

Abstract
Activation of macrophage inflammasomes leads to interleukin (IL)-1β and IL-18 secretion and promotes atherosclerosis and its complications in mice and humans. However, the specific role and underlying mechanisms of the inflammasome in atherogenesis are topics of active research. Several studies in hyperlipidemic mouse models found that the NOD-like receptor protein 3 (NLRP3) inflammasome contributes to atherosclerosis, but recent work suggests that a second hit, such as defective cholesterol efflux or accumulation of oxidized mitochondrial DNA, may be required for significant inflammasome activation. Cholesterol crystal uptake or formation in lysosomes may damage membranes and activate NLRP3 inflammasomes. Alternatively, plasma or ER membrane cholesterol accumulation may condition macrophages for inflammasome activation in the presence of danger-associated molecular patterns, such as oxidized LDL. Inflammasome activation in macrophages or neutrophils leads to gasdermin-D cleavage that induces membrane pore formation, releasing IL-1β and IL-18, and eventuating in pyroptosis or neutrophil extracellular trap formation (NETosis). In humans, inflammasome activation and NETosis may contribute to atherosclerotic plaque erosion and thrombosis, especially in patients with type 2 diabetes, chronic kidney disease, or clonal hematopoiesis. Suppression of the inflammasome by activation of cholesterol efflux or by direct inhibition of inflammasome components may benefit patients with CVD and underlying susceptibility to inflammasome activation.
Keywords: adenosine 5′-triphosphate binding cassette transporters, high density lipoprotein, oxidized lipids, atherosclerosis, macrophages
Graphical Abstract
Atherosclerotic CVD arises from a macrophage-driven inflammatory response to modified LDL in the arterial wall. This view has received strong support from the positive outcome of the CANTOS trial (Canakinumab Antiinflammatory Thrombosis Outcome Study), involving administration of an interleukin (IL)-1β antibody to patients with elevated levels of C-reactive protein (CRP) (1). IL-1β is a key inflammatory cytokine that promotes monocyte and neutrophil entry into sites of inflammation. IL-1β is synthesized as a pro-form that undergoes proteolytic cleavage by CASPASE-1 in the inflammasome, a protein complex assembled in the cytosol of macrophages in response to pathogen-associated molecular patterns or danger-associated molecular patterns, leading to secretion of the active form of IL-1β. The pro-form of IL-18 is similarly processed by inflammasomes resulting in IL-18 secretion (2). Together with preclinical studies (3–7), CANTOS points to a role of inflammasomes in atherothrombotic disease.
This review will discuss the role of inflammasomes in atherosclerosis and the mechanisms underlying inflammasome activation in response to cholesterol accumulation in macrophages and neutrophils. Typical for an emerging area, several aspects of these studies are controversial. We will also discuss a potential link between inflammasomes and plaque neutrophil extracellular trap formation (NETosis). NETosis involves the release of chromatin and granule contents from neutrophils, giving rise to large extracellular webs containing DNA, proteases, and myeloperoxidase that help to trap and inactivate pathogens (8). NETosis has been implicated in atherothrombosis, notably in plaque erosion and thrombosis (9, 10), a process that may be increasingly important in acute coronary syndromes (11).
MECHANISMS OF INFLAMMASOME ACTIVATION
The NOD-like receptor protein 3 (NLRP3) inflammasome is activated by a wide variety of microbial and metabolic signals. This involves a priming step mediated by Toll-like receptors (TLRs) that leads to increased expression of Il-1β, and the inflammasome components, Caspase-1 and Nlrp3, followed by an activation step in which the components of the inflammasome assemble in the cytoplasm and CASPASE-1 is cleaved (2). A variety of stimuli, including extracellular ATP, silica particles, uric acid crystals, or cholesterol crystals, can activate the NLRP3 inflammasome. Activation involves a sensor (NLRP3) that assembles with an adaptor [adaptor protein apoptosis-associated speck-like protein containing CARD (ASC)] and forms a filamentous structure that provides a platform for CASPASE-1 cleavage (12). The NLRP3 inflammasome seems to sense membrane damage (13), which may lead to K+ efflux and mitochondrial reactive oxygen species (ROS) generation. Recent studies have shown that priming [in response to lipopolysaccharide (LPS)] involves the induction of mitochondrial DNA synthesis, including the enzyme mitochondrial deoxyribonucleotide kinase [uridine/cytidine monophosphate kinase 2 (UMP-CMPK2)], while activation in response to ATP or nigericin leads to the oxidation and release of mitochondrial DNA that binds and activates the NLRP3 inflammasome (14). However, it is not yet clear that this represents a universal mechanism of NLRP3 inflammasome activation. NLRP3 inflammasome activation can occur without evident mitochondrial ROS generation or lysosomal damage (7, 13), and, moreover, in some models, mitochondrial ROS generation is prevented by NLRP3 deficiency (15), suggesting that ROS can be generated downstream of inflammasome activation. Cytosolic double-stranded DNA introduced by microbes or formed in response to mitochondrial damage activates the NLRP3 inflammasome in human myeloid cells (16) and the absent in melanoma 2 (AIM2) inflammasome in mice (17). Deficiency of 25-hydroxycholesterol, a suppressor of cholesterol biosynthesis, triggers release of DNA from mitochondria, AIM2 inflammasome activation, and secretion of IL-1β and IL-18 (18). The noncanonical inflammasome is responsible for mortality during LPS-induced sepsis (13). This involves activation of cytosolic CASPASE-11, possibly in response to direct binding of LPS or oxidized lipids (19). Active CASPASE-11 can induce NLRP3-mediated CASPASE-1 cleavage (13), likely as a consequence of membrane damage and, thus, indirectly promote IL-1β and IL-18 cleavage.
INFLAMMASOMES, PYROPTOSIS, AND NETosis
Inflammasome activation can lead to pyroptosis, an inflammatory mode of cell death involving osmotic swelling, cell necrosis, and release of IL-1β and IL-18 as well as various danger-associated molecular patterns, such as IL-1α, high-mobility group box 1 (HMGB1) proteins, and ATP. Activated CASPASE-1 or CASPASE-11 cleave gasdermin-D (GSDMD), releasing an N-terminal fragment that forms membrane pores facilitating release of the aforementioned molecules, and likely as pores grow larger, eventuating in pyroptosis (20). Remarkably, a similar process in neutrophils, requiring inflammasome activation and GSDMD cleavage, leads to granule membrane dissolution, chromatin decondensation, plasma membrane leakiness, and expulsion of DNA and proteases, a process apparently identical to NETosis (21, 22).
INFLAMMASOMES AND ATHEROSCLEROSIS
The role of the NLRP3 inflammasome in atherosclerosis was first explored by Duewell et al. (3). They found a major impact of the NRLP3 inflammasome on early lesion area in Western-type diet (WTD)-fed Ldlr−/− mice that had been transplanted with bone marrow (BM) deficient in the key inflammasome components, Asc or Nlrp3. In contrast, Menu et al. (23) did not find any impact of whole-body deficiency of Asc, Nlrp3, or Caspase-1/11 on the size of advanced atherosclerotic lesions in WTD-fed Apoe−/− mice. While some subsequent studies appeared to confirm the report of Duewell et al. (3) (see 4, 6, 24, 25), our own (7) and other studies (5) found no impact of deletion of the key inflammasome components, Nlrp3 or Caspase-1/11, on the area or morphology of either early or advanced lesions in WTD-fed Ldlr−/− mice. The reason for the discrepant results is unknown; although unlike the studies by Duewell et al. (3), we did not find signs of inflammasome activation in Ldlr−/− mice (7). When additional mutations that caused macrophage inflammasome activation, such as myeloid deficiency of ABCA1 and ABCG1 or hematopoietic deficiency of 8-oxoguanine glycosylase, were introduced into Ldlr−/− mice, the NLRP3 inflammasome was clearly activated, as shown by increased CASPASE-1 cleavage, and did contribute to lesion area and macrophage content (5, 7). ABCA1 and ABCG1 are the principal transporters mediating cholesterol efflux from macrophages (26), indicating a role of defective cholesterol efflux in inflammasome activation. The 8-oxoguanine glycosylase is the main enzyme mediating repair of mitochondrial oxidized DNA that accumulates in atherosclerotic lesions and may directly activate the NLRP3 inflammasome (27). The AIM2 inflammasome may also promote atherogenesis: double-stranded DNA was found in lesional cells, and deficiency of hematopoietic AIM2 resulted in an increase in smooth muscle cells, collagen, and fibrous cap thickness, and a decrease in the necrotic area of advanced lesions in Apoe−/− mice (28).
Duewell et al. (3) showed that cholesterol crystals could induce macrophage NLRP3 inflammasome activation in vitro and related their in vivo findings to tiny cholesterol crystals detected in early foam cell lesions by confocal reflectance microscopy. However, prior studies by Small and Shipley (29) based on lipid phase behavior and observation of fresh plaques by polarized microscopy under temperature-controlled conditions suggested that early foam cell lesions did not contain cholesterol crystals. Rather, such lesions contained liquid or liquid crystalline cholesteryl esters that undergo artifactual crystal formation when cooled below body temperature (29). Thus, while cholesterol crystals in advanced lesions may be involved in inflammasome activation, in our opinion, a role of cholesterol crystals in early foam cell lesions is questionable.
NETosis AND ATHEROSCLEROSIS
The formation of neutrophil extracellular traps (NETs) promotes venous and arterial thrombosis in mice (30–33). NETs promote atherosclerosis and carotid thrombosis in Apoe−/− mice, as shown using chloramidine, a chemical inhibitor of NET formation (34). A recent study using mice with knockouts of peptidyl arginine deiminase 4 (PAD4), an essential enzyme in histone citrullination, suggested no impact of NETs on lesion area or macrophage content in early foam cell lesions in WTD-fed Ldlr−/− mice, even though NETs were detected in lesions (9). In the same study, deficiency of PAD4 led to decreased neutrophil adherence, arterial injury, and thrombosis in the setting of disturbed carotid arterial flow, consistent with a role of NETosis in plaque erosion (9). These findings may have relevance to humans because NETs have been associated with unstable human atherosclerotic plaques, especially in regions of superficial erosion (10). In contrast to these studies, myeloid PAD4 deficiency did have an impact on lesion area in Apoe−/− mice (35). Thus, like inflammasome activation, NETs seem to contribute to plaque development and complications under specific experimental conditions. As noted above, inflammasomes and NETs may be mechanistically interdependent. One study reported that cholesterol crystals could promote NET release that in turn promoted macrophage inflammasome activation (36). However, the conclusion that NETosis causes inflammasome activation has been questioned (33, 37). Our findings rather suggest that NETosis may be downstream of inflammasome activation in atherosclerosis (7). This conclusion is consistent with studies showing that inflammasome-dependent pyroptosis and NETosis are similar processes dependent on GSDMD (21, 22). We speculate that neutrophil inflammasome activation may induce pyroptosis/NETosis in murine atherosclerosis and perhaps contribute to plaque erosion and thrombosis in humans.
INFLAMMASOME ACTIVATION IN MICE WITH DEFECTIVE CHOLESTEROL EFFLUX PATHWAYS IN MYELOID OR DENDRITIC CELLS
To interrogate a potential role of defective cholesterol efflux pathways in macrophage inflammasome activation, we bred mice with myeloid knockout of Abca1 and Abcg1 using LysM-Cre transgenic mice. These MylABCDKO mice were bred with NLrp3−/− or Caspase1/11−/− mice and BM was transplanted into Ldlr−/− recipients (7). MylABCDKO BM-transplanted Ldlr−/− mice fed WTD showed prominently increased levels of plasma IL-18, a marker of inflammasome activation, increased caspase-1 cleavage, and IL-1β and IL-18 secretion by splenocytes. These findings were reversed by hematopoietic Nlrp3 or Caspase1/11 deficiency, indicating activation of the NLRP3 inflammasome in Ldlr−/− mice with myeloid Abca1/Abcg1 deficiency (7). Nlrp3 or Caspase-1/11 deficiency decreased atherosclerotic lesion size in female MylABCDKO BM-transplanted Ldlr−/− mice, particularly in early lesions (7).
Unexpectedly, there was marked neutrophil accumulation in early plaques of Ldlr−/− mice with myeloid Abca1/Abcg1 deficiency and extensive NETosis (shown by coincident staining of neutrophil markers, myeloperoxidase, and citrullinated histones). Neutrophil accumulation and NETosis were reversed by hematopoietic Nlrp3 or Caspase-1/11 deficiency, indicating that inflammasome activation promotes neutrophil recruitment and NETosis in early atherosclerotic plaques. These findings are consistent with evidence that neutrophils contribute to early plaque formation (33, 38). The genetic dependence of plaque NETosis on the NLRP3 inflammasome and caspase-1/11 in MylABCDKO mice might be due to inflammasome activation in neutrophils. Notably, Abca1/Abcg1-deficient neutrophils showed increased cholesterol content and increased cleavage of caspase-1 and caspase-11 (7), which could lead to GSDMD cleavage and pyroptosis/NETosis. We were not able to detect increased Caspase-1 or -11 cleavage in cells isolated from plaques of MylABCDKO BM-transplanted Ldlr−/− mice (7). This may reflect technical limitations and the lack of authentic reagents for detection of inflammasome activation in tissues. Alternatively, the effects of the inflammasome could be mediated through systemic effects, for example by the impact of IL-1β derived from macrophages on neutrophil activation and entry into plaques (39).
In contrast to myeloid Abca1/Abcg1 deficiency, we found that dendritic cell deficiency of Abca1/Abcg1 (DCABCDDKO mice) in normolipidemic chow-fed mice markedly promoted NLRP3 inflammasome activation, with a distinct phenotype of auto-immune inflammation and a lupus-like syndrome (40). Similar to cholesterol-25-hydroxylase-deficient mice that also show increased autoimmunity (41), DCABCDDKO mice showed prominent induction of Th17 cells (40), probably secondary to release of inflammatory cytokines from dendritic cells, including IL-1β. Thus, inflammasome activation due to disturbances of cholesterol homeostasis in dendritic cells can connect innate and acquired immune systems and promote auto-immunity.
MECHANISMS OF INFLAMMASOME ACTIVATION BY CHOLESTEROL
On a mechanistic level, myeloid Abca1/Abcg1 deficiency led to increased expression of Nlrp3, Caspase-1, and Il-1β in CD11b+ splenocytes, consistent with earlier findings showing plasma membrane cholesterol enrichment and increased TLR4 signaling in Abca1/Abcg1-deficient macrophages (42) and indicative of inflammasome priming. There was prominent accumulation of cholesterol in lysosomes and a small increase in refractile material inside these cells, as detected by confocal reflectance microscopy (7). Sheedy et al. (43) showed that CD36-mediated uptake of oxidized LDL by macrophages can lead to formation of cholesterol crystals in lysosomes leading to lysosomal damage and NLRP3 inflammasome activation. However, in MylABCDKO macrophages, there was no evidence of lysosomal damage or increased mitochondrial ROS in splenic monocytes, macrophages, and neutrophils. Searching for alternative mechanisms to explain NLRP3 inflammasome activation, we discovered that myeloid Abca1/Abcg1 deficiency also activated the noncanonical inflammasome (7). Moreover, there was increased susceptibility to LPS-induced death in MylABCDKO mice, which was rescued by Caspase-1/11 deficiency but not by Nlrp3 deficiency, a signature of noncanonical inflammasome activation (7). Activation of the noncanonical inflammasome can lead to NLRP3 inflammasome activation (44), providing a potential mechanism to explain NLRP3 inflammasome activation. However, this is unlikely to be a complete explanation for the dramatic activation of the NLRP3 inflammasome in MylABCDKO mice.
A NEW MODEL TO EXPLAIN STEROL-DEPENDENT INFLAMMASOME ACTIVATION
Recent studies have shown that macrophages from Niemann-Pick C1 (Npc1)-deficient mice, that display prominent lysosomal cholesterol accumulation similar to MylABCDKO macrophages, are protected from NLRP3 inflammasome activation; these studies suggested that ER rather than lysosomal cholesterol accumulation promotes inflammasome activation (45) (Fig. 1). Our earlier studies in Abca1/Abcg1-deficient macrophages suggested that these cells have increased ER cholesterol content, as shown by reduced expression of the SREBP2 target genes, Hmgcr and Ldlr (46). Moreover, macrophages deficient in cholesterol-25-hydroxylase that likely have increased ER cholesterol content due to derepression of SREBP2 processing and increased cholesterol biosynthesis show increased NLRP3, AIM2, and NLRC4 inflammasome activation (41). Together these observations suggest that ER or plasma membrane cholesterol accumulation may promote the assembly of different inflammasome sensors with ASC leading to inflammasome formation. It is likely that an additional activation signal is required to produce NLRP3 inflammasome activation. We speculate that, in atherosclerosis, this may be dependent on the uptake or recognition of lipoprotein-derived oxidized phospholipids or oxysterols by macrophages.
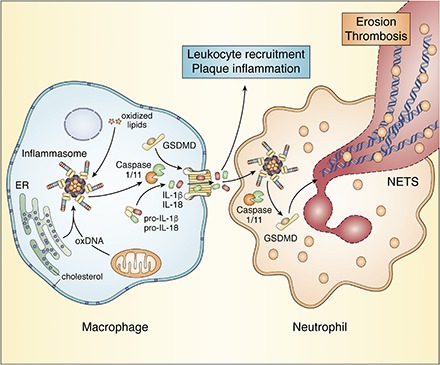
Fig. 1.

Inflammasome activation by lipids in macrophages and neutrophils. Step 1: NLRP3 inflammasome priming. Oxidized LDL (oxLDL) activates the TLR4, leading to activation of NF-κB and transcription of NLRP3 and pro-IL-1β. Step 2: Inflammasome activation. OxLDL is taken up via the scavenger receptor CD36 and hydrolyzed in the lysosome. Oxidized lipids that enter the cytosol activate the noncanonical inflammasome resulting in caspase-11 cleavage. In the absence of Abca1 and Abcg1, cholesterol accumulates in the plasma membrane and is then transported to the ER. ER cholesterol accumulation activates the NLRP3 inflammasome, resulting in caspase-1 cleavage and subsequent cleavage of pro-IL-1β and pro-IL-18. Step 3: GSDMD cleavage. The active (cleaved) forms of caspase-1 and caspase-11 cleave GSDMD, and its N-terminal form (GSDMD-NT) stimulates membrane pore formation. The NLRP3 inflammasome is also activated downstream of caspase-11 cleavage as a result of membrane pore formation. In addition, pore formation leads to pyroptosis, NETosis, and IL-1β and IL-18 secretion.
HUMAN RELEVANCE
Whole-body Abca1 deficiency induced NLRP3 inflammasome activation in Ldlr−/− mice, while myeloid deficiency of Abca1 did not (7). Only the former is associated with low HDL levels (26), indicating that myeloid Abca1 deficiency combined with low HDL levels is sufficient to induce inflammasome activation. This is presumably because the low HDL causes defective cholesterol efflux via non-ABCA1 pathways such as ABCG1. Tangier disease (TD) patients who are homozygous for a loss-of-function of the ABCA1 gene displayed elevated IL-18 plasma levels, showing human relevance (7). This suggests that low HDL, defective apoA-1, and reduced expression of ABCA1/ABCG1 in monocyte/macrophages may be sufficient to induce inflammasome activation in humans. Such changes occur commonly in patients with poorly controlled type 2 diabetes and chronic kidney disease and with ageing (47–53). TD patients sometimes present with premature atherosclerotic CVD (54); however, the more consistent phenotype among adult TD patients is peripheral neuropathy (55). A recent study has shown that defective myelin clearance due to microglial Abca1/Abcg1 deficiency promotes inflammasome activation and limits remyelination following a neuronal injury in aged mice (56). Together with our findings, this suggests that macrophage inflammasome activation may be involved in the pathogenesis of peripheral neuropathy in TD and conceivably type 2 diabetes.
Clonal hematopoiesis involving variants in several genes that predispose to hematological malignancies, including loss-of-function epigenetic modifiers, such as TET2, or gain-of-function JAK/STAT signaling (JAK2V617F), has recently emerged as a major risk factor for coronary heart disease, especially in the elderly (57, 58). Studies in mice with myeloid Tet2 deficiency have shown macrophage inflammasome activation leading to increased IL-1β production and accelerated atherosclerosis (58, 59). NLRP3 inflammasome activation is also prominent in splenic myeloid cells in JAK2V617F BM-transplanted Ldlr−/− mice that have accelerated atherosclerosis with increased necrotic cores (60). NLRP3 inflammasome activation may promote atherosclerosis and thrombosis in JAK2V617F patients with clonal hematopoiesis or myeloproliferative neoplasms.
PERSPECTIVES FOR FUTURE STUDIES
New mechanistic and genetic studies may help to clarify the upstream signals and molecules involved in inflammasome activation and their relevance in metabolic diseases. There is a need to develop sensitive authentic reagents for detection of inflammasome activation in mouse and human tissues. This may help to distinguish local versus systemic effects of inflammasomes in atherogenesis and to evaluate the role of inflammasome activation and pyroptosis/NETosis in plaque erosion and atherothrombosis. That different underlying risk factors for CVD (such as type 2 diabetes, chronic kidney disease, and clonal hematopoiesis) may mechanistically link to atherothrombosis via inflammasome activation could be evaluated in human observational studies, using plasma IL-18 levels or tissue samples to measure inflammasome activation. While the CANTOS suggests a role for inflammasome-derived IL-1β in human CVD, the magnitude of the benefit was moderate, and there was an excess of infections associated with treatment (1), perhaps due to decreased neutrophil levels. It may be informative to determine whether elevated IL-18 levels or the presence of clonal hematopoiesis mutations help to define subgroups of patients who particularly benefitted from treatment. This may help to stratify patients in future clinical studies targeting NLRP3, noncanonical, or AIM2 inflammasomes. On a therapeutic level, removal of cholesterol from macrophages and neutrophils by infusion of reconstituted HDL particles, which are under clinical evaluation in a phase 3 clinical study (Aegis-II, NCT03473223), may alleviate inflammasome activation when administered after an acute coronary syndrome (61). LXR activators, perhaps targeted to myeloid cells in nanoparticles (62), could reduce inflammasome activation both by upregulating ABCA1/ABCG1 and by direct suppression of Il-lβ (63). Recent progress in inflammasome research suggests that molecules upstream of IL-1β secretion, such as NLRP3, caspase-1/11, or CMPK2, may provide additional therapeutic targets for preventing CVD in patients with evidence of underlying inflammasome activation.
Footnotes
Abbreviations:
- AIM2
- absent in melanoma 2
- ASC
- adaptor protein apoptosis-associated speck-like protein containing CARD
- BM
- bone marrow
- GSDMD
- gasdermin-D
- IL
- interleukin
- LPS
- lipopolysaccharide
- NET
- neutrophil extracellular trap
- NETosis
- neutrophil extracellular trap formation
- NLRP3
- NOD-like receptor protein 3
- PAD4
- peptidyl arginine deiminase 4
- ROS
- reactive oxygen species
- TD
- Tangier disease
- TLR
- Toll-like receptor
- WTD
- Western-type diet
This work was supported by National Heart, Lung, and Blood Institute Grant R01HL107653 (to A.R.T.); Netherlands Organisation for Scientific Research VIDI-Grant 917.15.350 (to M.W.); and a Rosalind Franklin Fellowship from the University Medical Center Groningen (to M.W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. A.R.T. reports being a consultant to Amgen, CSL, Janssen Pharmaceutica, Staten Biotech, and Fortico Biotech. M.W. reports no conflicts of interest.
REFERENCES
- 1.Ridker P. M., Everett B. M., Thuren T., MacFadyen J. G., Chang W. H., Ballantyne C., Fonseca F., Nicolau J., Koenig W., Anker S. D., et al. 2017. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377: 1119–1131. [DOI] [PubMed] [Google Scholar]
- 2.Martinon F., Burns K., and Tschopp J.. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 10: 417–426. [DOI] [PubMed] [Google Scholar]
- 3.Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nunez G., Schnurr M., et al. 2010. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 464: 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gage J., Hasu M., Thabet M., and Whitman S. C.. 2012. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can. J. Cardiol. 28: 222–229. [DOI] [PubMed] [Google Scholar]
- 5.Tumurkhuu G., Shimada K., Dagvadorj J., Crother T. R., Zhang W., Luthringer D., Gottlieb R. A., Chen S., and Arditi M.. 2016. Ogg1-dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circ. Res. 119: e76–e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Usui F., Shirasuna K., Kimura H., Tatsumi K., Kawashima A., Karasawa T., Hida S., Sagara J., Taniguchi S., and Takahashi M.. 2012. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 425: 162–168. [DOI] [PubMed] [Google Scholar]
- 7.Westerterp M., Fotakis P., Ouimet M., Bochem A. E., Zhang H., Molusky M. M., Wang W., Abramowicz S., la Bastide-van Gemert S., Wang N., et al. 2018. Cholesterol efflux pathways suppress inflammasome activation, NETosis and atherogenesis. Circulation. 138: 898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jorch S. K., and Kubes P.. 2017. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 23: 279–287. [DOI] [PubMed] [Google Scholar]
- 9.Franck G., Mawson T. L., Folco E. J., Molinaro R., Ruvkun V., Engelbertsen D., Liu X., Tesmenitsky Y., Shvartz E., Sukhova G. K., et al. 2018. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implications for superficial erosion. Circ. Res. 123: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quillard T., Araujo H. A., Franck G., Shvartz E., Sukhova G., and Libby P.. 2015. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur. Heart J. 36: 1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasterkamp G., den Ruijter H. M., and Libby P.. 2017. Temporal shifts in clinical presentation and underlying mechanisms of atherosclerotic disease. Nat. Rev. Cardiol. 14: 21–29. [DOI] [PubMed] [Google Scholar]
- 12.Schroder K., and Tschopp J.. 2010. The inflammasomes. Cell. 140: 821–832. [DOI] [PubMed] [Google Scholar]
- 13.Kayagaki N., Warming S., Lamkanfi M., Vande Walle L., Louie S., Dong J., Newton K., Qu Y., Liu J., Heldens S., et al. 2011. Non-canonical inflammasome activation targets caspase-11. Nature. 479: 117–121. [DOI] [PubMed] [Google Scholar]
- 14.Zhong Z., Liang S., Sanchez-Lopez E., He F., Shalapour S., Lin X. J., Wong J., Ding S., Seki E., Schnabl B., et al. 2018. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 560: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu J., Nagasu H., Murakami T., Hoang H., Broderick L., Hoffman H. M., and Horng T.. 2014. Inflammasome activation leads to caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA. 111: 15514–15519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaidt M. M., Ebert T. S., Chauhan D., Ramshorn K., Pinci F., Zuber S., O’Duill F., Schmid-Burgk J. L., Hoss F., Buhmann R., et al. 2017. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell. 171: 1110–1124.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D. R., Latz E., and Fitzgerald K. A.. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 458: 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dang E. V., McDonald J. G., Russell D. W., and Cyster J. G.. 2017. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. 171: 1057–1071.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang R., Zeng L., Zhu S., Xie Y., Liu J., Wen Q., Cao L., Xie M., Ran Q., Kroemer G., et al. 2018. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 24: 97–108.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V. G., Wu H., and Lieberman J.. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 535: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sollberger G., Choidas A., Burn G. L., Habenberger P., Di Lucrezia R., Kordes S., Menninger S., Eickhoff J., Nussbaumer P., Klebl B., et al. 2018. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 3: doi:10.1126/sciimmunol.aar6689. [DOI] [PubMed] [Google Scholar]
- 22.Chen K. W., Monteleone M., Boucher D., Sollberger G., Ramnath D., Condon N. D., von Pein J. B., Broz P., Sweet M. J., and Schroder K.. 2018. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 3: doi:10.1126/sciimmunol.aar6676. [DOI] [PubMed] [Google Scholar]
- 23.Menu P., Pellegrin M., Aubert J. F., Bouzourene K., Tardivel A., Mazzolai L., and Tschopp J.. 2011. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2: e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng F., Xing S., Gong Z., Mu W., and Xing Q.. 2014. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014: 507208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hendrikx T., Jeurissen M. L., van Gorp P. J., Gijbels M. J., Walenbergh S. M., Houben T., van Gorp R., Pottgens C. C., Stienstra R., Netea M. G., et al. 2015. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. 282: 2327–2338. [DOI] [PubMed] [Google Scholar]
- 26.Westerterp M., Bochem A. E., Yvan-Charvet L., Murphy A. J., Wang N., and Tall A. R.. 2014. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ. Res. 114: 157–170. [DOI] [PubMed] [Google Scholar]
- 27.Shimada K., Crother T. R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V. K., Wolf A. J., Vergnes L., Ojcius D. M., et al. 2012. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 36: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulin N., Viola J. R., Maas S. L., de Jong R., Fernandes-Alnemri T., Weber C., Drechsler M., Doring Y., and Soehnlein O.. 2018. Double-strand DNA sensing Aim2 inflammasome regulates atherosclerotic plaque vulnerability. Circulation. 138: 321–323. [DOI] [PubMed] [Google Scholar]
- 29.Small D. M., and Shipley G. G.. 1974. Physical-chemical basis of lipid deposition in atherosclerosis. Science. 185: 222–229. [DOI] [PubMed] [Google Scholar]
- 30.Fuchs T. A., Brill A., and Wagner D. D.. 2012. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 32: 1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Brühl M. L., Stark K., Steinhart A., Chandraratne S., Konrad I., Lorenz M., Khandoga A., Tirniceriu A., Coletti R., Kollnberger M., et al. 2012. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209: 819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massberg S., Grahl L., von Bruehl M. L., Manukyan D., Pfeiler S., Goosmann C., Brinkmann V., Lorenz M., Bidzhekov K., Khandagale A. B., et al. 2010. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 16: 887–896. [DOI] [PubMed] [Google Scholar]
- 33.Döring Y., Soehnlein O., and Weber C.. 2017. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ. Res. 120: 736–743. [DOI] [PubMed] [Google Scholar]
- 34.Knight J. S., Luo W., O’Dell A. A., Yalavarthi S., Zhao W., Subramanian V., Guo C., Grenn R. C., Thompson P. R., Eitzman D. T., et al. 2014. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 114: 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y., Carmona-Rivera C., Moore E., Seto N. L., Knight J. S., Pryor M., Yang Z. H., Hemmers S., Remaley A. T., Mowen K. A., et al. 2018. Myeloid-specific deletion of peptidylarginine deiminase 4 mitigates atherosclerosis. Front. Immunol. 9: 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warnatsch A., Ioannou M., Wang Q., and Papayannopoulos V.. 2015. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 349: 316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soehnlein O., Ortega-Gomez A., Doring Y., and Weber C.. 2015. Neutrophil-macrophage interplay in atherosclerosis: protease-mediated cytokine processing versus NET release. Thromb. Haemost. 114: 866–867. [DOI] [PubMed] [Google Scholar]
- 38.Döring Y., Drechsler M., Soehnlein O., and Weber C.. 2015. Neutrophils in atherosclerosis: from mice to man. Arterioscler. Thromb. Vasc. Biol. 35: 288–295. [DOI] [PubMed] [Google Scholar]
- 39.Kirii H., Niwa T., Yamada Y., Wada H., Saito K., Iwakura Y., Asano M., Moriwaki H., and Seishima M.. 2003. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23: 656–660. [DOI] [PubMed] [Google Scholar]
- 40.Westerterp M., Gautier E. L., Ganda A., Molusky M. M., Wang W., Fotakis P., Wang N., Randolph G. J., D’Agati V. D., Yvan-Charvet L., et al. 2017. Cholesterol accumulation in dendritic cells links the inflammasome to acquired immunity. Cell Metab. 25: 1294–1304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reboldi A., Dang E. V., McDonald J. G., Liang G., Russell D. W., and Cyster J. G.. 2014. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science. 345: 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yvan-Charvet L., Welch C., Pagler T. A., Ranalletta M., Lamkanfi M., Han S., Ishibashi M., Li R., Wang N., and Tall A. R.. 2008. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 118: 1837–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheedy F. J., Grebe A., Rayner K. J., Kalantari P., Ramkhelawon B., Carpenter S. B., Becker C. E., Ediriweera H. N., Mullick A. E., Golenbock D. T., et al. 2013. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14: 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kayagaki N., Stowe I. B., Lee B. L., O’Rourke K., Anderson K., Warming S., Cuellar T., Haley B., Roose-Girma M., Phung Q. T., et al. 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 526: 666–671. [DOI] [PubMed] [Google Scholar]
- 45.de la Roche M., Hamilton C., Mortensen R., Jeyaprakash A. A., Ghosh S., and Anand P. K.. 2018. Trafficking of cholesterol to the ER is required for NLRP3 inflammasome activation. J. Cell Biol. 217: 3560–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang N., Ranalletta M., Matsuura F., Peng F., and Tall A. R.. 2006. LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler. Thromb. Vasc. Biol. 26: 1310–1316. [DOI] [PubMed] [Google Scholar]
- 47.Sene A., Khan A. A., Cox D., Nakamura R. E., Santeford A., Kim B. M., Sidhu R., Onken M. D., Harbour J. W., Hagbi-Levi S., et al. 2013. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 17: 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ganda A., Yvan-Charvet L., Zhang Y., Lai E. J., Regunathan-Shenk R., Hussain F. N., Avasare R., Chakraborty B., Febus A. J., Vernocchi L., et al. 2017. Plasma metabolite profiles, cellular cholesterol efflux, and non-traditional cardiovascular risk in patients with CKD. J. Mol. Cell. Cardiol. 112: 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mauldin J. P., Nagelin M. H., Wojcik A. J., Srinivasan S., Skaflen M. D., Ayers C. R., McNamara C. A., and Hedrick C. C.. 2008. Reduced expression of ATP-binding cassette transporter G1 increases cholesterol accumulation in macrophages of patients with type 2 diabetes mellitus. Circulation. 117: 2785–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang C., Kanter J. E., Bornfeldt K. E., Leboeuf R. C., and Oram J. F.. 2010. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J. Lipid Res. 51: 1719–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daffu G., Shen X., Senatus L., Thiagarajan D., Abedini A., Hurtado Del Pozo C., Rosario R., Song F., Friedman R. A., Ramasamy R., et al. 2015. RAGE suppresses ABCG1-mediated macrophage cholesterol efflux in diabetes. Diabetes. 64: 4046–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel D. C., Albrecht C., Pavitt D., Paul V., Pourreyron C., Newman S. P., Godsland I. F., Valabhji J., and Johnston D. G.. 2011. Type 2 diabetes is associated with reduced ATP-binding cassette transporter A1 gene expression, protein and function. PLoS One. 6: e22142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kashyap S. R., Osme A., Ilchenko S., Golizeh M., Lee K., Wang S., Bena J., Previs S. F., Smith J. D., and Kasumov T.. 2018. Glycation reduces the stability of ApoAI and increases HDL dysfunction in diet-controlled type 2 diabetes. J. Clin. Endocrinol. Metab. 103: 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abdel-Razek O., Sadananda S. N., Li X., Cermakova L., Frohlich J., and Brunham L. R.. 2018. Increased prevalence of clinical and subclinical atherosclerosis in patients with damaging mutations in ABCA1 or APOA1. J. Clin. Lipidol. 12: 116–121. [DOI] [PubMed] [Google Scholar]
- 55.Schaefer E. J., Santos R. D., and Asztalos B. F.. 2010. Marked HDL deficiency and premature coronary heart disease. Curr. Opin. Lipidol. 21: 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cantuti-Castelvetri L., Fitzner D., Bosch-Queralt M., Weil M. T., Su M., Sen P., Ruhwedel T., Mitkovski M., Trendelenburg G., Lutjohann D., et al. 2018. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science. 359: 684–688. [DOI] [PubMed] [Google Scholar]
- 57.Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P. V., Mar B. G., Lindsley R. C., Mermel C. H., Burtt N., Chavez A., et al. 2014. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371: 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaiswal S., Natarajan P., Silver A. J., Gibson C. J., Bick A. G., Shvartz E., McConkey M., Gupta N., Gabriel S., Ardissino D., et al. 2017. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377: 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fuster J. J., MacLauchlan S., Zuriaga M. A., Polackal M. N., Ostriker A. C., Chakraborty R., Wu C. L., Sano S., Muralidharan S., Rius C., et al. 2017. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 355: 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang W., Liu W., Fidler T., Wang Y., Tang Y., Woods B., Welch C., Cai B., Silvestre-Roig C., Ai D., et al. 2018. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in Jak2V617F mice. Circ. Res. 123: e35–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michael Gibson C., Korjian S., Tricoci P., Daaboul Y., Yee M., Jain P., Alexander J. H., Steg P. G., Lincoff A. M., Kastelein J. J., et al. 2016. Safety and tolerability of CSL112, a reconstituted, infusible, plasma-derived apolipoprotein A-I, after acute myocardial infarction: the AEGIS-I trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation. 134: 1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu M., Amengual J., Menon A., Kamaly N., Zhou F., Xu X., Saw P. E., Lee S. J., Si K., Ortega C. A., et al. 2017. Targeted nanotherapeutics encapsulating liver X receptor agonist GW3965 enhance antiatherogenic effects without adverse effects on hepatic lipid metabolism in Ldlr(-/-) mice. Adv. Healthc. Mater. 6: doi:10.1002/adhm.201700313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas D. G., Doran A. C., Fotakis P., Westerterp M., Antonson P., Jiang H., Jiang X-C., Gustafsson J-A., Tabas I., and Tall A. R.. 2018. LXR suppresses inflammatory chromatin and neutrophil migration through a cis-repressive activity. Cell Rep. 25: 3774–3785.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]