

In this study, Shah et al. show that Tgifs, which repress gene expression by binding directly to DNA or interacting with transforming growth factor β (TGFβ)-responsive SMADs, promote adenoma growth in the context of mutant Apc (adenomatous polyposis coli). Their findings suggest that Tgifs play an important role in regulating basic energy metabolism in normal cells and that this function of Tgifs is amplified in some cancers.
Keywords: TGFβ, Tgif, Wnt, cancer, colorectal cancer, gene expression, metabolism, repressor
Abstract
Tgif1 (thymine–guanine-interacting factor 1) and Tgif2 repress gene expression by binding directly to DNA or interacting with transforming growth factor (TGF) β-responsive SMADs. Tgifs are essential for embryogenesis and may function in tumor progression. By analyzing both gain and loss of Tgif function in a well-established mouse model of intestinal cancer, we show that Tgifs promote adenoma growth in the context of mutant Apc (adenomatous polyposis coli). Despite the tumor-suppressive role of TGFβ signaling, transcriptome profiling of colon tumors suggests minimal effect of Tgifs on the TGFβ pathway. Instead, it appears that Tgifs, which are up-regulated in Apc mutant colon tumors, contribute to reprogramming metabolic gene expression. Integrating gene expression data from colon tumors with other gene expression and chromatin-binding data identifies a set of direct Tgif target genes encoding proteins involved in acetyl CoA and pyruvate metabolism. Analysis of both tumor and nontumor tissues indicates that these genes are targets of Tgif repression in multiple settings, suggesting that this is a core Tgif function. We propose that Tgifs play an important role in regulating basic energy metabolism in normal cells, and that this function of Tgifs is amplified in some cancers.
Colorectal cancer (CRC) is among the most frequently diagnosed cancers and is the third leading cause of cancer deaths in adults (Miller et al. 2016). The adenomatous polyposis coli (APC) gene, which is mutated in up to 80% of sporadic CRC (Kinzler and Vogelstein 1996; Segditsas and Tomlinson 2006), encodes a scaffold-like protein that assembles a complex of proteins including β-catenin, Axins, and GSK3β (Sancho et al. 2004; Clevers and Nusse 2012). In the absence of Wnt ligands, β-catenin is phosphorylated and degraded. Wnt signaling prevents phosphorylation and destruction of β-catenin, allowing it to accumulate and activate gene expression. Mutation or deletion of APC results in constitutive β-catenin-dependent gene activation, primarily via interactions with the TCF/LEF transcription factors (Polakis 1995; Sancho et al. 2004). Inactivation of one allele of the mouse Apc gene by germ-line mutation (Moser et al. 1990; Fodde et al. 1994; Oshima et al. 1995) or by CRE-mediated excision of a loxP-flanked exon (Shibata et al. 1997; Colnot et al. 2004; Hinoi et al. 2007) causes multiple adenomas, due to sporadic inactivation of the remaining allele.
In response to transforming growth factor β (TGFβ) ligands, the type I and type II receptors form a complex, resulting in phosphorylation and activation of the SMAD transcription factors (Feng and Derynck 2005; Massagué et al. 2005). TGFβ, Activin, or Nodal signaling results primarily in activation of SMAD2 and SMAD3, which, in association with SMAD4, accumulate in the nucleus to activate gene expression. TGFβ signaling is tumor suppressive in many cancers, in part due to the antiproliferative effects of the pathway (Levy and Hill 2006; Massagué 2008). Inactivating mutations in the TGFBR2 gene, encoding the type II receptor, are found in around 25% of CRC (Markowitz et al. 1995; Grady et al. 1999). Loss of heterozygosity of a region of chromosome 18 that includes both SMAD2 and SMAD4 is seen in almost 70% of CRC but is less frequently observed in colorectal adenomas (Vogelstein et al. 1988). Conditional inactivation of the mouse Tgfbr2 gene in combination with an Apc mutation results in progression to invasive adenocarcinoma, whereas homozygous Tgfbr2 deletion alone in the intestine has no effect (Muñoz et al. 2006).
TGIF1 (thymine–guanine-interacting factor) and the closely related TGIF2 are homeodomain transcription factors that are members of the TALE (three-amino-acid loop extension) superfamily (Bertolino et al. 1995; Melhuish et al. 2001; Hyman et al. 2003). Other TALE homeodomain proteins include the Meis and Pbx families that activate gene expression (Bürglin and Affolter 2016). In contrast, TGIF1 and TGIF2 are transcriptional repressors that interact with multiple general corepressors, including mSin3 and histone deacetylases (Wotton et al. 1999a; Melhuish et al. 2001). In addition, TGIF1 recruits CtBP1/2 corepressors via a conserved interaction motif (Melhuish and Wotton 2000). Tgifs limit the response to TGFβ signaling by recruiting corepressors to the SMAD transcription factors (Wotton et al. 1999a; Melhuish et al. 2001). In addition to SMAD interaction, other mechanisms for TGFβ pathway inhibition have been suggested, including promoting SMAD2 ubiquitylation and degradation or preventing SMAD2 phosphorylation in response to TGFβ signaling (Seo et al. 2004, 2006). Loss-of-function TGIF1 mutations are associated with holoprosencephaly (HPE), a severe genetic disease affecting forebrain development (Wotton and Taniguchi 2018). Mouse models of Tgif1 and Tgif2 loss of function suggest they play a redundant, but essential role in early embryogenesis (Powers et al. 2010). Conditional mutants survive to mid-gestation with multiple developmental abnormalities, including HPE (Taniguchi et al. 2012, 2017).
Although developmental defects in embryos lacking Tgif1 and Tgif2 can be partly rescued by reducing TGFβ family signaling through mutation of Nodal (Powers et al. 2010; Taniguchi et al. 2012, 2017), transcriptome profiling of early embryos or primary mouse embryo fibroblasts (MEFs) lacking Tgifs suggests that the majority of gene expression changes are unlikely to be due to altered TGFβ family signaling (Zerlanko et al. 2012; Anderson et al. 2017). TGIF1 was first identified by its ability to bind a retinoid response element of the Rbp2 gene and reduce activation by RXR nuclear receptors (Bertolino et al. 1995). TGIFs can bind directly to DNA and repress transcription via a well-defined consensus site, cTGTCAa, where the central five bases are most important (Bertolino et al. 1995; Wotton et al. 1999b). Direct repression via this consensus site has been shown for a small number of Tgif target genes (Anderson et al. 2017; Taniguchi et al. 2017). Recent genome-wide analysis identified a large number of potential Tgif1-binding sites, with enrichment for the known TGIF consensus element (Lee et al. 2015).
Increased Tgif levels have been implicated in ovarian, esophageal, and lung cancer among others (Imoto et al. 2000; Nakakuki et al. 2002; Wang et al. 2015). Tgif1 promoted breast cancer progression in a mouse model, independent of effects on TGFβ signaling (Zhang et al. 2015). The TGIF1 gene was shown to be a direct β-catenin/TCF transcriptional target that is activated by Wnt/β-catenin signaling, and the possibility that TGIF1 sequesters Axins to activate Wnt/β-catenin signaling was also suggested as a mechanism to explain its protumorigenic function (Zhang et al. 2015). Recent work with human CRC cell lines suggested a role for TGIF1 in CRC progression and also implicated TGIF1 in controlling the output of the Wnt/β-catenin pathway, although this appeared to be independent of effects on Axins (Wang et al. 2017). Thus, Tgifs can promote tumorigenesis, but questions regarding mechanisms of action and potential overlapping roles of Tgif1 and Tgif2 remain.
We used genetically engineered mouse models to address the function of Tgifs in intestinal epithelial neoplasms and to identify downstream Tgif target genes. Overexpression of TGIF1 in intestinal epithelial cells increased the size and number of adenomas in the small intestine, and deletion of Tgif1 and Tgif2 reduced tumor size in both the small intestine and colon. Transcriptional profiling of colon tumors from these mice revealed little effect of Tgifs on either Wnt/β-catenin or TGFβ signaling. Instead, we found that deleting Tgifs from colon tumors caused changes in expression of genes affecting multiple metabolic pathways. Integrating these data with additional gene expression profiling results suggests that Tgifs play a fundamental role in regulating energy metabolism, and may contribute to the reprogramming of metabolic gene expression that occurs in CRC.
Results
Increased Tgif expression in colorectal tumors
Analysis of TCGA colorectal data showed elevated TGIF1 and TGIF2 in adenocarcinomas (Fig. 1A). Similar results were seen with additional CRC data sets, and in comparison of paired tumor and normal samples, TGIF1 expression was higher in tumors in all cases (Supplemental Fig. S1). Mouse models of intestinal cancer, based on genetic alterations found in human cancers or treatment with chemical carcinogens, have been analyzed by gene expression array (Kaiser et al. 2007). In these analyses, Tgif1 expression was significantly higher in the azoxymethane (AOM) and Apc mutant models but not in one based on inactivation of Smad3, a component of the TGFβ signaling pathway (Supplemental Fig. S2A). Similar results were found with Tgif2 expression in this data set, although the signal in the AOM samples was too variable to reach statistical significance.
Figure 1.
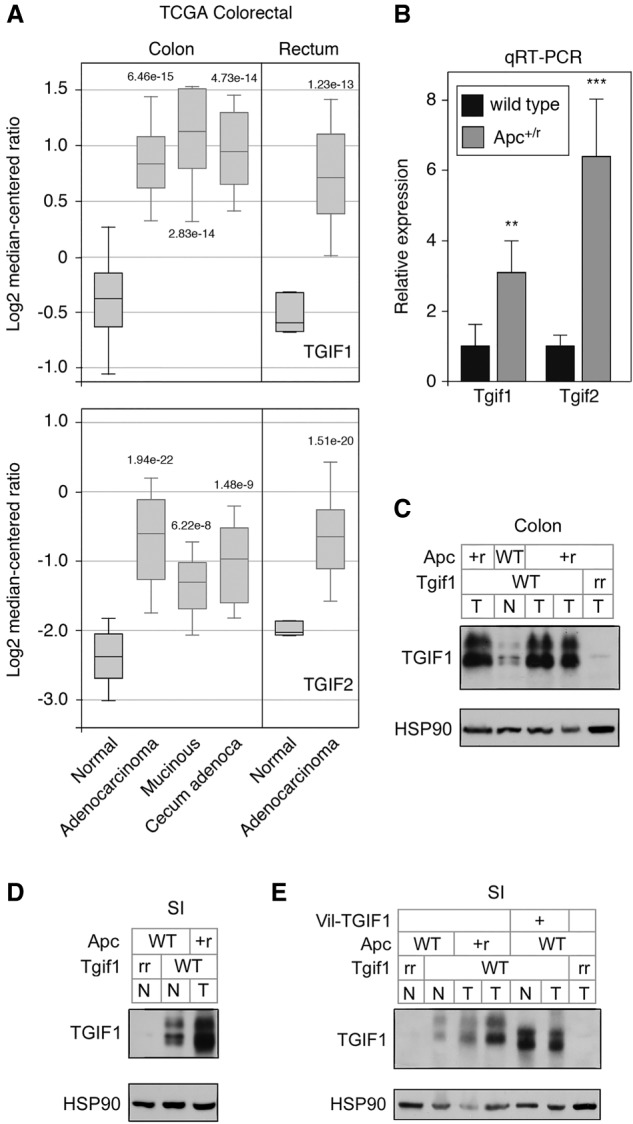
Increased expression of Tgifs in intestine tumors. (A) Log2 median centered expression data for TGIF1 and TGIF2 in the TCGA colorectal data set (analysis from Oncomine, with upper and lower quartiles and 10th and 90th percentiles), for normal and the indicated tumor types. P-values for comparisons with normal are shown. (B) Relative Tgif1 and Tgif2 expression (mean + SD of quadruplicate samples) determined by qRT-PCR, from wild-type (normal) colon or from Apc mutant colon tumors. (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001. (C) Expression of Tgif1 was analyzed by Western blot (with Hsp90 as a loading control) from normal colon (N) and tumor (T) from tissue of the indicated genotypes. (+) Wild type; (r) recombined allele. (D) Tgif1 expression from the small intestine (normal or tumor, as in C). (E) Expression of Tgif1 in normal (N) and tumor (T) tissue from mice of the indicated genotypes is shown by western blot with a TGIF1-specific antiserum and HSP90 as a loading control. Note the transgenic TGIF1 migrates slightly faster than the endogenous mouse Tgif1.
To test expression of Tgifs in Apc mutant mouse colon tumors, we combined a Villin-Cre transgene with a loxP-flanked allele of Apc isolated normal colon and colon tumors at 12 wk of age and analyzed gene expression by qRT-PCR. We observed a significant increase in expression of both Tgif1 and Tgif2 in colon tumors compared with normal tissue (Fig. 1B). Western blot analysis of similar 12-wk tumors showed increased Tgif1 protein expression in tumors compared with normal (Fig. 1C, note that Tgif1 migrates as a doublet due to MAPK mediated phosphorylation; Lo et al. 2001). An increase in Tgif1 expression in small intestine tumors compared with normal tissue was also observed (Fig. 1D). We also analyzed tumors in which Tgif1 was deleted specifically from epithelial cells, by including homozygous conditional loxP flanked alleles of Tgif1. Little or no Tgif1 signal was detectable in these samples, suggesting that the majority of Tgif1 present in normal colon and its increase in tumors were due to expression in the intestinal epithelium (Fig. 1C). In support of this, analysis of Tgif1 expression in normal intestinal crypts by immunofluorescence (IF) suggests that Tgif1 is expressed in the majority of epithelial cells within the crypt (Supplemental Fig. S3). Thus, expression of both Tgif1 and Tgif2 is higher in human CRC and in Apc mutant intestinal tumors in mice.
Overexpression of TGIF1 in intestinal epithelium
To test effects of increased TGIF1 expression in intestine, we generated a transgene in which T7 epitope-tagged human TGIF1 was expressed from the Villin promoter. Analysis of transgene expression in a panel of tissues by Western blot showed robust expression in the intestine, with no detectable expression in any other tissue examined (Supplemental Fig. S2B). Within the small intestine, we observed readily detectable expression in the proximal, middle, and distal thirds, with much lower expression in the colon and none in nontransgenic tissue (Supplemental Fig. S2C; Parini et al. 2018). To generate mice lacking both Tgif1 and Tgif2 in the intestinal epithelium, we used Vil-Cre to delete loxP-flanked Tgif1 (as previously described) and loxP-flanked Tgif2, derived from a knockout first allele from EUCOMM. Mice lacking both Tgifs from the intestinal epithelium were viable and grossly normal. Similarly, Vil-TGIF1 transgenic mice were normal and viable to at least 150 d.
To compare expression of the Vil-TGIF1 transgene with the endogenous Tgif1 in Apc mutant tumors we performed Western blots with a TGIF1 antiserum that recognizes both human and mouse Tgif1. There was an increase in endogenous Tgif1 expression in regions of the small intestine with tumors, compared with wild-type tissue (Fig. 1E). The levels of expression of transgenic TGIF1 were similar in both tumor and normal and, while higher than the expression of mouse Tgif1 in normal tissue, were quite similar to the increased level of endogenous Tgif1 in tumors (Fig. 1E). The transgenic TGIF1 migrates more rapidly on SDS-PAGE than mouse Tgif1, and it appears that expression of the transgene effectively reduces expression of endogenous Tgif1, as evidenced by the almost complete absence of the slower migrating Tgif1 band in the transgenic samples (Fig. 1E). Thus, Vil-TGIF1 is overexpressed to a level similar to that of the elevated endogenous expression seen in tumors.
Altered tumor burden in the intestine
To test effects of Tgifs on tumorigenesis, we combined conditional alleles of Tgif1, both Tgif1 and Tgif2, or the Vil-TGIF1 transgene with Vil-Cre and a heterozygous loxP-flanked Apc allele. At 12 wk of age, small intestines were separated into proximal, middle, and distal thirds and opened along the length to identify tumors. Although the number of tumors per animal was quite variable, there was a significant reduction in tumor numbers in mice lacking both Tgif1 and Tgif2 and an increase in the Vil-TGIF1 mice (Fig. 2A). The number of tumors >1.5 mm in diameter was significantly lower in both the Tgif1 and Tgif1;Tgif2 mutants (Fig. 2A). The increase in larger tumors in the TGIF1-overexpressing mice was highly significant, whereas there were no significant differences in the number of smaller (<1.5-mm) tumors. Histological examination of tumors isolated from animals of all four genotypes revealed no clear differences in tumor morphology (Supplemental Fig. S4A). All tumors examined were adenomas, and we did not observe invasive carcinomas in these animals. Thus, increasing TGIF1 expression to a level similar to that seen in Apc mutant tumors enhances adenoma growth, but does not promote transition to invasive adenocarcinoma.
Figure 2.

Tgif expression promotes intestinal tumorigenesis. (A) The numbers of tumors per animal (at 12 wk) in the small intestine are shown (median, upper, and lower quartiles, 5th and 95th percentiles) for each genotype. Numbers are shown for all tumors and separately for those <1.5 mm or >1.5 mm in diameter. P-values for comparison with the Apc+/r mice are shown. (B) Number of colon tumors per mouse and tumor volume in mm3 are shown (median, upper, and lower quartiles, 5th and 95th percentiles). P-values for comparison with the Apc+/r mice are shown. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. (C) Representative images of hematoxylin and eosin (H&E)-stained colon tumors from the indicated genotypes are shown. Images were captured at 200× magnification.
In the middle and distal regions of the small intestine, we observed increased numbers of larger tumors in Vil-TGIF1 mice and a decrease in total tumor numbers in Tgif1;Tgif2 mice driven primarily by changes in the numbers of larger tumors (Supplemental Fig. S4B,C). Apc mutant mice lacking only Tgif1 had an intermediate phenotype between that of the Apc and mice lacking both Tgif1 and Tgif2. This was particularly evident when analyzing the proportion of tumors in each mouse that were >1.5 mm in diameter (Supplemental Fig. S4D). Although the Vil-Cre;Apc model primarily generates tumors in the small intestine, there are also colon tumors in these animals. Comparison of tumor number and size in the colon between Apc mice and those lacking Tgif1 did not reveal any significant differences (Fig. 2B). However, in the Apc;Tgif1;Tgif2 mice (referred to here as ATT), average tumor volume was significantly lower, despite the fact that the sizes were quite variable (Fig. 2B). Thus, it appears that further reducing overall Tgif levels by deleting Tgif1 and Tgif2 enhances the relatively mild effect of deletion of Tgif1 alone, implying redundant function. As with the SI tumors, there were no clear histological differences between the colon tumors from mice of each genotype (Fig. 2C). Together, these data suggest that Tgif1 and Tgif2 contribute to Apc mutant intestinal tumorigenesis, and that increasing TGIF1 expression drives adenoma growth.
Transcriptional changes in Tgif mutant tumors
To address how increased Tgif levels contribute to intestinal tumor growth we performed transcriptome profiling, comparing normal wild-type colon with colon tumors from Apc and ATT mice. RNA was isolated from five normal colon samples and from seven tumors from mice of each of the two genotypes from both males and females. The samples from each of the three genotypes clustered separately, although there was considerable spread among the tumors, and the two tumor genotypes clustered closer to each other than to the wild types (Fig. 3A). To identify genes that were differently expressed, we performed pairwise comparisons using a 0.5 log2 fold change and an adjusted P-value cutoff of <0.01. This identified close to 2000 genes that were differentially expressed between the two tumor genotypes, with 884 being higher in the ATT than in the Apc tumors and 1160 with lower expression (Supplemental Table S1). Hierarchical clustering of each of these two gene lists suggested that, among the genes with increased expression in the ATT compared with the Apc tumors, a small fraction was also more highly expressed in wild-type colon (Fig. 3B). This is consistent with these genes being Tgif targets that are repressed in Apc tumors by increased Tgif expression. Among the genes with lower expression in ATT tumors, many are increased in the Apc tumors compared with wild type, consistent with loss of Tgifs reversing at least part of the Apc mutant gene expression program (Fig. 3C). Despite the presence of some genes that decrease in the Apc tumor compared with wild type and increase in ATT tumors, there was minimal enrichment for this class among genes that are significantly differently expressed in both the Apc to wild type and ATT to Apc comparisons (Fig. 3D). However, more genes with higher expression in ATT tumors had reduced expression in Apc tumors compared with wild type (124/392; 31.6%) than had higher expression (72/392; 18.4%). Thus, it appears that there is a subset of differentially expressed genes that fit with being Tgif targets, but there is also significant tumor to tumor variability and a larger number of genes that do not fit a simple direct Tgif target model.
Figure 3.

Gene expression changes in Tgif mutant colon tumors. (A) Principle component analysis of RNA sequencing (RNA-seq) data from normal wild-type mouse colon or from colon tumors isolated from Apc heterozygous mice (Apc) or Apc heterozygous mice with homozygous deletion of both Tgif1 and Tgif2 (ATT). Heat maps are shown for all genes with significantly (log2 fold change > 0.5, P-adjusted < 0.01) higher (B) or lower (C) expression in ATT than in Apc. (D) Venn diagrams indicating the overlap between genes that are significantly differently expressed between Apc versus wild-type and Apc versus ATT. (E) Gene set enrichment analysis (GSEA) indicates enrichment of epithelial-to-mesenchymal transition (EMT), KRAS signaling, glycolysis, and glutamine metabolism in Apc tumors compared with ATT. The nominal enrichment score (NES) and false discovery rate q-value are shown.
Tgifs are well characterized as repressors of TGFβ-responsive transcription (Wotton et al. 1999a; Wotton and Taniguchi 2018) and have been suggested to promote Wnt-responsive gene expression (Zhang et al. 2015; Wang et al. 2017). We therefore examined expression of genes that are known targets of these pathways. For a panel of well-characterized TGFβ targets (Smad7, Skil, Serpine1, Cdkn1a, and Cdkn2b) and some additional genes that respond to TGFβ in LS1034 CRC cells (Labbé et al. 2007) there was no consistent pattern in expression differences between normal colon and Apc tumors, and deletion of Tgifs had minimal effect (Supplemental Fig. S5A). Overlapping gene expression changes in ATT compared with Apc tumors with expression array data from mouse Apc colon tumors or Apc tumors lacking the TGFβ type 2 receptor (GSE82133) (Miguchi et al. 2016) revealed minimal overlap (Supplemental Fig. S5B). qRT-PCR analysis of canonical TGFβ target genes in a set of ATT and Apc colon tumors did not show significant increases in expression in the Tgif mutants (Supplemental Fig. S5C).
To examine Wnt signaling we looked at expression of canonical targets of the pathway. These genes were clearly activated in Apc tumors compared with wild-type colon but were not further activated by deletion of Tgifs (Supplemental Fig. S5D). Comparing a set of β-catenin activated or repressed target genes (Herbst et al. 2014) with our data showed limited overlap with expression differences between ATT and Apc tumors, whereas there was clear enrichment for these target genes in the comparison between Apc and wild-type tissue (Supplemental Fig. S5E). This was supported by qRT-PCR analysis showing increased expression of Axin2, Lgr5, and Lef1 in Apc mutant tumors, but no decrease in ATT tumors, as would be expected if Tgif1 promotes β-catenin-activated gene expression (Supplemental Fig. S5F). Thus, it appears that in the context of colon tumors in mice, Tgifs do not play a major role in regulating either TGFβ or Wnt/β-catenin signaling.
Altered metabolic gene expression in tumors lacking Tgifs
To identify functional groups among the gene expression changes, we performed gene set enrichment analysis (GSEA). Gene sets indicative of epithelial-to-mesenchymal transition (EMT) and KRAS signaling were among the most significantly enriched in the Apc compared with ATT tumors (Fig. 3E). Surprisingly, glycolysis was also one of the most significantly enriched gene sets in the Apc tumors, and other metabolic signatures were enriched in Apc compared with ATT tumors (Fig. 3E). Comparing ATT with Apc tumors, one of the most down-regulated glycolytic genes was Slc2a1, encoding Glut1, the major glucose transporter in intestine. For most glycolytic enzymes there was a more modest reduction in expression in ATT tumors (Fig. 4A). Examining expression of genes encoding proteins that function to generate glucose from pyruvate revealed that these genes were generally slightly more highly expressed in the ATT tumors. Summing the relative expression for each tumor for a panel of glycolysis or gluconeogenesis-specific genes revealed a clear separation of Apc and ATT tumors, consistent with the GSEA result, despite the relatively minimal changes in expression of most components of these pathways (Fig. 4A).
Figure 4.

Altered metabolic gene expression in Tgif mutant colon tumors. (A) Heat maps are shown indicating fold-change (comparing ATT with Apc tumors) for the glycolytic pathway and for genes involved specifically in the conversion of pyruvate to glucose. The plot to the right shows summed Z-scores for a panel of genes involved only in glycolysis or in gluconeogenesis, plotted as gluconeogenesis versus glycolysis for each tumor. (B) IF analysis is shown for Glut1 and β-catenin in colon tumors (T) with adjacent normal tissue (N). Heat maps are shown for all genes in the purine and pyrimidine metabolic pathways (C) or amino acid metabolic pathways (D) that are significantly differently expressed comparing Apc with ATT tumors.
Since Slc2a1 was the most down-regulated glycolytic gene in ATT tumors, we examined expression of the Glut1 protein in colon tumors of each genotype by IF. Glut1 was expressed throughout normal colon and Apc mutant tumor tissue, with relatively little difference in expression between the two (Fig. 4B). In contrast, there was clearly lower expression of Glut1 in the ATT tumor tissue compared with adjacent normal tissue and with Apc mutant tumors (Fig. 4B). We also examined the RNA sequencing (RNA-seq) data for changes in other metabolic pathways by comparing all genes that were significantly differently expressed between Apc and ATT tumors to metabolic gene lists from Kyoto Encyclopedia of Genes and Genomes (KEGG). This analysis revealed reduced expression of multiple genes with links to purine and pyrimidine synthesis and amino acid metabolic pathways (Fig. 4C,D). Together, these analyses suggest that loss of Tgifs from Apc tumors results in widespread changes in metabolic gene expression.
Increased expression of acetyl CoA metabolism genes in Tgif mutant tumors
The majority of metabolic gene expression changes examined so far are decreases in expression in the absence of Tgifs, suggesting that they are unlikely to be direct Tgif targets. To identify Tgif target genes we overlapped gene expression changes found here with transcriptome profiling from wild-type and conditional Tgif1;Tgif2-null (conditional double knockout [cdKO]) mouse embryos (Anderson et al. 2017). There was relatively little overlap between these two data sets, but among the genes that changed in both, there was a significant enrichment for genes that increased with deletion of Tgifs from embryos and tumors (Fig. 5A). We reasoned that comparison of these two very different systems might allow us to identify Tgif-regulated genes with higher confidence by ruling out secondary tissue-specific effects. ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) analysis from mouse embryonic stem (ES) cells identified >12,000 potential Tgif1-bound regions across the genome (Lee et al. 2015). To enrich for higher-confidence targets, we considered only the top 40% of putative Tgif1-bound regions from this analysis, and overlapped this list with genes that were differently expressed in cdKO embryos and tumors lacking Tgifs (Fig. 5B). This revealed a greater overlap with genes that were activated by loss of Tgifs than with genes that had lower expression in the mutants (Fig. 5B,C). Among the genes with increased expression in both cdKO embryos and tumors almost 70% had high confidence ChIP peaks (Fig. 5C).
Figure 5.

Identification of putative Tgif target genes. (A) Differentially expressed genes from RNA-seq data from control or Tgif1;Tgif2-null (cdKO) day 9 mouse embryos (GSE78728) overlapped with genes that are significantly differently expressed in ATT versus Apc tumors. (B) Genes with significantly higher (left) or lower (right) expression in either dataset were overlapped with Tgif1 ChIP-seq data from mouse ES cells (GSE55404). (C) The percentage of genes from each of the indicated overlaps between expression data from embryos and tumors with ChIP-seq peaks is shown. (D) EnrichR analysis of the 125 genes with increased expression in embryos and tumors that also have ChIP-seq peaks is shown. (E) A pathway map for selected genes involved in acetyl CoA and pyruvate metabolism is shown, with mitochondrial and cytoplasmic compartments shown separately. Black arrows indicate metabolic reactions, green arrows translocations, and the dashed arrows links to additional metabolic pathways. For each gene shown (boxed), the three colored squares represent fold changes in comparisons of Apc with wild-type (left), ATT with Apc tumor (center), and cdKO with control embryo (right). Larger boxes indicate significant change (P-adjusted < 0.01). Smaller boxes are not significant at this cutoff.
Analysis of the 125 genes with ChIP-seq peaks and higher expression in both RNA-seq data sets revealed a significant enrichment for a MEIS1 consensus site (which is identical to a TGIF site) associated with these genes, consistent with the idea that they are direct Tgif targets (Fig. 5D). Propanoate metabolism and acetyl CoA biosynthetic process were the most significantly enriched pathways, and among the 125 gene list were three genes encoding enzymes that synthesize acetyl CoA: Acss1, Acss2, and Mlycd. To place these changes in context, we visualized expression changes for genes encoding a number of enzymes involved in acetyl CoA metabolism as part of a metabolic pathway map. Acss2 was significantly increased in both cdKO embryos and in tumors lacking Tgifs and decreased in Apc tumors compared with wild-type colon (Fig. 5E). Similarly, the mitochondrial Acss1 was increased in Tgif mutant embryos and tumors and decreased in the Apc tumors. Other genes that showed this pattern included Mlycd, which encodes a cytosolic enzyme that converts malonyl CoA to acetyl CoA, and Acat1 which generates acetoacetyl CoA from acetyl CoA in the mitochondria as the first step of ketone synthesis (Fig. 5E). As there was some increase in expression of genes associated with the early stages of pyruvate metabolism (Fig. 4A) and Mpc1 and Pcx expression was increased in cdKO embryos, we also examined some changes in this pathway. Mpc1 expression showed a similar pattern to the acetyl CoA synthetic genes, as did Pcx, although the increase in Pcx expression in ATT tumors was not statistically significant (Fig. 5E). This analysis is consistent with the idea that Tgifs directly repress multiple genes involved in acetyl CoA metabolism and suggests they may also play a similar function for pyruvate metabolic genes.
IF analysis of colon tissue from Apc and ATT mice indicated that Acss2 expression was reduced in Apc mutant tumors compared with adjacent normal colon, and that expression was higher in both normal and tumor tissue in the ATT mice (Fig. 6A). In both small intestine and colon, we observed higher Acss2 expression, with more evident nuclear localization in the Tgif1;Tgif2 mice compared with wild type (Supplemental Fig. S6A,B). In support of this, Western blot of colon tumors indicated higher Acss2 expression in ATT than in Apc tumors, and Pcx expression was also higher in the tumors lacking Tgifs (Fig. 6B). We also analyzed expression of the mitochondrial Acat1. Expression was clearly higher in ATT tumors than in the Apc and in normal small intestine lacking both Tgifs (Fig. 6C; Supplemental Fig. S6C). As with Acss2, expression of Acat1 and Mlycd was increased in ATT compared with Apc colon tumors (Fig. 6D), consistent with the idea that loss of Tgifs in colon tumors results in higher expression of both mitochondrial and nuclear/cytoplasmic metabolic regulators. To further explore this potential Tgif function we analyzed human tumor gene expression data sets. Separating the human The Cancer Genome Atlas (TCGA) CRC data set by TGIF1 expression levels revealed higher expression of ACSS1 and ACSS2 in the quartile of tumors with lowest TGIF1 expression compared with the upper quartile (Supplemental Fig. S7A). Furthermore, GSEA comparing the upper and lower 10% of tumors from the TCGA PanCancer colon cancer data set showed enrichment for glycolysis in the TGIF1 high and enrichment for bile acid and fatty acid metabolism in the lowest 10% by TGIF1 expression (Supplemental Fig. S7B). Comparison of ACSS2 levels in multiple CRC data sets suggests that levels are reduced in CRC and that lower expression correlates with poor survival, a pattern that is opposite to that seen for Tgifs (Supplemental Fig. S7C–E). This suggests that similar metabolic gene expression differences are seen in human tumors stratified by TGIF1 expression levels to those seen in our transcriptome profiling of Apc and ATT tumors.
Figure 6.

Increased Acss2 expression in Tgif mutant tumors. (A) IF analysis is shown for Acss2 and β-catenin in colon tumors (T) with adjacent normal (N) tissue from Apc mutants and ATT mice. (B) Western blot analysis of colon tumors from Apc and ATT mice showing expression of Acss2 and Pcx, together with Hsp90 and γ-tubulin loading controls. (C) IF analysis is shown for Acat1 and β-catenin in colon tumors with adjacent normal tissue from Apc and ATT mice. (D) Western blot analysis of Apc and ATT colon tumors showing expression of Acat1 and Mlycd, together with Hsp90 and γ-tubulin loading controls. Molecular weight markers are shown for B and D.
Direct Tgif-mediated repression of metabolic gene expression
To address the possibility that genes involved in acetyl CoA and pyruvate metabolism are direct Tgif targets in multiple cell types, we tested expression of a panel of these genes by qRT-PCR in normal small intestine and in primary MEFs. All three acetyl CoA synthetic genes and Acat1 were significantly more highly expressed in Tgif1;Tgif2 null small intestine than in wild-type tissue (Fig. 7A). Similarly, expression of Pcx and Mpc1 was also higher in the mutant. We observed a similar pattern for five of the six genes in primary MEFs (Fig. 7B). Acss1 expression did not increase in MEFs, but its expression is very low in cultured cells, including primary MEFs. Analysis of metabolite levels in normal small intestine isolated from wild-type mice or those lacking both Tgifs suggested that acetate levels were decreased in the absence of Tgifs consistent with increased metabolism by higher levels of Acss1 and Acss2 (Fig. 7C). Propionate levels were also lower, but did not quite reach significance due to the variability among samples. In contrast, other metabolites were essentially unchanged in Tgif mutant small intestine compared with the normal (Fig. 7C). Together with the gene expression changes seen in both embryos and tumors, this is consistent with the possibility that the regulation of acetate metabolism is a conserved core function of Tgifs.
Figure 7.

Tgif1 regulation of metabolic gene expression in normal tissue. (A) Expression of the indicated genes was analyzed by qRT-PCR from normal small intestine from wild-type or cdKO (Tgif1;Tgif2 cdKO) mice. (B) Expression of the same genes was analyzed in wild-type and cdKO primary MEFs. Expression is plotted relative to the wild-type mean (+SD) of four and three replicates for intestine and MEFs. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, for comparison with wild type. (C) Analysis of metabolites in normal small intestine from wild-type and cdKO mice (mean + SEM). Relative levels of the indicated metabolites are shown with the wild-type level for each set to 1. (*) P < 0.05 for comparison with wild type.
We next examined the sequences of the ChIP-seq peaks associated with each of the six genes analyzed in small intestine and primary MEFs. In each case the potential Tgif1-bound region overlapped the transcriptional start site, and for all but Acss1, at least two TGIF consensus sites were present (cTGTCA or TGTCAa) (Fig. 8A; Supplemental Fig. S8A). To test Tgif1 recruitment, we performed ChIP-qPCR for the five genes that had ChIP-seq peaks with consensus TGIF sites. For Mpc1, we amplified two regions as the predicted peak was quite broad and had consensus sites close to each end (Fig. 8A). In chromatin from wild-type small intestine, we observed significant enrichment of the putative Tgif1-binding regions from all five genes compared with a negative control region, and similar results were obtained from primary MEFs (Fig. 8B,C). We observed significant enrichment for Acss2 in the wild-type chromatin compared with chromatin from Tgif1;Tgif2 mutant MEFs, indicating the specificity of this TGIF1 antiserum, as previously demonstrated (Supplemental Fig. S8B; Anderson et al. 2017; Taniguchi et al. 2017). Transient knockdown of both TGIF1 and TGIF2 in the human CRC cell line, HCT116, resulted in increased expression of all five metabolic genes, consistent with this being a relatively rapid effect of reduced TGIF levels rather than a longer term response to sustained deletion of Tgif genes (Fig. 8D). To test direct target gene regulation, we amplified the proximal promoter regions from the Acss2 and Pcx genes and created transcriptional reporters. We chose Acss2 because it has a cluster of conserved TGIF consensus sites and might be more sensitive to TGIF levels. Pcx, which has only a single conserved site in the proximal promoter, facilitated the generation of a simple mutant version of the reporter in which this site was disrupted (Fig. 8E). Since manipulating TGIF levels affected expression of ACSS2 and PC (human Pcx) in HCT116 cells, we analyzed Acss2 and Pcx reporter activity by transient transfection into HCT116. Both the Acss2 and Pcx transcriptional reporters were significantly repressed by coexpression of increasing amounts of TGIF1 (Fig. 8F,G). Analysis of a deletion mutant of the Accs2 reporter lacking all TGIF sites and of the Pcx mutant in which the single site was disrupted revealed significantly less repression by TGIF1 than seen with the wild-type reporters. In addition, mutant versions of TGIF1, with single amino acid changes within the homeodomain that prevent DNA binding failed to repress either the Acss2 or Pcx reporter (Supplemental Fig. S8C). These data suggest that DNA binding by TGIF1 is required to repress expression of Acss2 and Pcx, and that this repression is mediated at least in part by conserved consensus TGIF sites. Taken together, these data suggest that Tgifs are direct transcriptional repressors of a set of genes involved in acetyl CoA and pyruvate metabolism.
Figure 8.

Tgif1-mediated repression of Acss2 and Pcx. (A) The positions of the ChIP-seq peaks, qPCR amplicons, and TGIF consensus sites for each to the five genes tested are shown using University of California at Santa Cruz genome browser views. A 4-kb region, centered on the transcriptional start, is shown for each mouse gene, with similarity to human below. (B) Tgif1 binding to each peak region was analyzed by ChIP-qPCR from normal wild-type small intestine. (C) Tgif1 binding in primary MEFs was analyzed by ChIP-qPCR. Chromatin was precipitated with a TGIF1 antiserum or preimmune serum (pre-I). Data is mean + SD of triplicates and is plotted in arbitrary units with the TGIF1 IP for the negative control region (fbpk3) set equal to 1. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, for comparison with fbpk3. (D) TGIF1 and TGIF2 were transiently knocked down in HCT116 cells, and expression of the indicated genes analyzed by qRT-PCR. Expression is plotted relative to the nontargeting control knockdown mean (+SD) of 3 replicates. (*) P < 0.05; (**) P < 0.01, for comparison with control. (E) The Acss2 and Pcx luciferase reporters are shown schematically. Nucleotide positions relative to the transcriptional start site are shown. Black bars are TGIF sites, with the mutant site indicated in white. Gray represents 5′- untranslated sequence, and white is proximal promoter. (F,G) HCT116 cells were transiently transfected with the indicated luciferase reporters together with increasing amounts of a TGIF1 expression vector (or a control plasmid). Luciferase activity is shown (normalized to a Renilla-luc transfection control), with the activity of the Acss2-525-luc (F) and the wild-type Pcx-368-luc (G) set to 1. (#) P < 0.01; (##) P < 0.001, for comparison with no TGIF1 transfection. (*) P < 0.01; (**) P < 0.001; (***) P < 0.0001, for comparison with fold-repression of the wild-type reporter.
Discussion
Using an intestinal cancer model as a starting point to analyze TGIF function, we provide evidence that TGIF transcription factors directly regulate genes involved in acetyl CoA metabolism. This function of TGIFs does not appear to be limited to intestinal tumor or normal tissue and may represent a key unexpected function of these transcription factors, independent of the other pathways they are known to regulate.
TGIFs are thought to be oncogenic by limiting the antiproliferative effects of TGFβ signaling. Mutating the Tgfbr2 gene in concert with an Apc mutation drives the transition from adenoma to invasive adenocarcinoma (Muñoz et al. 2006). Expression of both TGIF1 and TGIF2 is higher in CRC than in normal tissue, and Tgif expression is increased in Apc mutant intestinal tumors. Such increased Tgif levels might be expected to dampen the TGFβ response and promote tumor growth. Indeed, we show that Tgif expression promotes growth of early adenomas. However, transcriptome profiling of colon tumors revealed almost no overlap with changes in TGFβ-responsive gene expression, suggesting that at least in this model Tgifs are not major regulators of TGFβ signaling. This is in contrast to the effect of Arkadia (Rnf111), a ubiquitin E3 that promotes TGFβ signaling by driving degradation of the Ski and Skil (SnoN) SMAD corepressors. Rnf111 deletion increased tumor numbers in a mouse colon cancer model, increased Skil expression, and reduced the TGFβ response (Sharma et al. 2011). Thus, altered SMAD corepressor levels can affect CRC tumor progression, although it remains possible that other Arkadia substrates contribute.
TGIF1 was proposed to activate Wnt signaling by sequestering Axins, allowing activation of Wnt/β-catenin target genes (Zhang et al. 2015). In our transcriptome data there is no consistent decrease in expression of canonical Wnt target genes in the absence of Tgifs, as would be predicted by this model. Although it remains possible that any effect on Wnt signaling is a cell type-specific function of Tgif1, our results suggest the Wnt pathway is not a major Tgif1 target in colon tumors. It is possible that Tgif1 promotes β-catenin function, but its effect is masked by the overriding pathway activation caused by Apc mutation. However, this still argues against a Tgif effect on tumor promotion via β-catenin in this mouse model. A further link to Wnt signaling is the demonstration that TGIF1 is directly activated by Wnt/β-catenin signaling (Zhang et al. 2015). Our data are consistent with this in that Tgif1 and Tgif2 expression is increased in Apc mutant tumors compared with normal tissue, although we do not know whether Tgif genes are β-catenin targets in the intestine. Thus, it appears that Tgifs promote intestinal tumor growth independent of effects on the two most likely pathways.
Genome-wide analysis of Tgif1 binding to chromatin in mouse ES cells suggests that a large part of Tgif function is mediated by direct binding to DNA (Lee et al. 2015). In agreement with this, structural studies show that, unlike many other homeodomain proteins, TGIF1 binds with high specificity and relatively high affinity to its cognate site (Guca et al. 2018). Given the large numbers of gene expression changes observed in tumors lacking Tgifs we attempted to identify higher confidence core Tgif target genes by integrating ChIP-seq data with gene expression data sets from very divergent systems. Surprisingly, pathway analysis of this high confidence gene set identified acetyl CoA metabolism as the most significantly enriched biological process. In addition, analysis of all gene expression changes between Apc tumors and Apc tumors lacking both Tgifs revealed changes in multiple metabolic pathways, further supporting a role for Tgifs as regulators of metabolism. Increased expression of acetyl CoA metabolic genes is not cell-type specific, occurring in early mouse embryos, primary MEFs, normal small intestine and colon tumors, suggesting that this may be a fundamental, yet unexpected, role of Tgifs. We validated a panel of genes with links to acetyl CoA and pyruvate metabolism as direct Tgif1 targets in normal small intestine and primary MEFs, and demonstrated direct repression of both the Acss2- and Pcx-proximal promoters by TGIF1. Full repression was dependent on the presence of conserved TGIF consensus sites within the promoters, and was abolished by single amino acid changes in TGIF1 that prevent DNA binding. This is consistent with the notion that the repression of expression of a subset of metabolic genes is a core Tgif function conserved across multiple cell types that occurs via direct DNA binding to cognate TGIF sites. This further supports recent work suggesting that while Tgifs may limit TGFβ-activated gene expression during embryogenesis, much of the Tgif transcriptional function is mediated by direct DNA binding.
Although we observed extensive changes in expression of metabolic genes, relatively few were increased in the Tgif mutant tumors, suggesting that the majority of changes may be indirect effects. Attempting to place potential direct Tgif target genes in context indicates that Tgifs repress anabolic metabolism rather than catabolism. For example, Tgif repression of mitochondrial Acss1 and Acat1 would be expected to limit ketone synthesis and the utilization of acetate by both Acss1 and Acss2 would be reduced by Tgif-mediated repression. In support of this, we show that in the absence of Tgifs acetate levels are lower in the small intestine, consistent with increased Acss1 and Acss2 activity. However, more extensive analyses of metabolic intermediates is clearly required in both normal and tumor tissue to fully validate Tgifs as metabolic regulators. In the absence of Tgifs, anabolic metabolism may favor synthesis of ketones and sterols, and the utilization of pyruvate to generate other metabolic intermediates. This proposed function of Tgifs is reminiscent of the reprogramming of metabolic gene expression that is recognized as one of the hallmarks of cancer. In addition to an increased reliance on glycolysis, termed the Warburg effect (Warburg 1956), there is extensive rewiring of energy metabolism in cancer cells (Hanahan and Weinberg 2011; Pavlova and Thompson 2016).
Recent work suggests that metabolic reprogramming occurs at the adenoma stage of CRC (Satoh et al. 2017), and it appears that this is downstream from activation of oncogenes, such as KRAS or BRAF, and requires high MYC expression (Hutton et al. 2016; Satoh et al. 2017). Our data suggest that Tgifs play a role in regulating metabolic gene expression in both normal and tumor tissues and may mediate part of the metabolic reprogramming that occurs in colon adenomas. A role for TGIF1 in regulating metabolism in human CRC is supported by GSEA comparing colon tumors separated by TGIF1 expression levels. Interestingly, one of the best-correlated Tgif target metabolic genes in CRC is ACSS2, which we show is directly repressed by TGIF1. Several recent studies have linked ACSS2 expression levels to tumor progression (Comerford et al. 2014; Mashimo et al. 2014; Schug et al. 2015). In contrast to our data, high levels of ACSS2 expression correlate with poor prognosis in other cancer types, raising the possibility that ACSS2 may function differently in CRC. This possibility is supported by analysis of CRC gene expression data, suggesting that unlike for other tumor types, a reduction in ACSS2 expression is observed in CRC and that high expression correlates with better patient prognosis.
One difference that should be noted in this context is the unusual metabolic environment in which colon tumors develop. The metabolic activity of gut bacteria results in high levels of short chain fatty acids, including acetate, which constitute a major fuel source for colonocytes (Donohoe et al. 2011). This difference compared with most other tissues in the body may impose differing metabolic pressures on developing tumors. For example, a reduction in short chain fatty acid metabolism might facilitate the switch to glycolysis. Therefore, it will be of interest to test functionally whether decreased expression of ACSS2 or other Tgif targets can promote CRC progression. In addition to direct effects of Tgif expression on genes involved in acetate metabolism, one of the most dramatically affected genes encodes the major glucose transporter, Glut1. High Tgif levels appear to favor Glut1 expression specifically in tumors, which may contribute to a switch away from short chain fatty acid metabolism. However, since expression of Slc2a1 increased in the absence of Tgifs, this is likely not a direct target for Tgif regulation and may be a response to other alterations in metabolism.
In summary, our data suggest a model in which Tgifs function in multiple cell types to limit expression of a core set of acetyl CoA metabolic genes. In some cancers, including CRC, where Tgif levels increase, this normal Tgif function may be co-opted by the tumor as part of the metabolic reprogramming.
Materials and methods
Mice
All animal procedures were approved by the Animal Care and Use Committee of the University of Virginia, which is fully accredited by the AAALAC. Conditional alleles with loxP flanked exons are referred to here as “f” for loxP flanked, or “r” for recombined (null). Mice were maintained on a C57BL/6 background. Conditional Apc mice were from the NCI (01XAA; B6.Cg-Apctm2Rak/Nci), and the Vilin-Cre line was from Jax [B6.Cg-Tg(Vil1-cre)1000Gum/J; 021504] (Madison et al. 2002). Conditional Tgif2 mice were generated from targeted ES cells from EUCOMM [Tgif2tm1a(EUCOMM)Wtsi; International Knockout Mouse Consortium (IKMC) project 24492; C57BL/6N ES cells] and crossed to a conditional Tgif1 line that had been back-crossed for six generations to C57BL/6 (Powers et al. 2010). Villin-TGIF1 transgenic mice were generated at the UVA GEMM Core and were backcrossed to C57BL/6 for at least three generations prior to tumor analysis. The human TGIF1 cDNA with an amino-terminal T7 epitope tag was inserted into the Villin promoter plasmid (12.4kbVillin-ΔATG), which was a gift from Deborah Gumucio (Addgene plasmid 19358; Madison et al. 2002). Germ line transmission was verified by PCR and expression by Western blot.
Tumor analysis, IF, and histology
Tissues were fixed in zinc-formalin, paraffin-embedded, sectioned at 5 µm and stained with hematoxylin and eosin (H&E) or prepared for immunostaining as described (Hao et al. 2018). Images were captured with 10×, 20×, or 40× objectives using a Nikon Eclipse NI-U with a DS-QI1 or DS-Ri1 camera and NIS Elements software and adjusted in Adobe Photoshop. For IF, antibodies were as follows: rabbit anti-Acss2 (Abcam, 66038), mouse anti-TGIF1 (Santa Cruz Biotechnology, SC-17000), rabbit anti-Acat1 (Proteintech, 16215-1-AP), rabbit anti-Slc2a1 (Millipore 07-1401), and mouse anti-β-catenin (BD Transduction Labs, 610153).
RNA isolation and qRT-PCR
RNA from snap-frozen tissue was isolated and purified using Absolutely RNA kit (Agilent) and quality-checked by Bioanalyzer. cDNA was generated using SuperScript III (Invitrogen) and analyzed by real-time PCR using a Bio-Rad MyIQ cycler and Sensimix Plus SYBRgreen plus FITC mix (Bioline) with intron-spanning primer pairs selected using Primer3 (http://frodo.wi.mit.edu). Expression was normalized to Rpl4 and Cyclophilin using the ΔCt method.
RNA-seq and analysis
Poly-A RNA-seq libraries generated with Illumina barcodes were sequenced (NextSeq 500 at the University of Virginia Genome Analysis and Technology Core) to at least 25 million single-end 75-bp reads per sample. Data were analyzed using the Galaxy server (https://usegalaxy.org). Transcript quantification was performed using Salmon (Patro et al. 2017) to map to the mm10 mouse genome build, and DESeq2 (Love et al. 2014) within the Galaxy site was used for normalizing count data, estimating dispersion, fitting a negative binomial model for each gene, and comparing expression between groups. A cutoff of ±0.5 log2 and an adjusted P-value of <0.01 were considered significant. Enrichment was analyzed with ENRICHR (http://amp.pharm.mssm.edu/Enrichr) (Chen et al. 2013; Kuleshov et al. 2016), and heat maps were generated with Heatmapper (http://www2.heatmapper.ca/expression) (Babicki et al. 2016). Gene set enrichment was by GSEA software from the Broad Institute (Mootha et al. 2003; Subramanian et al. 2005). RNA-seq data have been deposited at Gene Expression Omnibus (GSE116578).
Western blot and metabolite analysis
Tissues were ground in PBS followed by addition of NP-40 to 1%, lysates were separated by SDS-PAGE and transferred to Immobilon-P (Millipore), and proteins were visualized using ECL (Pierce). Primary antibodies were against Acss2 (Abcam, 66038), TGIF1 (Wotton et al. 1999a), Acat1 (Proteintech, 16215-1-AP), Mlycd (Proteintech, 15265-1-AP), γ-tubulin (Sigma, T6557), and HSP90 (Cell Signaling, 4874). Pcx was detected using Neutravidin-conjugated HRP (Thermo Fisher). Metabolites from normal small intestine (wild type [N = 4] or lacking Tgif1 and Tgif2 [N = 6]) were analyzed by mass spectrometry: Samples were prepared by derivitization with 3-NPH and analyzed essentially as described (Han et al. 2015). A Waters I-class Acquity chromatography system inline with a Waters TQS mass spectrometer was used for the separation and detection. Metabolite levels were normalized to total protein content in the sample.
ChIP
Chromatin was cross-linked for 20 min in 1% formaldehyde and sonicated to 200–1000 bp using a Branson digital sonifier, with microtip as described (Bjerke et al. 2011). Immunoprecipitation was carried out using 10 μL of polyclonal TGIF1 antiserum (Wotton et al. 1999a) or preimmune serum. Bound and input fractions were analyzed by qPCR on a Bio-Rad MyIQ cycler using Sensimix Plus SYBR Green plus FITC mix (Bioline).
Cell culture and siRNA knockdown
HCT116 cells were cultured in RPMI (Invitrogen) with 10% fetal bovine serum (Hyclone). Primary MEFs were cultured as described (Zerlanko et al. 2012). For knockdown, cells were plated in six-well plates and transfected with Dharmacon SMARTpool oligonucleotides against human TGIF1 and TGIF2 (as in Anderson et al. 2017), or with a control siGENOME pool using DharmaFECT reagent 1.
DNA constructs and luciferase assays
All luciferase reporter constructs were generated in pGL3 basic (Promega) by PCR from genomic DNA. Cells were transfected with firefly luciferase reporters and a phCMVRLuc control (Promega), with pCMV5 TGIF1 as indicated (2, 6, 18 ng per well), using PEI. The two mutant TGIF1 constructs encode TGIF1 with changes in conserved DNA-binding residues in the homeodomain: either Arg91 altered to methionine (R91M) or Asp88 altered to serine (N88S) (Bjerke et al. 2011). After 48 h, activity was assayed with luciferase assay reagent (Biotium) using a Berthold LB953 luminometer. Results were normalized using Renilla luciferase activity and assayed with 0.09 μM coelenterazine (Biosynth) as in Hyman-Walsh et al. (2010). Results of replicate transfections are shown (N = 3; mean + standard deviation) normalized to the RLuc transfection control.
Supplementary Material
Acknowledgments
We thank members of the Wotton laboratory for discussions, and Dr. M. Mayo for advice. This work was supported by grants from the National Institute of Neurological Disorders and Stroke (NS077958) and the National Institute of General Medical Sciences (GM099853) to D.W.
Author contributions: A.S., T.A.M., and T.E.F. performed the experiments. A.S., T.A.M., T.E.F., H.F.F., and D.W. analyzed the data. A.S. and D.W. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.320127.118.
References
- Anderson AE, Taniguchi K, Hao Y, Melhuish TA, Shah A, Turner SD, Sutherland AE, Wotton D. 2017. Tgif1 and Tgif2 repress expression of the RabGAP Evi5l. Mol Cell Biol 37: e00527–00516. 10.1128/MCB.00527-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, Wishart DS. 2016. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44: W147–W153. 10.1093/nar/gkw419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino E, Reimund B, Wildt-Perinic D, Clerc R. 1995. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J Biol Chem 270: 31178–31188. 10.1074/jbc.270.52.31178 [DOI] [PubMed] [Google Scholar]
- Bjerke GA, Hyman-Walsh C, Wotton D. 2011. Cooperative transcriptional activation by Klf4, Meis2, and Pbx1. Mol Cell Biol 31: 3723–3733. 10.1128/MCB.01456-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürglin TR, Affolter M. 2016. Homeodomain proteins: an update. Chromosoma 125: 497–521. 10.1007/s00412-015-0543-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma'ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, Nusse R. 2012. Wnt/β-catenin signaling and disease. Cell 149: 1192–1205. 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- Colnot S, Niwa-Kawakita M, Hamard G, Godard C, Le Plenier S, Houbron C, Romagnolo B, Berrebi D, Giovannini M, Perret C. 2004. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest 84: 1619–1630. 10.1038/labinvest.3700180 [DOI] [PubMed] [Google Scholar]
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. 2014. Acetate dependence of tumors. Cell 159: 1591–1602. 10.1016/j.cell.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, Bultman SJ. 2011. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13: 517–526. 10.1016/j.cmet.2011.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. 2005. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol 21: 659–693. 10.1146/annurev.cellbio.21.022404.142018 [DOI] [PubMed] [Google Scholar]
- Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM, et al. 1994. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci 91: 8969–8973. 10.1073/pnas.91.19.8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady WM, Myeroff LL, Swinler SE, Rajput A, Thiagalingam S, Lutterbaugh JD, Neumann A, Chang J, Kim S-J, Kinzler KW, et al. 1999. Mutational inactivation of transforming growth factor b receptor type II in microsatellite stable colon cancers. Cancer Res 59: 320–324. [PubMed] [Google Scholar]
- Guca E, Suñol D, Ruiz L, Konkol A, Cordero J, Torner C, Aragon E, Martin-Malpartida P, Riera A, Macias MJ. 2018. TGIF1 homeodomain interacts with Smad MH1 domain and represses TGF-β signaling. Nucleic Acids Res 46: 9220–9235. 10.1093/nar/gky680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lin K, Sequeira C, Borchers CH. 2015. An isotope-labeled chemical derivatization method for the quantitation of short-chain fatty acids in human feces by liquid chromatography-tandem mass spectrometry. Anal Chim Acta 854: 86–94. 10.1016/j.aca.2014.11.015 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hao Y, Bjerke GA, Pietrzak K, Melhuish TA, Han Y, Turner SD, Frierson HF Jr, Wotton D. 2018. TGFβ signaling limits lineage plasticity in prostate cancer. PLoS Genet 14: e1007409 10.1371/journal.pgen.1007409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Göke B, Kolligs FT. 2014. Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signaling. BMC Genomics 15: 74 10.1186/1471-2164-15-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, Cho KR, Fearon ER. 2007. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res 67: 9721–9730. 10.1158/0008-5472.CAN-07-2735 [DOI] [PubMed] [Google Scholar]
- Hutton JE, Wang X, Zimmerman LJ, Slebos RJ, Trenary IA, Young JD, Li M, Liebler DC. 2016. Oncogenic KRAS and BRAF drive metabolic reprogramming in colorectal cancer. Mol Cell Proteomics 15: 2924–2938. 10.1074/mcp.M116.058925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman CA, Bartholin L, Newfeld SJ, Wotton D. 2003. Drosophila TGIF proteins are transcriptional activators. Mol Cell Biol 23: 9262–9274. 10.1128/MCB.23.24.9262-9274.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman-Walsh C, Bjerke GA, Wotton D. 2010. An autoinhibitory effect of the homothorax domain of Meis2. FEBS J 277: 2584–2597. 10.1111/j.1742-4658.2010.07668.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto I, Pimkhaokham A, Watanabe T, Saito-Ohara F, Soeda E, Inazawa J. 2000. Amplification and overexpression of TGIF2, a novel homeobox gene of the TALE superclass, in ovarian cancer cell lines. Biochem Biophys Res Commun 276: 264–270. 10.1006/bbrc.2000.3449 [DOI] [PubMed] [Google Scholar]
- Kaiser S, Park YK, Franklin JL, Halberg RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG, et al. 2007. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol 8: R131 10.1186/gb-2007-8-7-r131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. 1996. Lessons from hereditary colorectal cancer. Cell 87: 159–170. 10.1016/S0092-8674(00)81333-1 [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé E, Lock L, Letamendia A, Gorska AE, Gryfe R, Gallinger S, Moses HL, Attisano L. 2007. Transcriptional cooperation between the transforming growth factor-β and Wnt pathways in mammary and intestinal tumorigenesis. Cancer Res 67: 75–84. 10.1158/0008-5472.CAN-06-2559 [DOI] [PubMed] [Google Scholar]
- Lee BK, Shen W, Lee J, Rhee C, Chung H, Kim KY, Park IH, Kim J. 2015. Tgif1 counterbalances the activity of core pluripotency factors in mouse embryonic stem cells. Cell Rep 13: 52–60. 10.1016/j.celrep.2015.08.067 [DOI] [PubMed] [Google Scholar]
- Levy L, Hill CS. 2006. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev 17: 41–58. 10.1016/j.cytogfr.2005.09.009 [DOI] [PubMed] [Google Scholar]
- Lo RS, Wotton D, Massagué J. 2001. Epidermal growth factor signaling via Ras controls the Smad transcriptional co-repressor TGIF. EMBO J 20: 128–136. 10.1093/emboj/20.1.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. 2002. cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem 277: 33275–33283. 10.1074/jbc.M204935200 [DOI] [PubMed] [Google Scholar]
- Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. 1995. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 268: 1336–1338. 10.1126/science.7761852 [DOI] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. 2014. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159: 1603–1614. 10.1016/j.cell.2014.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. 2008. TGFβ in cancer. Cell 134: 215–230. 10.1016/j.cell.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D. 2005. Smad transcription factors. Genes Dev 19: 2783–2810. 10.1101/gad.1350705 [DOI] [PubMed] [Google Scholar]
- Melhuish TA, Wotton D. 2000. The interaction of the carboxyl terminus-binding protein with the Smad corepressor TGIF is disrupted by a holoprosencephaly mutation in TGIF. J Biol Chem 275: 39762–39766. 10.1074/jbc.C000416200 [DOI] [PubMed] [Google Scholar]
- Melhuish TA, Gallo CM, Wotton D. 2001. TGIF2 interacts with histone deacetylase 1 and represses transcription. J Biol Chem 276: 32109–32114. 10.1074/jbc.M103377200 [DOI] [PubMed] [Google Scholar]
- Miguchi M, Hinoi T, Shimomura M, Adachi T, Saito Y, Niitsu H, Kochi M, Sada H, Sotomaru Y, Ikenoue T, et al. 2016. Gasdermin C is upregulated by inactivation of transforming growth factor β receptor type II in the presence of mutated Apc, promoting colorectal cancer proliferation. PLoS One 11: e0166422 10.1371/journal.pone.0166422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. 2016. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 66: 271–289. 10.3322/caac.21349 [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. 2003. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273. 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, Dove WF. 1990. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247: 322–324. 10.1126/science.2296722 [DOI] [PubMed] [Google Scholar]
- Muñoz NM, Upton M, Rojas A, Washington MK, Lin L, Chytil A, Sozmen EG, Madison BB, Pozzi A, Moon RT, et al. 2006. Transforming growth factor β receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by Apc mutation. Cancer Res 66: 9837–9844. 10.1158/0008-5472.CAN-06-0890 [DOI] [PubMed] [Google Scholar]
- Nakakuki K, Imoto I, Pimkhaokham A, Fukuda Y, Shimada Y, Imamura M, Amagasa T, Inazawa J. 2002. Novel targets for the 18p11.3 amplification frequently observed in esophageal squamous cell carcinomas. Carcinogenesis 23: 19–24. 10.1093/carcin/23.1.19 [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. 1995. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci 92: 4482–4486. 10.1073/pnas.92.10.4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parini P, Melhuish TA, Wotton D, Larsson L, Ahmed O, Eriksson M, Pramfalk C. 2018. Overexpression of transforming growth factor β induced factor homeobox1 represses NPC1L1 and lowers markers of intestinal cholesterol absorption. Atherosclerosis 275: 246–255. 10.1016/j.atherosclerosis.2018.06.867 [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. 2016. The emerging hallmarks of cancer metabolism. Cell Metab 23: 27–47. 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. 1995. Mutations in the APC gene and their implications for protein structure and function. Curr Opin Genet Dev 5: 66–71. 10.1016/S0959-437X(95)90055-1 [DOI] [PubMed] [Google Scholar]
- Powers SE, Taniguchi K, Yen W, Melhuish TA, Shen J, Walsh CA, Sutherland AE, Wotton D. 2010. Tgif1 and Tgif2 regulate Nodal signaling and are required for gastrulation. Development 137: 249–259. 10.1242/dev.040782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho E, Batlle E, Clevers H. 2004. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol 20: 695–723. 10.1146/annurev.cellbio.20.010403.092805 [DOI] [PubMed] [Google Scholar]
- Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, Akamoto S, Tabata S, Saitoh K, Kato K, Sato S, et al. 2017. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci 114: E7697–E7706. 10.1073/pnas.1710366114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. 2015. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer cell 27: 57–71. 10.1016/j.ccell.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segditsas S, Tomlinson I. 2006. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 25: 7531–7537. 10.1038/sj.onc.1210059 [DOI] [PubMed] [Google Scholar]
- Seo SR, Lallemand F, Ferrand N, Pessah M, L'Hoste S, Camonis J, Atfi A. 2004. The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J 23: 3780–3792. 10.1038/sj.emboj.7600398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SR, Ferrand N, Faresse N, Prunier C, Abécassis L, Pessah M, Bourgeade MF, Atfi A. 2006. Nuclear retention of the tumor suppressor cPML by the homeodomain protein TGIF restricts TGF-β signaling. Mol Cell 23: 547–559. 10.1016/j.molcel.2006.06.018 [DOI] [PubMed] [Google Scholar]
- Sharma V, Antonacopoulou AG, Tanaka S, Panoutsopoulos AA, Bravou V, Kalofonos HP, Episkopou V. 2011. Enhancement of TGF-β signaling responses by the E3 ubiquitin ligase Arkadia provides tumor suppression in colorectal cancer. Cancer Res 71: 6438–6449. 10.1158/0008-5472.CAN-11-1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, Matsumoto H, Takano H, Akiyama T, Toyoshima K, et al. 1997. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 278: 120–123. 10.1126/science.278.5335.120 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Anderson AE, Sutherland AE, Wotton D. 2012. Loss of Tgif function causes holoprosencephaly by disrupting the SHH signaling pathway. PLoS Genet 8: e1002524 10.1371/journal.pgen.1002524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Anderson AE, Melhuish TA, Carlton AL, Manukyan A, Sutherland AE, Wotton D. 2017. Genetic and molecular analyses indicate independent effects of TGIFs on Nodal and Gli3 in neural tube patterning. Eur J Hum Genet 25: 208–215. 10.1038/ejhg.2016.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. 1988. Genetic alterations during colorectal-tumor development. N Engl J Med 319: 525–532. 10.1056/NEJM198809013190901 [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang H, Gao H, Xu B, Zhai W, Li J, Zhang C. 2015. Elevated expression of TGIF is involved in lung carcinogenesis. Tumour Biol 36: 9223–9231. 10.1007/s13277-015-3615-8 [DOI] [PubMed] [Google Scholar]
- Wang JL, Qi Z, Li YH, Zhao HM, Chen YG, Fu W. 2017. TGFβ induced factor homeobox 1 promotes colorectal cancer development through activating Wnt/β-catenin signaling. Oncotarget 8: 70214–70225. 10.18632/oncotarget.19603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. 1956. On the origin of cancer cells. Science 123: 309–314. 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- Wotton D, Taniguchi K. 2018. Functions of TGIF homeodomain proteins and their roles in normal brain development and holoprosencephaly. Am J Med Genet C Semin Med Genet 178: 128–139. 10.1002/ajmg.c.31612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massagué J. 1999a. A Smad transcriptional corepressor. Cell 97: 29–39. 10.1016/S0092-8674(00)80712-6 [DOI] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Swaby LA, Massagué J. 1999b. Multiple modes of repression by the smad transcriptional corepressor TGIF. J Biol Chem 274: 37105–37110. 10.1074/jbc.274.52.37105 [DOI] [PubMed] [Google Scholar]
- Zerlanko BJ, Bartholin L, Melhuish TA, Wotton D. 2012. Premature senescence and increased TGFβ signaling in the absence of Tgif1. PLoS One 7: e35460 10.1371/journal.pone.0035460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MZ, Ferrigno O, Wang Z, Ohnishi M, Prunier C, Levy L, Razzaque M, Horne WC, Romero D, Tzivion G, et al. 2015. TGIF governs a feed-forward network that empowers Wnt signaling to drive mammary tumorigenesis. Cancer Cell 27: 547–560. 10.1016/j.ccell.2015.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.