

Abstract
Drugs are discovered through the biological screening of collections of compounds, followed by optimization toward functional endpoints. The properties of screening collections are often balanced between diversity, physicochemical favorability, intrinsic complexity and synthetic tractability.1 Whereas natural product (NP) collections excel in the first three attributes, NPs suffer a disadvantage on the last point. Academic total synthesis research has worked to solve this problem by devising syntheses of NP leads, diversifying late-stage intermediates or derivatizing the NP target. This work has led to the discovery of reaction mechanisms, the invention of new methods and the development of FDA-approved drugs. Few drugs, however, are themselves NPs; instead NP-analogs predominate. Here we highlight past examples of NP analog development and successful NP-derived drugs. More recently, chemists have explored how NP analogs alter the retrosynthetic analysis of complex scaffolds, merging structural design and synthetic design. This strategy maintains the intrinsic complexity of the NP but can alter the physicochemical properties of the scaffold, like core instability that renders the NP a poor chemotype. Focused libraries based on these syntheses may exclude the NP but maintain the molecular properties that distinguish NP-space from synthetic space,2 properties that have statistical advantages in clinical progression.3,4 Research that expedites synthetic access to NP-motifs can prevent homogeneity of chemical matter available for lead discovery. Easily accessed, focused libraries of NP scaffolds can fill empty but active gaps in screening sets and expand the molecular diversity of synthetic collections.
Introduction
The “marketplace of ideas” refers to a symbolic open exchange where beliefs compete for uptake. This metaphor arose from Supreme Court opinions attempting to uphold free-speech rights enshrined by the First Amendment to the US Constitution.5 Justice William Brennan’s rationale6 behind an economic analogy points out that a ‘purchase’ of truth might only occur if the supply of ideas remains unsuppressed. The “marketplace” emphasizes the practical importance of diversity—diversity as a search parameter. If maximized, diversity increases the probability that a search is successful. In the search for truth, diversity of thought ensures that truth is available at all.
In the search for new medicines, molecular diversity within chemical libraries increases the probability that useful leads are identified.7 Put negatively, modest diversity limits the range of chemotypes identified as leads in a high-throughput screen. Low diversity reduces the probability of lead identification at all.8 What is molecular diversity? We were invited by JACS to provide a Perspective from our area of research: natural product (NP) synthesis.
Through the lens of chemical informatics, molecular diversity describes the distribution of a molecular set in an N-dimensional chemical space defined by molecular descriptors N.9 Characterization of diversity within and between chemical libraries depends on which combination of the existing ~3,100 molecular descriptors are used to define a reference chemical space: formula weight, total polar surface area, fraction sp3 (Fsp3), hydrogen bond donor number, etc.9 A chemical library is said to be diverse if the distribution of molecules is broad relative to the chosen molecular descriptor(s) and their values. Hence a productive discussion of molecular diversity is contingent upon choice of meaningful molecular descriptors.10 In this Perspective, we emphasize molecular descriptors that differentiate NP space from synthetic collections.
When screening for drug leads, biological activity becomes a required molecular descriptor. In this context, a chemical library is diverse if it possesses a large number of target-active compound clusters (chemotypes), with a minimum number of compounds per cluster.11 At the extremes (Figure 1), all compounds are active, but clustered (similar, non-diverse); or all the compounds are active and unclustered (unique, diverse). Using this definition, it’s easy to see why mere increase in library size only ‘increases the chances of not finding leads.’11b To put it another way: low-diversity combinatorial chemistry doesn’t find more needles, it creates more hay.12
Figure 1.
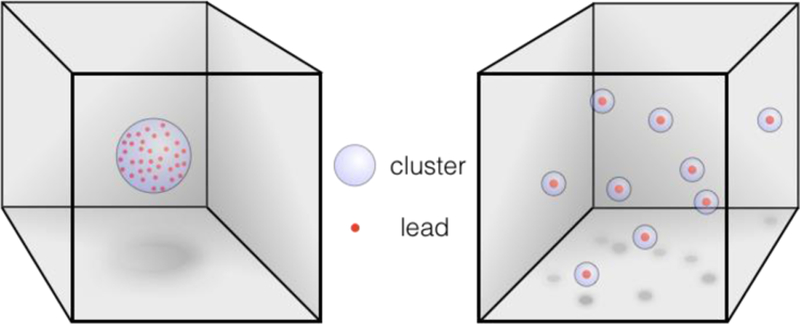
Extremes of non-diverse vs. diverse leads (chemical targets) in a molecular collection sorted by chemotype.
Characterization of lead quality does not depend on targeted activity alone. Instead, biological activity is one important molecular descriptor among others. If an active lead is covered in related patent space, its value is low.11a If a lead possesses poor physicochemical properties, its optimization begins from a disadvantaged starting point. Falling into this ‘local minimum’ can prove costly: on average, only small changes occur during optimization between lead and drug candidate,10 and failure in clinical trials is expensive. On the other hand, if a lead is structurally advantaged but synthetically intractable, its quick advance through medicinal chemistry optimization is unlikely.
Natural products have served historically as leads for drug development, either through anecdotal reports of active extracts, traditional medicine or high-throughput screens. Yet NPs inhabit the tension between favorable properties and synthetic intractability. They occupy areas of chemical space unpopulated by commercial, synthetic molecules and drugs (Figure 3), as defined by high Fsp3, high stereochemical content, high oxygen content, high ring content, and low aromaticity.16,13 Many of these molecular descriptors have been earmarked as desirable since they correlate with increased progression through clinical trials—NP space may represent a series of clusters with statistical advantages.14,15 NPs have been pre-optimized through evolution to bind biomolecules and penetrate cells.16 NPs contain ‘constellations’ of functional groups, stereogenic centers, and ring motifs in high density.17 Properties imparted by these constellations of features have been described as ‘emergent’ and therefore hard to design de novo.18 Inclusion of NPs, NP-analogs, or NP-like compounds in traditional synthetic or combinatorial sets increases the diversity of these collections because it fills unoccupied chemical space.2 Yet the synthetic complexity of NPs limits the representation of NP-space in synthetic commercial libraries and limits NP uptake in medicinal chemistry campaigns.19
Figure 3.
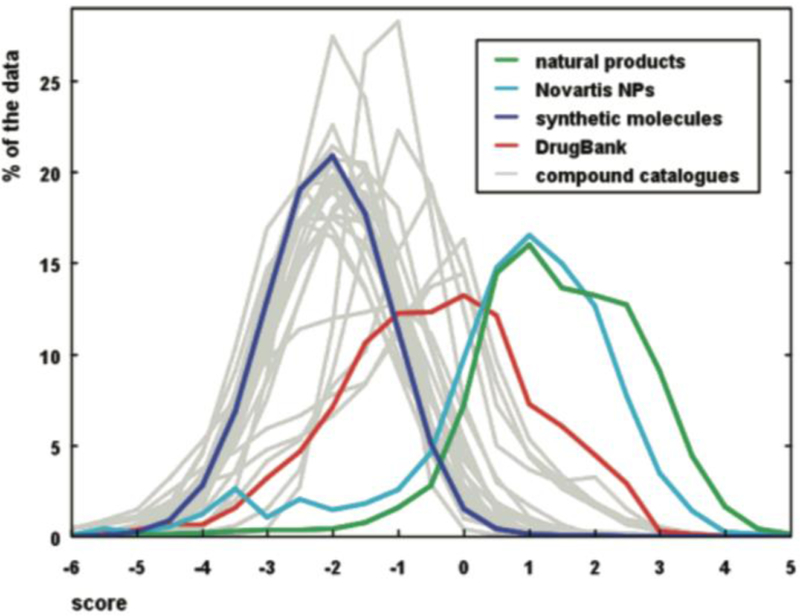
Used from Ref. 16 (Ertl, Roggo and Schuffenhauer, J. Chem. Inf. Model. 2008, 48, 68). Natural products differ from most synthetic molecules, but overlap with drugs, which have been optimized for biocompatibility. NPs are preoptimized.
One solution to the problem of synthetic intractability has been to target a structurally-simplified version of a NP that retains its activity. This approach relies on a general correlation between intrinsic complexity and synthetic complexity: the more complex a structure, the more challenging its synthesis.20 These attributes are related, but non-identical: a complex molecule may be the product of spontaneous assembly from simple precursors (Figure 4).21 For example, Heathcock discovered that a simple squalene-related dialdehyde undergoes polycyclization to the core of the daphniphylene alkaloids when heated with methylamine and acid. Although the atom count (80) does not change at all, the information content22 (see paragraph below) increases 140% — a reflection of increased intrinsic complexity, comparable to adding 400 pages to a 1000-page book.
Figure 4.
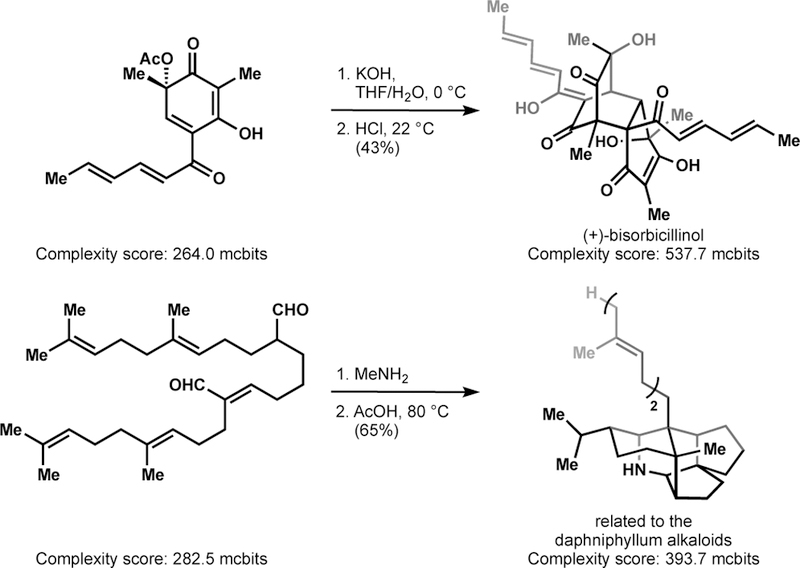
Structural complexity and synthetic complexity are related, but not identical.
Intrinsic complexity, a fixed structural property, is easy to determine, whereas synthetic complexity is hard to define a priori and is time dependent: synthetic technology advances every day.20,23 Intrinsic complexity can be measured by structural topology,24 heteroatomic and stereochemical content,23 or as information content according to, for example, Böttcher’s method.22 This latter method, used in the sections below, accounts for atom count, connectivity, stereochemistry, heteroatomic content and symmetry. Its ease of calculation makes Böttcher’s method appealing (Excel tables can be found in the SI).
Reduction of intrinsic complexity has been correlated with loss of specificity.25 Reduced intrinsic complexity can remove a structure from NP space and push it towards more populated areas of synthetic chemical space. This approach rapidly arrives at function and can enable discovery (Function-Oriented Synthesis, FOS, see below),20 but sometimes at the cost of diversity and the physicochemical benefits associated with NP space.
Yet NPs themselves, narrowly defined by structure, are not highly represented among approved drugs. Instead, NP-derived analogs predominate. From 1981–2014, a period when 385 NPs or NP-analogs became clinically approved new chemical entities (NCEs), NP-derived analogs outpaced NPs 316:69. The ratio was higher (83:13) among anti-infective drugs (antibacterial, antifungal, antiparasitic, and antiviral).26 Many of these NP-analogs retain all the complexity of the NP itself and reside squarely in NP-space (see Figure 2). These approved drugs are not direct products of biosynthesis. Given the predominance of NP-analogs over NPs, why recapitulate what nature has already made? To only target a single metabolite and its congeners is to strip a crucial design element from the synthetic chemist.27 Instead, the interplay between structure, synthesis, and function benefits from the powerful tools of chemistry.
Figure 2.
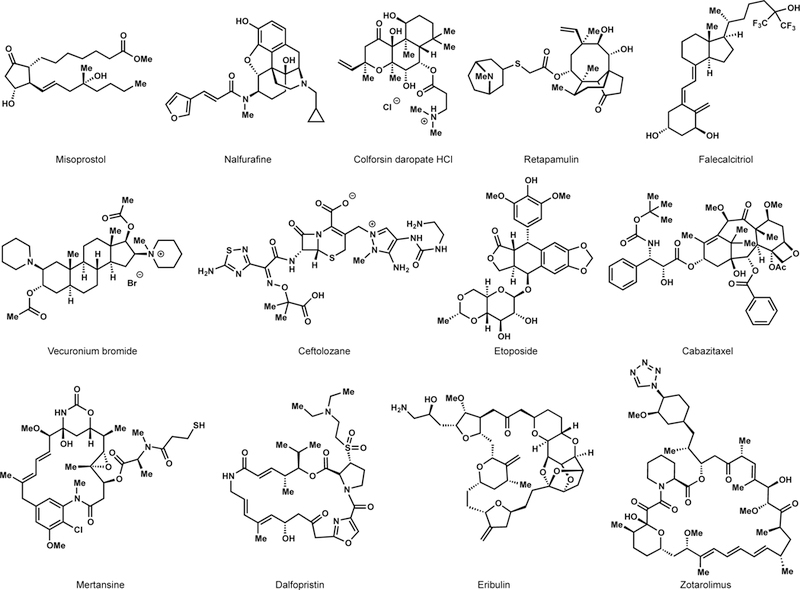
FDA-approved NP analogs.
Synthetic chemists have been tinkering with, altering, and studying NPs for decades. However, semisynthetic modification of NPs remains the major source of FDA approved NP analogs. Fully synthetic approaches leading to an approved NP analog are rare. In the following section, we highlight fully synthetic (de novo) routes to therapeutically relevant NP analogs. First, we highlight fully synthetic NP analogs that are approved therapeutics or are under clinical investigation. Second, we highlight fully synthetic NP analogs that are based on FDA approved NPs. As evidenced by Tanimoto indices28 and intrinsic complexity scores,22 these NP-analogs retain structural features of NP space without compromising intrinsic complexity/reducing information content. Both metrics are easy to calculate and provide some basis to evaluate deviation from the NP. Third, we highlight fully synthetic NP analogs that possess a promising path for future advancement. Finally, we cover recent examples of structural designs of NP analogs that shorten or alter synthetic routes compared to the NP itself. Progress in this area will expand the coverage of NP chemical space by enabling the synthesis of focused libraries of NP analogs with favorable properties for advance as clinical candidates. We conclude with a look to the future based on advances in molecular biology.
Selected Examples of Fully Synthetic Natural Product Analogs
Tetracycline.
Since the discovery of chlorotetracycline in 1948, the tetracyclines have engendered widespread use as broad spectrum antibiotics.29 Characterized by four linearly fused six-membered carbon rings, tetracyclines inhibit protein synthesis by binding to the 30S ribosomal subunit thereby preventing attachment of aminoacyl-tRNA to the ribosomal acceptor (A) site.30 Like most β-lactam and macrolide antibiotics, nearly all tetracyclines approved for clinical use are derived via fermentation or through semisynthesis from fermentation products.
Despite this abundance, tetracyclines have attracted notable attention from synthetic chemists. Woodward’s first total synthesis of a tetracycline, a non-natural semisynthetic analog with superior stability to tetracyline, sancycline, featured a “left to right” D to A ring construction strategy.31 This D to A approach would serve as the basic strategy toward this class until crystal structures of tetracycline bound to the 30S ribosomal subunit suggested the D-ring would be a desirable site for modification.29 Since D to A ring construction was not ideally suited to rapidly access D-ring functionalized tetracyclines, the Myers group developed a concise synthesis in which the C ring was constructed via convergent coupling of functionalized D- and AB-ring precursors.32
Using this D plus AB strategy, Tetraphase pharmaceuticals, a company established in 2006 to commercialize the Myers group’s tetracycline synthetic platform, was able to construct over 3,000 tetracycline analogs, many of which would be inaccessible via semisynthesis.33 One promising analog, Eravacycline (2), exhibited improved broad-spectrum antibiotic activity against multidrug-resistant bacteria (MIC90 = 0.008 – 2 µg/mL), relative to approved tetracyclines.34 Eravacycline’s superior activity is linked to its decreased susceptibility to tetracycline-specific resistance mechanisms such as ribosomal protection and efflux.35 Furthermore, Eravacycline is highly active against vancomycin resistant E. faecalis and penicillin and macrolide resistant S. pneumonia. Eravacycline was recently approved by the FDA for the treatment of Complicated Intra-abdominal Infections and is currently the only fully synthetic tetracycline analog to achieve FDA approval.
Vorapaxar.
Himbacine (3), a piperdine alkaloid characterized by its linearly fused tricyclic lactone core, was isolated from the bark of the Australian magnolias, Galbulimima baccata, in the early 1960’s.36 Himbacine initially attracted attention as a potential lead for Alzheimer’s disease (AD) owing to its potent muscarinic receptor antagonist activity against M1 and M2 subtypes.37 Subtype selective antagonism of M2 receptors enhances synaptic acetylcholine levels—counteracting synaptic acetylcholine deficiencies observed in the cortical and hippocampal brain regions of dementia afflicted AD patients. To improve receptor selectivity and potency, Chackalamannil and coworkers developed a concise synthesis of himbacine and analogs.38
Although the AD therapeutic target was abandoned, the focused library of himbacine derivatives yielded a potent antagonist of PAR-1, a G-protein coupled receptor (GPCR) that mediates thrombin-induced platelet aggregation.39 Selective antagonism of PAR-1 was attractive for suppression of thrombin-induced platelet activation present in diseased states such as arterial thrombosis, without inhibition of other critical thrombin-dependent processes such as the formation of fibrin-based blood clots.40 SAR identified by the synthetic route resulted in the discovery of analog 4, also known as Vorapaxar, an antithrombotic agent approved in 2014 for the treatment of acute coronary syndrome and secondary prevention of cardiovascular events.41 Vorapaxar is an FDA approved natural product analog that was derived from fully synthetic efforts. From an AD lead to an approved drug to treat acute coronary syndrome, himbacine and its analogs exemplify the privileged activity possessed by NPs. The therapeutic endpoint can change, but the privileged nature of NP space remains constant.
Epothilone B.
Epothilone B (EpoB, 5), a 16-membered macrocyclic lactone isolated from the myxobacterium Sorangium cellulosum strain 90, was originally disclosed as a potential antifungal agent in 1987.42 Merck later rediscovered EpoB in 1995 as an agent possessing superior activity to paclitaxel in tubulin polymerization assays.43 Evaluation confirmed that EpoB binds to tubulin at the paclitaxel binding site and hyperstabilizes microtubules, resulting in mitosis inhibition and apoptosis.42 Unlike most antitumor agents, EpoB is much less susceptible to Pgp-mediated efflux and as a result maintains significant potency against multi-drug-resistant cancer cell lines.42
The superior bioactivity of EpoB and its unique structural features captured the attention of multiple research groups in industry and academia. Kosan Biosciences and Bristol-Myers Squibb were among the first to develop fermentation processes to access epothilone B, with the goal to improve the pharmacological profile via semisynthesis.44 However, semisynthetic modification of the core proved challenging and the pool of available transformations was limited. These restrictions ignited intense competition to establish a fully synthetic route that could enable SAR exploration and optimization.
By 1997, Nicolaou and Danishefsky each established synthetic routes that proved instrumental to understand which structural elements were critical to bioactivity.45 The remarkable SAR data supplied by these two labs culminated in the discovery of several clinical candidates.46 One promising analog disclosed by Danishefsky, iso-fludelone (6) was shown to possess superior potency and bioavailability, a larger therapeutic window, and reduced nonspecific cytotoxicity relative to epothilone B (Figure 7).47 Specifically, removal of the epoxide decreased nonspecific cytotoxicity, unsaturation of C9-C10 improved potency and metabolic stability, incorporation of CF3 at C12 decreased cytotoxicity and broadened the therapeutic window, and replacement of the thiazole with isoxazole increased potency and solubility.48 Importantly, 6 achieved complete remission and cures in several xenograft mouse models including: extra-large MX-1, ovarian carcinoma SK-OV-3, and lung carcinoma A549.47 Iso-fludelone is currently in phase I clinical trials to examine dose escalation and pharmacokinetics in patients with advanced solid tumors.49 Despite significant fully synthetic efforts, Ixabepilone, a semisynthetic EpoB analog developed at Bristol-Myers Squibb, remains the only epothilone analog approved by the FDA.44b
Figure 7.
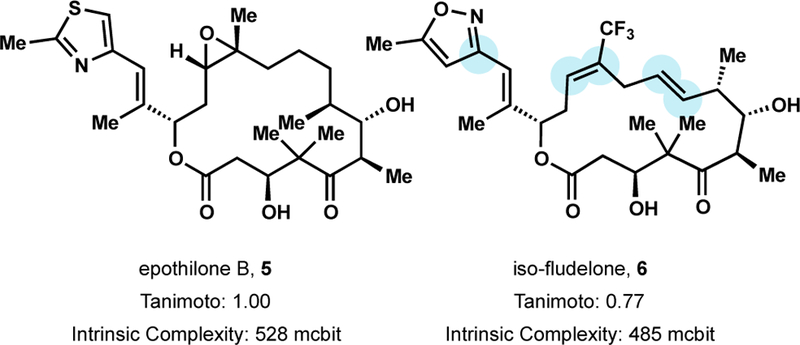
Isofludelone, a fully synthetic epothilone B analog under clinical investigation.
Erythromycin.
Isolated from a Philippine soil sample in 1949, erythromycin (7) is the forerunner natural product of clinically approved macrolide antibiotics, a class characterized by 14-, 15-, and 16-membered macrocyclic lactones with one or more pendant glycosidic residues.50 Erythromycin’s antibacterial activity results from protein synthesis inhibition via reversible binding to the 50S ribosomal subunit.51 Although erythromycin was the first clinically approved macrolide (1952), the undesired gastrointestinal side-effects derived from its acid instability inspired the search for second-generation macrolides.52 However, erythromycin’s complex scaffold proved challenging for early synthetic efforts and the production of novel erythromycin- based antibiotics was mostly limited to semisynthetic modification of erythromycin. Clarithromycin (1991), azithromycin (1991), and telithromycin (2004) are FDA approved semisynthetic analogs derived from erythromycin in 6-, 4-, and 12-steps respectively.53 Semisynthesis from fermentation-derived material can provide clear advantages from a supply perspective but semisynthesis can also limit the diversity of accessible analogs. Selective modification of a complex NP is challenging and usually only a few positions can be effectively modified.54
The limitations of semisynthesis coupled with growing resistance to approved macrolide antibiotics prompted the Myers group to develop a highly convergent and fully synthetic route to erythromycin analogs.55 From 8 simple building blocks (Figure 8a) 14- and 15-membered azaketolides, and 14-membered ketolides were assembled in 10–14 steps (LLS). The efficiency of the route enabled the synthesis of more than 300 erythromycin analogs, many inaccessible via semisynthesis. 83% of these fully synthetic erythromycin analogs exhibited a minimum inhibitory concentration (MIC) ≤ 4.0 µg/mL against wild-type S. pneumoniae. One of the more promising analogs, analog 8, (Figure 8b) is more potent than all clinically approved macrolides in challenging strains such as: S. pneumoniae expressing both ermB and mefA genes (MIC ≤ 0.03 µg/mL), vancomycin-resistant Enterococcus expressing ermB (1.0 µg/mL), methicillin-resistant Staphylococcus aureus (16.0 µg/mL), and Pseudomonas aeruginosa (16.0 µg/mL). Analog 8 also possessed superior potency relative to clinically approved macrolides in Gram-negative bacteria. Though many erythromycin analogs such as 8 would require preclinical development, the versatility of Myer’s convergent fully synthetic route broadened the coverage of erythromycin’s local chemical space and enabled rapid access to unprecedented erythromycin analogs.
Figure 8.
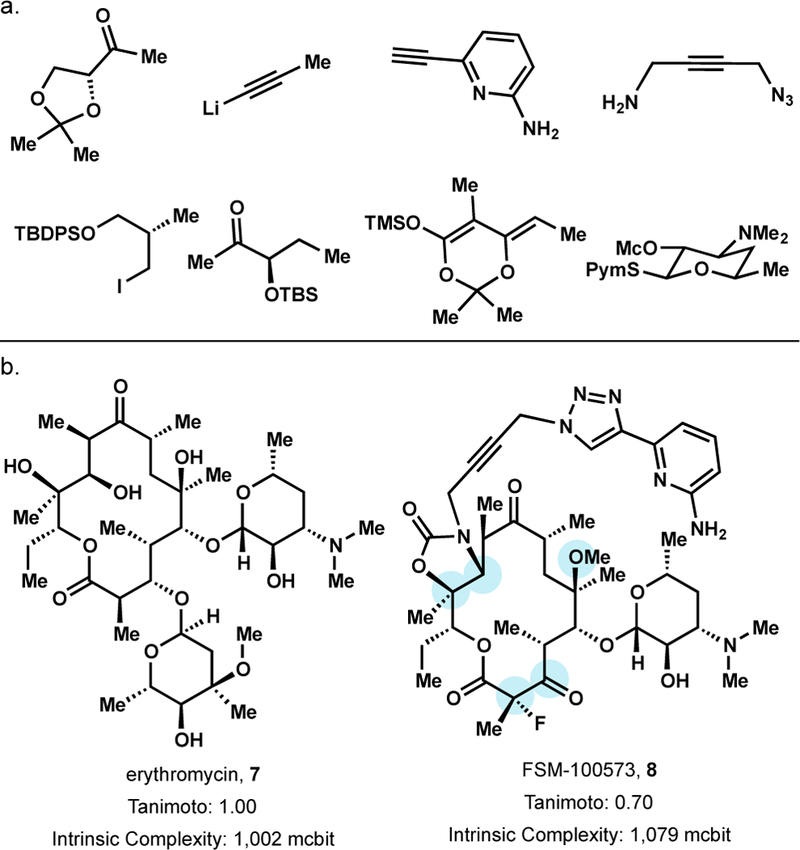
Myers’ erythromycin / solithromycin analog FSM-100573.
Vancomycin.
The flagship member of glycopeptide antibiotics, vancomycin (9), was discovered by Eli Lilly in 1952, and has long been used as a drug of last resort for the treatment of resistant and lethal infections.56 Characteristic of glycopeptide antibiotics, vancomycin inhibits bacterial cell wall synthesis by binding and sequestering the peptidoglycan peptide terminus D-alanine-D-alanine (D-Ala-D-Ala).57,58
Sequestration of this key precursor prevents transpeptidation and cell wall cross-linking, which results in cell lysis.
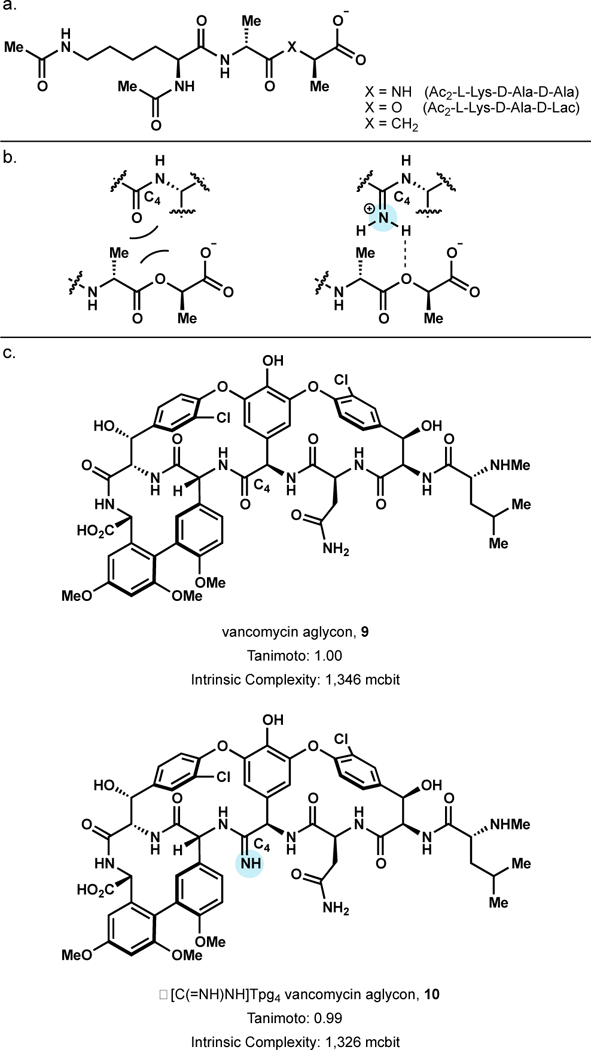
In response to vancomycin treatment, resistant bacteria such as VanA and VanB enterococcal, modify the precursor peptidoglycan terminus D-Ala-D-Ala to D-Ala-D-Lac.59 This elegant resistance mechanism—exchange an amide linkage for an ester linkage—results in a 1000-fold decrease in binding affinity to vancomycin. Analysis of vancomycin complexed with D-Ala-D-X (X = Alanine, Lactic acid, or isobutyric acid) revealed that loss of a central H-bonding interaction and a repulsive lone pair interaction between C4 carbonyl and D-Ala-D-Lac ester oxygen were likely responsible for the loss in binding affinity—the repulsive lone pair interactions predominating (Figure 9a).60 Since selective semisynthetic modification of vancomycin at C4 was not yet possible, a divergent total synthesis was developed by Boger to access a series of C4 functionalized vancomycin aglycons.61 Notably, replacement of C4 amide with an amidine was hypothesized to bind D-Ala-D-Lac and retain affinity for D-Ala-D-Ala by both removing destabilizing electrostatic interactions and providing an additional handle for H-bonding (Figure 9b).
Figure 9.
Activity against VanA-resistant bacteria restored by Boger’s vancomycin amidine analog.
Indeed, C4 amidine analog 10 (Figure 9c) not only exhibits a 600-fold increase in binding affinity to D-Ala-D-Lac (Ka = 6.9 × 104 M−1) over that of vancomycin aglycon (Ka = 1.2 × 102 M−1), 10 also retains binding to D-Ala-D-Ala (Ka = 7.3 × 104 M−1).62 Furthermore, 10 possessed significant activity (MIC = 0.31 µg/mL) against VanA resistant E. faecalis (VanA VRE, BM4166), one of the most challenging strains of vancomycin-resistant bacteria. Importantly, 10 maintains significant activity against vancomycin sensitive bacteria (MIC = 0.30—2.0 µg/mL)—equipotent to vancomycin and vancomycin aglycon. These efforts exemplify how synthetic route development toward a rationally designed NP analog can be useful in both exploration of a biologically interesting hypothesis and improvement of the activity of the parent natural product.
Vinblastine.
Isolated from Catharanthus roseus in the late 1950’s, vinblastine (11), a Vinca alkaloid dimer containing velbanamine and vindoline-derived subunits, was among the first small molecules shown to bind to tubulin and inhibit microtubule formation and mitosis.63 Despite its introduction as an anti-cancer therapeutic in 1965, vinblastine remains efficacious and is used in combination therapies to treat Hodgkin’s disease, testicular, ovarian, breast, and head and neck cancer. However, prolonged use of vinblastine leads to development of phosphoglycoprotein (Pgp)-mediated resistance. Identification of vinblastine analogs that address Pgp resistance is of paramount importance and a primary focus in the Boger group.
To develop vinblastine analogs with activity against Pgp overexpressing cancers, the Boger lab leveraged their previously developed Fe(III)-promoted oxidative coupling between vindoline and catharanthine and late stage Fe(III)/NaBH4-mediated radical alkene oxidation at C20’.64 These C20’ vinblastine analogs included N3, NH2,CN, SCN, NHCONH2 Cl, and F substitution using their in-house hydrofunctionalization methods.64c Particularly noteworthy were C20’ urea-based analogs, which displayed both increased potency over vinblastine and increased activity against a Pgp overexpressing, vinblastine resistant tumor cell line.65 Further exploration of urea analogs resulted in the discovery of 12.65b Urea 12 was found to be, on average, 30-fold more potent than vinblastine across a panel of 15 tumor cell lines (avg. IC50 = 200 pM) and 100–200-fold more active against the clinically relevant vinblastine resistant HCT116/VM46 cell line (IC50 = 3.5 nM)–improved affinity to tubulin confirmed increased target binding correlated to enhanced potency.65b Further exploration revealed amides, not only ureas, to exhibit high on-target (tubulin) affinity but low off-target (Pgp) affinity and exceptional potency against vinblastine-sensitive and vinblastine resistant cell lines.65c
Boger’s contributions to vinblastine analogs exemplify how the drive to produce and improve modern medicines can lead to the development of new and far reaching synthetic methodology. The significant progress in HAT chemistry observed in recent years66 is indebted to the pursuit of vinblastine and analogs thereof.
Diazonamide
A. A poly-heteroaromatic macrocyclic marine metabolite and potent cytotoxin, diazonamide A (DZA, 13), was isolated from the colonial ascidian Diazona Chinesis in the late 1980’s.67 Though DZA possessed potent in vitro activity against human colon and melanoma cancer cell lines, the mechanism of action was initially unclear. Like vinca alkaloids and taxanes, DZA inhibited cell division during mitosis, and caused a loss in spindle microtubules.68 Yet direct interaction with traditional tubulin binding sites was not observed in early reports, indicating DZA either possessed a unique binding interaction with tubulin or perturbed microtubule dynamics indirectly through interaction with an alternative biological target.67 Evidence to support the latter hypothesis was proposed when ornithine δ-amino transferase was discovered to both directly interact with DZA and play a role in regulating mitotic cell division.69
The unique architecture of DZA, its potent antitumor activity, and seemingly unique mechanism of action inspired extensive investigations from many, including the Harran lab.70 Following completion of their total synthesis in 2003, Harran and co-workers began to explore the mechanism of action through the construction of diazonamide analogs. During these investigations it was found that unlike most microtubule-targeting agents (MTAs), diazonamide treatment did not cause weight loss and neutropenia at doses sufficient to regress human tumor xenografts in nude mice.71 Neutropenia (decreased neutrophil count) is a primary dose-limiting toxicity during treatment with MTAs such as vinca alkaloids and taxanes, and often leads to discontinuation of drug or reduction in dose.72 The clinical implications of the observed lack of overt toxicity by DZA triggered the establishment of Joyant pharmaceuticals in 2005, a company that leveraged Harran’s established route to gain key insights into diazomamide SAR.
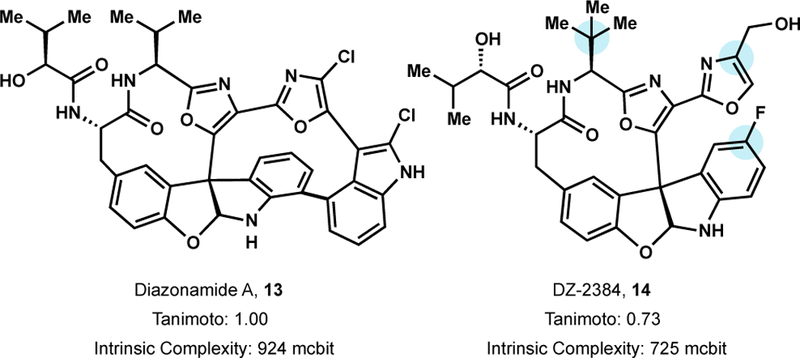
Critical knowledge gained from these studies included: 1) introduction of a fluorine atom to the indoline ring and replacement of the isopropyl group with a tert-butyl group improved the pharmacokinetic profile 2) The parent bis oxazole framework outcompeted other oxazole-heterocycle combinations and 3) The eastern macrocycle was not necessary for activity.73 The above insights led to the discovery of DZ-2384 (14), an analog chosen for preclinical development at Diazon pharmaceuticals.
DZ-2384 is efficacious in several preclinical cancer models including metastatic triple-negative breast cancer and subcutaneous xenograft models of pancreatic adenocarcinoma and colon cancer.74 Furthermore, DZ-2384 was shown to have a much wider safety margin than FDA approved vinorelbine. Notably, peripheral neuropathy, a major toxicity issue for MTAs, was not observed at therapeutically relevant doses. Detailed mechanistic investigations revealed DZ-2384 binds to tubulin at the vinca alkaloid site and inhibits the growth rate of microtubules. However, unlike vinca alkaloids, DZ-2384 preserves the microtubule network in interphase cells and primary cortical neurons. This preservation is proposed to arise from the pronounced straightening in the curvature and pitch of microtubule protofilaments upon formation of DZ-2384-tubulin complexes relative to vinblastine-tubulin complexes. The straightening of tubulin dimers is necessary for incorporation into the microtubule lattice and may be responsible for DZ-2384’s higher microtubule rescue frequency.74 Taken together, DZ-2384, a complex natural product analog of diazonamide A, is a safety enhanced MTA that inhibits microtubule growth through a novel mechanism of action.
Pleuromutilin.
First isolated from the Basidiomycetes Pleurotus mutilus and P. Passeckerianus in 1951, pleuromutilin (15) is a potent antibacterial that inhibits protein synthesis by binding to the peptidyl transferase center (PTC) of the 50S ribosomal subunit.75 Pleuromutilin’s tricyclic core forms key interactions with residues essential to RNA transfer binding and induces structural rearrangements that tightly lock the binding pocket.75b Resistance to pleuromutilin through spontaneous mutation is slow to occur due to a binding site and pose where multiple residue changes are required to reduce affinity yet maintain PTC function. Exploration of pleuromutilin analogs has mostly been limited to semi-synthetic modification of the C-14 sidechain. Extensive investigation of C-14 derivatives led to the discovery of Retapamulin, an FDA approved topical antibiotic used to treat methicillin resistant Staphylococcus aureus (MRSA) infections, and Lefamulin, currently under clinical investigation (NDA filed December 2018) for the treatment of community acquired bacterial pneumonia.76 Until recently, most mutilin analogs were explored for topical application only, since first-pass liver metabolism oxidizes the mutilin core to an inactive form.77 Nabriva delivered a major breakthrough in the discovery of a C-14 sidechain that reduced P450 binding and thus provided the potential for systemic treatment.78
Five groups have reported syntheses of the mutilin core, of which three reports have occurred in just the last six years (summarized in Figure 12).79 While the strategies and step counts differ, the overall yields to 15 are similar and the potential for discovery of interesting analogs are equally high. Notably, the Herzon group also accessed 12-epi-pleuromutilin (16) (19 steps, 1.1% overall yield), which was previously shown by researchers at Nabriva to possess extended-spectrum activity.80 Given the inevitability of bacterial resistance, the development of an arsenal of mutilin analogs is crucial. The systemic compatibility imparted by Nabriva’s sidechain and the new synthetic developments of Figure 12 make useful and diverse analogs more possible than ever.
Figure 12.
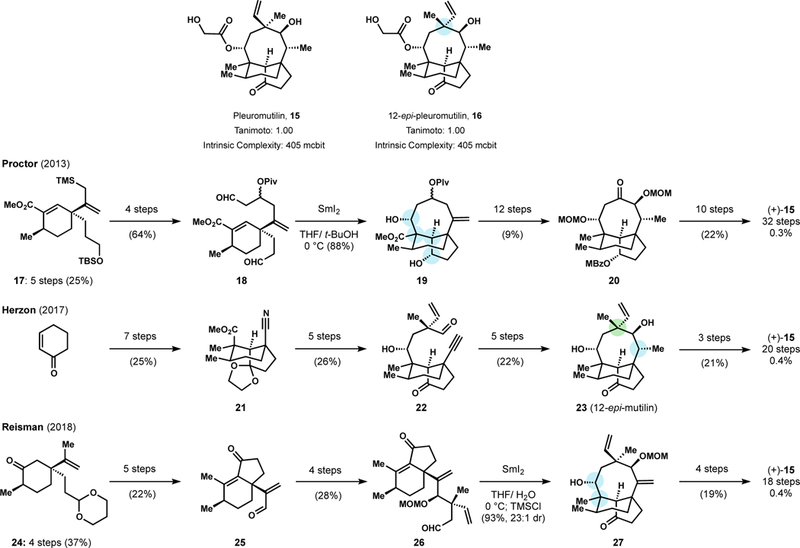
Recent total syntheses of pleuromutilin.
Analogs targeted to change synthetic design.
The compounds described above are not themselves NP, but NP-analogs that remain in NP-chemical space. As demonstrated by Tanimoto indices and complexity scores, these compounds provide to total synthesis all the challenges and opportunities for innovation as NPs. But they also provide opportunities to reach a functional endpoint: resistant strain potency, increased stability, increased selectivity or decreased toxicity. Notably, the compounds in Figures 5–12 arose from derivatization of a NP total synthesis intermediate: divergent total synthesis.81 However, perturbation of NP structure, i.e. analog design, can alter the main strategy. Using Corey’s nomenclature, the EX-TGT tree108 changes: some branches disappear, and others emerge.
Figure 5.
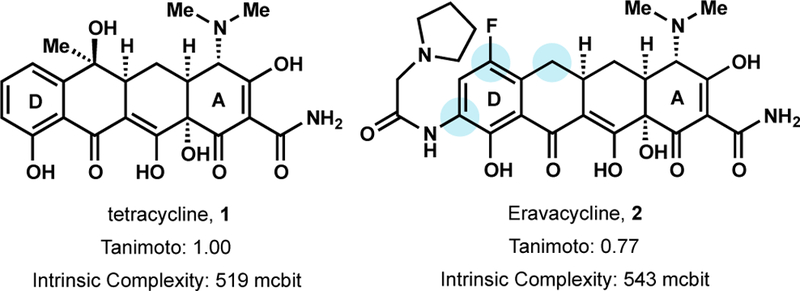
Eravacycline, an analog of tetracycline from Tetraphase.
Waldmann, Ertl, and others have used cheminformatics to organize existing NP scaffolds according to bioactivity and identify theoretical scaffolds within this hierarchy. In this case, NPs represent biologically-active nodes in chemical space about which analogs are clustered. These clusters can be reasonably predicted to contain multiple, biologically active compounds—experimentation has indeed produced novel scaffolds targeting ERα, pyruvate kinase, and monoamine oxidase A.82 By organizing these clusters in a hierarchy according to intrinsic complexity, scaffolds can be identified that may retain activity, but reduce synthetic burden. This method can be transformative for identifying fragments or simple scaffolds, but at the cost of NP-attributes like complexity. It is interesting to consider that movement along NP-based scaffold chains from high to low structural complexity resembles retrosynthetic analysis where the synthetic target (TGT) is dynamic not static.
A similar strategy might be used to identify analogs that retain intrinsic complexity but reduce synthetic complexity. While intrinsic complexity can be measured by several methods,22–23,24,25 there are fewer computational packages capable of reasonably evaluating synthetic complexity (tractability) of novel compounds. SciFinder has for years delivered full synthetic routes to known compounds by compiling literature reports. De novo design of syntheses, however, is still in its infancy. Retrosynthesis software in development may provide a way forward, but it must incorporate the ‘fuzzy logic’ of methods development or the predictive rigor of transition state calculation. Empiricism seems unavoidable. In the meantime, some synthesis groups have stepped in to bridge the gap.
Artemisinin.
Artemisinin (34), a tricyclic sesquiterpene lactone featuring a structurally unique endoperoxide bridge, was discovered in the early 1970’s after Chinese scientists performed a low temperature ether extraction of the herb, Artemesia annua.83 Artemisinin possesses potent anti-malaria activity—killing the parasite at most of its asexual development stages in human blood.84 Although artemisinin’s mechanism of action is not fully understood, oxidative stress induced parasite death and non-specific protein modification, initiated via heme-mediated homolytic cleavage of the endoperoxide bridge, remains a widely accepted mechanism.85
Since its debut in 1987, artemisinin and more soluble semisynthetic analogs artemether (1987), artesunate (1987), and artether (2000) have been deployed in artemisinin-based combination therapies (ACT) to treat malaria. As of 2016, ACT is the first-line treatment policy in 80 countries with 409 million treatment courses reported in 2016.86 Reports from the World Health Organization highlighting the progression of ACT resistant P. falciparum, have motivated some to develop artemisinin analogs as alternative therapeutic agents and biological probes.
Despite notable synthetic achievements, including Avery’s diverted total synthesis and Cook’s concise 9-step total synthesis, artemisinin analogs are predominately supplied via semisynthesis.87 Functional groups susceptible to semisynthetic modification, however, are limited; mostly the D-ring lactone has been modified.88 To uncover the little explored effects of C-ring functionalization, Oguri and co-workers designed a concise synthesis of 6-aza-artemisin analogs.89 Strategically targeting an aza-artemisinin analog lacking a C6 carbon enabled the exploration of a deep-seated core modification. More importantly, this C to N perturbation changed the available retrosynthetic transforms and opened a new strategy.
Leveraging the newly available disconnections, Oguri and co-workers designed a modular 4-step sequence to the anti-malarial tetracyclic scaffold from three simple building blocks: an amine, aldehyde, and alkyne (Figure 13). Simple Cu-catalyzed condensation using Carreira’s catalyst (CuBr/(R,M)-PINAP) assembled the three component linear precursor in high yield (92%) and high ee (93%).90 Next, Ti(II)-mediated cyclization of the ene-yne followed by protonation afforded the desired piperidine (29) as a single diastereomer. Finally, the tetra-cyclic scaffold was constructed via a one-pot procedure featuring: ketal deprotection and formation of the ammonium salt, [3+2] cycloaddition from the less hindered face to form the molozonide, migration of the silyl group to form the aldehyde and trimethyl silyl peroxide, which in the presence of additional trifluoroacetic acid cyclized to form trioxane 32.
Figure 13.
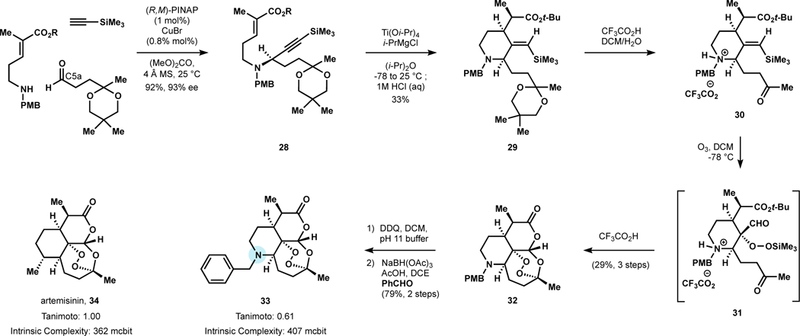
Substitution of C6 carbon with nitrogen enables the design of a 4-step route to artemisinin chemical space.
Efficient access to 32 enabled the synthesis of N-substituted analogs via reductive amination with a variety of aldehydes. One 6-aza-artemisinin analog, 33, was more potent than artemisinin in the P. berghei rodent malaria model and comparable in potency to the first-line semisynthetic drug artesunate.
Strategic perturbation of artemisinin’s chemical structure permitted access to unprecedented artemisinin analogs and new retrosynthetic disconnections. Elemental substitution17 from C6 to N6 enabled the convergent assembly of three linear building blocks at C5a, a position that is minimally involved in previous artemisinin total syntheses. At 7 total chemical transformations, Oguri’s synthesis is among the most concise fully synthetic routes to artemisinin’s local chemical space. The consequences of intrinsic complexity in this space has yet to be explored, but comparison to the simpler OZT endoperoxides may prove instructive.
Spongistatin 1.
Isolated in 1993, spongistatin 1 (35) is a complex marine macrolide with remarkably potent antitumor activity against the NCI panel of 60 human cancer cell lines—boasting an average GI50 value of 0.12 nM.91 Like vinblastine, spongistatin 1’s antitumor activity derives from mitosis inhibition via binding to the Vinca alkaloid domain of tubulin.92 Despite interest as a potential payload for antibody-drug conjugates, limited supply via isolation has prohibited clinical development. The synthetic community has attempted to solve the supply problem but streamlined access to a 42-membered macrolide ring with 24 stereocenters, two pyranyl subunits, and two spiroketals is no simple task.93 The particularly challenging CD-ring spiroketal exists as a thermodynamically less stable singly anomeric stereoisomer. Attempts to acquire the desired spiroketal from dihydroxy ketone precursors requires iterative separations from the major doubly anomeric product.93 The high intrinsic complexity (1,504 mcbits) led to total syntheses of spongistatin 1 that require >30 steps in the longest linear sequence (LLS) and >105 total steps.93
To develop a more concise route to spongistatin 1 functional chemical space, the Leighton group designed a new synthetic target that they hypothesized would retain potency and intrinsic complexity but decrease synthetic complexity (Figure 14).94 In a brilliant insight, the singly-anomeric CD spiroketal was identified as a source of synthetic complexity and instability, but biological irrelevance. Stereochemical inversion to the stabilized, doubly-anomeric spiroketal could occur via CH2→O interchange, which necessitated removal of the C25 hydroxyl. The newly designed CD spiroketal was hypothesized to be isostructural to spongistatin 1 and hence retain potency. Importantly, however, spiroketalization could be thermodynamically controlled by simple acid-catalyzed dehydration of a ketodiol—a synthetically simplifying maneuver. Such a structural perturbation might prove advantageous in the context of ADC development since the increased acid stability of the spiroketal might render it resistant to isomerization in the low-pH cellular microenvironments of endosomes and lysosomes.
Figure 14.
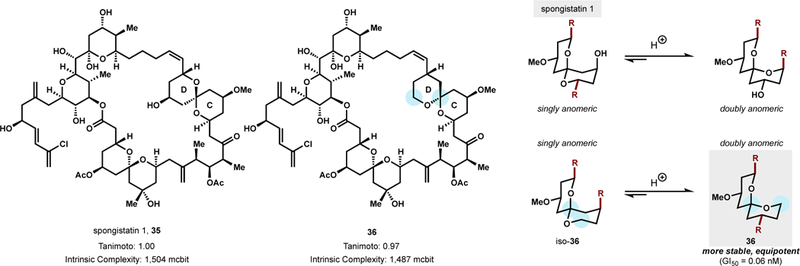
CD spiroketal redesign decreases synthetic complexity and enables the discovery of a thermodynamically more stable and equipotent analog, 36.
Analog 36 was prepared in 37 fewer steps than the shortest synthesis of spongistatin 1 or 2 and shown to possess comparable potency to spongistatin 1 (GI50 = 0.06 nM, 1A9 ovarian cells; 33x more potent than taxol). Its synthetic complexity is lower, but its intrinsic complexity is nearly identical (1487 vs. 1504 mcbits). Up to 40 g of key fragments in the synthesis could be prepared.95
Salvinorin A.
The neo-clerodane diterpene, salvinorin A (SalA, 37), was identified as the primary psychoactive component of the hallucinogenic herb, Salvia divinorum.96 Unlike prior hallucinogenic natural products, SalA did not target the serotonin 5-HT2A receptor. Instead, SalA was discovered to be a potent and selective agonist of the kappa-opioid receptor (κ-OR), a GPCR expressed throughout the central and peripheral nervous system that modulates consciousness, mood, and pain.97 Selective agonism of κ-ORs has garnered significant attention as a promising pain-relieving strategy because antinociceptive activity is observed in the absence of euphoria and respiratory suppression. The latter activity, or lack thereof, is significant since the majority of antinociceptive drugs that target µ-opioid and δ-opioid receptors induce respiratory suppression—the cause of death in opioid overdose.98
The potency and selectivity of SalA, coupled with the need to remove hallucinogenic side-effects, have stimulated semisynthetic and fully-synthetic efforts toward the development of SalA analogs.99 While most SalA analogs are made via semisynthesis, core instability limits the pool of available chemical transformation, and hence the diversity of semisynthetic SalA analogs. Specifically, C8 epimerization occurs under basic and acidic conditions to favor the less potent 8-epi-SalA (154–356 fold potency loss).100 The driving force for the trans- to cis-ring fusion had not been identified. Our group hypothesized that C20 axial strain was the driving force responsible for core instability.
To stabilize the core and attenuate epimerization, our group deleted the C20 methyl from the target (Figure 15a, 38).101 This new structure not only probed our strain driven epimerization hypothesis but also changed the available retrosynthetic disconnections. Deletion of the C20 methyl stabilized decalone intermediates and significantly enhanced access to simple starting materials, resulting in a 10-step synthesis, which compares favorably to the 20–29-step syntheses of SalA itself. The equilibrium of 20-nor-SalA and its C8 epimer is reversed from SalA and 8-epi-SalA (Figure 15b). An additional modification to change the C-ring lactone to a cyclohexanone completely suppressed epimerization. Notably, this work identified a new carboxylate-directed Heck reaction to install aryl rings on the SalA scaffold, providing paths forward for method development, analog development, and SAR discovery. This carboxylate directivity enables the first example of intermolecular Heck reactions with simple, tetrasubstituted alkenes.102 Modification of NP structure to ease synthesis does not preclude the design of new methods, rather it opens new retrosynthetic paths.
Figure 15.
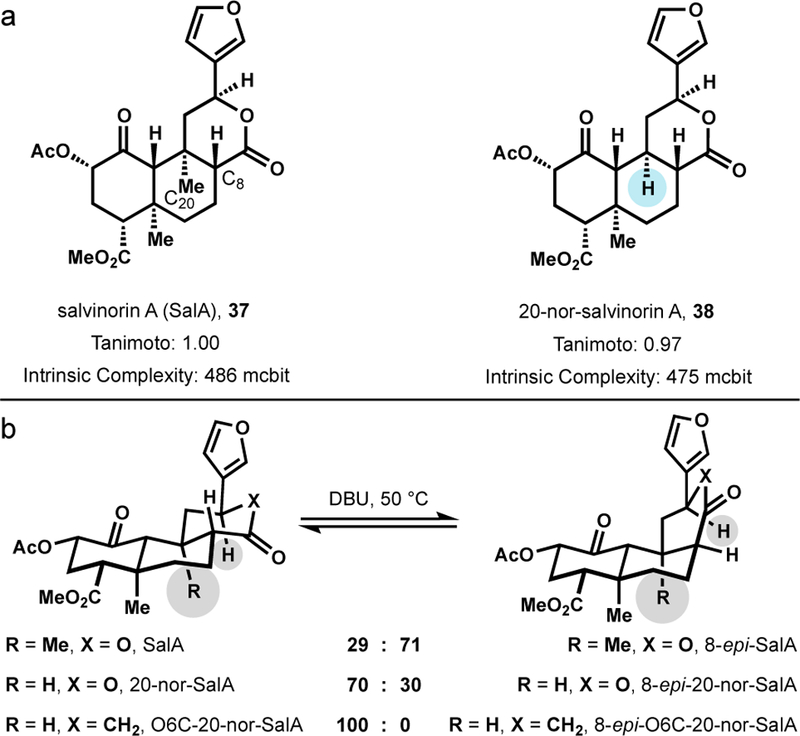
Methyl deletion and oxygen substitution stabilize the SalA scaffold, maintain potency and selectivity, and enable a short synthesis.
Discussion.
Molecular diversity describes the coverage of active clusters (chemotypes) within a chemical space. The greater the number of chemotypes in a library, the more likely a useful molecular lead will be identified. One privileged subset of chemical space, natural product chemical space, possesses properties that have been shown to be more successful in early screening and in the clinic. However, interrogation of this privileged area of chemical space is limited by synthetic access. The ~385 NP/NP-derived analog new chemical entities are primarily supplied by isolation/fermentation and semisynthesis. Fully synthetic campaigns are rare.
If a goal of synthesis is to enable rapid access to useful molecules, the target of synthesis campaigns need not be the NP itself. The target could be the functional NP space around the NP. A dynamic approach to retrosynthesis changes the TGT from a constant to an independent variable. The structure of the TGT is no longer static and hence can be changed to influence the retrosynthesis. Discoveries in the forward synthesis can influence what structural modifications can and should be made to the dynamic target.
This approach complements current strategies to access NP analogs. Divergent81 or diverted103 total synthesis relies on developing a route to the static NP target and diversifying late stage intermediates to NP analogs. Function oriented synthesis (FOS) relies on simplifying the NP target such that only the component necessary for biological activity is retained.20 In some cases, these changes also enable scaffold stabilization by removal of a structural liability,104 or significant reductions in synthetic burden.105 In other cases, FOS moves the target from privileged NP space towards areas of chemical space that are already populated.106 FOS is particularly attractive for large, highly complex structures (halichondrin→eribulin107). For small but dense NPs, a dynamic approach may prove helpful, and analogy to the heuristics of retrosynthetic analysis makes a good starting point.
Alteration of structure to change synthetic complexity is a fundamental aspect of retrosynthetic analysis.108 From a static target, retrosynthetic transforms are applied to iteratively reduce intrinsic complexity. Transforms may also strategically increase intrinsic complexity if the net result is to reduce synthetic complexity. A dynamic approach adds the constraint of biological function but removes the constraint of the transform. That is, change of final TGT structure need not correspond to a chemical reaction: ‘nonsense’ transforms like replacement of CH2 with NH in artemisinin are allowable. Otherwise, the ideas are the same as Corey’s hierarchies of transforms108: perturbation of structure can have a positive (+1), negative (−1) or no effect (0) on intrinsic complexity (see Figure 16), yet each may reduce synthetic complexity by opening a new retrosynthetic path. The added constraints of biological function and proximity in chemical space to the NP help guide structural changes. Like the heuristics of retrosynthetic analysis, these ideas are meant to encourage others to discover NP alterations and strategies for themselves, not dictate rules.
Figure 16.
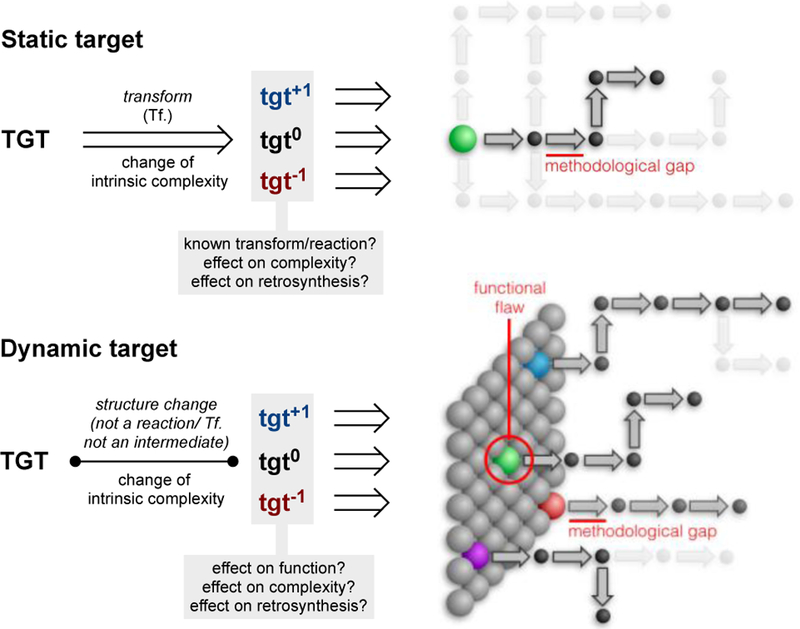
Dynamic retrosynthetic analysis expands the basis set of retrosynthesis to include structural perturbations.
A dynamic retrosynthetic analysis may be most useful when an active NP is limited by its chemical or metabolic instability. Peripheral instability may not impact retrosynthesis, but instability at the core requires redesign. Crystal structures of natural products bound to proteins or other biomolecular targets of therapeutic interest are available in the literature. Known binding sites and poses of a NP with its target can inspire and guide structure editing. These data are not necessary to modify a target and measure the functional effects of a revised structure, but they may provide the clearest path forward.
Conclusion.
Over the last few decades, biology has become increasingly molecular. How has synthesis changed? In some ways, it has not and should not: the goals of efficient, economical and benign synthetic processes remain significant and open-ended. In other ways, changes have come quickly, especially at the interface of biology and medicine. Spectroscopy, crystallography and microscopy have characterized protein structure to expedite the optimization of drug candidates, in some cases designed from small fragments.109 In the absence of known structure, homology modeling using genomic data has aided medicinal chemistry synthesis campaigns.110 High-throughput screens111 including activity-based protein profiling112 have provided big-data applications for large molecular libraries, some generated by combinatorial synthesis.
The underlying theory behind natural product total synthesis has remained untouched by these trends. We wonder if an interface between molecular biology and the heuristics of retrosynthetic analysis might provide a useful direction.
The synthetic chemist already evaluates how strategic structural changes expedite synthesis in the context of retrosynthetic analysis. Molecular biology allows this theory to be extended towards functional goals. Diversity of NP structure perturbation and the diversity of synthetic strategies it imparts can lead to new ways of looking at NP leads, even well-investigated targets. This Perspective is not a declaration but an invitation: the creative editing of natural product structure invites discovery.
Supplementary Material
Figure 6.
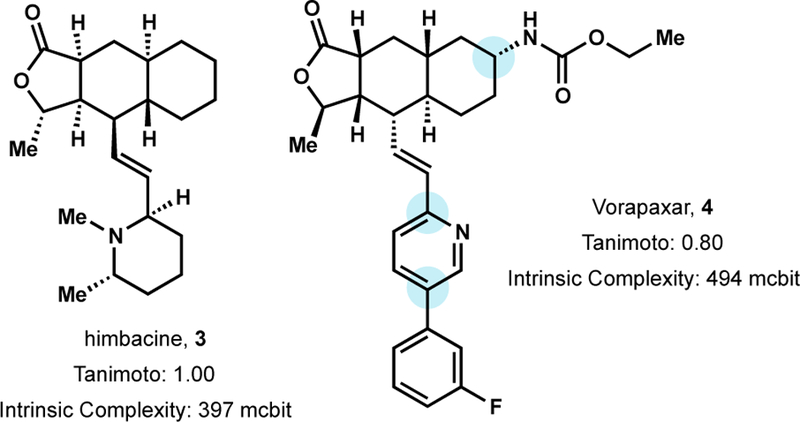
A screen of synthetic himbacine analogs enabled the discovery of Vorapaxar.
Figure 10.
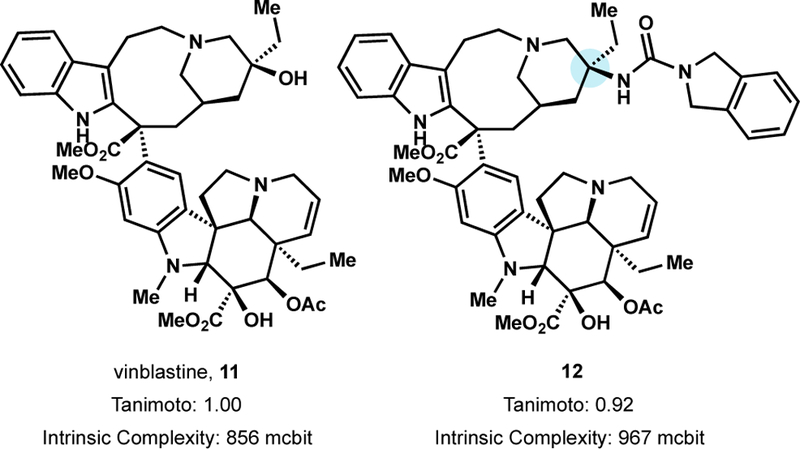
A urea side-chain restores potency of vinblastine alkaloids against resistant cell lines.
Figure 11.
DZ-2384 inhibits microtubule dynamics via a novel mechanism of action.
ACKNOWLEDGMENT
We thank Professor Dale Boger for his support and kind proof-reading of this manuscript.
Funding Sources
Generous support was provided by the National Science Foundation (GRFP to B. J. H.) and the National Institutes of Health (R35 GM122606).
ABBREVIATIONS
- NP
natural product
- FDA
United States Food and Drug Administration
- TGT
synthetic target
- FOS
function-oriented synthesis
- SAR
structure-activity relationship(s)
- DZA
diazonamide A
Footnotes
ASSOCIATED CONTENT
Supporting Information
Data files
Tanimoto scores were calculated using DataWarrior (SkelSpheres Descriptor).
Intrinsic complexity calculations (.xls)
The Supporting Information is available free of charge on the ACS Publications website.
Contributor Information
Benjamin J. Huffman, Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States
Ryan A. Shenvi, Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States.
REFERENCES
- 1.Huggins DJ; Venkitaraman AR; Spring DR “Rational Methods for the Selection of Diverse Screening Compounds.” ACS Chem. Biol 2011, 6, 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stratton CF; Newman DJ; Tan DS “Cheminformatic comparison of approved drugs from natural product versus synthetic origins.” Bioorg. Med. Chem. Lett 2015, 25, 4802–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luker T; Alcaraz L; Chohan KK; Blomberg N; Brown DS; Butlin RJ; Elebring T; Griffin AM; Guile S; St-Gallay S; Swahn BM; Swallow S; Waring MJ; Wenlock MC; Leeson PD “Strategies to improve in vivo toxicology outcomes for basic candidate drug molecules Bioorg. Med. Chem. Lett 2011, 21, 5673. [DOI] [PubMed] [Google Scholar]
- 4.Ritchie TJ; Macdonald SJF “The impact of aromatic ring count on compound developability – are too many aromatic rings a liability in drug design?” Drug Disc. Today 2009, 14, 1011. [DOI] [PubMed] [Google Scholar]
- 5.Brazeal Gregory, “How Much Does a Belief Cost?: Revisiting the Marketplace of Ideas” (March 26, 2012). Southern California Interdisciplinary Law Journal, Vol. 21, 2011. Available at SSRN: https://ssrn.com/abstract=1691952 [Google Scholar]
- 6. Lamont v. Postmaster Gen., 381 U.S. 301, 308 (1965) (Brennan, J., concurring)
- 7.Schreiber SL “Organic Chemistry: Molecular Diversity by Design” Nature, 2009, 457, 153. [DOI] [PubMed] [Google Scholar]
- 8.Schreiber SL “Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery” Science, 2000, 287, 1964. [DOI] [PubMed] [Google Scholar]
- 9.Maldonado AG; Doucet JP; Petitjean M Fan B. “Molecular Similarity and diversity in cheminformatics: from theory to applications” Molecular Diversity, 2006, 10, 39–79 [DOI] [PubMed] [Google Scholar]
- 10.Bleicher KH; Böhm H-J; Müller K; Alanine AI “Hit and Lead Generation: Beyond High-Throughput Screening” Nat. Rev. Drug. Disc 2003, 2, 369–378. [DOI] [PubMed] [Google Scholar]
- 11.a) Harper G; Pickett SD; Green DVS. “Design of a Compound Screening Collection for use in High Throughput Screening” Combi. Chem. High Throughput Screening 2004, 7, 63–71; [DOI] [PubMed] [Google Scholar]; b) Gorse A-D. “Diversity in Medicinal Chemistry Space” Curr. Top. Med. Chem 2006, 6, 3–18. [DOI] [PubMed] [Google Scholar]
- 12.Hillisch A; Hilgenfeld R eds. “Modern Methods of Drug Discovery” [DOI] [PubMed] [Google Scholar]
- 13.Lachance H; Wetzel S; Kumar K; Waldmann H “Charting, Navigating, and Populating Natural Product Chemical Space for Drug Discovery” J. Med. Chem 2012, 55, 5989–6001. [DOI] [PubMed] [Google Scholar]
- 14.Lovering F; Bikker J; Humbolt C “Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success” J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 15.Lovering F “Escape from Flatland 2: Complexity and Promiscuity” Med. Chem. Comm 213, 4, 515–519. [Google Scholar]
- 16.Ertl P; Roggo S; Schuffenhauer A “Natural Product-likeness Score and Its Application for Prioritization of Compound Libraries” J. Chem. Inf. Model 2008, 48, 68–74. [DOI] [PubMed] [Google Scholar]
- 17.a) Boger DL. “The Difference a Single Atom Can Make: Synthesis and Design at the Chemistry−Biology Interface” J. Org. Chem 2017, 82, 11961; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Alleman O; Brutsch M; Lukesh JC III; Brody DM; Boger DL. “Synthesis of a Potent Vinblastine: Rationally Designed Added Benign Complexity” J. Am. Chem. Soc 2016, 138, 8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herzon SB “Emergent Properties of Natural Products” SynLett 2018, 29, 1823. [Google Scholar]
- 19. One referee noted that “If NPs are on the decline, we must then accept that truth has prevailed, and it is not in favor of NPs.” We note that in Ref 3,4,14,15 and 25 stereochemically-rich, high-sp3 scaffolds—hallmarks of NPs—are deemed valuable for drug discovery, which may signal a coming ascendency for NP space.
- 20.Wender PA; Verma VA; Paxton TJ; Pillow TH “Function-Oriented Synthesis, Step Economy, and Drug Design” Acc. Chem. Res 2008, 41, 40–49. [DOI] [PubMed] [Google Scholar]
- 21.Nicolaou KC; Vassilikogiannakis G; Simonsen KB; Baran PS; Zhong YL; Vidali VP; Pitsinos EN; Couladouros EA “Biomimetic total synthesis of bisorbicillinol, bisorbibutenolide, trichodimerol, and designed analogues of the bisorbicillinoids” J. Am. Chem. Soc 2000, 122, 3071. [DOI] [PubMed] [Google Scholar]
- 22.Böttcher T “An Additive Method of Molecular Complexity” J. Chem. Inf. Model 2016, 56, 462–470. [DOI] [PubMed] [Google Scholar]
- 23.Li J; Eastgate MD “Current complexity: a tool for assessing the complexity of organic molecules” Org. Biomol. Chem 2015, 13, 7164–7176. [DOI] [PubMed] [Google Scholar]
- 24.Bertz SH “The First General Index of Molecular Complexity” J. Am. Chem. Soc 1981, 103, 3599–3601. [Google Scholar]
- 25.Clemons PA; Bodycombe NE; Carrinski HA; Wilson JA; Shamji AF; Wagner BK; Keohler AN; Schreiber SL “Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles” Proc. Natl. Acad. Sci 2010, 107, 18787–18792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newman DJ; Cragg GM “Natural products as sources of new drugs from 1981–2014” J. Nat. Prod 2016, 79, 629–661 [DOI] [PubMed] [Google Scholar]
- 27. If there is no good reason to interrogate the actual metabolite, a cultural bias against not reaching the target can still obstruct progress. To enforce a culture of form over function loses sight of the main target. In economic terms, ‘any observed statistical regularity will tend to collapse once pressure is placed upon it for control purposes,’ [Goodhart’s Law]. In other words, a measure that becomes a target ceases to be a good measure, as paraphrased by Marilyn Strathern. In this case, simply making a molecule may not be the best measure of success.
- 28.a) Bajusz D; Racs A; Heberger K. “Why is Tanimoto Index an Appropriate Choice for Fingerprint-based Similarity Calculations” Journal of Cheminformatics 2015, 7, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sander T. Freyss J, Korff M,; Rufener C. “DataWarrior: An Open Source Program For Chemistry Aware Data Visualization and Analysis” J. Chem. Inf. Model 2015, 55, 460–473 [DOI] [PubMed] [Google Scholar]
- 29.Duggar BM “Aureomycin: a product of the continuing search for new antibiotics”, Ann. NY. Acad. Sci 1948, 51, 177−181. [DOI] [PubMed] [Google Scholar]
- 30.Brodersen DE; Ramakrishnan V “The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit”, Cell, 2000, 103, 1143–1154. [DOI] [PubMed] [Google Scholar]
- 31.Woodward RB; Conover LH; Butler K; Johnston JD; Korst JJ “The Total Synthesis of 6-Demethyl-6-deoxytetracycline” 1962, 84, 3222. [Google Scholar]
- 32.a) Charest MG; Lerner CD; Brubaker JD; Siegel DR; Myers AG. “A Convergent Enantioselective Route to Structurally Diverse 6-Deoxytetracycline Antibiotics”, Science, 2005, 308, 395–398 [DOI] [PubMed] [Google Scholar]; b) Sun C; Myers AG. “A Robust Platform for the Synthesis of New Tetracycline Antibiotics” J. Am. Chem. Soc 2008, 130, 17913–17927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu F; Myers AG “Development of a Platform for the Discovery and Practical Synthesis of New Tetracycline Antibiotics” Curr. Opin. Chem. Biol 2016, 32, 48–57. [DOI] [PubMed] [Google Scholar]
- 34.a) Xiao X-Y; Hunt DK; Zhou J; Clark RB; Dunwoody N; Fyfe C; Grossman TH; O’Brien WJ; Plamondon L; Rönn M; Sun C; Zhang W-Y; Sutcliffe JA. “7-Fluoro-9-pyrrolidinoacetamido-6-demethyl-6-deoxytetracycline: A Potent, Broad Spectrum Antibacterial Agent” J. Med. Chem 2012, 55, 597–605. [DOI] [PubMed] [Google Scholar]; b) Sutcliffe J; O’Brien WJ; Fyfe C; Grossman TH. “Antibacterial Activity of Eravacycline (TP-434), a Novel Fluorocycline, Against Hospital and Community Pathogen” 2013, 57, 5548–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grossman TH; Starosta AL; Fyfe C; Rothstein DM; Mikolajka A; Wilson DN; Sutcliffe JA. “Target- and Resistance-Based Mechanistic Studies with TP-434, A Novel Fluorocycline Antibiotic” Antimicrob Agents Chemother 2012, 56, 2559–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinhey JT; Ritchie E; Taylor WC “The Chemical Constituents of Himantandra (Galbulimima)Species. IV. The Structures of Himbacine, Himbeline, Himandravine, and Himgravine” Aust. J. Chem 1961, 14, 106 [Google Scholar]; (b) Brown RFC; Drummond R; Fogerty AC; Hughes GK; Pinhey JT; Ritchie E; Taylor WC. “The chemical constituents of Himantandra species. II. The isolation of the alkaloids of Himantandra baccata Bail. and H. belgraveana F.Muell” Aust. J. Chem 1956, 9, 283. [Google Scholar]
- 37.a) Malaska MJ; Fauq AH; Kozikowski AP; Aagaard PJ; McKinney M.”Chemical modification of ring c of himbacine: Discovery of a pharmacophoric element for M2-selectivity” Bioorg. Med. Chem. Lett 1995, 5, 61 [Google Scholar]; b) Kozikowski AP; Fauq AH; Miller JH; McKinney M. “Alzheimer’s therapy: an approach to novel muscarinic ligands based upon the naturally occurring alkaloid himbacine.” Bioorg. Med. Chem. Lett 1992, 2, 797 [Google Scholar]; c) Darroch SA; Taylor WC; Choo LK; Mitchelson F. “Structure-activity relationships of some Galbulimima alkaloids related to himbacine” Eur. J. Pharmacol 1990, 182, 131. [DOI] [PubMed] [Google Scholar]
- 38.a) Chackalamannil S; Davies RJ; Asberom T; Doller D; Leone D. “A Highly Efficient Total Synthesis of (+)-Himbacine” J. Am. Chem. Soc 1996, 118, 9812–9813 [Google Scholar]; b) Chackalamannil S; Davies RJ; Wang Y; Asberom T; Doller D; Wong J; Leone D. “Total Synthesis of (+)-Himbacine and (+)-Himbeline” J. Org. Chem 1999, 64, 1932–1940. [DOI] [PubMed] [Google Scholar]
- 39.Chackalamannil S; Xia Y; Greenlee WJ; Clasby M; Doller D; Tsai H; Asberom T; Czarniecki M; Ahn H-S; Boykow G; Foster C; Agans-Fantuzzi J; Bryant M; Lau J; Chintala M “Discovery of Potent Orally Active Thrombin Receptor (Protease Activated Receptor 1) Antagonists as Novel Antithrombotic Agents” J. Med. Chem 2005, 48, 5884–5887 [DOI] [PubMed] [Google Scholar]
- 40.Alberelli MA; Candia E “Functional Role of Protease Activated Receptors in Vascular Biology” Vascular Pharmacology 2014, 62, 72–81. [DOI] [PubMed] [Google Scholar]
- 41.Chackalamannil S; Wang Y; Greenlee WJ; Hu Z; Xia Y; Ahn H; Boykow G; Hsieh Y; Palamanda J; Agans-Fantuzzi J; Kurowski S; Graziano M; Chintala M “Discovery of a Novel, Orally Active Himbacine-Based Thrombin Receptor Antagonist (SCH 530348) with Potent Antiplatelet Activity” J. Med. Chem 2008, 51, 3061–3064. [DOI] [PubMed] [Google Scholar]
- 42.Gerth K; Bedorf N; Hofle G; Irschik H; Reichenbach H “Epothilons A and B: antifungal and cytotoxic compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties” J. Antibiot 1996, 49, 560–563. [DOI] [PubMed] [Google Scholar]
- 43.Bollag DM; Mcqueney PA; Zhu J; Hensens O; Koupal L; Liesch J; Goetz M; Lazarides E; Woods CM “Epothilones, a New Class of Microtubule-Stabilizing Agents with a Taxol-like Mechanism of Action” Cancer Res 1995, 55, 2325–2333. [PubMed] [Google Scholar]
- 44.a) Altmann K-H; Höfle G; Müller R; Mulzer J; Prantz K. “The Epothilones: An Outstanding Family of Anti-Tumor Agents: Progress in the Chemistry of Organic Natural Products” Springer: Berlin, 2009; Vol. 90. [PubMed] [Google Scholar]; b) Borzilleri RM; Zheng X; Schmidt RJ; Johnson JA; Kim S; DiMarco JD; Fairchild CR; Gougoutas JZ; Lee FYF; Long BH; Vite GH. “A Novel Application of a Pd (0) -Catalyzed Nucleophilic Substitution Reaction to the Regio- and Stereoselective Synthesis of Lactam Analogues of the Epothilone Natural Products” J. Am. Chem. Soc 2000, 122, 8890–8897. [Google Scholar]
- 45.a) Balog A; Meng D; Kamenecka T; Bertinato P; Su D-S; Sorensen EJ; Danishefsky SJ. “Total Synthesis of (−)-Epothilone A” Angew. Chem., Int. Ed. Engl 1996, 35, 2801–2803 [Google Scholar]; b) Yang Z; He Y; Vourloumis D; Vallberg H; Nicolaou KC. “Total Synthesis of Epothilone A: The Olefin Metathesis Approach” Angew. Chem., Int. Ed. Engl 1997, 36, 166–168. [Google Scholar]
- 46.a) Altmann K; Pfeiffer B; Arseniyadis S; Pratt BA; Nicolaou KC. “The Chemistry and Biology of Epothilones—The Wheel Keeps Turning” ChemMedChem 2007, 2, 396–423 [DOI] [PubMed] [Google Scholar]; </j>b) Su D; Balog A; Meng D; Bertinato P; Danishefsky SJ; Zheng Y; Chou T; He L; Horwitz SB. “Structure−Activity Relationship of the Epothilones and the First In Vivo Comparison with Paclitaxel” Angew. Chem., Int. Ed. Engl 1997, 36, 2093–2096 [Google Scholar]; c) Harris CR; Danishefsky SJ. “Complex Target-Oriented Synthesis in the Drug Discovery Process: A Case History in the dEpoB Series” J. Org. Chem 1999, 64, 8434–8456 [Google Scholar]; d) Nicolaou KC; Chen JS; Dalby SM. “From nature to the laboratory and into the clinic” Bioorg. Med. Chem 2009, 2290–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivkin A; Chou T; Danishefsky SJ “On the Remarkable Antitumor Properties of Fludelone: How We Got There” Angew. Chem. Int. Ed 2005, 44, 2838–2850. [DOI] [PubMed] [Google Scholar]
- 48.Chou T; Zhang X; Zhong ZY; Li Y; Feng L; Eng S; Myles DR; Johnson R Jr.; Wu N; Yin YI; Wilson RM; Danishefsky SJ “Therapeutic effect against human xenograft tumors in nude mice by the third generation microtubule stabilizing epothilones” Proc. Natl. Acad. Sci 2008, 105, 13157–13162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. https://clinicaltrials.gov/ct2/show/NCT01379287.
- 50.Mcguire JM; Bunch RL; Anderson RC; Boaz HE; Flynn EH; Powell HM; Smith JW “llotycin, a new antibiotic” Antibiot. Chemother 1952, 2, 281–283. [PubMed] [Google Scholar]
- 51.Kanoh S; Rubin BK “Mechanisms of action and clinical application of macrolides as immunomodulatory medications” Clin. Microbiol. Rev 2010, 23, 590–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaynor M; Mankin AS “Macrolide Antibiotics: Binding Site, Mechanim of Action, Resistance” Frontiers in Medicinal Chemistry 2005, 2, 21–35. [DOI] [PubMed] [Google Scholar]
- 53.a) Bright GM. “Synthesis, in vitro and in vivo activity of novel 9-deoxo-9a-aza-9a-homoerythromycin A derivatives–a new class of macrolide antibiotics, the azalides” J. Antibiot. (Tokyo) 1988, 41, 1029–1047. [DOI] [PubMed] [Google Scholar]; b) Morimoto S; Takahashi Y; Watanabe Y; Omura S. “Chemical modification of erythromycins. 1. Synthesis and antibacterial activity of 6-O-methylerythromycins-A” J. Antibiot. (Tokyo) 1984, 37, 187–189. [DOI] [PubMed] [Google Scholar]
- 54.Fischback MA; Walsh CT “Antibiotics for emerging pathogens” Science 2009, 325, 1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seiple IB; Zhang Z; Jakubec P; Langlois-mercier A; Wright PM; Hog DT; Yabu K; Rao Allu1 S; Fukuzaki T; Carlsen PN; Kitamura Y; Zhou X; Condakes ML; Szczypiński FT; Green WD; Myers AG. “A Platform for the Discovery of New Macrolide Antibiotics” Nature 2016, 533, 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levine DP “Vancomycin: A History” Clinical Infectious Diseases 2006, 42, S5–S12 [DOI] [PubMed] [Google Scholar]
- 57.Perkins HR “Vancomycin and Related Antibiotics” Pharmacol. Ther 1982, 16, 181–197. [DOI] [PubMed] [Google Scholar]
- 58.Williams DH; Bardsley B “The Vancomycin Group of Antibiotics and the Fight against Resistant Bacteria” Angew. Chem. Int. Ed 1999, 38, 1172–1193. [DOI] [PubMed] [Google Scholar]
- 59.Bugg TDH, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, and Walsh CT “Molecular basis of vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA” Biochemistry 1991, 30, 10408–10415. [DOI] [PubMed] [Google Scholar]
- 60.McComas CC; Crowley BM; Boger DL “Partitioning the Loss in Vancomycin Binding Affinity for d-Ala-d-Lac into Lost H-Bond and Repulsive Lone Pair Contributions” J. Am. Chem. Soc 2003, 125, 9314–9315. [DOI] [PubMed] [Google Scholar]
- 61.a) Boger DL; Miyazaki S; Kim SH; Wu JH; Loiseleur O; Castle SL. “Diastereoselective Total Synthesis of the Vancomycin Aglycon with Ordered Atropisomer Equilibrations” J. Am. Chem. Soc 1999, 121, 3226. [Google Scholar]; b) Boger DL; Miyazaki S; Kim SH; Wu JH; Castle SL; Loiseleur O; Jin Q. “Total Synthesis of the Vancomycin Aglycon” J. Am. Chem. Soc 1999, 121, 10004 [Google Scholar]; c) Crowley BM; Boger DB. “Total Synthesis and Evaluation of [Ψ[CH2NH]Tpg4]Vancomycin Aglycon: Reengineering Vancomycin for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding”, J. Am. Chem. Soc 2006, 128, 2885–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie J; Pierce JG; James RC; Okano A; Boger DL “A Redesigned Vancomycin Engineered for Dual D-Ala-D-Ala and D-Ala-D-Lac Binding Exhibits Potent Antimicrobial Activity Against Vancomycin-Resistant Bacteria” J. Am. Chem. Soc 2011, 133, 13946–13949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.a) Noble RL; Beer CT; Cutts JH. “Role of chance observation in chemotherapy: Vinca Rosea” Ann. N.Y. Acad. Sci 1958, 76, 882–894. [DOI] [PubMed] [Google Scholar]; b) Owellen RI; Hartke CA; Dickerson RM; Haines FO. “Inhibition of Tubulin-Microtubule Polymerization by Drugs of the Vinca Alkaloid Class” Cancer. Res 1976, 36, 1499. [PubMed] [Google Scholar]
- 64.a) Vukovic J; Goodbody AE; Kutney JP; Misawa M “Productionof 3’,4’-anhydrovinblastine:a unique chemical synthesis” Tetrahedron 1988, 44, 325–331. [Google Scholar]; b) Ishikawa H; Colby DA; Boger DL “Direct coupling of catharanthine and vindoline to provide vinblastine: total synthesis of (+)- and ent-(−)-vinblastine” J. Am. Chem. Soc 2008, 130, 420–421. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Leggans EK; Barker TJ; Duncan KK; Boger DL “Iron(III)/NaBH4-mediated additions to unactivated alkenes: synthesis of novel 20′-vinblastine analogues” Org. Lett 2012, 14, 1428–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Barker TJ; Boger DL “Fe(III)/NaBH4-mediated free radical hydrofluorination of unactivated alkenes” J. Am. Chem. Soc 2012, 134, 13588–13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.a) Leggans EK; Duncan KK; Barker TJ; Schleicher KD; Boger DL. “A remarkable series of vinblastine analogues displaying enhanced activity and an unprecedented tubulin binding steric tolerance: C20′ urea derivatives.” J. Med. Chem 2013, 56, 628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Barker TJ; Duncan KK; Otrubova K; Boger DL. “Potent vinblastine C20′ ureas displaying additionally improved activity against a vinblastine-resistant cancer cell line.” ACS Med. Chem. Lett 2013, 4, 985–988; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lukesh JC III; Carney DW; Dong H; Cross M; Shukla V; Duncan KK; Yang S; Brody DM; Brütch MM; Radakovic A; Boger DL. “Vinblastine 20’ Amides: Synthetic Analogues That Maintain or Improve Potency and Simultaneously Overcome Pgp-Derived Efflux and Resistance” J. Med. Chem 2017, 60, 7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crossley SWM; Martinez RM; Obradors C; Shenvi RA “Mn, Fe, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins” Chem. Rev 2016, 116, 8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindquist N; Fenical W; Van Duyne GD; Clardy J “Isolation and Structure Determination of Diazonamides A and B, Unusual Cytotoxic Metabolites from the Marine Ascidian Diazona Chinesis” J. Am. Chem. Soc 1991, 113, 2303–2304. [Google Scholar]
- 68.Cruz-Monserrate Z; Vervoort HC; Bai R; Newman DJ; Howell SB; Los G; Mullaney JT; Williams MD; Pettit GR; Fenical W; Hamel E “Diazonamide A and a Synthetic Structural Analog: Disruptive Effects on Mitosis and Cellular Microtubules and Analysis of Their Interactions with Tubulin” Mol Pharmacol, 2003, 63, 1723–1280. [DOI] [PubMed] [Google Scholar]
- 69.Wang G; Shang L; Burgett AWG; Harran PG; Wang X “Diazonamide Toxins Reveal an Unexpected Function for Ornithine Gamma-Amino Transferase in Mitotic Cell Division.” Proc. Natl. Acad. Sci 2007, 104, 2068–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.a) Nicolaou KC; Bella M; Chen DYK; Huang X; Ling T; Snyder SA. “Total Synthesis of Diazonamide A” Angew. Chem., Int. Ed 2002, 41, 3495–3499. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC; Chen DYK; Huang X; Ling T; Bella M; Snyder SA. “Chemistry and Biology of Diazonamide A: First Total Synthesis and Confirmation of the True Structure” J. Am. Chem. Soc 2004, 126, 12888–12896. [DOI] [PubMed] [Google Scholar]; c) Knowles RA; Carpenter J; Blakey SB; Kayano A; Mangion IK; Sinz CJ; MacMillan DWC. “Total Synthesis of Diazonamide A” Chem. Sci 2011, 2, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Burgett AWG; Li Q; Wei Q; Harran PG. “A Concise and Flexible Total Synthesis of (−)-Diazonamide A” Angew. Chem., Int. Ed 2003, 42, 4961–4966. [DOI] [PubMed] [Google Scholar]
- 71.Williams NS; Burgett AWG; Atkins AS; Wang X; Patrick G Harran PG; McKnight SL. “Therapeutic anticancer efficacy of a synthetic diazonamide analog in the absence of overt toxicity” Proc. Natl. Acad. Sci 2008, 104, 2074–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.a) Conroy T. “Activity of vinorelbine in gastrointestinal cancers” Crit. Rev. Oncol. Hematol 2002, 42, 173–178 [DOI] [PubMed] [Google Scholar]; b) Luck H-J, Roche H. “Weekly paclitaxel: an effective and well-tolerated treatment in patients with advanced breast cancer” Crit. Rev. Oncol. Hematol 2002, 44, S15–S30 [DOI] [PubMed] [Google Scholar]
- 73.a) Wei Q; Zhou M; Xu X; Caldwell C; Harran S; Wang L. “Diazonamide Analogs for the Treatment of Cancer and Other Proliferative Disorders” U.S. Patent 20120270841A1 2012; b) Ding H; DeRoy PL; Perreault C; Larivee A; Siddiqui A; Caldwell CG; Harran S; Harran PG. “Electrolytic Macrocyclizations: Scalable Synthesis of a Diazonamide-Based Drug Development Candidate” Angew. Chem., Int. Ed 2015, 54, 4818–4822. [DOI] [PubMed] [Google Scholar]
- 74.Wieczorek M; Tcherkezian J; Bernier C; Prota AE; Chaaban S; Rolland Y; Godbout C; Hancock MA; Arezzo JC; Ocal O; Rocha C; Olieric N; Hall A; Ding H; Bramoulle A; Annis MG; Zogopoulos G; Harran PG; Wilkie TM; Brekken RA; Siegel PM; Steinmetz MO; Shore GC; Brouhard GJ; Roulston A “The Synthetic Diazonamide DZ-2384 Has Distinct Effects on Microtubule Curvature and Dynamics without Neurotoxicity” Sci. Transl. Med 2016, 8, 365RA159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kavanagh F; Hervey A; Robbins WJ “Antibiotic Substances from Basidiomycetes” Proc. Natl. Acad. Sci. U.S.A 1951, 37, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Davidovich C; Bashan A; Auerbach-Nevo T; Yaggie RD; Gontarek RR; Yonath A. “Induced-fit Tightens Pleuromutilins binding to Ribosomes and Remoter Interactions Enable Their Selectivity” Proc. Natl. Acad. Sci. U.S.A 2007, 104, 4291–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jones RN; Fritsche TR; Sader HS; Ross JE “Activity of Retapamulin (SB-275833), a Novel Pleuromutilin, Against Selected Resistant Gram-Positive Cocci” Antimicrob. Agents. Chemother 2006, 50, 2583–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun F; Zhang H; Gonzales GB; Zhou J; Li Y; Zhang J; Jin Y; Wang Z; Li Y; Cao Z; Zhang S; Yang S “Unraveling the Metabolic Routes of Retapamulin: Insights into Drug Development of Pleuromutilins” Antimicrob. Agents. Chemother 2018, 62, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Veve MP; Wagner JL “Lefamulin: Review of a Promising Novel Pleuromutilin Antibiotic” 2018, 38, 935–946. [DOI] [PubMed] [Google Scholar]
- 79.a) Murphy SK; Zeng M; Herzon SB. “A modular and enantioselective synthesis of the pleuromutilin antibiotics” Science 2017, 356, 956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Farney EP; Feng SS; Schäfer F; Reisman SE. “Total Synthesis of (+)-Pleuromutilin” J. Am. Chem. Soc 2018, 140, 1267–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fazakerly NJ; Helm MD; Procter DJ. “Total Synthesis of (+)-Pleuromutilin” Chem. Eur. J 2013, 19, 6718–6723. [DOI] [PubMed] [Google Scholar]; d) Boeckman RK Jr.; Springer DM; Alessi TR. “Synthetic Studies Directed Toward Naturally Occurring Cyclooctanoids. 2. A Stereocontrolled Assembly of (+/−)-Pleuromutilin via a Remarkable Sterically Demanding Oxy-Cope Rearrangement 1989, 111, 8284–8286. [Google Scholar]; e) Gibbons EG. “Total Synthesis of (+/−)-Pleuromutilin” J. Am. Chem. Soc 1982, 104, 1767–1769 [Google Scholar]
- 80.Thirring K; Heilmayer W; Riedl R; Kollmann H; Ivezic-Schoenfeld Z; Wolfgang W; Paukner S; Strickmann D. U.S. Patent WO2015110481A1, 2015.
- 81.Boger DL; Brotherton CE “Total Synthesis of Azafluoranthene Alkaloids: Rufescine and Imeluteine” J. Org. Chem 1984, 49, 4050. [Google Scholar]
- 82.a) Möcklinghoff S; von Otterlo WA; Rose R; Fuchs S; Zimmerman TJ; Dominguez Seoane M; Waldmann H; Ottman C; Brunsveld L. “Design and Evaluation of Fragment-Like Estrogen Receptor Tetrahydroisoquinoline Ligands from a Scaffold-Detection Approach” J. Med. Chem 2011, 54, 2005–2011. [DOI] [PubMed] [Google Scholar]; b) Inglese J; Austin CP. “Quantitative High-Throughput Screening: A Titration-Based Approach that Efficiently Identifies Biological Activities in Large Chemical Libraries” Proc. Natl. Acad. Sci. U. S. A, 103, 11473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wetzel S; Wilk W; Chammaa S; Sperl B; Roth AG; Yektaoglu A; Renner S; Berg T; Arenz C; Giannis A; Oprea TI; Rauh D; Kaiser M; Waldmann H. “A Scaffold-Tree-Merging Strategy for Prospective Bioactivity Annotation of γ-Pyrones” Angew. Chem. Int. Ed 2010, 49, 3660–3670. [DOI] [PubMed] [Google Scholar]
- 83.White NJ “Qinghaosu (Artemisinin): The Price of Success” Science, 2008, 320, 330–334. [DOI] [PubMed] [Google Scholar]
- 84.Terkuile F; White NJ; Holloway P; Pasvol G; Krishna S “Plasmodium falciparum: in vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria” Exp. Parasitol 1993, 76, 86–95. [DOI] [PubMed] [Google Scholar]
- 85.O’Niell PM; Posner GH “A Medicinal Chemistry Perspective on Artemisinin and Related Endoperoxides” J. Med. Chem 2004, 47, 2945–2964 [DOI] [PubMed] [Google Scholar]
- 86. http://www.who.int/malaria/areas/treatment/overview/en/
- 87.a) Zhu C; Cook SP. “A Concise Synthesis of (+)-Artemisinin” J. Am. Chem. Soc 2012, 134, 13577–13579. [DOI] [PubMed] [Google Scholar]; b) Avery MA; Gao F; Chong WKM; Mehrotra S; Milhous WK. “Structure-Activity Relationships of the Antimalarial Agent Artemisinin. 1. Synthesis and Comparative Molecular Field Analysis of C-9 Analogs of Artemisinin and 10-Deoxoartemisinin” J. Med. Chem 1993, 36, 4264–4275. [DOI] [PubMed] [Google Scholar]
- 88.O’Neill PM; Posner GH “A Medicinal Chemistry Perspective on Artemisinin and Related Endoperoxides” J. Med. Chem 2004, 47, 2945–2964. [DOI] [PubMed] [Google Scholar]
- 89.Bonepally KR; Hiruma T; Mizoguchi H; Ochiai K; Suzuki S; Oikawa H; Ishiyama A; Hokari R; Iwatsuki M; Otoguro K; Ōmura S; Oguri H “Design and De Novo Synthesis of 6‑Aza-artemisinins” Org. Lett 2018, 20, 4667–4671. [DOI] [PubMed] [Google Scholar]
- 90.Knöpfel TF; Aschwanden P; Ichikawa T; Watanabe T; Carreira EM. “Readily Available Biaryl P,N Ligands for Asymmetric Catalysis” Angew. Chem., Int. Ed 2004, 43, 5971–5973. [DOI] [PubMed] [Google Scholar]
- 91.a) Pettit GR; Chicacz ZA,; Gao F; Herald CL; Boyd MR; Schmidt JM; Hooper JNA. “Antineoplastic Agents. 257 Isolation and Structure of Spongistatin 1” J Org Chem 1993; 58, 1302–1304. [Google Scholar]; b) Bai R; Cichacz ZA; Herald CL; Pettit GR; Hamel E. “Spongistatin 1, a Highly Cytotoxic, Sponge Derived, Marine Natural Product that Inhibits Mitosis, Microtubule Assembly, and the Binding of Vinblastine to Tubulin” Mol Pharmacol 1993; 44, 757–766. [PubMed] [Google Scholar]
- 92.Bai R; Taylor GF; Cichacz ZA; Herald CL; Kepler JA; Pettit GR; Hamel E. “The Spongistatins, Potently Cytotoxic Inhibitors of Tubulin Polymerization, Bind in a Distinct Region of the Vinca Domain” Biochemistry, 1995, 34, 9714–9721. [DOI] [PubMed] [Google Scholar]
- 93.a) Guo J; Duffy KJ; Stevens KL; Dalko PI; Roth RM; Hayward MM; Kishi Y. “Total Synthesis of Altohyrtin A (Spongistatin 1): Part 1” Angew. Chem. Int. Ed 1998, 37, 187–192. [Google Scholar]; b) Hayward MM; Roth RM; Duffy KJ; Dalko PI; Stevens KL; Guo J; Kishi Y; “Total Synthesis of Altohyrtin A (Spongistatin 1): Part 2” Angew. Chem. Int. Ed 1998, 37, 192–196. [Google Scholar]; c) Smith AB III; Doughty VA; Lin Q; Zhuang L; McBriar MD; Boldi AM; Moser WH; Murase N; Nakayama K; Sobukawa M. “The Spongistatins: Architecturally Complex Natural Products—Part One: A Formal Synthesis of (+)‐Spongistatin 1 by Construction of an Advanced ABCD Fragment” Angew. Chem. Int. Ed 2001, 40, 191–195. [DOI] [PubMed] [Google Scholar]; d) Smith AB III; Sfouggatakis C; Risatti CA; Sperry JB; Zhu W; Doughty VA; Tomioka T; Gotchev DB; Bennett DS; Sakamoto S; Atasoylu O; Shirakami S; Bauer D; Takeuchi M; Koyanagi J; Sakamoto Y. “Spongipyran synthetic studies. Evolution of a scalable total synthesis of (+)-spongistatin 1” Tetrahedron 2009, 65, 6489–6509. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Paterson I; Chen D; Coster MJ; Aceña JL; Bach J; Gibson KR; Keown LE; Oballa RM; Trieselmann T; Wallace DJ; Hodgson AP; Norcross RD. “Stereocontrolled Total Synthesis of (+)‐Altohyrtin A/Spongistatin 1” Angew. Chem. Int. Ed 2001, 40, 4055–4060. [DOI] [PubMed] [Google Scholar]; f) Crimmins MT; Katz JD; Washburn DG; Allwein SP; McAtee LF. “Asymmetric Total Synthesis of Spongistatins 1 and 2” J. Am. Chem. Soc 2002, 124, 5661–5663. [DOI] [PubMed] [Google Scholar]; g) Ball M; Gaunt MJ; Hook DF; Jessiman AS; Kawahara S; Orsini P; Scolaro A; Talbot AC; Tanner HR; Yamanoi S; Ley SV. “Total Synthesis of Spongistatin 1: A Synthetic Strategy Exploiting Its Latent Pseudo‐Symmetry” Angew. Chem. Int. Ed 2005, 44, 5433–5438. [DOI] [PubMed] [Google Scholar]
- 94.Suen LM; Tekle-Smith MA; Williamson KS; Infantine JR; Reznik SK; Tanis PS; Cassleman TD; Sackett DL; Leighton J “Design and 22-step synthesis of highly potent D-ring modified and linker-equipped analogs of spongistatin 1” Nature Communications, 2018, 9, 4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reznik SK; Leighton JL. “Toward a More Step-Economical and Scalable Synthesis of Spongistatin 1 to Facilitate Cancer Drug Development efforts” Chem. Sci 2013, 4, 1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.a) Valdes LJ; J. “Salvia Divinorum and the Unique Diterpene Hallucinogen, Salvinorin (Divinorin) A” Psychoactive Drugs 1994, 26, 277–283; [DOI] [PubMed] [Google Scholar]; b) Bryan L. Roth BL; Baner K; Westkaemper R; Siebert D; Rice KC; Steinberg S; Ernsberger P; Rothman RB; Roth BL; “Salvinorin A: a potent naturally occurring nonnitrogenous κ opioid selective agonist” Proc. Natl. Acad. Sci. U. S. A 2002, 99, 11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu H. Wacker D; Mileni M; Katritch V; Han GW; Vardy E; Liu W; Thompson AA; Huang X; Carroll FI; Mascarella SW; Westkaemper RB; Mosier PD; Roth BL; Cherezov1 V; Stevens RC. “Structure of the human κ-opioid receptor in complex with JDTic” Nature 2012, 485, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vanderah TW. “Delta and kappa opioid receptors as suitable drug targets for pain” Clin. J. Pain 2010, 26, S10–S15. [DOI] [PubMed] [Google Scholar]
- 98. http://www.who.int/substance_abuse/information-sheet/en/
- 99.a) Prisinzano TE; Rothman RB. “Salvinorin A analogs as probes in opioid pharmacology” Chem. Rev 2008, 108, 1732–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sherwood AM; Crowley RS. Paton KF; Biggerstaff A. Neuenswander B; Day VW; Bronwyn M. Kivell BM; Prisinzano TE. “Addressing Structural Flexibility at the A- Ring on Salvinorin A: Discovery of a Potent Kappa-Opioid Agonist with Enhanced Metabolic Stability” J. Med. Chem 2017, 60, 3866–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Scheerer JR; Lawrence JF; Wang GC; Evans DA. “Asymmetric Synthesis of Salvinorin A, a Potent κ-Opioid Agonist” J. Am. Chem. Soc 2007, 129, 8968–8969; [DOI] [PubMed] [Google Scholar]; d) Nozawa M; Suka Y; Hoshi T; Suzuki T; Hagiwara H. “Total Synthesis of the Hallucinogenic Neoclerodane Diterpenoid Salvinorin A” Org. Lett 2008, 10, 1365–1368; [DOI] [PubMed] [Google Scholar]; e) Hagiwara H; Suka Y; Nojima T; Hoshi T; Suzuki T. Second-generation Synthesis of Salvinorin A” Tetrahedron 2009, 65, 4820–4825; [Google Scholar]; f) Line NJ; Burns AC; Butler SC; Casbohm J; Forsyth CJ. “Total Synthesis of (−)-Salvinorin A” Chem. - Eur. J 2016, 22, 17983–17986. [DOI] [PubMed] [Google Scholar]
- 100.Béguin C; Richards MR; Li JG; Wang Y; Xu W; Lie-chen LY; Carlezon WA; Cohen BM. “Synthesis and in Vitro Evaluation of Salvinorin A Analogues: Effect of Configuration at C(2) and Substitution at C(18)” Bioorg. Med. Chem. Lett 2006, 16, 4679–4685. [DOI] [PubMed] [Google Scholar]
- 101.Roach JJ; Sasano Y; Schmid CL; Zaidi S; Katritch V; Stevens RC; Bohn LM; Shenvi RA; “Dynamic Strategic Bond Analysis Yields a Ten-Step Synthesis of 20- nor-Salvinorin A, a Potent κ‑OR Agonist” ACS Cent. Sci 2017, 3, 1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huffman TR; Wu Y; Emmerich A; Shenvi RA “Intermolecular Heck Coupling with Hindered Alkenes Directed by Potassium Carboxylates” Angew. Chem. Int. Ed DOI: 10.1002/anie.201813233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Njardson JT; Gaul C; Shan D; Huang X-Y; Danishefsky SJ “Discovery of Potent Cell Migration Inhibitors through Total Synthesis: Lessons from Structure−Activity Studies of (+)-Migrastatin” J. Am. Chem. Soc 2004, 126, 1038–1040. [DOI] [PubMed] [Google Scholar]
- 104.Wender PA; Hegde SG; Hubbard RD; Zhang L; Mooberry SL “Synthesis and Biological Evaluation of (−)-Laulimalide Analogues” Org. Lett 2003, 5, 3507–3509. [DOI] [PubMed] [Google Scholar]
- 105.Wender PA; Baryza JL; Bennett CE; Bi C; Brenner SE; Clarke MO; Horan JC; Kan C; Lacote E; Lippa B; Nell PG; Turner TM “The Practical Synthesis of a Novel and Highly Potent Analogue of Bryostatin” J. Am. Chem. Soc 2002, 124, 13648–13649. [DOI] [PubMed] [Google Scholar]
- 106.Wender PA; Koehler KF; Sharkey NA; Dellaquila ML; Blumberg PM “Analysis of the Phorbol Ester Pharmacophore on Protein Kinase C as a Guide to the Rational Design of New Classes of Analogs” Proc. Natl. Acad. Sci. U.S.A 1986, 83, 4214–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yu MJ; Zheng W; Seletsky BM; Littlefield BA; Kishi Y “Case History: Discovery of Eribulin (Halaventm), a Halichondrin B Analogue That Prolongs Overall Survival in Patients with Metastatic Breast Cancer” Ann. Rep. Med. Chem 2011, 46, 227–241. [Google Scholar]
- 108.Corey EJ; Cheng X-M “The Logic of Chemical Synthesis” Wiley: New York, 1995. [Google Scholar]
- 109.Hajduk PJ; Greer J “A decade of fragment-based drug design: strategic advances and lessons learned” Nature Rev. Drug. Disc 2007, 6, 211–219. [DOI] [PubMed] [Google Scholar]
- 110.Cavasotto CN; Phatak SS “Homology modeling in drug discovery: current trends and applications” Drug Disc. Today 2009, 14, 676–683. [DOI] [PubMed] [Google Scholar]
- 111.Kearney SE et al. “Canvass: A Crowd-Sourced, Natural-Product Screening Library for Exploring Biological Space” ACS Cent. Sci 2018, 4, 1727–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nomura DK; Dix MM; Cravatt BF “Activity-based Protein Profiling for biochemical pathway discovery in cancer” Nature Rev. Cancer 2010, 10, 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.