

Abstract
The discovery of the phosphatidylinositol-3-kinase (PI3K) pathway was a major advance in understanding growth factor signaling. The high frequency of PI3K pathway mutations in many cancers has encouraged a new field targeting cancer driver mutations. Although there have been many successes, targeting PI3K itself has proven challenging, in part because of its multiple isoforms with distinct roles. Despite promising preclinical results, development of PI3K inhibitors as pharmacologic anticancer agents has been limited by modest single-agent efficacy and significant adverse effects. If we could overcome these limitations, PI3K inhibitors would be a powerful cancer-fighting tool. Data from phase III clinical trials yields insight into some of the problems with PI3K inhibitors. Recent advances have shed light on the mechanisms of tumor resistance to PI3K inhibitors via feedback pathways that cause elevated insulin levels that then activate the same PI3K pathways that are the targets of inhibition. Improving our understanding of the complex regulatory feedback pathways that activate in response to PI3K inhibition will reveal ways to increase the efficacy of PI3K inhibitors and reduce adverse effects, increasing the usefulness of this class as a treatment option for multiple cancer types.
Keywords: phosphoinositide, insulin signaling, insulin resistance, phosphoinositides, lipid kinases, receptor tyrosine kinases, signal transduction, targeted therapies
Graphical Abstract
PI3K SIGNALING PATHWAYS
Cell growth and proliferation in higher organisms such as humans normally depends on instructive signals provided by growth factors. These signals are transduced across the plasma membrane through receptors such as the insulin receptor (InsR), insulin-like growth factor receptor, epidermal growth factor receptor, and platelet-derived growth factor receptor. The receptors serve to activate intracellular signaling pathways through phosphatidylinositol-3-kinase (PI3K). PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PI-4,5-P2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3). Phosphatidylinositols (PIs) are amphipathic lipids composed of two acyl chains fused to a glycerol (making diacylglycerol) fused to a six-carbon inositol headgroup. This headgroup can be phosphorylated on the 3-, 4-, or 5-position and the location of phosphates determines how the PI-phosphate (phosphoinositide or PIP) interacts with proteins. PI3K is a general term for a kinase that phosphorylates a phosphoinositide on the 3-position. There are three classes of PI3Ks: class I PI3Ks convert PI-4,5-P2 to PIP3 and include the isoforms most frequently mutated in cancer. These are the subject of the following discussion. Additionally, class II PI3Ks convert PI4P to PI-3,4-P2, another important signaling phospholipid expressed on early endosomes and involved in AKT signaling (1, 2). Class III PI3Ks convert PI to PI3P, a major phospholipid in autophagy and vesicular trafficking (3, 4).
The class I PI3Ks are composed of a catalytic subunit (p110) encoded by four genes, PIK3CA, PIK3CB, PIK3CG, and PIK3CD, encoding, respectively, the p110α, p110β, p110γ, and p110δ isoforms. They are constitutively bound to a regulatory subunit encoded by PIK3R1, PIK3R2, PIK3R3, PIK3R5, and PIK3R6, encoding, respectively, p85α, p85β, p55γ, p101, and p87. The p85/p55 subunits heterodimerize with p110α, p110β, or p110δ, forming complexes that are regulated primarily by receptor tyrosine kinases (RTKs). The p101 and p87 subunits heterodimerize with p110γ, forming complexes that are regulated primarily by G protein-coupled receptors. Complexes containing p110β are also activated by G protein-coupled receptors. The p85/p55 regulatory subunits contain two SH2 domains and an inter-SH2 (iSH2) coiled coil domain that mediates the interaction with the catalytic subunit. The SH2 domains bind to the pY-X-X-M amino acid motif of activated RTKs or RTK adaptor proteins, recruiting PI3K to the plasma membrane, where its substrate, PI-4,5-P2, is abundant, and triggering a conformational change that enhances PI3K activity (5, 6).
PIK3CA and PIK3CB are broadly expressed across tissue types. PIK3CG and PIK3CD are expressed more specifically in hematopoietic cells. As an example of the role of PI3K in growth factor signaling, when a RTK is activated, such as the InsR, it recruits insulin receptor substrate 1, which undergoes tyrosine phosphorylation on multiple Y-X-X-M motifs that in turn interact with the SH2 domains of p85 to change PI3K conformation, while recruiting it to the substrate-rich plasma membrane, resulting in robust synthesis of PIP3 (Fig. 1). AKT binds directly to PIP3, stimulating AKT’s protein kinase activity and thus activating downstream growth and survival pathways. The PIP3 signal is turned off by phosphatases: PIP3 is returned to PI-4,5-P2 by PTEN or converted to PI-3,4-P2 by Ship2. PI3K activation initiates a cascade of downstream signals that support growth and proliferation of the cell via pathways including glucose uptake (e.g., GLUT1 and TXNIP) (7), cell growth (e.g., TSC2 and PRAS40, activating mTOR complex 1), and survival (e.g., FOXO) (8). As such, there has been great interest in targeting this pathway with novel targeted therapeutics.
Fig. 1.

PI3K signaling pathway: growth factors such as insulin stimulate tyrosine kinase receptors resulting in their autophosphorylation. In the case of insulin, this recruits IRS-1 that activates PI3K by binding to the SH2 domain of p85. Active PI3K then rapidly converts PI-4,5-P2 to PI-3,4,5-P3, which recruits PDK and AKT via their PH domains leading to AKT kinase activation. AKT phosphorylates many downstream substrates that regulate cell growth, metabolism, and survival.
PI3K AS AN ONCOGENE
Mutations that increase PI3K activity drive cell growth and proliferation independently of growth factors, resulting in unregulated tissue expansion. The PI3K pathway is one of the most commonly mutated pathways in cancer. PIK3CA, PIK3R1, PTEN, and AKT combined are mutated in about 1/3 of all solid tumors with mutation rates over 90% in some tumor types, such as uterine, and over 50% of breast cancers (TCGA PanCancer Atlas, https://cancergenome.nih.gov). If one includes additional downstream targets and regulators, such as TSC1, TSC2, mTOR, and LKB1, this number increases further and includes >50% of lung, ovarian, and some gastrointestinal tumors (Fig. 2). In addition to the high proportion of cancers with PI3K pathway mutations, PI3K activity has been implicated in mediating signals from other driver mutations including RAS (9–11) and RTKs, such as the human epidermal growth factor receptor, HER2 (12, 13). PI3K activation has also been suggested as a mechanism of tumor escape from HER2-targeted therapies (14–16), and combination inhibition of PI3K with HER2 has improved efficacy in preclinical models leading to clinical trials of the combination (17). The first clinical trial to report on this combination used buparlisib, a pan-PI3K inhibitor, with trastuzumab, a HER2 inhibitor, and was only able to resensitize trastuzumab-resistant breast cancers in 10% of cases (18). However, in this and another trial combining buparlisib with lapatinib as a HER2/epidermal growth factor receptor inhibitor, each had one complete response (2–4%) as well as evidence of clinical response in several patients, suggesting activity in select patients (19). These mixed responses reinforce the need to determine which patients will respond to PI3K inhibitors and how to increase the likelihood of successful treatment. Possible explanations for these varied responses include differences in underlying PI3K pathway mutations and differences in the metabolic environment in the patients.
Fig. 2.

The PI3K pathway is commonly mutated in many cancer types. Shown is the mutation frequency observed in The Cancer Genome Atlas (TCGA) PanCancer Atlas by cancer type for the set of PI3K pathway genes that includes PIK3CA, PIK3R1, PTEN, AKT1, AKT2, MTOR, LKB1 (STK11), TSC1, and TSC2.
While obesity has been known to be a risk factor for some types of cancer for decades (20), the mechanism is still not clearly established and is likely multifactorial. Studies have repeatedly shown that for some cancers, such as uterine and breast cancer, BMI is an independent risk factor for development or recurrence of cancer (21), while several additional types of cancer have worse outcomes and increased risk of death in patients with higher BMI, including cervical, kidney, pancreas, and esophageal cancer (22). One explanation for these observations is that insulin resistance, often associated with obesity, results in high circulating insulin levels that activate PI3K pathways in tumors, independent of PIK3CA mutations. As discussed later, high insulin levels may also be a mechanism of escape from treatment with PI3K inhibitors.
The most common PI3K pathway mutation in cancers is in PIK3CA, encoding the p110α isoform. This isoform is chiefly responsible for mediating signaling by RTKs, making it an appealing therapeutic target. The importance of this signaling pathway across cell types is evident by the toxicities observed in clinical use of PI3K-p110α-specific inhibitors. The ideal drug would target the mutant form, allowing maximal cancer-specific benefit, while avoiding the broad toxicities.
PI3K INHIBITORS IN CLINICAL USE
A massive drug development effort has produced many different PI3K inhibitors. Some act on all PI3K isoforms, while others target individual isoforms. In spite of promising preclinical data and dozens of clinical trials, PI3K inhibitors have been slow to make it to clinical use. An added wrinkle comes from the high homology between the lipid kinase domain of PI3K and the protein kinase domain of mTOR, allowing for inhibitors that target both PI3K and mTOR simultaneously, with markedly increased toxicity.
Only three PI3K inhibitors currently have US Food and Drug Administration (FDA) approval, all for treating non-Hodgkin lymphoma by targeting the p110δ isoform. It was initially thought that by sparing the more broadly expressed α and β isoforms, the toxicities of these inhibitors would be more tolerable. Predictably, side effects of p110δ inhibitors include immunosuppression and cytopenias due to on-target effects of blocking PI3Kδ activity in healthy hematopoietic cells; however, other side effects including colitis, hepatitis, and pneumonitis suggest that there may also be inappropriate immune activation, possibly through suppression of regulatory T cell activity.
The first FDA-approved PI3K inhibitor was idelalisib. Idelalisib is a p110δ-specific inhibitor that was approved in 2014 for second line therapy in combination with rituximab for chronic lymphocytic leukemia (CLL). This therapy yields improvement in progression-free survival (PFS) from 5.5 months to >14 months (not reached in initial publication) and improved overall survival at 1 year from 80% to 92% (23). It was also approved that same year for third line treatment of follicular lymphoma (FL) and small lymphocytic lymphoma (SLL) (24). In 2016, six phase III trials of idelalisib in combination with other cancer drugs were stopped by regulatory agencies due to excess deaths in the treatment arms due mainly to infection, pneumonitis, or sepsis. Now, use of idelalisib requires concurrent prophylaxis against pneumocystis jiroveci pneumonia due to the immunosuppression that results from its use.
The second FDA-approved PI3K inhibitor was copanlisib. Copanlisib targets the p110α and p110δ isoforms of PI3K. It was approved in 2017 for third line treatment of CLL/SLL and FL based on a phase II clinical trial and contingent on the phase III trial supporting the initial results (25, 26). As copanlisib also targets the p110α isoform, active outside the immune system, it may have efficacy in other tumor types. Based on preclinical data suggesting that HER2-driven tumors are especially sensitive to PI3K inhibition (12, 17), phase II trials are underway using copanlisib in combination with trastuzumab for HER2+ breast cancer. Notably, additional inhibition of the p110α isoform does not markedly alter the side effect profile, suggesting that many of the severe side effects were due to p110δ inhibition. However, as expected, there is a higher incidence of hyperglycemia with p110α inhibition.
The most recently approved PI3K inhibitor is duvelisib. Duvelisib targets both hematopoietic isoforms, p110γ and p110δ. It was approved in 2018 for third line treatment of FL, CLL, and SLL. The DUO phase III clinical trial data compared duvelisib to ofatumumab (monoclonal antibody against CD20) for relapsed or refractory CLL/SLL. Duvelisib improved PFS from 9.9 to 13.3 months and improved objective overall response rate (ORR) from 45% to 74% (27). Side effect profiles were similar to other PI3K inhibitors and postmarketing data is still being collected.
CHALLENGES OF DEVELOPING PI3K-p110α INHIBITORS
There is evidence emerging from clinical trials that PI3K inhibitors may be more active in PIK3CA mutant tumors, where PI3K is driving tumor growth (28). As such, specific inhibitors of PIK3CA have garnered interest. However, development of inhibitors that block PIK3CA activity has been limited by only modest benefits and significant side effects in clinical trials. While targeting PIK3CA expands the number of sensitive tumor types, it likely also increases the side effects. For example, two recent PIK3CA inhibitors, taselisib and buparlisib, were both tested in randomized placebo-controlled phase III clinical trials in combination with fulvestrant for treating breast cancer. While both drugs met their primary endpoints of improved PFS, development of both drugs was stopped after data analysis showed only modest increases in PFS with markedly increased toxicity. Buparlisib crosses the blood-brain barrier and causes unacceptable depression and anxiety while failing to significantly improve overall survival over fulvestrant with placebo (29).
Alpelisib, on the other hand, is another p110α-specific inhibitor that recently had a positive phase III clinical trial treating breast cancer in combination with fulvestrant, showing a significantly improved PFS of 11 versus 5.7 months and an ORR of 36% versus 16% (30). The difference in trial outcome is at least partially due to trial design. The alpelisib trial included a prespecified analysis specifically in patients with PIK3CA mutations, which was the group that demonstrated benefit. The PFS and ORR differences disappear when evaluating all-comers. This is in notable contrast to the p110δ-specific inhibitors, which provide benefit regardless of mutation status and suggests that the p110α-specific inhibitor is specifically targeting a driver mutation.
One of the challenges highlighted by the examples above is efficacy. PI3K inhibitors are very promising in preclinical data where conditions are more controlled, but when used in diverse cancer patient populations, we find that there is great variability in their efficacy that was not predicted from preclinical data. One reason could be variations in downstream PI3K pathway mutations that render PI3K inhibitors less effective. Mutation in PTEN, AKT, or mTOR may lead to pathway activation without dependence on PI3K signaling and co-occurrence is not unusual. This hypothesis could be tested by post hoc analysis comparing PI3K pathway mutation responders and nonresponders so that subsequent clinical trials may benefit from treating a target population known to have a vulnerable mutational profile. Changes in regulation of PI-4,5-P2, the substrate of PI3K, abundance or acyl chain composition have also been shown to affect PI3K activity (31, 32), and variation in these factors between patients may also affect response to PI3K inhibitors.
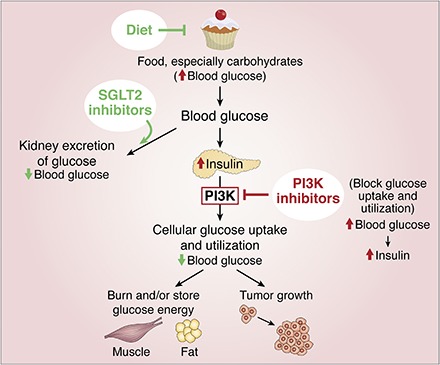
Another reason for limited efficacy could be due to the hyperglycemic response noted as a frequent side effect in the trials. Hyperglycemia is a predictable on-target effect of PI3K-p110α inhibitors (see the supplemental Graphical Abstract). When signaling through the InsR is blocked, glucose is not taken up by cells and blood sugar goes up. This triggers increased insulin release from the pancreas, and subsequently very high circulating insulin levels, which can activate insulin-driven growth pathways that undermine the efficacy of the inhibitor (33). In clinical trials so far, metformin was commonly used to treat hyperglycemia. However, as shown in Hopkins et al. (33), metformin was insufficient to prevent activation of this feedback pathway, resulting in hyperinsulinemia and tumor growth in mice. By contrast, alternative strategies to treat hyperglycemia, such as use of SGLT2 inhibitors (to promote glucose excretion by the kidney) or adoption of a very low carbohydrate (ketogenic) diet, were effective. The key point to take away from these data is that improving hyperglycemic control and the resulting hyperinsulinemia, either through judiciously chosen medication or diet, improves tumor sensitivity to PI3K-p110α inhibitors, suggesting a path for future clinical trials.
The other challenge to overcome is the significant side effect profile of PI3K inhibitors. Severe and frequent toxicities (grade III or IV in >10% of patients) include hyperglycemia, diarrhea, hypertension, rash, pneumonitis, transaminitis, cytopenias, and, more rarely, intestinal perforation. One approach to overcome toxicity of targeting the ubiquitous p110α isoform would be the development of mutation-specific inhibitors. This approach has proven to be difficult because the most common mutations, H1047R and E545K, provide little change to specifically target. Another approach might be to use combination therapies to allow effective tumor killing at lower doses of PI3K inhibitor. Addressing these challenges will be critical to move PI3K inhibitors forward in treating more types of cancer for broader clinical benefit.
PI3K-p110γ INHIBITORS IN CANCER
Finally, a completely different approach to treating solid malignancies that avoids many of the challenges described above is to use a PI3K-p110γ-specific inhibitor, such as IPI-549, which is currently in expanded phase Ib clinical trials for several cancer types. The p110γ-specific inhibitors work at the overlap of targeted therapy and immunotherapy. They do not target solid tumors directly, which do not express PIK3CG, but rather they alter macrophage responses from the wound-healing (M2) phenotype to the inflammatory (M1) type. This effect is measured by increased interferon-γ levels and results in reinvigoration of exhausted T cells to increase tumor killing or prevent metastasis (34). The recent successes of immunotherapies in treating many different tumor types make this an exciting area for future study.
Footnotes
Abbreviations:
- CLL
- chronic lymphocytic leukemia
- FDA
- Food and Drug Administration
- FL
- follicular lymphoma
- HER2
- human epidermal growth factor receptor 2
- InsR
- insulin receptor
- ORR
- overall response rate
- PFS
- progression-free survival
- PI
- phosphatidylinositol
- PI3K
- phosphatidylinositol-3-kinase
- PIP
- phosphoinositide
- PIP3
- phosphatidylinositol-3,4,5-trisphosphate
- PI-4
- 5-P2, phosphatidylinositol-4,5-bisphosphate
- RTK
- receptor tyrosine kinase
- SLL
- small lymphocytic lymphoma
This work was supported by National Institutes of Health Grants R01 GM120055 and R01 CA201303 (S.J.F.), National Cancer Institute Grant R35 CA197588 (L.C.C.), a Burroughs Wellcome Fund Career Award in the Biomedical Sciences (S.J.F.), and a Scholar-Innovator Award from the Harrington Discovery Institute (S.J.F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. L.C.C. is a founder and member of the Scientific Advisory Board of Agios Pharmaceuticals and of Petra Pharmaceuticals. These companies are developing novel therapies for cancer. L.C.C.’s laboratory (including M.N.P.) also receives some financial support from Petra Pharmaceuticals. The other author declares that there are no additional conflicts of interest with the contents of this article.
REFERENCES
- 1.Braccini L., Ciraolo E., Campa C. C., Perino A., Longo D. L., Tibolla G., Pregnolato M., Cao Y., Tassone B., Damilano F., et al. 2015. PI3K-C2gamma is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 6: 7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franke T. F., Kaplan D. R., Cantley L. C., and Toker A.. 1997. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 275: 665–668. [DOI] [PubMed] [Google Scholar]
- 3.Whitman M., Downes C. P., Keeler M., Keller T., and Cantley L.. 1988. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 332: 644–646. [DOI] [PubMed] [Google Scholar]
- 4.Wallroth A., and Haucke V.. 2018. Phosphoinositide conversion in endocytosis and the endolysosomal system. J. Biol. Chem. 293: 1526–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Songyang Z., Shoelson S. E., Chaudhuri M., Gish G., Pawson T., Haser W. G., King F., Roberts T., Ratnofsky S., Lechleider R. J., et al. 1993. SH2 domains recognize specific phosphopeptide sequences. Cell. 72: 767–778. [DOI] [PubMed] [Google Scholar]
- 6.Cantley L. C., and Songyang Z.. 1994. Specificity in recognition of phosphopeptides by src-homology 2 domains. J. Cell Sci. Suppl. 18: 121–126. [DOI] [PubMed] [Google Scholar]
- 7.Waldhart A. N., Dykstra H., Peck A. S., Boguslawski E. A., Madaj Z. B., Wen J., Veldkamp K., Hollowell M., Zheng B., Cantley L. C., et al. 2017. Phosphorylation of TXNIP by AKT mediates acute influx of glucose in response to insulin. Cell Reports. 19: 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manning B. D., and Cantley L. C.. 2007. AKT/PKB signaling: navigating downstream. Cell. 129: 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eser S., Reiff N., Messer M., Seidler B., Gottschalk K., Dobler M., Hieber M., Arbeiter A., Klein S., Kong B., et al. 2013. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 23: 406–420. [DOI] [PubMed] [Google Scholar]
- 10.Castellano E., Sheridan C., Thin M., Nye E., Spencer-Dene B., Diefenbacher M., Moore C., Kumar M., Murillo M., Grönroos E., et al. 2013. Requirement for Interaction of PI3-Kinase p110α with RAS in Lung Tumor Maintenance. Cancer Cell. 24: 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wee S., Jagani Z., Xiang K. X., Loo A., Dorsch M., Yao Y. M., Sellers W. R., Lengauer C., and Stegmeier F.. 2009. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 69: 4286–4293. [DOI] [PubMed] [Google Scholar]
- 12.Ruiz-Saenz A., Dreyer C., Campbell M. R., Steri V., Gulizia N., and Moasser M. M.. 2018. HER2 amplification in tumors activates PI3K/Akt signaling independent of HER3. Cancer Res. 78: 3645–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kebenko M., Drenckhan A., Gros S. J., Jücker M., Grabinski N., Ewald F., Grottke A., Schultze A., Izbicki J. R., Bokemeyer C., et al. 2015. ErbB2 signaling activates the Hedgehog pathway via PI3K-Akt in human esophageal adenocarcinoma: identification of novel targets for concerted therapy concepts. Cell. Signal. 27: 373–381. [DOI] [PubMed] [Google Scholar]
- 14.Nagata Y., Lan K. H., Zhou X., Tan M., Esteva F. J., Sahin A. A., Klos K. S., Li P., Monia B. P., Nguyen N. T., et al. 2004. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 6: 117–127. [DOI] [PubMed] [Google Scholar]
- 15.Berns K., Horlings H. M., Hennessy B. T., Madiredjo M., Hijmans E. M., Beelen K., Linn S. C., Gonzalez-Angulo A. M., Stemke-Hale K., Hauptmann M., et al. 2007. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 12: 395–402. [DOI] [PubMed] [Google Scholar]
- 16.Rimawi M., Ferrero J. M., de la Haba-Rodriguez J., Poole C., De Placido S., Osborne C. K., Hegg R., Easton V., Wohlfarth C., Arpino G., et al. 2018. First-line trastuzumab plus an aromatase inhibitor, with or without pertuzumab, in human epidermal growth factor receptor 2-positive and hormone receptor-positive metastatic or locally advanced breast cancer (PERTAIN): a randomized, open-label phase II trial. J. Clin. Oncol. 36: 2826–2835. [DOI] [PubMed] [Google Scholar]
- 17.Rexer B. N., Chanthaphaychith S., Dahlman K., and Arteaga C. L.. 2014. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res. 16: R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pistilli B., Pluard T., Urruticoechea A., Farci D., Kong A., Bachelot T., Chan S., Han H. S., Jerusalem G., Urban P., et al. 2018. Phase II study of buparlisib (BKM120) and trastuzumab in patients with HER2+ locally advanced or metastatic breast cancer resistant to trastuzumab-based therapy. Breast Cancer Res. Treat. 168: 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerin M., Rezai K., Isambert N., Campone M., Autret A., Pakradouni J., Provansal M., Camerlo J., Sabatier R., Bertucci F., et al. 2017. PIKHER2: A phase IB study evaluating buparlisib in combination with lapatinib in trastuzumab-resistant HER2-positive advanced breast cancer. Eur. J. Cancer. 86: 28–36. [DOI] [PubMed] [Google Scholar]
- 20.Carroll K. K. 1998. Obesity as a risk factor for certain types of cancer. Lipids. 33: 1055–1059. [DOI] [PubMed] [Google Scholar]
- 21.Bhaskaran K., Douglas I., Forbes H., dos-Santos-Silva I., Leon D. A., and Smeeth L.. 2014. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5·24 million UK adults. Lancet. 384: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calle E. E., Rodriguez C., Walker-Thurmond K., and Thun M. J.. 2003. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N. Engl. J. Med. 348: 1625–1638. [DOI] [PubMed] [Google Scholar]
- 23.Furman R. R., Sharman J. P., Coutre S. E., Cheson B. D., Pagel J. M., Hillmen P., Barrientos J. C., Zelenetz A. D., Kipps T. J., Flinn I., et al. 2014. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 370: 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gopal A. K., Kahl B. S., de Vos S., Wagner-Johnston N. D., Schuster S. J., Jurczak W. J., Flinn I. W., Flowers C. R., Martin P., Viardot A., et al. 2014. PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 370: 1008–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dreyling M., Morschhauser F., Bouabdallah K., Bron D., Cunningham D., Assouline S. E., Verhoef G., Linton K., Thieblemont C., Vitolo U., et al. 2017. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann. Oncol. 28: 2169–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dreyling M., Santoro A., Mollica L., Leppa S., Follows G. A., Lenz G., Kim W. S., Nagler A., Panayiotidis P., Demeter J., et al. 2017. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J. Clin. Oncol. 35: 3898–3905. [DOI] [PubMed] [Google Scholar]
- 27.Flinn I. W., Hillmen P., Montillo M., Nagy Z., Illes A., Etienne G., Delgado J., Kuss B. J., Tam C. S., Gasztonyi Z., et al. 2018. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood. 132: 2446–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer I. A., Abramson V. G., Formisano L., Balko J. M., Estrada M. V., Sanders M. E., Juric D., Solit D., Berger M. F., Won H. H., et al. 2017. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2-metastatic breast cancer. Clin. Cancer Res. 23: 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campone M., Im S. A., Iwata H., Clemons M., Ito Y., Awada A., Chia S., Jagiełło-Gruszfeld A., Pistilli B., Tseng L. M., et al. 2018. Buparlisib plus fulvestrant versus placebo plus fulvestrant for postmenopausal, hormone receptor-positive, human epidermal growth factor receptor 2-negative, advanced breast cancer: Overall survival results from BELLE-2. Eur. J. Cancer. 103: 147–154. [DOI] [PubMed] [Google Scholar]
- 30.André F., Ciruelos E. M., Rubovszky G., Campone M., Loibl S., Rugo H. S., Iwata H., Conte P., Mayer I. A., Kaufman B., et al. 2018. LBA3_PR alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the phase III SOLAR-1 trial. Ann. Oncol. 29: doi:10.1093/annonc/mdy424.010. [Google Scholar]
- 31.Saito K., Tolias K. F., Saci A., Koon H. B., Humphries L. A., Scharenberg A., Rawlings D. J., Kinet J. P., and Carpenter C. L.. 2003. BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity. 19: 669–678. [DOI] [PubMed] [Google Scholar]
- 32.Clark J., Anderson K. E., Juvin V., Smith T. S., Karpe F., Wakelam M. J., Stephens L. R., and Hawkins P. T.. 2011. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat. Methods. 8: 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hopkins B. D., Pauli C., Du X., Wang D. G., Li X., Wu D., Amadiume S. C., Goncalves M. D., Hodakoski C., Lundquist M. R., et al. 2018. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. 560: 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sullivan R. J., Hong D. S., Tolcher A. W., Patnaik A., Shapiro G., Chmielowski B., Ribas A., Brail L. H., Roberts J., Lee L., et al. 2018. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J. Clin. Oncol. 36: 3013. [Google Scholar]