

Abstract
Von Hippel–Lindau (VHL) disease is an autosomal dominant disorder characterized by various endocrine, nonendocrine, benign, and malignant tumors in various organs. VHL tumor suppressor gene, located on short arm of chromosome 3 is responsible for this. Pheochromocytoma (PCC) is one of the important endocrine manifestations that needs to be ruled out in case of VHL suspicion. In this review, we summarize the endocrine manifestations of VHL disease and their management while giving case history of five such cases.
Keywords: Neuroendocrine tumor, pheochromocytoma, von Hippel–Lindau Disease
INTRODUCTION
Von Hippel–Lindau (VHL) disease is an autosomal dominant disorder, which was first described in von Hippel's literature in 1911 and Lindau's literature in 1926.[1,2] The disease is characterized by various endocrine, nonendocrine, benign, and malignant tumors in various organs. VHL tumor suppressor gene, located on short arm of chromosome 3 is responsible for this. The identification of VHL gene has made diagnosis of VHL disease easier by direct mutational analysis. Pancreatic Neuroendocrine tumors (NET) and pheochromocytomas (PCCs)/paragangliomas are important endocrine manifestations of VHL disease. They display characteristic clinical, biochemical, and histopathologic features. Pancreatic NETs to emerge as a significant source of metastatic disease. PCC is one of the important endocrine manifestations that needs to be ruled out in case of VHL suspicion. VHL is clinically classified into two types based on the presence or absence of PCC. Type 1 is without PCC whereas type 2 has PCC. Type 2 is further subclassified into type 2A (no RCC), type 2B (all tumors), type 2C (only PCC).
In this review, we summarize the endocrine manifestations of VHL disease and their management while giving case history of five such cases [Table 1].
Table 1.
Table summarizing the five cases
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | |
|---|---|---|---|---|---|
| Age/Sex | 11/F | 7/M | 16/M | 26/M | 30/F |
| Clinical presentation | hypertension | hypertension | Sudden quadriparesis | Hemicranial headache | Progressive spastic quadriparesis |
| Tumors | |||||
| Pheochromocytoma | + | + | + | + | -- |
| Renal Cell Carcinoma | - | - | - | - | + |
| Hemangioblastoma | - | - | + | + | + |
| Others | - | - | Pancreatic and renal cysts | Pancreatic neuroendocrine tumor | Pancreatic cyst |
| Mutation | + | + | + | + | - |
| Type of VHL | 2C | 2C | 2A | 2A | I |
| Family history | + | + | + | + | + |
| Surgery performed | Staged b/l adrenalectomy | Bilateral adrenalectomy | Bilateral adrenalectomy | Unilateral adrenalectomy Pancreaticoduodenectomy | Radical nephrectomy |
| Excision of hemangioblastoma | Excision of hemangioblastoma | Excision of hemangiobalstoma | |||
| Histopathology | Pheochromocytoma without hyperplasia | Pheochromocytoma without hyperplasia | Pheochromocytoma without hyperplasia | Pheochromocytoma without hyperplasia NET with unncertain malignant potential | |
| Outcome | Asymptomatic | Asymptomatic | Post-operative mortality | Asymptomatic | Asymptomatic |
Case 1
An 11-year-old female was admitted with complaints of episodes of headache, sweating, palpitation along with history of vomiting, and blurring vision; and examination revealed tachycardia, hypertension (230/130 mmHg). Subsequent evaluation revealed elevated NMN (2959 μg/ml). CECT revealed a right adrenal mass 3 × 3 cm and hence she was provisionally diagnosed as a case of right PCC with probability of MEN 2A or VHL in view of young age. However, there was no family history and no evidence of medullary thyroid cancer or HPT in her or her siblings. She underwent laparoscopic right adrenalectomy after adequate alpha blockade. HPE confirmed PCC without hyperplasia of normal adrenal tissue. She was subsequently followed up. She then presented 6 years later with history of paroxysms and one episode of hypertension. On examination her blood pressure was found to be 180/110 on standing and 150/110 on lying down. Her UNMN was 6,620 μg/ml at 24 h (0–600). PET CT revealed a 5.8 × 5.5 cm left adrenal PCC. She was then put on alpha blockade and underwent a left laparoscopic adrenalectomy under steroid cover. Postop period was uneventful and HPE revealed PCC without adrenal hyperplasia. Her postoperative UMN returned to normal levels. She was discharged on and is doing well in follow-up. It was also found that her cousin had undergone bilateral adrenalectomy with us (case 2) during her period of follow-up and he was found to be having a positive VHL gene mutation; hence she was also tested for VHL mutation and was found to be harboring VHL gene mutation also. (NM_000551.3 [VHL]:c.382C>Tin exon 2. Protein change: leucine to phenylalanine).
Case 2
A 7-year-old male with history of sweating and palpitations for 2 years and enuresis was evaluated elsewhere where on ultrasonography was found to have adrenal mass and referred to our institute. On history taking he had one paternal cousin who was operated for PCC. His blood pressure was found high during paroxysm. With clinical suspicion of PCC he was evaluated for same. His 24-h urinary normetanephrines were found to be high (4,027 mcg/day [normal is <600]).
CECT abdomen [Figure 1a] showed heterogeneously enhancing left adrenal mass measuring 6.8 × 3.2 cm with small heterogeneously enhancing right adrenal mass measuring 1.4 × 1.3 cm.
Figure 1.
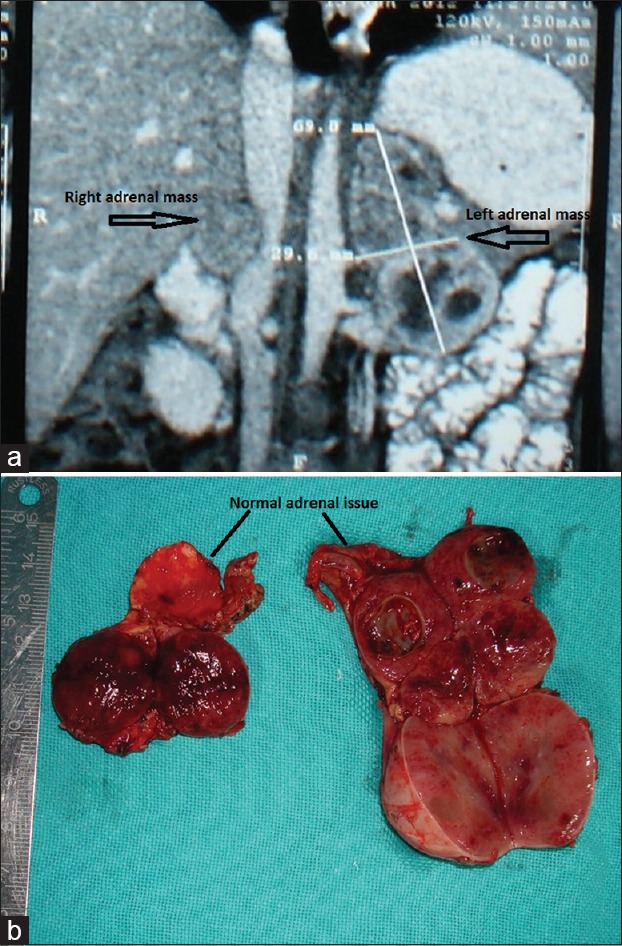
(a) CECT of case 2 showing bilateral adrenal pheochromocytomas. (b) Cut section of the same patient showing normal adrenal tissue attached to the tumor
Other investigations for MEN work up were negative. Serum calcium, intact PTH, S calcitonin, and USG neck were normal.
MIBG scan showed uptake in bilateral adrenals. Hence, patient was planned for b/l adrenalectomy [Figure 1b]. The highest recorded blood pressure intraoperatively was 230/140 mm Hg and lowest was 70/40 mm Hg. He was found to be having positive VHL gene mutation (NM_000551.3 [VHL]:c. 382C>Tin exon 2. Protein change: leucin to phenyl alanine). Histopathology showed b/l PCC.
Case 3
A 16-year-old male developed holocranial headache with projectile vomiting 3 years ago and subsequently developed sudden quadriparesis. Subsequent CECT revealed a posterior fossa fourth ventricle tumor with hydrocephalus. He underwent a suboccupalcraniatectomy with CI laminectomy with biopsy of the tumor along with VP shunt and then was referred to SGPGIMS. He underwent DSA embolization in view of hypervascular tumor and in preparation of excision. Subsequently, as his biopsy was suggestive of hemangioblastoma and he had a strong family history (his grandfather died of a brain tumor at the age of 45, his cousin brother was operated for a bilateral PCC and his paternal uncle was diagnosed with hemangioblastoma), he was suspected of VHL and CECT abdomen was done that revealed bilateral adrenal lesions and pancreatic and renal cysts; subsequently, elevated urinary MN/NMN (5,476 mcg/ml) confirmed bilateral PCC and he was taken up for bilateral adrenalectomy after adequate alpha blockade. HPE of adrenal lesion revealed bilateral tumors without hyperplasia. He was then transferred to neurosurgery unit where he underwent re-exploration and complete excision of brainstem hemangioblastoma. However, in postop period he gradually deteriorated and later developed septic shock and eventually succumbed.
Genetic mutation revealed heterozygous VHL: C277G known pathogenic variant (NM_000551.3 [VHL]:c.277G>Cin exon 1. Protein change glycine to arginine [p. Gly93Arg]).
Case 4
A 26-year-old male with history of hemicranial intermittent severe headache with vomiting, sweating, and palpitations since 6 years. Patient also had intermittent abdominal pain with no focal neurological deficits or seizures. He consulted a neurosurgeon for these complaints and MRI brain was done that showed cerebellar hemangioblastoma. Patient underwent surgery for same. Patient was lost to follow up after the surgery and then after 4 years consulted our institute for persistence of similar complaints. Patient was evaluated keeping syndromic association in mind because of strong family history of cerebellar hemangioblastoma.
On examination patient was found to be hypertensive with postural fall. On hormonal evaluation he was found to have high levels of 24 h urinary normetanephrines. CECT abdomen [Figure 2a] showed enlarged right adrenal gland heterogeneous (red), pancreatic head solid mass situated posteriorly (blue), and cyst in pancreatic tail (green). In order to rule out other NET and tumor in contralateral adrenal functional imaging was done. FDG PET scan showed avid uptake in right adrenal and pancreatic head. Hence, a clinical diagnosis of Vhl was made. With adequate alpha blockade patient was planned for right adrenalectomy and pancreatic surgery. Open transperitoneal anterior adrenalectomy with IOUSG and bidigital palpation of pancreas—tumor located posteriorly, close to main pancreatic duct as enucleation not possible hence decision was taken for pancreaticoduodenectomy [Figure 2b]. Postoperative course was uneventful. Postoperative urinary normetanephrines were normal on 10th day. Histopathology showed right adrenal PCC and pancreatic NET with uncertain malignant potential. Mutation analysis revealed positive mutation in exon 3 of VHL gene at 167th position of amino acid Arg (arginine) is replaced by Gln (glutamine) happens due to CGG-CAG a single nucleotide change.
Figure 2.
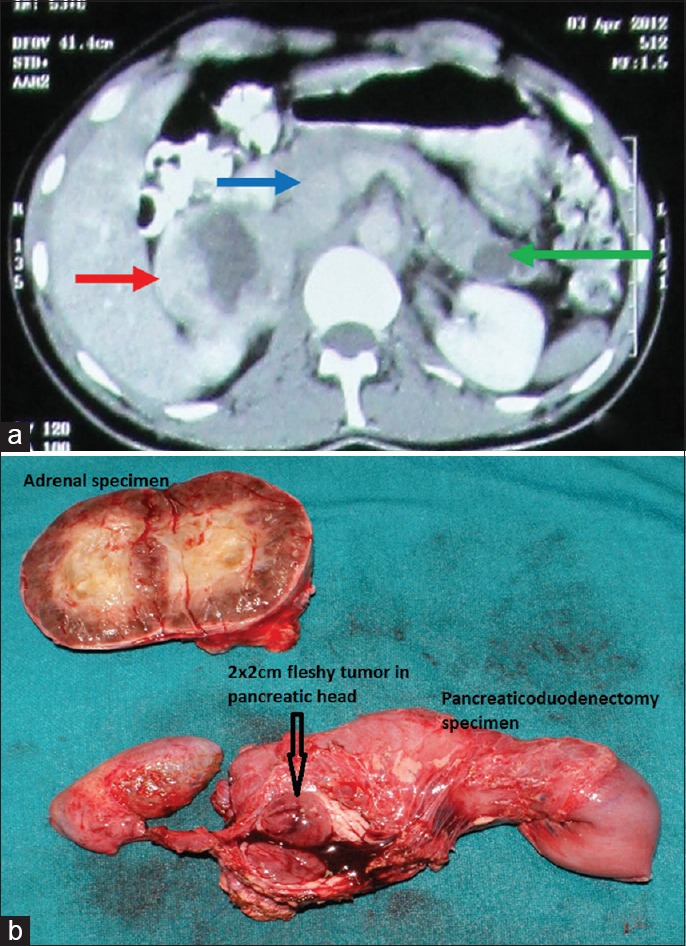
(a) CECT abdomen showed enlarged right adrenal gland heterogeneous (red) Pancreatic head solid mass situated posteriorly (blue) and Cyst in pancreatic tail (green), (b) Right Adrenalectomy and Pancreaticoduodenectomy specimen of the same patient
Case 5
A 30-year-old married female with two children with a family history of renal cell carcinoma (RCC) in mother who died at the age of 45 presented to us with history of progressive spastic quadriparesis, with history of hematuria, dysuria as well as severe constipation. MRI of cervical spine revealed cervical hemangioblastoma extending from CVJ to C4 level. CECT abdomen revealed a 3 × 2.1 cm SOL in upper pole of right kidney located posteriorly, along with pancreatic cysts and 2 × 1.8 cm soft tissue lesion above UB suggestive of metastasis. Direct opthalmoscopy revealed retinal angiomas. Patient was evaluated by endocrinologist to rule out PCC in view of a suspicion of VHL. However, 24 h MN/NMN were normal. She underwent right lap radical nephrectomy with excision of pelvic mass. HPE was clear cell RCC (grade II) and the mesenteric mass show metastases. She subsequently underwent suboccipital craniectomy with C1–C2 laminectomy with excision of tumor. HPE was suggestive of cervicomedullary junction lesion consistent with hemangioblastoma. There was improvement in spasticity and she was discharged. However, on genetic studies for VHL mutation no pathogenic variation was identified in the coding region of VHL gene.
DISCUSSION
VHL disease is an autosomal dominant multiorgan inherited neoplastic disorder that is characterized by development of highly vascular benign and malignant tumors and cyst. VHL tumor suppressor gene, located on short arm of chromosome 3 is responsible for this.[3,4] VHL gene is a short gene as protein-coding region is contained in 3 exons only. The major cause underlying the development of VHL tumors is inactivation and loss of function of the tumor suppressor VHL protein and the elongin B, C complex (VBC complex). This results in failure of the degradation and ubiquitination of HIF as well as development of pseudohypoxic conditions; accumulation of HIF (hypoxia inducible factor) and other proteins lead to increased transcription of pro-tumorigenic molecules like VEGF, PDGF, and TGF-alpha that results in development of highly vascular tumors characteristic of VHL.[5,6,7] In 80% cases it is inherited as a germ-line mutation whereas in 20% it is due to de novo mutation. If there is confirmed family history of VHL, a diagnosis of VHL disease can be made by finding a single major VHL tumor (CNS hemangiobalstoma, retinal hemangioblastoma, clear cell RCC, PCC, pancreatic endocrine tumors or endolymphatic sac tumor) in an at-risk relative. In cases of sporadic (or a nonfamilial) event, a clinical diagnosis of VHL disease in a patient without family history require presence of two tumors[8] (e.g., two HB or a HB with a visceral tumor).
CNS hemangioblastomas
It is the presenting feature in 40% cases and patients present with symptoms of raised ICT (intra cranial tension) and with limb or truncal ataxia as happened in all three of our cases who had HB.[9] Generally the results of surgery for solitary cerebellar HB are excellent but surgery of brain stem and spinal HB is challenging.[10] The other dilemma in patients with associated PCC is to operate on which lesion first? If HB is operated first, there is a substantial risk of HDI due to excessive catecholamine release. If PCC is operated first there is a risk of significant rise of ICT during surgery. Further, it has become clear that HB and its postoperative morbidity are major causes of physical disability in VHL patients. In our two patients who had PCC with HB, bilateral adrenalectomy was done first followed by excision of HB. In our patient with spinal HB the outcome was bad compared to those two patients who had a cerebellar HB and spinal HB.
Pheochromoctoma
Genotype-phenotype correlation studies are useful in predicting risk of PCC. VHL is clinically classified into two types based on the presence or absence of PCC. Type 1 is without PCC whereas type 2 has PCC. Type 2 is further subclassified into type 2A (no RCC), type 2B (all tumors), type 2C (only PCC). PCC occurs in association with specific alleles, usually due to missense mutations rather than deletions; the mutation is usually at nucleotide 238 in exon 3.[11,12,13] The cause of development of PCC is threefold.[14] One mechanism is the increased production of tyrosine hydroxylase by HIF and subsequent overproduction of catecholamines. The other mechanism is accumulation of atypical protein kinase gamma due to dysfunction of VBC complex. This results in overproduction of B-jun for inhibition of apoptosis of neural crest cells in the medulla of adrenal gland. The third mechanism of PCC formation is the binding of pVHL with fibronectin. It has been suggested that loss or change of fibronectin matrix assembly may be responsible for development of PCC.[14] Mean age of presentation is <30 years. Our four cases were all under 30 years. As they are usually diagnosed early, laparoscopic surgery is feasible. Also because it is usually a single tumor and that too without hyperplasia of the rest of the gland, unlike that in MEN2 PCC it is possible to do a cortical preserving adrenalectomy with low recurrence rates in long term follow-up. The risk of malignancy is very low (<5%) in PCC associated with VHL. Of note is the fact that in PCC there is a strong genotype-biochemical-phenotype correlation. They have three distinct features:
VHL-related paragangliomas/PCCs show increased levels of dopamine and norepinephrine. This might be due to a reduced expression of tyrosine hydroxylase and phenylethanolamine N-methyltransferase (norepinephrine to epinephrine converting enzyme) in VHL-related PCCs.[15] It has also been postulated to be one of the reasons why patients with VHL are less likely to present with hypertension (especially of the paroxysmal type) than patients with MEN2. Measurement of plasma normetanephrine levels is reported to be the most sensitive test for detecting pheochromocytoma
In a series of 14 VHL-related PCCs, distinctive histopathologic features consisted of a thick vascular tumor capsule[16]
In contrast to multiple endocrineneoplasia type 2 (MEN2)-related PCCs, adrenal medullary hyperplasia is not associated with VHL related PCCs[17]
Also due to sustained nature of catecholamine secretion from tumors in VHL patients, they are usually asymptomatic compared to MEN2 patients[15,18]
The identification of mutation in proband allows identification of mutation carriers among family members who do not yet exhibit any clinical manifestations of VHL disease. There is also a codon based correlation. There was a 62% risk for PCC associated with the arg238trp and arg238In mutations.[19,20]
Renal cell carcinoma
It is present in type 1 and type 2B VHL disease. Mean age at diagnosis is 40 years but routine screening may detect asymptomatic tumors.[21] Two important histological features of RCC in VHL are as follows: it has a conventional or clear cell appearance and there are associated multiple microscopic foci along with the main tumor.[8] There is no place for radical nephrectomy for VHL associated RCC and nephron—sparing approach is the most accepted form of treatment for tumors approaching 3 cm.[22,23] For tumors less than 3 cm careful observation/surveillance is preferable. Benign renal cysts are also commonly present in patients with VHL but they are usually asymptomatic and if there is no solid component in the cysts, they do not require any treatment.
Retinal hemangioblastomas
It occurs in both type 1 and type 2 diseases and are seen in as many as 60% of VHL patients.[24] They are usually asymptomatic and their age of onset is 21 years, lowest among other manifestations of VHL. They are usually detected by opthalmoscopy or fluorescin angiography. They resemble CNS HB grossly and histologically. Laser photocoagulation is effective for most RHB and complications like blindness or sever visual defects are seen only in <1% cases. However, screening examination should be conducted annually beginning at age 1 year or less.
Pancreatic cysts and tumors
There are three manifestations in pancreas in VHL disease: benign multiple cysts (70%), serous micro cystadenoms (10%), and NET (20%).[25] Pancreatic cysts are usually asymptomatic but sometimes may cause pain due to intrapancreatic expansion. Islet cell tumors are usually nonsecretory but can be malignant (<10%). The mean age at which pancreatic NETs are diagnosed in patients with VHL disease is lower than in sporadic cases (35 vs 58 years).[26] VHL-related pancreatic NETs characteristically contain clear cells or multivacuolated lipid-rich cells in varying proportion along with nuclear features characteristic of NETs. Clear cells in these tumors frequently coexist with lipid-containing foamy cells. This phenomenon is also attributed to the activation of HIF1-a, leading to a status of pseudohypoxia resulting in lipid and glycogen accumulation in tumor cells.[27] Regardless of the size or grade of NETs, pancreatic NETs are malignant neoplasms. Estimation of chromogranin A may be useful for biochemical diagnosis. Endoscopic ultrasonography is considered one of the most sensitive modalities to image pancreatic NETs. VHL-related NET tumors differ from sporadic NETs by their tendency to be multiple, asymptomatic, with overall indolent behavior.[28,29,30] Therefore, noninterventional options might be desirable to reduce mortality and morbidity, such as potential loss of pancreatic function. If surgery is chosen they are treated by either excision, or Whipple's or distal pancreatectomy depending upon the size and location of the lesion. In tumors lying close to the duct, a resection may be safer than enucleation, because the risk of fistula is higher in this setting. In our case no. 3, Whipple's procedure had to be performed precisely because of this reason.
CONCLUSION
Management of VHL-associated tumors requires multidisciplinary approach. Advances in genetic testing have allowed early diagnosis of this syndrome and may lead to early intervention and improved outcome. Management of some aspects of VHL disease, especially brain-stem HB is highly complex, challenging and is the main cause of mortality in VHL disease. Besides surgical approach, with advances in understanding the molecular biology of VHL disease, several anti-angiogeneic therapies are currently available in treating patients with common manifestions such as RCC.[31]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.von Hippel E. Die anatomische Grundlage der von mir beschriebenen ‘sehr seltenen Erkrankung der Netzhaut’. Graefes Arch Ophthalmol. 1911;79:350–77. [Google Scholar]
- 2.Lindau A. Studienuber Kleinhirncysten.Bau, Pathogenese und Bezoejimgemzur Angiomatosis retinae. Acta Pathol Microbiol Scand. 1926;(Suppl 1):1–128. [Google Scholar]
- 3.Woodward ER, Buchberger A, Clifford SC, Hurst LD, Affara NA, Maher ER. Comparative sequence analysis of the VHL tumor suppressor gene. Genomics. 2000;65:253–65. doi: 10.1006/geno.2000.6144. [DOI] [PubMed] [Google Scholar]
- 4.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 5.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumor suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 6.Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–61. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 7.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 8.Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet. 2011;19:617–23. doi: 10.1038/ejhg.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richard S, David P, Marsot-Dupuch K, Giraud S, Beroud C, Resche F. Central nervous system hemangioblastomas, endolymphatic sac tumours, and von Hippel-Lindau disease. Neurosurg Rev. 2000;23:1–22. doi: 10.1007/s101430050024. discussion 23-4. [DOI] [PubMed] [Google Scholar]
- 10.Lonser RR, Weil RJ, Wanebo JE, DeVroom HL, Oldfield EH. Surgical management of spinal cord haemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98:106–16. doi: 10.3171/jns.2003.98.1.0106. [DOI] [PubMed] [Google Scholar]
- 11.Neumann HPH, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, et al. The FreiburgWarsaw-Columbus pheochromocytoma Study Group Germ-line mutations in nonsyndromicpheochromocytoma. N Engl J Med. 2002;346:1459–66. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 12.Lamiell JM, Salazar FG, Hsia YE. von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine (Baltimore) 1989;68:1–29. doi: 10.1097/00005792-198901000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: Correlations with phenotype. Hum Mutat. 1995;5:66–75. doi: 10.1002/humu.1380050109. [DOI] [PubMed] [Google Scholar]
- 14.Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG., Jr von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001;10:1019–27. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- 15.Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. JClinEndocrinolMetab. 2001;86:1999–2008. doi: 10.1210/jcem.86.5.7496. [DOI] [PubMed] [Google Scholar]
- 16.Koch CA, Mauro D, Walther MM, Linehan WM, Vortmeyer AO, Jaffe R, et al. Pheochromocytoma in von HippelLindau disease: Distinct histopathologic phenotype compared to pheochromocytoma in multiple endocrine neoplasia type 2. Endocr Pathol. 2002;13:17–27. doi: 10.1385/ep:13:1:17. [DOI] [PubMed] [Google Scholar]
- 17.Mete O, Tischler AS, de Krijger R, McNicol AM, Eisenhofer G, Pacak K, et al. Protocol for the examination of specimens from patients with pheochromocytomas and extra-adrenal paragangliomas. Arch Pathol Lab Med. 2014;138:182–8. doi: 10.5858/arpa.2012-0551-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinato DJ, Ramachandran R, Toussi ST, Vergine M, Ngo N, Sharma R, et al. Immunohistochemical markers of the hypoxic response can identify malignancy in pheochromocytomas and paragangliomas and optimize the detection of tumors with VHL germline mutations. Br J Cancer. 2013;108:429–37. doi: 10.1038/bjc.2012.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glenn GM, Linehan WM, Hosoe S, Latif F, Yao M, Choyke P, et al. Screening for von Hippel-Lindau disease by DNA polymorphism analysis. JAMA. 1992;267:1226–31. [PubMed] [Google Scholar]
- 20.Crossey PA, Richards FM, Foster K, Green JS, Prowse A, Latif F, et al. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet. 1994;3:1303–8. doi: 10.1093/hmg/3.8.1303. [DOI] [PubMed] [Google Scholar]
- 21.Ong KR, Woodward ER, Killick P, Lim C, Macdonald F, Maher ER. Genotype-phenotype correlations in von Hippel Lindau disease. Hum Mut. 2007;28:143–9. doi: 10.1002/humu.20385. [DOI] [PubMed] [Google Scholar]
- 22.Rupert M, Hopirtean V, Mejean A, Thiounn N, Dufour B, Chretien Y, et al. Nephron sparing surgery for renal cell carcinoma and von Hippel-Lindau's disease: A single center experience. JUrol. 2003;170:1752–5. doi: 10.1097/01.ju.0000092780.85876.de. [DOI] [PubMed] [Google Scholar]
- 23.Steinbach F, Novick AC, Zincke H, Miller DP, Williams RD, Lund G, et al. Treatment of Renal-Cell Carcinoma in von Hippel-Lindau Disease - a Multicenter Study. JUrol. 1995;153:1812–6. [PubMed] [Google Scholar]
- 24.Dollfus H1, Massin P, Taupin P, Nemeth C, Amara S, Giraud S, et al. Ocular manifestations in von Hippel-Lindau disease: A clinical and molecular study. Invest Ophthalmol Vis Sci. 2002;43:3067–74. [PubMed] [Google Scholar]
- 25.Hammel PR, Vilgrain V, Terris B, Penfornis A, Sauvanet A, Correas JM, et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d′Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000;119:1087–95. doi: 10.1053/gast.2000.18143. [DOI] [PubMed] [Google Scholar]
- 26.Blansfield JA, Choyke L, Morita SY, Choyke PL, Pingpank JF, Alexander HR, et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs) Surgery. 2007;142:814–8. doi: 10.1016/j.surg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gucer H, Szentgyorgi E, Ezzat S, Asa SL, Mete O. Inhibin-expressing clear cell neuroendocrine tumor of the ampulla: An unusual presentation of Von Hippel-Lindau Disease. Virchows Arch. 2013;463:593–7. doi: 10.1007/s00428-013-1465-6. [DOI] [PubMed] [Google Scholar]
- 28.Charlesworth M, Verbeke CS, Falk GA, Walsh M, Smith AM, Morris-Stiff G. Pancreatic lesions in von Hippel-Lindau disease: A systematic review and metasynthesis of the literature. JGastrointest Surg. 2012;16:1422–8. doi: 10.1007/s11605-012-1847-0. [DOI] [PubMed] [Google Scholar]
- 29.Burns WR, Edil BH. Neuroendocrine pancreatic tumors: Guidelines for management and update. Curr Treat Options Oncol. 2012;13:24–34. doi: 10.1007/s11864-011-0172-2. [DOI] [PubMed] [Google Scholar]
- 30.Weisbrod AB, Zhang L, Jain M, Barak S, Quezado MM, Kebebew E. Altered PTEN, ATRX, CHGA, CHGB, and TP53 expression are associated with aggressive VHL-associated pancreatic neuroendocrine tumors. Horm Cancer. 2013;4:165–75. doi: 10.1007/s12672-013-0134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009;373:1119–32. doi: 10.1016/S0140-6736(09)60229-4. [DOI] [PubMed] [Google Scholar]