed. CBF changes might occur as a response to pathophysiological changes such as neuronal hyperexcitability and autoregulatory impairment. GWAS studies, investigating the susceptibility to develop migraine, revealed suggestive candidate genes related to vascular diseases/mechanisms. One might postulate that some headache attacks are linked to underlying undiscovered vascular diseases. On the other hand, neuronal abnormalities have an effect on vascular mechanisms and function. Even though the original vascular theory is considered to be secondary in nature, these findings still emphasize the importance of the vasculature in primary headaches.
To summarize, this review gives rise to the following connections between primary headaches and CBF: (i) headache attacks start in the CNS and result in activated and sensitized ganglia and neurotransmitter release (e.g. CGRP, PACAP), (ii) the intracranial cerebral vasculature (large cerebral arteries and arterioles on the surface of the brain) is supplied with perivascular nerve fibers originating from the sensory, sympathetic and parasympathetic nervous systems, (iii) neurovascular activation putatively gives rise to vasoconstrictive/vasodilative phases and altered CBF and metabolism in the affected brain regions, (iv) neurotransmitter release seems to affect cAMP levels followed by inflammatory responses, but also plasma–protein extravasation in certain areas such as the dura mater (if SP is released), and (v) inflammation might reinforce neuronal hyperexcitability, pathological alterations and cellular dysfunctions through a feed-forward loop and thus contribute to chronification from the episodic state.
Finally, from a treatment perspective, it is important to detect and treat primary headache disorders in time in order to avoid chronification. Furthermore, new treatment strategies are required, which take into account that some individuals are genetically predisposed to develop primary headaches while others, primarily, might develop it as a response to environmental stimuli (e.g. stress).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by grants from the Lundbeck foundation (Denmark), Swedish research council (grant no 5958) and Heart-Lung foundation.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Pressman A, Jacobson A, Eguilos R, et al. Prevalence of migraine in a diverse community – electronic methods for migraine ascertainment in a large integrated health plan. Cephalalgia 2016; 36: 325–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.IHC. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013; 33: 629–808. [DOI] [PubMed] [Google Scholar]
- 3.Manzoni GC, Taga A, Russo M, et al. Age of onset of episodic and chronic cluster headache – a review of a large case series from a single headache centre. J Headache Pain 2016; 17: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magon S, May A, Stankewitz A, et al. Morphological abnormalities of thalamic subnuclei in migraine: a multicenter MRI study at 3 Tesla. J Neurosci 2015; 35: 13800–13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olesen J, Burstein R, Ashina M, et al. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol 2009; 8: 679–690. [DOI] [PubMed] [Google Scholar]
- 6.Hodkinson DJ, Veggeberg R, Wilcox SL, et al. Primary somatosensory cortices contain altered patterns of regional cerebral blood flow in the interictal phase of migraine. PLoS One 2015; 10: e0137971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodkinson DJ, Veggeberg R, Kucyi A, et al. Cortico-cortical connections of primary sensory areas and associated symptoms in migraine. eNeuro 2016; 3(6). DOI: 10.1523/ENEURO.0163-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schytz HW, Barlose M, Guo S, et al. Experimental activation of the sphenopalatine ganglion provokes cluster-like attacks in humans. Cephalalgia 2013; 33: 831–841. [DOI] [PubMed] [Google Scholar]
- 9.Felisati G, Arnone F, Lozza P, et al. Sphenopalatine endoscopic ganglion block: a revision of a traditional technique for cluster headache. Laryngoscope 2006; 116: 1447–1450. [DOI] [PubMed] [Google Scholar]
- 10.Sarchielli P, Alberti A, Codini M, et al. Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia 2000; 20: 907–918. [DOI] [PubMed] [Google Scholar]
- 11.Gallai V, Sarchielli P, Floridi A, et al. Vasoactive peptide levels in the plasma of young migraine patients with and without aura assessed both interictally and ictally. Cephalalgia 1995; 15: 384–390. [DOI] [PubMed] [Google Scholar]
- 12.Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 1993; 33: 48–56. [DOI] [PubMed] [Google Scholar]
- 13.Edvinsson L, Uddman R. Neurobiology in primary headaches. Brain Res Brain Res Rev 2005; 48: 438–456. [DOI] [PubMed] [Google Scholar]
- 14.Edvinsson L, Villalon CM, MaassenVanDenBrink A. Basic mechanisms of migraine and its acute treatment. Pharmacol Ther 2012; 136: 319–333. [DOI] [PubMed] [Google Scholar]
- 15.Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol 2010; 6: 573–582. [DOI] [PubMed] [Google Scholar]
- 16.Anttila V, Stefansson H, Kallela M, et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet 2010; 42: 869–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacchelli E, Cainazzo MM, Cameli C, et al. A genome-wide analysis in cluster headache points to neprilysin and PACAP receptor gene variants. J Headache Pain 2016; 17: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leo AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol 1944; 7: 359–390. [DOI] [PubMed] [Google Scholar]
- 19.Wolff HG. Headache and other head pain, New York: Oxford University Press, 1948. [Google Scholar]
- 20.Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol 1981; 9: 344–352. [DOI] [PubMed] [Google Scholar]
- 21.Shevel E. The extracranial vascular theory of migraine: an artificial controversy. J Neural Transm 2011; 118: 525–530. [DOI] [PubMed] [Google Scholar]
- 22.Amin FM, Asghar MS, Hougaard A, et al. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol 2013; 12: 454–461. [DOI] [PubMed] [Google Scholar]
- 23.MaassenVanDenBrink A, Ibrahimi K, Edvinsson L. Intracranial and extracranial arteries in migraine. Lancet Neurol 2013; 12: 847–848. [DOI] [PubMed] [Google Scholar]
- 24.Costa C, Tozzi A, Rainero I, et al. Cortical spreading depression as a target for anti-migraine agents. J Headache Pain 2013; 14: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Genizi J, Khourieh Matar A, Zelnik N, et al. Frequency of pediatric migraine with aura in a clinic-based sample. Headache 2016; 56: 113–117. [DOI] [PubMed] [Google Scholar]
- 26.Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol 2013; 75: 365–391. [DOI] [PubMed] [Google Scholar]
- 27.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 2014; 15: 379–393. [DOI] [PubMed] [Google Scholar]
- 28.Moskowitz MA. Pathophysiology of headache – past and present. Headache 2007; April(47 Suppl 1): S58–S63. [DOI] [PubMed]
- 29.Goadsby PJ, Edvinsson L, Ekman R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol 1988; 23: 193–196. [DOI] [PubMed] [Google Scholar]
- 30.Moskowitz MA, Buzzi MG, Sakas DE, et al. Pain mechanisms underlying vascular headaches. Progress Report 1989. Rev Neurol 1989; 145: 181–193. [PubMed] [Google Scholar]
- 31.Moskowitz MA. Defining a pathway to discovery from bench to bedside: the trigeminovascular system and sensitization. Headache 2008; 48: 688–690. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen AN, Fabricius M, Lauritzen M. Scanning laser-Doppler flowmetry of rat cerebral circulation during cortical spreading depression. J Vasc Res 2000; 37: 513–522. [DOI] [PubMed] [Google Scholar]
- 33.Woods RP, Iacoboni M, Mazziotta JC. Brief report: bilateral spreading cerebral hypoperfusion during spontaneous migraine headache. N Engl J Med 1994; 331: 1689–1692. [DOI] [PubMed] [Google Scholar]
- 34.Olesen J, Lauritzen M, Tfelt-Hansen P, et al. Spreading cerebral oligemia in classical- and normal cerebral blood flow in common migraine. Headache 1982; 22: 242–248. [DOI] [PubMed] [Google Scholar]
- 35.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015; 95: 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maniyar FH, Sprenger T, Monteith T, et al. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain 2014; 137: 232–241. [DOI] [PubMed] [Google Scholar]
- 37.Schulte LH, May A. The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain 2016; 139: 1987–1993. [DOI] [PubMed] [Google Scholar]
- 38.Weiller C, May A, Limmroth V, et al. Brain stem activation in spontaneous human migraine attacks. Nat Med 1995; 1: 658–660. [DOI] [PubMed] [Google Scholar]
- 39.Akerman S, Holland PR, Goadsby PJ. Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci 2011; 12: 570–584. [DOI] [PubMed] [Google Scholar]
- 40.Jullienne A, Badaut J. Molecular contributions to neurovascular unit dysfunctions after brain injuries: lessons for target-specific drug development. Future Neurol 2013; 8: 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol 2006; 100: 1059–1064. [DOI] [PubMed] [Google Scholar]
- 42.Aaslid R, Lindegaard KF, Sorteberg W, et al. Cerebral autoregulation dynamics in humans. Stroke 1989; 20: 45–52. [DOI] [PubMed] [Google Scholar]
- 43.Paulson OB, Strandgaard S, Edvinsson L. Cerebral autoregulation. Cerebrovasc Brain Metab Rev 1990; 2: 161–192. [PubMed] [Google Scholar]
- 44.Skinhoj E. Regulation of cerebral blood flow as a single function of the interstitial pH in the brain. A hypothesis. Acta Neurol Scand 1966; 42: 604–607. [PubMed] [Google Scholar]
- 45.Shapiro W, Wasserman AJ, Patterson JL., Jr Mechanism and pattern of human cerebrovascular regulation after rapid changes in blood CO2 tension. J Clin Invest 1966; 45: 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Azevedo E, Rosengarten B, Santos R, et al. Interplay of cerebral autoregulation and neurovascular coupling evaluated by functional TCD in different orthostatic conditions. J Neurol 2007; 254: 236–241. [DOI] [PubMed] [Google Scholar]
- 47.Edvinsson L, Krause DN. Cerebral blood flow and metabolism, 2nd ed Philadelphia, PA: Lippincott Williams & Wilkins, 2002. [Google Scholar]
- 48.Edward M. The cerebral circulation. Br J Anaesth 2001; 1: 5. [Google Scholar]
- 49.Olesen J, Friberg L, Olsen TS, et al. Timing and topography of cerebral blood flow, aura, and headache during migraine attacks. Ann Neurol 1990; 28: 791–798. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez del Rio M, Bakker D, Wu O, et al. Perfusion weighted imaging during migraine: spontaneous visual aura and headache. Cephalalgia 1999; 19: 701–707. [DOI] [PubMed] [Google Scholar]
- 51.Loehrer E, Vernooij MW, van der Lugt A, et al. Migraine and cerebral blood flow in the general population. Cephalalgia 2015; 35: 190–198. [DOI] [PubMed] [Google Scholar]
- 52.Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab 2009; 29: 1517–1527. [DOI] [PubMed] [Google Scholar]
- 53.Chang JC, Shook LL, Biag J, et al. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain 2010; 133: 996–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willis T. Cerebri Anatome: cui accessit nervorum descriptio et usus. London: Londini: typis Ja. Flesher, impensis Jo. Martyn & Ja. Allestry apud insigne Campanae in Ciemeterio D. Pauli, 1664, p.538.
- 55.Nielsen KC, Owman C. Adrenergic innervation of pial arteries related to the circle of Willis in the cat. Brain Res 1967; 6: 773–776. [DOI] [PubMed] [Google Scholar]
- 56.Edvinsson L, MacKenzie ET. Amine mechanisms in the cerebral circulation. Pharmacol Rev 1976; 28: 275–348. [PubMed] [Google Scholar]
- 57.Edvinsson LM ET, McCulloch J. Cerebral blood flow and metabolism, New York: Raven Press, 1993. [Google Scholar]
- 58.Harper AM, Lassen NA, MacKenzie ET, et al. Proceedings: the upper limit of ‘autoregulation’ of cerebral blood flow in the baboon. J Physiol 1973; 234: 61p–62p. [PubMed] [Google Scholar]
- 59.Larsson LI, Edvinsson L, Fahrenkrug J, et al. Immunohistochemical localization of a vasodilatory polypeptide (VIP) in cerebrovascular nerves. Brain Res 1976; 113: 400–404. [DOI] [PubMed] [Google Scholar]
- 60.Goadsby PJ, Edvinsson L. Human in vivo evidence for trigeminovascular activation in cluster headache. Neuropeptide changes and effects of acute attacks therapies. Brain 1994; 117(Pt 3): 427–434. [DOI] [PubMed] [Google Scholar]
- 61.McCulloch J, Uddman R, Kingman TA, et al. Calcitonin gene-related peptide: functional role in cerebrovascular regulation. Proc Natl Acad Sci USA 1986; 83: 5731–5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whittaker JR, Driver ID, Bright MG, et al. The absolute CBF response to activation is preserved during elevated perfusion: implications for neurovascular coupling measures. Neuroimage 2016; 125: 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moulton EA, Becerra L, Borsook D. An fMRI case report of photophobia: activation of the trigeminal nociceptive pathway. Pain 2009; 145: 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baeres FM, Moller M. Origin of PACAP-immunoreactive nerve fibers innervating the subarachnoidal blood vessels of the rat brain. J Cereb Blood Flow Metab 2004; 24: 628–635. [DOI] [PubMed] [Google Scholar]
- 65.Mayberg M, Langer RS, Zervas NT, et al. Perivascular meningeal projections from cat trigeminal ganglia: possible pathway for vascular headaches in man. Science 1981; 213: 228–230. [DOI] [PubMed] [Google Scholar]
- 66.Uddman R, Hara H, Edvinsson L. Neuronal pathways to the rat middle meningeal artery revealed by retrograde tracing and immunocytochemistry. J Auton Nerv Syst 1989; 26: 69–75. [DOI] [PubMed] [Google Scholar]
- 67.Uddman R, Edvinsson L. Neuropeptides in the cerebral circulation. Cerebrovasc Brain Metab Rev 1989; 1: 230–252. [PubMed] [Google Scholar]
- 68.Edvinsson L, Hara H, Uddman R. Retrograde tracing of nerve fibers to the rat middle cerebral artery with true blue: colocalization with different peptides. J Cereb Blood Flow Metab 1989; 9: 212–218. [DOI] [PubMed] [Google Scholar]
- 69.Liu Y, Zhang M, Broman J, et al. Central projections of sensory innervation of the rat superficial temporal artery. Brain Res 2003; 966: 126–133. [DOI] [PubMed] [Google Scholar]
- 70.Liu Y, Broman J, Edvinsson L. Central projections of sensory innervation of the rat superior sagittal sinus. Neuroscience 2004; 129: 431–437. [DOI] [PubMed] [Google Scholar]
- 71.Liu Y, Broman J, Edvinsson L. Central projections of the sensory innervation of the rat middle meningeal artery. Brain Res 2008; 1208: 103–110. [DOI] [PubMed] [Google Scholar]
- 72.Uddman R, Edvinsson L, Ekman R, et al. Innervation of the feline cerebral vasculature by nerve fibers containing calcitonin gene-related peptide: trigeminal origin and co-existence with substance P. Neurosci Lett 1985; 62: 131–136. [DOI] [PubMed] [Google Scholar]
- 73.Edvinsson L, Elsas T, Suzuki N, et al. Origin and co-localization of nitric oxide synthase, CGRP, PACAP, and VIP in the cerebral circulation of the rat. Microsc Res Tech 2001; 53: 221–228. [DOI] [PubMed] [Google Scholar]
- 74.Uddman R, Tajti J, Moller S, et al. Neuronal messengers and peptide receptors in the human sphenopalatine and otic ganglia. Brain Res 1999; 826: 193–199. [DOI] [PubMed] [Google Scholar]
- 75.Tajti J, Uddman R, Moller S, et al. Messenger molecules and receptor mRNA in the human trigeminal ganglion. J Auton Nerv Syst 1999; 76: 176–183. [DOI] [PubMed] [Google Scholar]
- 76.Samsam M, Covenas R, Csillik B, et al. Depletion of substance P, neurokinin A and calcitonin gene-related peptide from the contralateral and ipsilateral caudal trigeminal nucleus following unilateral electrical stimulation of the trigeminal ganglion; a possible neurophysiological and neuroanatomical link to generalized head pain. J Chem Neuroanat 2001; 21: 161–169. [DOI] [PubMed] [Google Scholar]
- 77.Samsam M, Covenas R, Ahangari R, et al. Simultaneous depletion of neurokinin A, substance P and calcitonin gene-related peptide from the caudal trigeminal nucleus of the rat during electrical stimulation of the trigeminal ganglion. Pain 2000; 84: 389–395. [DOI] [PubMed] [Google Scholar]
- 78.Tuka B, Helyes Z, Markovics A, et al. Peripheral and central alterations of pituitary adenylate cyclase activating polypeptide-like immunoreactivity in the rat in response to activation of the trigeminovascular system. Peptides 2012; 33: 307–316. [DOI] [PubMed] [Google Scholar]
- 79.Eftekhari S, Salvatore CA, Johansson S, et al. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res 2015; 1600: 93–109. [DOI] [PubMed] [Google Scholar]
- 80.Edvinsson L, Ekman R, Jansen I, et al. Peptide-containing nerve fibers in human cerebral arteries: immunocytochemistry, radioimmunoassay, and in vitro pharmacology. Ann Neurol 1987; 21: 431–437. [DOI] [PubMed] [Google Scholar]
- 81.Uddman R, Goadsby PJ, Jansen I, et al. PACAP, a VIP-like peptide: immunohistochemical localization and effect upon cat pial arteries and cerebral blood flow. J Cereb Blood Flow Metab 1993; 13: 291–297. [DOI] [PubMed] [Google Scholar]
- 82.Shankland WE., 2nd The trigeminal nerve. Part I: an over-view. Cranio 2000; 18: 238–248. [DOI] [PubMed] [Google Scholar]
- 83.Escott KJ, Beattie DT, Connor HE, et al. Trigeminal ganglion stimulation increases facial skin blood flow in the rat: a major role for calcitonin gene-related peptide. Brain Res 1995; 669: 93–99. [DOI] [PubMed] [Google Scholar]
- 84.Csati A, Tajti J, Tuka B, et al. Calcitonin gene-related peptide and its receptor components in the human sphenopalatine ganglion – interaction with the sensory system. Brain Res 2012; 1435: 29–39. [DOI] [PubMed] [Google Scholar]
- 85.Csati A, Tajti J, Kuris A, et al. Distribution of vasoactive intestinal peptide, pituitary adenylate cyclase-activating peptide, nitric oxide synthase, and their receptors in human and rat sphenopalatine ganglion. Neuroscience 2012; 202: 158–168. [DOI] [PubMed] [Google Scholar]
- 86.Pipolo C, Bussone G, Leone M, et al. Sphenopalatine endoscopic ganglion block in cluster headache: a reevaluation of the procedure after 5 years. Neurol Sci 2010; June(31 Suppl 1): S197–S199. [DOI] [PubMed]
- 87.Cady R, Saper J, Dexter K, et al. A double-blind, placebo-controlled study of repetitive transnasal sphenopalatine ganglion blockade with tx360((R)) as acute treatment for chronic migraine. Headache 2015; 55: 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cady RK, Saper J, Dexter K, et al. Long-term efficacy of a double-blind, placebo-controlled, randomized study for repetitive sphenopalatine blockade with bupivacaine vs. saline with the Tx360 device for treatment of chronic migraine. Headache 2015; 55: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hara H, Zhang QJ, Kuroyanagi T, et al. Parasympathetic cerebrovascular innervation: an anterograde tracing from the sphenopalatine ganglion in the rat. Neurosurgery 1993; 32: 822–827. discussion 7. [DOI] [PubMed] [Google Scholar]
- 90.Seylaz J, Hara H, Pinard E, et al. Effect of stimulation of the sphenopalatine ganglion on cortical blood flow in the rat. J Cereb Blood Flow Metab 1988; 8: 875–878. [DOI] [PubMed] [Google Scholar]
- 91.Levi H, Schoknecht K, Prager O, et al. Stimulation of the sphenopalatine ganglion induces reperfusion and blood-brain barrier protection in the photothrombotic stroke model. PLoS One 2012; 7: e39636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Steinberg A, Frederiksen SD, Blixt FW, et al. Expression of messenger molecules and receptors in rat and human sphenopalatine ganglion indicating therapeutic targets. J Headache Pain 2016; 17: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hara H, Jansen I, Ekman R, et al. Acetylcholine and vasoactive intestinal peptide in cerebral blood vessels: effect of extirpation of the sphenopalatine ganglion. J Cereb Blood Flow Metab 1989; 9: 204–211. [DOI] [PubMed] [Google Scholar]
- 94.Hardebo JE, Suzuki N, Ekblad E, et al. Vasoactive intestinal polypeptide and acetylcholine coexist with neuropeptide Y, dopamine-beta-hydroxylase, tyrosine hydroxylase, substance P or calcitonin gene-related peptide in neuronal subpopulations in cranial parasympathetic ganglia of rat. Cell Tissue Res 1992; 267: 291–300. [DOI] [PubMed] [Google Scholar]
- 95.Li AL, Zhang JD, Xie W, et al. Inflammatory changes in paravertebral sympathetic ganglia in two rat pain models. Neurosci Bull Epub ahead of print 22 May 2017. DOI: 10.1007/s12264-017-0142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bogduk N. The anatomical basis for cervicogenic headache. J Manipulative Physiol Ther 1992; 15: 67–70. [PubMed] [Google Scholar]
- 97.Calhoun AH, Ford S, Millen C, et al. The prevalence of neck pain in migraine. Headache 2010; 50: 1273–1277. [DOI] [PubMed] [Google Scholar]
- 98.Filosa JA, Morrison HW, Iddings JA, et al. Beyond neurovascular coupling, role of astrocytes in the regulation of vascular tone. Neuroscience 2016; 323: 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci 2007; 10: 1369–1376. [DOI] [PubMed] [Google Scholar]
- 100.Faraci FM. Effects of endothelin and vasopressin on cerebral blood vessels. Am J Physiol 1989; 257: H799–H803. [DOI] [PubMed] [Google Scholar]
- 101.Edvinsson L. Characterization of the contractile effect of neuropeptide Y in feline cerebral arteries. Acta Physiol Scand 1985; 125: 33–41. [DOI] [PubMed] [Google Scholar]
- 102.Nagasawa S, Handa H, Naruo Y, et al. Experimental cerebral vasospasm. Part 2. Contractility of spastic arterial wall. Stroke 1983; 14: 579–84. [DOI] [PubMed] [Google Scholar]
- 103.Kimball RW, Friedman AP, Vallejo E. Effect of serotonin in migraine patients. Neurology 1960; 10: 107–111. [DOI] [PubMed] [Google Scholar]
- 104.Kallela M, Farkkila M, Saijonmaa O, et al. Endothelin in migraine patients. Cephalalgia 1998; 18: 329–332. [DOI] [PubMed] [Google Scholar]
- 105.Uddman R, Tajti J, Cardell LO, et al. Endothelin ETA and ETB receptor expression in the human trigeminal ganglion. Neuro Endocrinol Lett 2006; 27: 345–349. [PubMed] [Google Scholar]
- 106.May A, Goadsby PJ. Pharmacological opportunities and pitfalls in the therapy of migraine. Curr Opin Neurol 2001; 14: 341–345. [DOI] [PubMed] [Google Scholar]
- 107.Nagata E, Shibata M, Hamada J, et al. Plasma 5-hydroxytryptamine (5-HT) in migraine during an attack-free period. Headache 2006; 46: 592–596. [DOI] [PubMed] [Google Scholar]
- 108.Deen M, Christensen CE, Hougaard A, et al. Serotonergic mechanisms in the migraine brain – a systematic review. Cephalalgia 2017; 37: 251–264. [DOI] [PubMed] [Google Scholar]
- 109.Daugaard D, Thomsen LL, Iversen HK, et al. Delayed migraine-like headache in healthy volunteers after a combination of acetazolamide and glyceryl trinitrate. Cephalalgia 2009; 29: 1294–300. [DOI] [PubMed] [Google Scholar]
- 110.Ashina M, Hansen JM, Olesen J. Pearls and pitfalls in human pharmacological models of migraine: 30 years’ experience. Cephalalgia 2013; 33: 540–553. [DOI] [PubMed] [Google Scholar]
- 111.Edvinsson L. Functional role of perivascular peptides in the control of cerebral circulation. Trends Neurosci 1985; 8: 126–131. [Google Scholar]
- 112.Edvinsson L, Fredholm BB, Hamel E, et al. Perivascular peptides relax cerebral arteries concomitant with stimulation of cyclic adenosine monophosphate accumulation or release of an endothelium-derived relaxing factor in the cat. Neurosci Lett 1985; 58: 213–217. [DOI] [PubMed] [Google Scholar]
- 113.Doods H, Arndt K, Rudolf K, et al. CGRP antagonists: unravelling the role of CGRP in migraine. Trends Pharmacol Sci 2007; 28: 580–587. [DOI] [PubMed] [Google Scholar]
- 114.Miyata A, Arimura A, Dahl RR, et al. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun 1989; 164: 567–574. [DOI] [PubMed] [Google Scholar]
- 115.Uddman R, Tajti J, Hou M, et al. Neuropeptide expression in the human trigeminal nucleus caudalis and in the cervical spinal cord C1 and C2. Cephalalgia 2002; 22: 112–116. [DOI] [PubMed] [Google Scholar]
- 116.Amin FM, Asghar MS, Guo S, et al. Headache and prolonged dilatation of the middle meningeal artery by PACAP38 in healthy volunteers. Cephalalgia 2012; 32: 140–149. [DOI] [PubMed] [Google Scholar]
- 117.Amin FM, Hougaard A, Schytz HW, et al. Investigation of the pathophysiological mechanisms of migraine attacks induced by pituitary adenylate cyclase-activating polypeptide-38. Brain 2014; 137: 779–794. [DOI] [PubMed] [Google Scholar]
- 118.Markovics A, Kormos V, Gaszner B, et al. Pituitary adenylate cyclase-activating polypeptide plays a key role in nitroglycerol-induced trigeminovascular activation in mice. Neurobiol Dis 2012; 45: 633–644. [DOI] [PubMed] [Google Scholar]
- 119.Guo S, Vollesen AL, Olesen J, et al. Premonitory and nonheadache symptoms induced by CGRP and PACAP38 in patients with migraine. Pain 2016; 157: 2773–2781. [DOI] [PubMed] [Google Scholar]
- 120.Asghar MS, Hansen AE, Kapijimpanga T, et al. Dilation by CGRP of middle meningeal artery and reversal by sumatriptan in normal volunteers. Neurology 2010; 75: 1520–1526. [DOI] [PubMed] [Google Scholar]
- 121.Bhatt DK, Gupta S, Olesen J, et al. PACAP-38 infusion causes sustained vasodilation of the middle meningeal artery in the rat: possible involvement of mast cells. Cephalalgia 2014; 34: 877–886. [DOI] [PubMed] [Google Scholar]
- 122.Kornieieva M, Hadidy A, Zhuravlova I. Variability of the middle meningeal artery subject to the shape of skull. J Neurol Surg B Skull Base 2015; 76: 451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Edvinsson L, Nilsson E, Jansen-Olesen I. Inhibitory effect of BIBN4096BS, CGRP(8-37), a CGRP antibody and an RNA-Spiegelmer on CGRP induced vasodilatation in the perfused and non-perfused rat middle cerebral artery. Br J Pharmacol 2007; 150: 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tuor UI, Edvinsson L, McCulloch J. Neuropeptide Y and cerebral blood flow. Lancet 1985; 1: 1271. [DOI] [PubMed] [Google Scholar]
- 125.Baun M, Hay-Schmidt A, Edvinsson L, et al. Pharmacological characterization and expression of VIP and PACAP receptors in isolated cranial arteries of the rat. Eur J Pharmacol 2011; 670: 186–194. [DOI] [PubMed] [Google Scholar]
- 126.Grande G, Labruijere S, Haanes KA, et al. Comparison of the vasodilator responses of isolated human and rat middle meningeal arteries to migraine related compounds. J Headache Pain 2014; 15: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Edvinsson L, Ekman R. Distribution and dilatory effect of vasoactive intestinal polypeptide (VIP) in human cerebral arteries. Peptides 1984; 5: 329–331. [DOI] [PubMed] [Google Scholar]
- 128.Schuh-Hofer S, Boehnke C, Reuter U, et al. A fluorescence-based method to assess plasma protein extravasation in rat dura mater using confocal laser scanning microscopy. Brain Res Brain Res Protoc 2003; 12: 77–82. [DOI] [PubMed] [Google Scholar]
- 129.Hoehlig K, Johnson KW, Pryazhnikov E, et al. A novel CGRP-neutralizing Spiegelmer attenuates neurogenic plasma protein extravasation. Br J Pharmacol 2015; 172: 3086–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bolay H, Reuter U, Dunn AK, et al. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 2002; 8: 136–142. [DOI] [PubMed] [Google Scholar]
- 131.Peitl B, Nemeth J, Szolcsanyi J, et al. Sensory nitrergic meningeal vasodilatation and non-nitrergic plasma extravasation in anaesthesized rats. Eur J Pharmacol 2004; 497: 293–299. [DOI] [PubMed] [Google Scholar]
- 132.Dreier JP, Jurkat-Rott K, Petzold GC, et al. Opening of the blood-brain barrier preceding cortical edema in a severe attack of FHM type II. Neurology 2005; 64: 2145–2147. [DOI] [PubMed] [Google Scholar]
- 133.Welsh JD, Muthard RW, Stalker TJ, et al. A systems approach to hemostasis: 4. How hemostatic thrombi limit the loss of plasma-borne molecules from the microvasculature. Blood 2016; 127: 1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Delepine L, Aubineau P. Plasma protein extravasation induced in the rat dura mater by stimulation of the parasympathetic sphenopalatine ganglion. Exp Neurol 1997; 147: 389–400. [DOI] [PubMed] [Google Scholar]
- 135.Markowitz S, Saito K, Moskowitz MA. Neurogenically mediated leakage of plasma protein occurs from blood vessels in dura mater but not brain. J Neurosci 1987; 7: 4129–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol 2005; 58: 698–705. [DOI] [PubMed] [Google Scholar]
- 137.Knotkova H, Pappagallo M. Imaging intracranial plasma extravasation in a migraine patient: a case report. Pain Med 2007; 8: 383–387. [DOI] [PubMed] [Google Scholar]
- 138.Ghabriel MN, Lu MX, Leigh C, et al. Substance P-induced enhanced permeability of dura mater microvessels is accompanied by pronounced ultrastructural changes, but is not dependent on the density of endothelial cell anionic sites. Acta Neuropathol 1999; 97: 297–305. [DOI] [PubMed] [Google Scholar]
- 139.Markowitz S, Saito K, Moskowitz MA. Neurogenically mediated plasma extravasation in dura mater: effect of ergot alkaloids. A possible mechanism of action in vascular headache. Cephalalgia 1988; 8: 83–91. [DOI] [PubMed] [Google Scholar]
- 140.Lee WS, Moskowitz MA. Conformationally restricted sumatriptan analogues, CP-122,288 and CP-122,638 exhibit enhanced potency against neurogenic inflammation in dura mater. Brain Res 1993; 626: 303–305. [DOI] [PubMed] [Google Scholar]
- 141.Diener HC, Gendolla A, Juptner M, et al. Emerging treatments in headache. Eur Neurol 1997; 38: 167–174. [DOI] [PubMed] [Google Scholar]
- 142.Tajti J, Szok D, Majlath Z, et al. Migraine and neuropeptides. Neuropeptides 2015; 52: 19–30. [DOI] [PubMed] [Google Scholar]
- 143.Cernuda-Morollon E, Martinez-Camblor P, Ramon C, et al. CGRP and VIP levels as predictors of efficacy of Onabotulinumtoxin type A in chronic migraine. Headache 2014; 54: 987–995. [DOI] [PubMed] [Google Scholar]
- 144.Cernuda-Morollon E, Martinez-Camblor P, Alvarez R, et al. Increased VIP levels in peripheral blood outside migraine attacks as a potential biomarker of cranial parasympathetic activation in chronic migraine. Cephalalgia 2015; 35: 310–316. [DOI] [PubMed] [Google Scholar]
- 145.Ashina M, Bendtsen L, Jensen R, et al. Evidence for increased plasma levels of calcitonin gene-related peptide in migraine outside of attacks. Pain 2000; 86: 133–138. [DOI] [PubMed] [Google Scholar]
- 146.Jang MU, Park JW, Kho HS, et al. Plasma and saliva levels of nerve growth factor and neuropeptides in chronic migraine patients. Oral Dis 2011; 17: 187–193. [DOI] [PubMed] [Google Scholar]
- 147.Komatsumoto S, Nara M. [Lower level of endothelin-1 in migraine with aura]. Rinsho Shinkeigaku 1995; 35: 1250–1252. [PubMed] [Google Scholar]
- 148.Cernuda-Morollon E, Riesco N, Martinez-Camblor P, et al. No change in interictal PACAP levels in peripheral blood in women with chronic migraine. Headache 2016; 56: 1448–1454. [DOI] [PubMed] [Google Scholar]
- 149.Rodriguez-Osorio X, Sobrino T, Brea D, et al. Endothelial progenitor cells: a new key for endothelial dysfunction in migraine. Neurology 2012; 79: 474–479. [DOI] [PubMed] [Google Scholar]
- 150.Tuka B, Helyes Z, Markovics A, et al. Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia 2013; 33: 1085–1095. [DOI] [PubMed] [Google Scholar]
- 151.Gangula PR, Zhao H, Supowit SC, et al. Increased blood pressure in alpha-calcitonin gene-related peptide/calcitonin gene knockout mice. Hypertension 2000; 35: 470–475. [DOI] [PubMed] [Google Scholar]
- 152.Zagami AS, Edvinsson L, Goadsby PJ. Pituitary adenylate cyclase activating polypeptide and migraine. Ann Clin Transl Neurol 2014; 1: 1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tuka B, Szabo N, Toth E, et al. Release of PACAP-38 in episodic cluster headache patients – an exploratory study. J Headache Pain 2016; 17: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schytz HW, Birk S, Wienecke T, et al. PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 2009; 132: 16–25. [DOI] [PubMed] [Google Scholar]
- 155.Cernuda-Morollon E, Larrosa D, Ramon C, et al. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology 2013; 81: 1191–1196. [DOI] [PubMed] [Google Scholar]
- 156.Edvinsson L, Goadsby PJ. Neuropeptides in migraine and cluster headache. Cephalalgia 1994; 14: 320–327. [DOI] [PubMed] [Google Scholar]
- 157.Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 1990; 28: 183–187. [DOI] [PubMed] [Google Scholar]
- 158.Warfvinge K, Edvinsson L. Pearls and pitfalls in neural CGRP immunohistochemistry. Cephalalgia 2013; 33: 593–603. [DOI] [PubMed] [Google Scholar]
- 159.Edvinsson L, Ekman R, Goadsby PJ. Measurement of vasoactive neuropeptides in biological materials: problems and pitfalls from 30 years of experience and novel future approaches. Cephalalgia 2010; 30: 761–766. [DOI] [PubMed] [Google Scholar]
- 160.Hoffmann J, Wecker S, Neeb L, et al. Primary trigeminal afferents are the main source for stimulus-induced CGRP release into jugular vein blood and CSF. Cephalalgia 2012; 32: 659–667. [DOI] [PubMed] [Google Scholar]
- 161.Jansen-Olesen I, Baun M, Amrutkar DV, et al. PACAP-38 but not VIP induces release of CGRP from trigeminal nucleus caudalis via a receptor distinct from the PAC1 receptor. Neuropeptides 2014; 48: 53–64. [DOI] [PubMed] [Google Scholar]
- 162.Bellamy JL, Cady RK, Durham PL. Salivary levels of CGRP and VIP in rhinosinusitis and migraine patients. Headache 2006; 46: 24–33. [DOI] [PubMed] [Google Scholar]
- 163.Arulmozhi DK, Veeranjaneyulu A, Bodhankar SL. Migraine: current concepts and emerging therapies. Vascul Pharmacol 2005; 43: 176–187. [DOI] [PubMed] [Google Scholar]
- 164.Gonzalez-Quintanilla V, Toriello M, Palacio E, et al. Systemic and cerebral endothelial dysfunction in chronic migraine. A case-control study with an active comparator. Cephalalgia 2016; 36: 552–560. [DOI] [PubMed] [Google Scholar]
- 165.Rajan R, Khurana D, Lal V. Interictal cerebral and systemic endothelial dysfunction in patients with migraine: a case-control study. J Neurol Neurosurg Psychiatry 2015; 86: 1253–1257. [DOI] [PubMed] [Google Scholar]
- 166.Jimenez Caballero PE, Munoz Escudero F. Peripheral endothelial function and arterial stiffness in patients with chronic migraine: a case-control study. J Headache Pain 2013; 14: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Cybulsky MI, Marsden PA. Effect of disturbed blood flow on endothelial cell gene expression: a role for changes in RNA processing. Arterioscler Thromb Vasc Biol 2014; 34: 1806–1808. [DOI] [PubMed] [Google Scholar]
- 168.Dirnagl U, Lindauer U, Villringer A. Role of nitric oxide in the coupling of cerebral blood flow to neuronal activation in rats. Neurosci Lett 1993; 149: 43–46. [DOI] [PubMed] [Google Scholar]
- 169.Sabri MR, Dehghan B, Yaghini O, et al. Endothelial dysfunction state in migraine headache and neutrally mediated syncope in children and young adults. J Res Med Sci 2015; 20: 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kallmann BA, Hummel V, Lindenlaub T, et al. Cytokine-induced modulation of cellular adhesion to human cerebral endothelial cells is mediated by soluble vascular cell adhesion molecule-1. Brain 2000; 123(Pt 4): 687–697. [DOI] [PubMed] [Google Scholar]
- 171.Weiss N, Miller F, Cazaubon S, et al. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta 2009; 1788: 842–857. [DOI] [PubMed] [Google Scholar]
- 172.Wang F, He Q, Ren Z, et al. Association of serum levels of intercellular adhesion molecule-1 and interleukin-6 with migraine. Neurol Sci 2015; 36: 535–540. [DOI] [PubMed] [Google Scholar]
- 173.Schankin CJ, Maniyar FH, Seo Y, et al. Ictal lack of binding to brain parenchyma suggests integrity of the blood-brain barrier for 11C-dihydroergotamine during glyceryl trinitrate-induced migraine. Brain 2016; 139: 1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Edvinsson L, Tfelt-Hansen P. The blood-brain barrier in migraine treatment. Cephalalgia 2008; 28: 1245–1258. [DOI] [PubMed] [Google Scholar]
- 175.Lundblad C, Haanes KA, Grande G, et al. Experimental inflammation following dural application of complete Freund’s adjuvant or inflammatory soup does not alter brain and trigeminal microvascular passage. J Headache Pain 2015; 16: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Saunders NR, Dziegielewska KM, Mollgard K, et al. Markers for blood-brain barrier integrity: how appropriate is Evans blue in the twenty-first century and what are the alternatives? Front Neurosci 2015; 9: 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Huber JD, Witt KA, Hom S, et al. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol 2001; 280: H1241–H1248. [DOI] [PubMed] [Google Scholar]
- 178.Huber JD, Hau VS, Borg L, et al. Blood-brain barrier tight junctions are altered during a 72-h exposure to lambda-carrageenan-induced inflammatory pain. Am J Physiol Heart Circ Physiol 2002; 283: H1531–H1537. [DOI] [PubMed] [Google Scholar]
- 179.Etame AB, Diaz RJ, Smith CA, et al. Focused ultrasound disruption of the blood-brain barrier: a new frontier for therapeutic delivery in molecular neurooncology. Neurosurg Focus 2012; 32: E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.DosSantos MF, Holanda-Afonso RC, Lima RL, et al. The role of the blood-brain barrier in the development and treatment of migraine and other pain disorders. Front Cell Neurosci 2014; 8: 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yang Y, Thompson JF, Taheri S, et al. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J Cereb Blood Flow Metab 2013; 33: 1104–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Goncalves FM, Martins-Oliveira A, Lacchini R, et al. Matrix metalloproteinase (MMP)-2 gene polymorphisms affect circulating MMP-2 levels in patients with migraine with aura. Gene 2013; 512: 35–40. [DOI] [PubMed] [Google Scholar]
- 183.Martins-Oliveira A, Goncalves FM, Speciali JG, et al. Specific matrix metalloproteinase 9 (MMP-9) haplotype affect the circulating MMP-9 levels in women with migraine. J Neuroimmunol 2012; 252: 89–94. [DOI] [PubMed] [Google Scholar]
- 184.Gursoy-Ozdemir Y, Qiu J, Matsuoka N, et al. Cortical spreading depression activates and upregulates MMP-9. J Clin Invest 2004; 113: 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Brown JA, Codreanu SG, Shi M, et al. Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit. J Neuroinflammation 2016; 13: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bolcskei H, Farkas B, Kocsis P, et al. Recent advancements in anti-migraine drug research: focus on attempts to decrease neuronal hyperexcitability. Recent Pat CNS Drug Discov 2009; 4: 14–36. [DOI] [PubMed] [Google Scholar]
- 187.Borkum JM. Migraine triggers and oxidative stress: a narrative review and synthesis. Headache 2016; 56: 12–35. [DOI] [PubMed] [Google Scholar]
- 188.Harle DE, Shepherd AJ, Evans BJ. Visual stimuli are common triggers of migraine and are associated with pattern glare. Headache 2006; 46: 1431–1440. [DOI] [PubMed] [Google Scholar]
- 189.Martin PR, Reece J, Forsyth M. Noise as a trigger for headaches: relationship between exposure and sensitivity. Headache 2006; 46: 962–972. [DOI] [PubMed] [Google Scholar]
- 190.Zhao YJ, Liu Y, Zhao YH, et al. Activation of satellite glial cells in the trigeminal ganglion contributes to masseter mechanical allodynia induced by restraint stress in rats. Neurosci Lett 2015; 602: 150–155. [DOI] [PubMed] [Google Scholar]
- 191.Macgregor EA, Rosenberg JD, Kurth T. Sex-related differences in epidemiological and clinic-based headache studies. Headache 2011; 51: 843–859. [DOI] [PubMed] [Google Scholar]
- 192.Manzoni GC. Gender ratio of cluster headache over the years: a possible role of changes in lifestyle. Cephalalgia 1998; 18: 138–142. [DOI] [PubMed] [Google Scholar]
- 193.Pavlovic JM, Allshouse AA, Santoro NF, et al. Sex hormones in women with and without migraine: evidence of migraine-specific hormone profiles. Neurology 2016; 87: 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Krejza J, Rudzinski W, Arkuszewski M, et al. Cerebrovascular reactivity across the menstrual cycle in young healthy women. Neuroradiol J 2013; 26: 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Madureira J, Castro P, Azevedo E. Demographic and systemic hemodynamic influences in mechanisms of cerebrovascular regulation in healthy adults. J Stroke Cerebrovasc Dis 2017; 26: 500–508. [DOI] [PubMed] [Google Scholar]
- 196.Xing CY, Tarumi T, Meijers RL, et al. Arterial pressure, heart rate, and cerebral hemodynamics across the adult life span. Hypertension 2017; 69: 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kozberg M, Hillman E. Neurovascular coupling and energy metabolism in the developing brain. Prog Brain Res 2016; 225: 213–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shu CY, Sanganahalli BG, Coman D, et al. New horizons in neurometabolic and neurovascular coupling from calibrated fMRI. Prog Brain Res 2016; 225: 99–122. [DOI] [PubMed] [Google Scholar]
- 199.Lundengard K, Cedersund G, Sten S, et al. Mechanistic mathematical modeling tests hypotheses of the neurovascular coupling in fMRI. PLoS Comput Biol 2016; 12: e1004971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gantenbein AR, Sandor PS, Fritschy J, et al. Sensory information processing may be neuroenergetically more demanding in migraine patients. Neuroreport 2013; 24: 202–205. [DOI] [PubMed] [Google Scholar]
- 201.de Vries B, Freilinger T, Vanmolkot KR, et al. Systematic analysis of three FHM genes in 39 sporadic patients with hemiplegic migraine. Neurology 2007; 69: 2170–2176. [DOI] [PubMed] [Google Scholar]
- 202.Pietrobon D. Familial hemiplegic migraine. Neurotherapeutics 2007; 4: 274–284. [DOI] [PubMed] [Google Scholar]
- 203.Terwindt GM, Ophoff RA, van Eijk R, et al. Involvement of the CACNA1A gene containing region on 19p13 in migraine with and without aura. Neurology 2001; 56: 1028–1032. [DOI] [PubMed] [Google Scholar]
- 204.Adams PJ, Rungta RL, Garcia E, et al. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc Natl Acad Sci USA 2010; 107: 18694–18699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Pelzer N, Blom DE, Stam AH, et al. Recurrent coma and fever in familial hemiplegic migraine type 2. A prospective 15-year follow-up of a large family with a novel ATP1A2 mutation. Cephalalgia 2017; 37: 737–755. [DOI] [PubMed] [Google Scholar]
- 206.MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 2017; 45: D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Anttila V, Winsvold BS, Gormley P, et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet 2013; 45: 912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gormley P, Anttila V, Winsvold BS, et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 2016; 48: 856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Baumber L, Sjostrand C, Leone M, et al. A genome-wide scan and HCRTR2 candidate gene analysis in a European cluster headache cohort. Neurology 2006; 66: 1888–1893. [DOI] [PubMed] [Google Scholar]
- 210.Weller CM, Wilbrink LA, Houwing-Duistermaat JJ, et al. Cluster headache and the hypocretin receptor 2 reconsidered: a genetic association study and meta-analysis. Cephalalgia 2015; 35: 741–747. [DOI] [PubMed] [Google Scholar]
- 211.Rainero I, Gallone S, Rubino E, et al. Haplotype analysis confirms the association between the HCRTR2 gene and cluster headache. Headache 2008; 48: 1108–1114. [DOI] [PubMed] [Google Scholar]
- 212.Perez MV, Pavlovic A, Shang C, et al. Systems genomics identifies a key role for hypocretin/orexin receptor-2 in human heart failure. J Am Coll Cardiol 2015; 66: 2522–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Demarquay G, Andre-Obadia N, Caclin A, et al. [Neurophysiological evaluation of cortical excitability in migraine: a review of the literature]. Rev Neurol 2013; 169: 427–435. [DOI] [PubMed] [Google Scholar]
- 214.Lai TH, Protsenko E, Cheng YC, et al. Neural plasticity in common forms of chronic headaches. Neural Plast 2015; 2015: 205985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Akerman S, Romero-Reyes M, Holland PR. Current and novel insights into the neurophysiology of migraine and its implications for therapeutics. Pharmacol Ther 2017; 172: 151–170. [DOI] [PubMed] [Google Scholar]
- 216.Roos-Araujo D, Stuart S, Lea RA, et al. Epigenetics and migraine; complex mitochondrial interactions contributing to disease susceptibility. Gene 2014; 543: 1–7. [DOI] [PubMed] [Google Scholar]
- 217.Tietjen GE, Buse DC, Collins SA. Childhood maltreatment in the migraine patient. Curr Treat Options Neurol 2016; 18: 31. [DOI] [PubMed] [Google Scholar]
- 218.Ochoa-Cortes F, Guerrero-Alba R, Valdez-Morales EE, et al. Chronic stress mediators act synergistically on colonic nociceptive mouse dorsal root ganglia neurons to increase excitability. Neurogastroenterol Motil 2014; 26: 334–345. [DOI] [PubMed] [Google Scholar]
- 219.Labruijere S, Stolk L, Verbiest M, et al. Methylation of migraine-related genes in different tissues of the rat. PLoS One 2014; 9: e87616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chabriat H, Pappata S, Ostergaard L, et al. Cerebral hemodynamics in CADASIL before and after acetazolamide challenge assessed with MRI bolus tracking. Stroke 2000; 31: 1904–1912. [DOI] [PubMed] [Google Scholar]
- 221.Eikermann-Haerter K, Yuzawa I, Dilekoz E, et al. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy syndrome mutations increase susceptibility to spreading depression. Ann Neurol 2011; 69: 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Tan RY, Markus HS. CADASIL: migraine, encephalopathy, stroke and their inter-relationships. PLoS One 2016; 11: e0157613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Hussain MB, Singhal S, Markus HS, et al. Abnormal vasoconstrictor responses to angiotensin II and noradrenaline in isolated small arteries from patients with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Stroke 2004; 35: 853–858. [DOI] [PubMed] [Google Scholar]
- 224.Ahnstedt H, Cao L, Krause DN, et al. Male-female differences in upregulation of vasoconstrictor responses in human cerebral arteries. PloS One 2013; 8: e62698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Takeda M, Takahashi M, Matsumoto S. Contribution of the activation of satellite glia in sensory ganglia to pathological pain. Neurosci Biobehav Rev 2009; 33: 784–792. [DOI] [PubMed] [Google Scholar]
- 226.Li J, Vause CV, Durham PL. Calcitonin gene-related peptide stimulation of nitric oxide synthesis and release from trigeminal ganglion glial cells. Brain Res 2008; 1196: 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.de Corato A, Capuano A, Curro D, et al. Trigeminal satellite cells modulate neuronal responses to triptans: relevance for migraine therapy. Neuron Glia Biol 2011; 7: 109–116. [DOI] [PubMed] [Google Scholar]
- 228.Ji RR, Berta T and Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain 2013; December(154 Suppl 1): S10–S28. [DOI] [PMC free article] [PubMed]
- 229.Poulsen JN, Warwick R, Duroux M, et al. Oxaliplatin enhances gap junction-mediated coupling in cell cultures of mouse trigeminal ganglia. Exp Cell Res 2015; 336: 94–99. [DOI] [PubMed] [Google Scholar]
- 230.Ardiles AO, Flores-Munoz C, Toro-Ayala G, et al. Pannexin 1 regulates bidirectional hippocampal synaptic plasticity in adult mice. Front Cell Neurosci 2014; 8: 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Han Y, Yu HX, Sun ML, et al. Astrocyte-restricted disruption of connexin-43 impairs neuronal plasticity in mouse barrel cortex. Eur J Neurosci 2014; 39: 35–45. [DOI] [PubMed] [Google Scholar]
- 232.Karatas H, Erdener SE, Gursoy-Ozdemir Y, et al. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013; 339: 1092–1095. [DOI] [PubMed] [Google Scholar]
- 233.May A. Update on the diagnosis and management of trigemino-autonomic headaches. J Neurol 2006; 253: 1525–1532. [DOI] [PubMed] [Google Scholar]
- 234.Harriott AM, Schwedt TJ. Migraine is associated with altered processing of sensory stimuli. Curr Pain Headache Rep 2014; 18: 458. [DOI] [PubMed] [Google Scholar]
- 235.Higbee D. Functional and anatomic relation of sphenopalatine ganglion to the autonomic nervous system. Arch Otolaryngol 1949; 50: 45–58. [DOI] [PubMed] [Google Scholar]
- 236.Burstein R, Yarnitsky D, Goor-Aryeh I, et al. An association between migraine and cutaneous allodynia. Ann Neurol 2000; 47: 614–624. [PubMed] [Google Scholar]
- 237.Edvinsson L, Goadsby PJ. Neuropeptides in the cerebral circulation: relevance to headache. Cephalalgia 1995; 15: 272–276. [DOI] [PubMed] [Google Scholar]
- 238.Goadsby PJ, Edvinsson L. Neuropeptide changes in a case of chronic paroxysmal hemicrania – evidence for trigemino-parasympathetic activation. Cephalalgia 1996; 16: 448–450. [DOI] [PubMed] [Google Scholar]
- 239.Perini F, D’Andrea G, Galloni E, et al. Plasma cytokine levels in migraineurs and controls. Headache 2005; 45: 926–931. [DOI] [PubMed] [Google Scholar]
- 240.Scher AI, Stewart WF, Ricci JA, et al. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 2003; 106: 81–89. [DOI] [PubMed] [Google Scholar]
- 241.Kandil MR, Hamed SA, Fadel KA, et al. Migraine in Assiut Governorate, Egypt: epidemiology, risk factors, comorbid conditions and predictors of change from episodic to chronic migraine. Neurol Res 2016; 38: 232–241. [DOI] [PubMed] [Google Scholar]
- 242.Galinski M, Sidhoum S, Cimerman P, et al. Early diagnosis of migraine necessary in children: 10-year follow-up. Pediatr Neurol 2015; 53: 319–323. [DOI] [PubMed] [Google Scholar]
- 243.Lukacs M, Haanes KA, Majlath Z, et al. Dural administration of inflammatory soup or Complete Freund’s Adjuvant induces activation and inflammatory response in the rat trigeminal ganglion. J Headache Pain 2015; 16: 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Blum E, Procacci P, Conte V, et al. Systemic inflammation alters satellite glial cell function and structure. A possible contribution to pain. Neuroscience 2014; 274: 209–217. [DOI] [PubMed] [Google Scholar]
- 245.Magni G, Merli D, Verderio C, et al. P2Y2 receptor antagonists as anti-allodynic agents in acute and sub-chronic trigeminal sensitization: role of satellite glial cells. Glia 2015; 63: 1256–1269. [DOI] [PubMed] [Google Scholar]
- 246.Takeda M, Tanimoto T, Kadoi J, et al. Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain 2007; 129: 155–166. [DOI] [PubMed] [Google Scholar]
- 247.Borsook D. Neurological diseases and pain. Brain 2012; 135: 320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Silberstein SD, Blumenfeld AM, Cady RK, et al. OnabotulinumtoxinA for treatment of chronic migraine: PREEMPT 24-week pooled subgroup analysis of patients who had acute headache medication overuse at baseline. J Neurol Sci 2013; 331: 48–56. [DOI] [PubMed] [Google Scholar]
- 249.Diener HC, Dodick DW, Goadsby PJ, et al. Utility of topiramate for the treatment of patients with chronic migraine in the presence or absence of acute medication overuse. Cephalalgia 2009; 29: 1021–1027. [DOI] [PubMed] [Google Scholar]
- 250.Phebus LA and Johnson KW. Dural inflammation model of migraine pain. Curr Protoc Neurosci 2001; Chapter 9: Unit9.1. [DOI] [PubMed]
- 251.Walker CS, Sundrum T, Hay DL. PACAP receptor pharmacology and agonist bias: analysis in primary neurons and glia from the trigeminal ganglia and transfected cells. Br J Pharmacol 2014; 171: 1521–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Schytz HW. Investigation of carbachol and PACAP38 in a human model of migraine. Dan Med Bull 2010; 57: B4223. [PubMed] [Google Scholar]
- 253.Garcia-Martin E, Martinez C, Serrador M, et al. Neuronal nitric oxide synthase (nNOS, NOS1) rs693534 and rs7977109 variants and risk for migraine. Headache 2015; 55: 1209–1217. [DOI] [PubMed] [Google Scholar]
- 254.Munno I, Marinaro M, Bassi A, et al. Immunological aspects in migraine: increase of IL-10 plasma levels during attack. Headache 2001; 41: 764–767. [DOI] [PubMed] [Google Scholar]
- 255.Forcelini CM, Dantas DC, Luz C, et al. Analysis of leukocytes in medication-overuse headache, chronic migraine, and episodic migraine. Headache 2011; 51: 1228–1238. [DOI] [PubMed] [Google Scholar]
- 256.Perry CJ, Blake P, Buettner C, et al. Upregulation of inflammatory gene transcripts in periosteum of chronic migraineurs: implications for extracranial origin of headache. Ann Neurol 2016; 79: 1000–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zhang X, Burstein R, Levy D. Local action of the proinflammatory cytokines IL-1beta and IL-6 on intracranial meningeal nociceptors. Cephalalgia 2012; 32: 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Fusayasu E, Kowa H, Takeshima T, et al. Increased plasma substance P and CGRP levels, and high ACE activity in migraineurs during headache-free periods. Pain 2007; 128: 209–214. [DOI] [PubMed] [Google Scholar]
- 259.Han X, Dong Z, Hou L, et al. Interictal plasma pituitary adenylate cyclase-activating polypeptide levels are decreased in migraineurs but remain unchanged in patients with tension-type headache. Clin Chim Acta 2015; 450: 151–154. [DOI] [PubMed] [Google Scholar]
- 260.Federico A, Di Donato I, Bianchi S, et al. Hereditary cerebral small vessel diseases: a review. J Neurol Sci 2012; 322: 25–30. [DOI] [PubMed] [Google Scholar]
- 261.Primo V, Graham M, Bigger-Allen AA, et al. Blood biomarkers in a mouse model of CADASIL. Brain Res 2016; 1644: 118–126. [DOI] [PubMed] [Google Scholar]
- 262.Verdura E, Herve D, Scharrer E, et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain 2015; 138: 2347–2358. [DOI] [PubMed] [Google Scholar]
- 263.Kaneko Y, Tachikawa M, Akaogi R, et al. Contribution of pannexin 1 and connexin 43 hemichannels to extracellular calcium-dependent transport dynamics in human blood-brain barrier endothelial cells. J Pharmacol Exp Ther 2015; 353: 192–200. [DOI] [PubMed] [Google Scholar]
- 264.Boulay AC, Cisternino S, Cohen-Salmon M. Immunoregulation at the gliovascular unit in the healthy brain: a focus on Connexin 43. Brain Behav Immun 2016; 56: 1–9. [DOI] [PubMed] [Google Scholar]
- 265.Jordan VK, Rosenfeld JA, Lalani SR, et al. Duplication of HEY2 in cardiac and neurologic development. Am J Med Genet A 2015; 167a: 2145–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Weber D, Wiese C, Gessler M. Hey bHLH transcription factors. Curr Top Dev Biol 2014; 110: 285–315. [DOI] [PubMed] [Google Scholar]
- 267.Weinsheimer S, Bendjilali N, Nelson J, et al. Genome-wide association study of sporadic brain arteriovenous malformations. J Neurol Neurosurg Psychiatry 2016; 87: 916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Yao Y, Yao J, Radparvar M, et al. Reducing Jagged 1 and 2 levels prevents cerebral arteriovenous malformations in matrix Gla protein deficiency. Proc Natl Acad Sci USA 2013; 110: 19071–19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Leblanc GG, Golanov E, Awad IA, et al. Biology of vascular malformations of the brain. Stroke 2009; 40: e694–e702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ruzali WA, Kehoe PG, Love S. LRP1 expression in cerebral cortex, choroid plexus and meningeal blood vessels: relationship to cerebral amyloid angiopathy and APOE status. Neurosci Lett 2012; 525: 123–128. [DOI] [PubMed] [Google Scholar]
- 271.Samanci B, Coban O, Baykan B. Late onset aura may herald cerebral amyloid angiopathy: a case report. Cephalalgia 2016; 36: 998–1001. [DOI] [PubMed] [Google Scholar]
- 272.Plein A, Fantin A, Ruhrberg C. Neuropilin regulation of angiogenesis, arteriogenesis, and vascular permeability. Microcirculation 2014; 21: 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Wang Y, Cao Y, Mangalam AK, et al. Neuropilin-1 modulates interferon-gamma-stimulated signaling in brain microvascular endothelial cells. J Cell Sci 2016; 129: 3911–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Zhang Q, Liu Y, Zhang D, et al. RNF213 as the major susceptibility gene for Chinese patients with moyamoya disease and its clinical relevance. J Neurosurg 2017; 126: 1106–1113. [DOI] [PubMed] [Google Scholar]
- 275.Bang OY, Chung JW, Cha J, et al. A polymorphism in RNF213 is a susceptibility gene for intracranial atherosclerosis. PLoS One 2016; 11: e0156607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kamada F, Aoki Y, Narisawa A, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet 2011; 56: 34–40. [DOI] [PubMed] [Google Scholar]
- 277.Smith KR, Leventer RJ, Mackay MT, et al. Identification of a novel RNF213 variant in a family with heterogeneous intracerebral vasculopathy. Int J Stroke 2014; 9: E26–E27. [DOI] [PubMed] [Google Scholar]
- 278.Jalloul AH, Szerencsei RT, Schnetkamp PP. Cation dependencies and turnover rates of the human K(+)-dependent Na(+)-Ca(2)(+) exchangers NCKX1, NCKX2, NCKX3 and NCKX4. Cell Calcium 2016; 59: 1–11. [DOI] [PubMed] [Google Scholar]
- 279.Citterio L, Simonini M, Zagato L, et al. Genes involved in vasoconstriction and vasodilation system affect salt-sensitive hypertension. PLoS One 2011; 6: e19620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Hellbach N, Weise SC, Vezzali R, et al. Neural deletion of Tgfbr2 impairs angiogenesis through an altered secretome. Hum Mol Genet 2014; 23: 6177–6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Choquet H, Pawlikowska L, Nelson J, et al. Polymorphisms in inflammatory and immune response genes associated with cerebral cavernous malformation type 1 severity. Cerebrovasc Dis 2014; 38: 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 2006; 7: 41–53. [DOI] [PubMed] [Google Scholar]