

In this study from Zhang et al., the authors found that H3K27me3 is strongly biased toward the maternal allele with some associated with DNA methylation–independent paternally expressed genes (PEGs) in human morulae. Their findings uncover the H3K27me3 landscape and establish a correlation between maternal-biased H3K27me3 and PEGs in human morulae.
Keywords: allele-specific gene expression, genomic imprinting, maternal-biased H3K27me3, DNA methylation, human morulae
Abstract
Genomic imprinting is an epigenetic mechanism by which genes are expressed in a parental origin–dependent manner. We recently discovered that, like DNA methylation, oocyte-inherited H3K27me3 can also serve as an imprinting mark in mouse preimplantation embryos. In this study, we found H3K27me3 is strongly biased toward the maternal allele with some associated with DNA methylation–independent paternally expressed genes (PEGs) in human morulae. The H3K27me3 domains largely overlap with DNA partially methylated domains (PMDs) and occupy developmental gene promoters. Thus, our study not only reveals the H3K27me3 landscape but also establishes a correlation between maternal-biased H3K27me3 and PEGs in human morulae.
Genomic imprinting is an epigenetic phenomenon that leads to parental allele-specific gene expression (Bartolomei and Ferguson-Smith 2011). Since the discovery of this phenomenon, allele-specific DNA methylation has been the only underlying mechanism (Bartolomei and Ferguson-Smith 2011). Allelic DNA methylation at the germline imprinting control regions (ICRs) is differentially established during gametogenesis and protected from the wave of global epigenetic reprogramming upon fertilization (Hanna and Kelsey 2014).
Recently, we discovered that oocyte-inherited H3K27me3 can also serve as a primary imprinting mark to regulate dozens of maternally imprinted genes, including Xist, in mouse preimplantation embryos (Inoue et al. 2017a,b). Oocyte-specific H3K27me3 domains are established during oogenesis by the Polycomb-repressive complex 2 (PRC2) (Cao et al. 2002) and are specifically maintained on the maternal allele, which can orchestrate paternal-specific gene expression in mouse early embryos (Inoue et al. 2018). Although the majority of the putative maternal H3K27me3-regulated imprinting genes exhibit transiently imprinted expression in mouse early embryos, at least five (i.e., Gab1, Phf17, Sfmbt2, Smoc1, and Slc38a4) maintain the imprinted expression to placenta and play an important role in placental development (Itoh et al. 2000; Sachs et al. 2000; Miri et al. 2013; Inoue et al. 2017a; Matoba et al. 2018).
To determine whether asymmetric H3K27me3 distribution is also correlated to allelic gene expression in human early embryos, we profiled both allelic gene expression and allelic H3K27me3 distribution in human morulae. The analysis revealed that human morula H3K27me3 is enriched at developmental gene promoters and tends to overlap with DNA partially methylated domains (PMDs). Importantly, H3K27me3 exhibits maternal-biased distribution and is enriched at promoters of several paternally expressed genes (PEGs) that do not contain germline differential DNA methylation. Taken together, our data suggest that maternal-biased H3K27me3 could regulate paternal-biased gene transcription in human early embryos.
Results and Discussion
H3K27me3 is enriched at developmental gene promoters in the human morula
To investigate the general distribution of H3K27me3 in the early human embryo, we profiled H3K27m3 using the cleavage under targets and release using nuclease (CUT&RUN) assay (Skene et al. 2018) with morulae pooled from two separate couples. In parallel, to determine whether any allelically expressed gene is regulated by H3K27me3 in human early embryos as in mice (Inoue et al. 2017a), we also performed single morula RNA sequencing (RNA-seq) using 15 morulae from five different couples (Fig. 1A). To infer the allelic information from the RNA-seq and the CUT&RUN data, maternal cumulus cells from the five couples were genotyped by whole-exome sequencing (WES) and/or whole-genome sequencing (WGS) (Fig. 1A; Supplemental Table S1).
Figure 1.
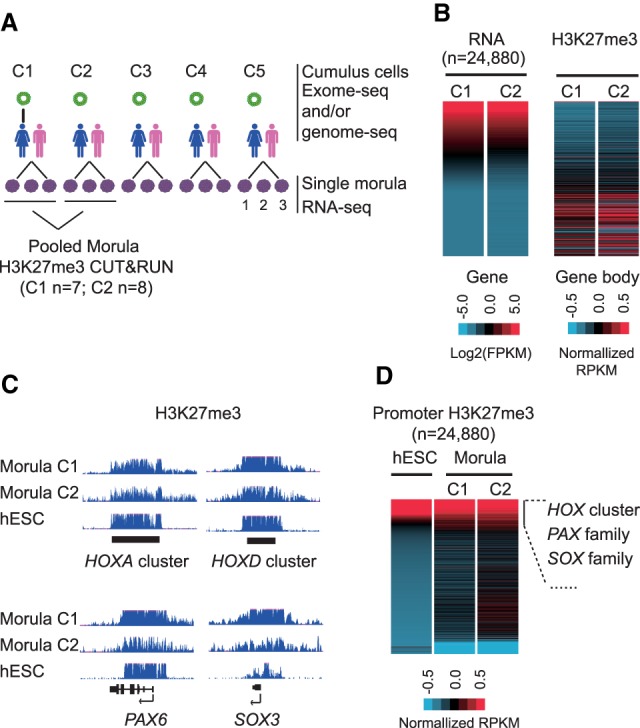
H3K27me3 is enriched at developmental gene promoters in the human morula. (A) A schematic summary of the data sets generated in this study. H3K27me3 CUT&RUN was performed using pooled morulae collected from couple 1 (C1; n = 7) and couple 2 (C2; n = 8). Three replicates for single morula RNA-seq were performed for each couple. (B) Heat map showing gene expression levels and H3K27me3 enrichment at the corresponding gene bodies in human morulae. RPKM values are Z-score normalized and log2-transform normalized for CUT&RUN and RNA-seq, respectively. (C) A genome browser view showing H3K27me3 enrichment at HOXA and HOXD clusters, PAX6, and SOX3 in both morula and human embryonic stem cells (hESCs). (D) Heat map showing enrichment of H3K27me3 at promoters (transcription start site ± 2.5 kb) in morulae (two replicates) and hESCs. RPKM was subject to Z-score normalization.
We first evaluated reproducibility of the two H3K27me3 CUT&RUN libraries generated using morulae from two pairs of couples. Even though only seven to eight morulae from each couple were available for the CUT&RUN assay, the two H3K27me3 CUT&RUN replicates showed high reproducibility (Supplemental Fig. S1A). Additionally, consistent with the notion that H3K27me3 is a transcription repressive histone modification (Martin and Zhang 2005), H3K27me3 enrichment is negatively correlated with gene expression (Fig. 1B). These results confirm the reliability of the H3K27me3 data sets and serve as the foundation for further analyses.
In mice, H3K27me3 is enriched at the promoters of developmental genes (e.g., Hox clusters, Pax and Sox families) in oocytes but is depleted upon fertilization and begins to be re-established at the blastocyst stage (Zheng et al. 2016). However, in contrast to mice (Supplemental Fig. S1B), classical Polycomb targets including the HOX clusters, PAX6, and SOX3 showed evident enrichment of H3K27me3 in human morulae (Fig. 1C). In addition, global analysis of H3K27me3 distribution revealed that H3K27me3 in human morulae, but not in mice (Supplemental Fig. S1C), is enriched at a subset of gene promoters, similar to that in embryonic stem cells (ESCs) (Fig. 1D; Liu et al. 2016; Zheng et al. 2016). The top 1500 genes with H3K27me3-occupied promoters are enriched for “developmental processes”–related gene ontology terms (Supplemental Fig. S1D). These findings are consistent with the known function of Polycomb proteins in regulating developmental genes and suggest that H3K27me3 at promoters may already begin to play a role in regulating gene transcription in human morulae.
Global H3K27me3 tends to overlap with PMDs in the human morula
In mice, although promoter H3K27me3 in oocytes is reprogrammed upon fertilization and becomes equalized with the paternal allele, maternal distal H3K27me3 domains are largely inherited, which leads to allele-specific H3K27me3 distribution during the preimplantation stage (Zheng et al. 2016). In addition, because de novo DNA methylation during oogenesis is transcription dependent (Chotalia et al. 2009; Veselovska et al. 2015), transcribed regions in oocytes overlap with DNA hypermethylated domains, whereas nontranscribed regions are associated with PMDs and H3K27me3 peaks (Zheng et al. 2016). Similar to these observations, H3K27me3 in human morulae also tends to overlap with PMDs exemplified by genome browser views (Fig. 2A). To gain further support for this initial observation, we identified PMDs (regions with length >10 kb and average DNA methylation level <40%) (see Supplemental Material) using the human morula whole-genome bisulfite sequencing (WGBS) data (Zhu et al. 2018) and found that, indeed, a subgroup of PMDs is enriched with H3K27me3 (Fig. 2B). To determine colocalization of H3K27me3 within PMDs, we identified H3K27me3 domains shared by both CUT&RUN data sets (Supplemental Fig. S2A,B; Supplemental Material) and found that the majority of H3K27me3 domains were located in morula PMDs (77.3%, 11,182 out of 14,455, vs. random regions: 49.7%, 7191 out of 14,455) (Fig. 2C). Collectively, these results suggest that, similar to mouse, human H3K27me3 enrichment in early embryos tends to overlap with PMDs, consistent with the reverse relationship between DNA methylation and H3K27me3 (Bogdanović et al. 2011; Zheng et al. 2016).
Figure 2.
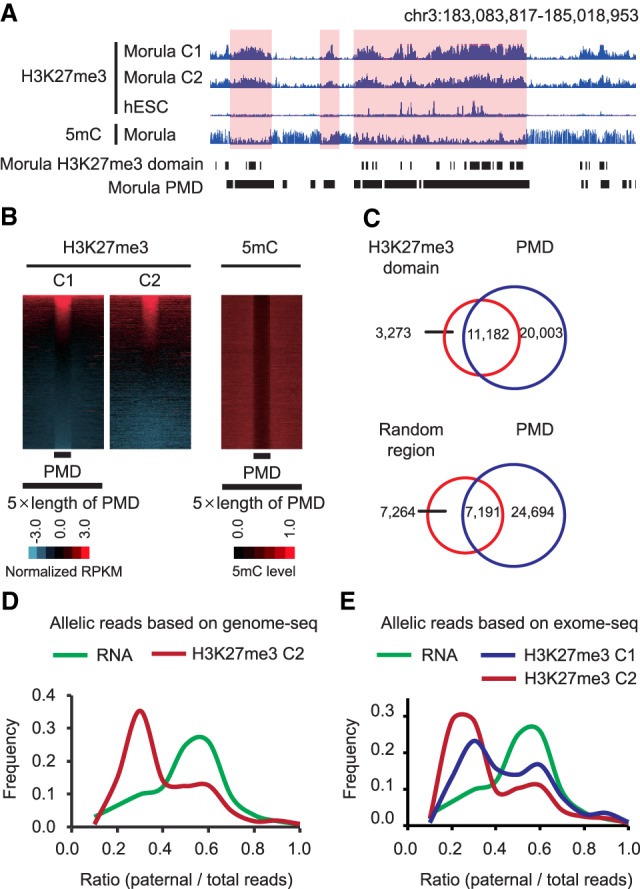
H3K27me3 tends to overlap with DNA PMDs and is biased toward the maternal allele in the human morula. (A) A representative genome browser view showing H3K27me3 signals and DNA methylation in morula (two replicates) and H3K27me3 distribution in hESCs. (B) Heat maps showing the H3K27me3 signals and DNA methylation levels (5mC/C) surrounding PMDs in morula (upstream and downstream region = 5 × length of PMD). (C) Venn diagram showing overlaps between H3K27me3 domains and PMDs in morula. Random regions within the same chromatin and same length for each H3K27me3 domain were used as controls. (D) Plot showing the distribution of paternal/total reads ratio at SNPs identified from morula H3K27me3 CUT&RUN data by corroborating WGS data of couple 2 cumulus cells. The x-axis represents the ratio of paternal/total reads. The y-axis represents the frequency of corresponding paternal/total reads ratio in the x-axis. (E) Plot showing distribution of paternal reads/total reads ratio at SNPs identified from morula H3K27me3 CUT&RUN data by corroborating WES data of couple 1 or couple 2 cumulus cells. The x-axis represents the ratio of paternal reads/total reads. The y-axis represents the frequency of corresponding paternal/total reads ratio in the x-axis.
H3K27me3 is biased toward the maternal allele in the human morula
To determine whether H3K27me3 in human morulae exhibits allelic-biased distribution, we identified parental-specific single-nucleotide polymorphisms (SNPs) in the H3K27me3 CUT&RUN data set from couple 2 by corroborating SNPs from the WGS data of cumulus cells (Fig. 1A; Supplemental Fig. S2C). Analysis of reliable SNPs with sufficient CUT&RUN reads coverage (with more than 10 monoclonal CUT&RUN reads; on average, 19.7) revealed that the majority of the qualified SNPs (5126 out of 9504) are located within H3K27me3 domains. Allele-specific enrichment analyses using total SNPs indicated that H3K27me3 exhibits a strong bias toward the maternal allele (Fig. 2D; Supplemental Table S2). Contrary to the maternal-biased H3K27me3, the median of the RNA-seq allelic reads ratio is ∼0.5 (Fig. 2D), which is consistent with the fact that most genes are already biallelically expressed in human morulae (Xue et al. 2013). We further confirmed this maternal-biased H3K27me3 distribution using another group of SNPs identified by corroborating the WES data of both couple 1 and couple 2 maternal cumulus cells (Fig. 2E; Supplemental Fig. S2C; Supplemental Table S2). Thus, we conclude that H3K27me3 exhibits a maternal-biased distribution in human morulae.
A subset of PEGs harbor germline DNA differentially methylated regions
The maternal-biased H3K27me3 distribution in human morulae prompted us to ask whether such asymmetric distribution may correlate with paternal-biased gene expression. To this end, we performed single morula RNA sequencing using 15 morulae from five different couples (three morulae per couple) (Fig. 1A). Pairwise comparative analysis indicated that these samples exhibit high correlation (Supplemental Fig. S3A; Supplemental Table S3). For identification of genes with paternal-biased expression, we first identified parental-specific SNPs in RNA-seq data sets by corroborating SNPs determined using WES data of the maternal cumulus cells and computed the allele-specific read counts at the gene level in each embryo (Supplemental Fig. S2C; Supplemental Table S4). To minimize the false positives potentially caused by cis-regulatory effects or random allelic variation, only genes that showed consistent paternal-biased expression in at least two embryos were considered as PEGs. Through these analyses, we identified a total of 44 PEGs in human morulae (Fig. 3A). Notably, several known maternally imprinted genes such as SNRPN, PEG10, and R3HCC1 were among the gene list (Court et al. 2014; Sanchez-Delgado et al. 2016), validating our approach.
Figure 3.
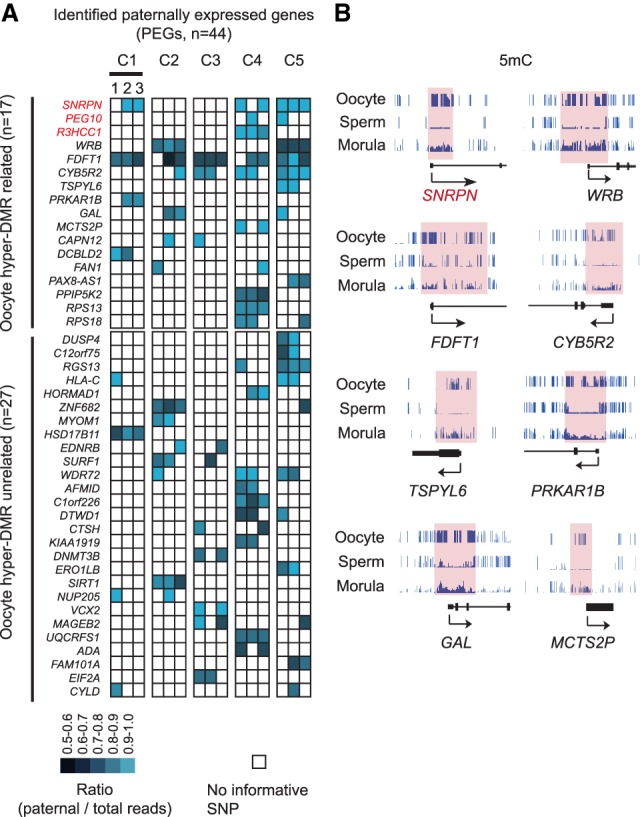
A subset of paternally expressed genes (PEGs) harbor germline differentially methylated regions (DMRs). (A) Heat map showing PEGs identified in this study. Each column represents one morula; a total of three morulae were analyzed for each couple. PEGs with oocyte hyper-DMR within 10 kb of the genes were considered as DMR-related (top panel). The remaining was considered as DMR unrelated (bottom panel). (B) Genome browser view showing examples of PEGs containing oocyte hyper-DMRs at promoter regions. DNA methylation in oocyte, sperm, and morula is shown.
To determine the mechanism underlying the paternal-biased expression, we first examined germline differentially methylated regions (DMRs) by comparing the sperm and oocyte DNA methylome and identified 8109 oocyte hyper-DMRs (Supplemental Fig. S2C). PEGs that are overlapped or are <10 kb from any DMRs are considered potentially regulated by allelic DNA methylation. We chose 10 kb as a cutoff because the majority of known mammalian germline DMRs are located within 10 kb of the nearby imprinted genes (Xie et al. 2012). Among the 44 PEGs, 17 are associated with maternal allele hyper-DMRs (Fig. 3A,B). Notably, some of these PEGs (e.g., FDFT1, CYB5R2, PRKAP1B, TSPYL6, GAL, and MCTS2P) had been previously reported to be associated with transient germline DMRs in human early embryos, as these germline DMRs become either hypermethylated or hypomethylated in later development (Sanchez-Delgado et al. 2016). Interestingly, WRB has been linked to comitant strabismus with a parent-of-origin effect in a GWAS study (Shaaban et al. 2018), and its expression level in placenta of IVF embryos has been linked to its promoter methylation level and subfertility (Litzky et al. 2017). Thus, our analysis not only confirmed the existence of imprinted expression regulated by transient DMRs in early human embryos, but also identified putative germline DNA methylation–independent PEGs.
A subset of putative DNA methylation–independent PEGs is associated with H3K27me3 domains
Next, we explored whether the remaining 27 putative oocyte DNA methylation–independent PEGs could be potentially regulated by H3K27me3. To this end, we asked whether these 27 PEGs harbor promoter H3K27me3 domains and found five genes (i.e., DUSP4, EDNRB, ERO1LB, FAM101A, and MAGEB2) have H3K27me3 domains within their promoters (±2.5 kb of the transcription start sites) (Fig. 4A; Supplemental Fig. S4A). Because of various limitations in obtaining allelic information of human embryos (see below), only the H3K27me3 domain at the FAM101A promoter has been covered by an informative SNP. Importantly, the CUT&RUN reads at the SNP exhibited a maternal bias (maternal/paternal reads ratio = 3.3), which reversely correlated with the paternal-biased expression (Fig. 3A), suggesting that the maternal-biased H3K27me3 distribution is likely involved in its paternal-biased expression. Thus, the H3K27me3 imprinting mechanism initially discovered in mice is at least partly conserved in early human embryos.
Figure 4.
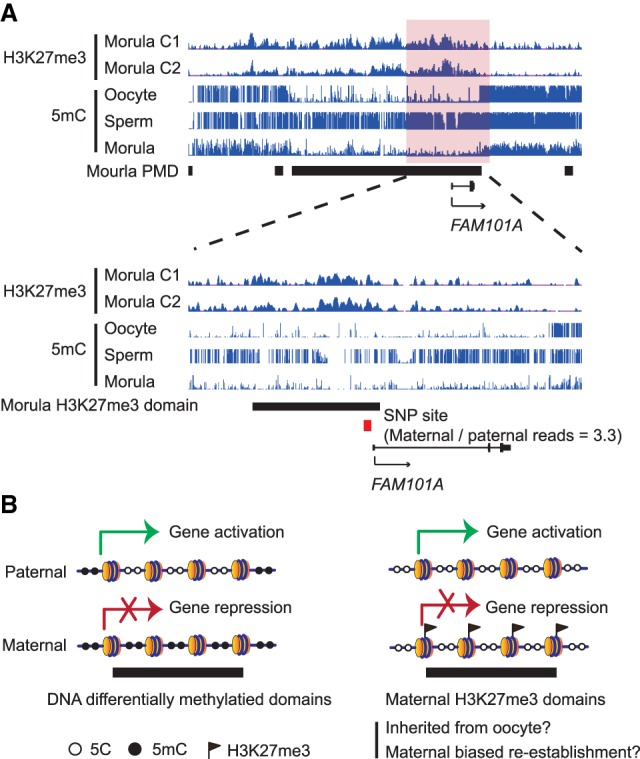
Putative H3K27m3 associated PEGs and proposed model for mechanisms underlying paternal-biased expression. (A) A genome browser view showing H3K27me3 enrichment in morula and DNA methylation levels in oocyte, sperm, and morula at FAM101A locus. Red square represents the SNP identified from H3K27me3 CUT&RUN data. (B) Schematic model showing paternal allelic gene expression can be regulated by either allelic DNA methylation or allelic H3K27me3 in human morulae.
Maternal-biased H3K27me3 in the human morula
In this study, we revealed that H3K27me3 domains in the human morula are largely overlapped with PMDs. We also showed that H3K27me3 at gene bodies is reversely correlated with transcriptional activity. These observations are consistent with previous reports in mouse embryos (Liu et al. 2016; Zheng et al. 2016). However, unlike mouse, in which H3K27me3 is generally depleted from Polycomb targets (i.e., HOXA cluster) upon fertilization (Zheng et al. 2016), there is evident enrichment of H3K27me3 at developmental gene promoters in human morulae. These data suggest that H3K27me3 reprogramming at specific loci can take place in a species-specific manner during early embryonic development, and H3K27me3 could play a more important role in gene regulation during early embryogenesis in human than that in mouse.
Because the morulae pooled for each H3K27me3 CUT&RUN experiment were from the same couple, we were able to identify parental-specific SNPs and reveal maternal-biased H3K27me3 enrichment in human morulae. As with mice, the maternal-biased H3K27me3 signals are also likely to be inherited from oocytes (Zheng et al. 2016). In support of this notion, H3K27me3 is localized specifically in the maternal pronuclei of human PN3 zygotes (van de Werken et al. 2014), indicating the inheritance of H3K27me3 from the oocyte to zygotes. Additionally, the persistence of maternal-biased H3K27me3 also parallels with the longer persistence of oocyte-derived DNA methylation in human preimplantation embryos compared with that in mice (Zhu et al. 2018). However, we cannot rule out the possibility that the maternal-biased H3K27me3 at the morula stage is the result of a complete reprogramming followed by an unknown mechanism of maternal-specific deposition.
Maternal-biased H3K27me3 correlates with paternal-specific gene expression
Integrative analyses of DNA methylome and allelic RNA-seq identified not only known canonical DNA methylation-dependent imprinted genes but also novel PEGs that do not contain germline DMRs. Of the five PEGs with promoter H3K27me3 domains, but not differential DNA methylation, FAM101A exhibited maternal-biased H3K27me3 enrichment and paternal-biased gene expression, suggesting that the maternal H3K27me3 is likely responsible for its paternal-biased expression. Unfortunately, unlike mice, which have a well-established SNP database generated by deep DNA sequencing of various inbred strains (Doran et al. 2016), humans are more heterogeneous, and it would have been extremely costly to perform deep sequencing to identify all the SNPs from all of the couples donating their embryos in this study. Because H3K27me3 presents at developmental gene promoters in human morulae, as with human embryonic stem cells (hESCs), extrapolating this result means that potentially more PEGs may be regulated by maternal-specific H3K27me3 in human morulae. Taken together, these data support the notion that gene silencing mediated by maternal-specific H3K27me3, possibly inherited from oocytes, may lead to paternal specific gene expression in human morulae.
Although the function of maternal-biased H3K27me3 in early human embryos remains unclear, the H3K27me3-associated imprinted genes identified in our study are known to play important roles in development. For example, FAM101A (also named CFM2) is involved in skeletal development. In mice, Cfm1 and Cfm2 double-knockouts showed severe skeletal malformation of the spine (Mizuhashi et al. 2014). In addition, mutation of EDNRB is the second most common genetic lesion associated with a congenital intestine malformation: Hirschsprung disease (Amiel et al. 2008). ERO1LB has a specific role in oxidative protein folding in pancreatic β cells and has been implicated in the regulation of glucose hemostasis (Zito et al. 2010). Further studies are warranted to determine whether the imprinted expression of these genes can persist in later development and to understand whether such imprinted expression is functionally significant.
Limitations in identifying allelic transcripts using human embryos
It should be noted that the PEGs identified in this study are far from comprehensive because of the challenges in identifying imprinted genes in noninbred organisms, such as the human. In this study, only 4.4%–8.5% of the total genes have sufficient allelic information in each morula embryo to allow allelic transcriptional analysis. In addition, we requested at least two out of the 15 embryos to show paternal-biased expression to be qualified as PEG, which further limited the genes been analyzed. This could partly explain why only three known maternal imprinted genes are identified. Furthermore, only 6.7% (970 out of 14, 455) of H3K27me3 domains contain SNP information, which further restricts identification of H3K27me3-relevant PEGs. The limited number of informative SNPs could be owing to the genetic nature between the couples, relatively rare mutations present in exons, and/or limitations in sequencing depth. Thus, we believe a lot more H3K27me3 imprinted genes should exist. Future studies using androgenetic and parthenogenetic embryos, which face the challenge of collecting large number of oocytes, should be able to address these issues for comprehensively identifying allelically expressed genes in humans.
Materials and methods
Human embryo and cumulus cell collection
Sample collection from patients undergoing IVF treatment at Brigham and Women's Hospital was approved by the Partners institutional review board under protocol number 2017P001711. All patients had signed a consent for donation of their otherwise discarded material for research. Morulae were generated by intracytoplasmic sperm injection (ICSI). 2-PN zygotes were cultured in Global Total medium (LifeGlobal) under 5% O2, 6%–7% CO2, and 88%–89% N2. Embryos that did not meet the criteria for clinical purposes but had developed to the morula stage were collected and stored at −80°C for molecular studies. Specifically, the morulae were collected on day 6, when normal embryos would grow into blastocyst stage. Despite the embryos were developmentally delayed, they were morphologically normal. Cumulus cells were collected from blood-free cumulus pieces cut from oocyte–cumulus complexes (three complexes per patient). After exposure of the pieces to 60 IU/mL hyaluronidase, dispersed cells were rinsed before being collected and stored at −80°C for subsequent exome-seq and/or WGS.
Data availability
Both RNA-seq and CUT&RUN data sets generated in this study have been deposited in the Gene Expression Omnibus under accession GSE123023. The DNA methylome data for human oocyte, sperm, and morula were obtained from GSE81233. The H3K27me3 chromatin immunoprecipitation (ChIP) combined with high-throughput sequencing (CHIP-seq) data for mouse morula and mouse ESCs were obtained from GSE73952. The H3K27me3 ChIP-seq data for hESCs were obtained from Epigenomics Roadmap Project (Roadmap Epigenomics Consortium et al. 2015).
Supplementary Material
Acknowledgments
We thank Dr. Luis Tuesta for critical reading of the manuscript. The protein A-MNase for CUT&RUN is a kind gift from Dr. Steven Henikoff. This project was supported by the National Institutes of Health (R01HD092465) and Howard Hughes Medical Institute. Y.Z. is an investigator of the Howard Hughes Medical Institute.
Author contributions: Y.Z. conceived the project; W.Z., Z.C., and Y.Z. designed the experiments; W.Z., Z.C., and Q.Y. performed the experiments; W.Z., and Z.C. analyzed the sequencing data sets; D.Z., and C.R. collected the samples; and W.Z., Z.C., and Y.Z. interpreted the data and wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.323105.118.
References
- Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, et al. 2008. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet 45: 1–14. 10.1136/jmg.2007.053959 [DOI] [PubMed] [Google Scholar]
- Bartolomei MS, Ferguson-Smith AC. 2011. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol 3: a002592 10.1101/cshperspect.a002592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanović O, Long SW, van Heeringen SJ, Brinkman AB, Gómez-Skarmeta JL, Stunnenberg HG, Jones PL, Veenstra GJ. 2011. Temporal uncoupling of the DNA methylome and transcriptional repression during embryogenesis. Genome Res 21: 1313–1327. 10.1101/gr.114843.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043. 10.1126/science.1076997 [DOI] [PubMed] [Google Scholar]
- Chotalia M, Smallwood SA, Ruf N, Dawson C, Lucifero D, Frontera M, James K, Dean W, Kelsey G. 2009. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev 23: 105–117. 10.1101/gad.495809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simon C, Moore H, Harness JV, et al. 2014. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res 24: 554–569. 10.1101/gr.164913.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran AG, Wong K, Flint J, Adams DJ, Hunter KW, Keane TM. 2016. Deep genome sequencing and variation analysis of 13 inbred mouse strains defines candidate phenotypic alleles, private variation and homozygous truncating mutations. Genome Biol 17: 167 10.1186/s13059-016-1024-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna CW, Kelsey G. 2014. The specification of imprints in mammals. Heredity 113: 176–183. 10.1038/hdy.2014.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Suzuki T, Zhang Y. 2017a. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547: 419–424. 10.1038/nature23262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Zhang Y. 2017b. Genomic imprinting of Xist by maternal H3K27me3. Genes Dev 31: 1927–1932. 10.1101/gad.304113.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Chen Z, Yin Q, Zhang Y. 2018. Maternal Eed knockout causes loss of H3K27me3 imprinting and random X inactivation in the extraembryonic cells. Genes Dev 32: 1525–1536. 10.1101/gad.318675.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Yoshida Y, Nishida K, Narimatsu M, Hibi M, Hirano T. 2000. Role of Gab1 in heart, placenta, and skin development and growth factor- and cytokine-induced extracellular signal-regulated kinase mitogen-activated protein kinase activation. Mol Cell Biol 20: 3695–3704. 10.1128/MCB.20.10.3695-3704.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litzky JF, Deyssenroth MA, Everson TM, Armstrong DA, Lambertini L, Chen J, Marsit CJ. 2017. Placental imprinting variation associated with assisted reproductive technologies and subfertility. Epigenetics 12: 653–661. 10.1080/15592294.2017.1336589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang C, Liu W, Li J, Li C, Kou X, Chen J, Zhao Y, Gao H, Wang H, et al. 2016. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537: 558–562. 10.1038/nature19362 [DOI] [PubMed] [Google Scholar]
- Martin C, Zhang Y. 2005. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6: 838–849. 10.1038/nrm1761 [DOI] [PubMed] [Google Scholar]
- Matoba S, Wang H, Jiang L, Lu F, Iwabuchi KA, Wu X, Inoue K, Yang L, Press W, Lee JT, et al. 2018. Loss of H3K27me3 imprinting in somatic cell nuclear transfer embryos disrupts post-implantation development. Cell Stem Cell 23: 343–354.e5. 10.1016/j.stem.2018.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miri K, Latham K, Panning B, Zhong Z, Andersen A, Varmuza S. 2013. The imprinted polycomb group gene Sfmbt2 is required for trophoblast maintenance and placenta development. Development 140: 4480–4489. 10.1242/dev.096511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuhashi K, Kanamoto T, Moriishi T, Muranishi Y, Miyazaki T, Terada K, Omori Y, Ito M, Komori T, Furukawa T. 2014. Filamin-interacting proteins, Cfm1 and Cfm2, are essential for the formation of cartilaginous skeletal elements. Hum Mol Genet 23: 2953–2967. 10.1093/hmg/ddu007 [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs M, Brohmann H, Zechner D, Müller T, Hülsken J, Walther I, Schaeper U, Birchmeier C, Birchmeier W. 2000. Essential role of Gab1 for signaling by the c-Met receptor in vivo. J Cell Biol 150: 1375–1384. 10.1083/jcb.150.6.1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Delgado M, Court F, Vidal E, Medrano J, Monteagudo-Sánchez A, Martin-Trujillo A, Tayama C, Iglesias-Platas I, Kondova I, Bontrop R, et al. 2016. Human oocyte-derived methylation differences persist in the placenta revealing widespread transient imprinting. PLoS Genet 12: e1006427 10.1371/journal.pgen.1006427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaaban S, MacKinnon S, Andrews C, Staffieri SE, Maconachie GDE, Chan WM, Whitman MC, Morton SU, Yazar S, MacGregor S, et al. 2018. Genome-wide association study identifies a susceptibility locus for comitant esotropia and suggests a parent-of-origin effect. Invest Ophthalmol Vis Sci 59: 4054–4064. 10.1167/iovs.18-24082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, Henikoff S. 2018. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13: 1006–1019. 10.1038/nprot.2018.015 [DOI] [PubMed] [Google Scholar]
- van de Werken C, van der Heijden GW, Eleveld C, Teeuwssen M, Albert M, Baarends WM, Laven JS, Peters AH, Baart EB. 2014. Paternal heterochromatin formation in human embryos is H3K9/HP1 directed and primed by sperm-derived histone modifications. Nat Commun 5: 5868 10.1038/ncomms6868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veselovska L, Smallwood SA, Saadeh H, Stewart KR, Krueger F, Maupetit-Méhouas S, Arnaud P, Tomizawa S, Andrews S, Kelsey G. 2015. Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol 16: 209 10.1186/s13059-015-0769-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, Ren B. 2012. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148: 816–831. 10.1016/j.cell.2011.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Z, Huang K, Cai C, Cai L, Jiang CY, Feng Y, Liu Z, Zeng Q, Cheng L, Sun YE, et al. 2013. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 500: 593–597. 10.1038/nature12364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Huang B, Zhang B, Xiang Y, Du Z, Xu Q, Li Y, Wang Q, Ma J, Peng X, et al. 2016. Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol Cell 63: 1066–1079. 10.1016/j.molcel.2016.08.032 [DOI] [PubMed] [Google Scholar]
- Zhu P, Guo H, Ren Y, Hou Y, Dong J, Li R, Lian Y, Fan X, Hu B, Gao Y, et al. 2018. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat Genet 50: 12–19. 10.1038/s41588-017-0007-6 [DOI] [PubMed] [Google Scholar]
- Zito E, Chin KT, Blais J, Harding HP, Ron D. 2010. ERO1-β, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J Cell Biol 188: 821–832. 10.1083/jcb.200911086 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Both RNA-seq and CUT&RUN data sets generated in this study have been deposited in the Gene Expression Omnibus under accession GSE123023. The DNA methylome data for human oocyte, sperm, and morula were obtained from GSE81233. The H3K27me3 chromatin immunoprecipitation (ChIP) combined with high-throughput sequencing (CHIP-seq) data for mouse morula and mouse ESCs were obtained from GSE73952. The H3K27me3 ChIP-seq data for hESCs were obtained from Epigenomics Roadmap Project (Roadmap Epigenomics Consortium et al. 2015).