

Abstract
Niacin, the first antidyslipidemic drug, has been at the center stage of lipid research for many decades before the discovery of statins. However, to date, despite its remarkable effects on lipid profiles, the clinical outcomes of niacin treatment on cardiac events is still debated. In addition to its historically well-defined interactions with central players of lipid metabolism, niacin can be processed by eukaryotic cells to synthesize a crucial cofactor, NAD+. NAD+ acts as a cofactor in key cellular processes, including oxidative phosphorylation, glycolysis, and DNA repair. More recently, evidence has emerged that NAD+ also is an essential cosubstrate for the sirtuin family of protein deacylases and thereby has an impact on a wide range of cellular processes, most notably mitochondrial homeostasis, energy homeostasis, and lipid metabolism. NAD+ achieves these remarkable effects through sirtuin-mediated deacetylation of key transcriptional regulators, such as peroxisome proliferator-activated receptor gamma coactivator 1-α, LXR, and SREBPs, that control these cellular processes. Here, we present an alternative point of view to explain niacin’s mechanism of action, with a strong focus on the importance of how this old drug acts as a control switch of NAD+/sirtuin-mediated control of metabolism.
Keywords: nicotinic acid, sirtuins, mitochondria, cholesterol, dyslipidemia, HDL, LDL, kidney disease, fatty acid oxidation, lipid synthesis
Graphical Abstract
Nicotinic acid was identified in the beginning of the 20th century by Conrad Elvehjem (1) as an effective treatment for pellagra, which at that time was endemic in the United States. The name of nicotinic acid was replaced by niacin in the 1940s to avoid any association with nicotine (2), and a decade later, the lipid-modulating effects of this molecule were described in patients by Rudolf Altschul (3), making niacin the oldest lipid-lowering drug. Despite the fact that several of the molecular mechanisms underlying its remarkable effects on lipid metabolism have been elucidated since then, the molecular mechanism of how niacin works remains elusive.
NIACIN, KNOWN MECHANISMS OF ACTION
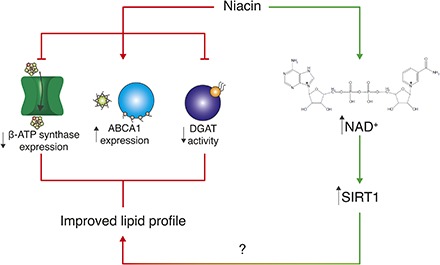
In pharmacological doses, niacin acts as a broad-spectrum lipid-modulating drug and increases the circulating levels of HDL (4) (Fig. 1). This class of lipoproteins, which is particularly enriched with ApoA-I and ApoA-II, are major players in reverse cholesterol transport. Most peripheral tissues indeed rely on HDL for cholesterol clearance and transport to the liver, where cholesterol is either processed or degraded (5). Thanks to this scavenging function, low HDL is considered, with few exceptions, an independent risk factor for coronary artery disease (6). Niacin increases HDL availability through different mechanisms (Fig. 1). First, it has a direct effect on ApoA-I stability and function. In fact, niacin boosts the expression of the membrane protein ABCA1, the main regulator of ApoA-I lipidation and consequent stabilization, through the LXR (7). Moreover, niacin prevents the surface expression of the hepatic HDL receptor β-chain ATP synthase (8). This inhibition decreases HDL uptake in the liver and consequently increases HDL availability for cholesterol scavenging in the blood (9).
Fig. 1.

Known mechanisms of action of niacin. Niacin increases circulating HDL particles for peripheral cholesterol scavenging by two described mechanisms: First, by reducing the surface expression of the hepatic HDL receptor β-ATP synthase, which is involved in the endocytosis of HDL particles in the liver, and, second, by increasing the expression of ABCA1, which promotes ApoA-I lipidation and stabilization and thus promotes HDL production. Moreover, niacin reduces the activity of DGAT, which leads to reduced TG synthesis, Apo B degradation, and reduced VLDL and LDL production and circulation.
The best-characterized effect of niacin on lipid metabolism is, however, the reduction of triglyceride (TG) and circulating FFA levels (Fig. 1). The first proposed mechanism explaining this outcome involves its inhibitory actions on adipocyte TG lipolysis, which would decrease FFA release and the availability of FFAs to stimulate liver TG synthesis (10). However, this hypothesis has been challenged when the niacin receptor, a G-protein-coupled receptor, termed GPR109A, was identified as the mediator of its antilipolytic effect. After niacin administration, Gpr109a-KO mice show the typical serum TG-lowering effect, despite the absence of adipocyte lipolysis. Moreover, GPR109A agonists were shown to inhibit lipolysis in patients with dyslipidemia in the absence of effects on circulating lipids (11), arguing that mechanisms independent from niacin’s antilipolytic function via activation of GPR109A are responsible for its beneficial effects on lipid homeostasis. The most recent hypothesis identifies the liver as the main contributor in the niacin-mediated modulation of serum lipids. More specifically, in vitro evidence suggests that the hepatic lipid-lowering effects of niacin are mediated by a noncompetitive direct interaction of niacin with the final enzyme of TG synthesis, diglyceride acyltransferase 2 (DGAT2) (Fig. 1) (12). In support of this, patients on niacin display a lipid-lowering response influenced by genetic DGAT2 polymorphisms (13). Moreover, the hepatic modulation of TG biosynthesis impacts on apo-B-containing VLDLs, and LDLs, the principal transporters of TG and cholesterol, respectively. These lipoproteins require TGs for their formation; therefore, reducing TG synthesis leads to destabilization of VLDL and LDL particles, decreasing circulating lipids. However, recent publications seem to quench the translation of these findings in humans, proving that the in vivo TG-lowering effects of DGAT2 inhibitors observed in murine models are not present in a primate model (14).
NIACIN, THE FIRST ANTIDYSLIPIDEMIC DRUG
The beneficial effects of niacin on lipid metabolism promoted several clinical trials in patients suffering from different cardiovascular conditions. However, the outcomes of these studies were controversial. The first large clinical study, the Coronary Drug Project, showed a reduced incidence of nonfatal reinfarction and all-cause mortality after 5 years in patients with documented myocardial infarction when compared with placebo (15). Following these initial positive results, niacin was tested in combination with other drugs to treat dyslipidemias. First, when it was coadministered with the bile acids sequestrant colestipol, niacin markedly increased HDL and atherosclerotic regression in two trials (16, 17). With the discovery of statins and their prominent beneficial effects on cholesterol metabolism, niacin has been investigated as a possible add-on to improve the therapeutic outcome of statins. In five different clinical trials, add-on of niacin to the statin treatment reduced the onset of cardiovascular events with a concomitant increase in HDL (18–22). The excitement for the potential application of niacin as an add-on to statin therapy has, however, been dampened when two recent, large clinical studies, the AIM-HIGH and the HPS2-THRIVE trials, failed in reproducing the benefits of previous studies. Niacin, in combination with simvastatin, did not improve the risk of major cardiovascular events; however, a significant improvement in HDL and TG levels remained (23, 24). The main difference from the previous clinical trials, besides the recruitment of a significantly larger group of patients (several thousand vs. hundreds), was the use of statins in the control conditions, as opposed to four out of the five prior studies in which no drug at all or double placebo were used. Interestingly, in AIM-HIGH, niacin treatment abolished the correlation between non-HDL cholesterol and cardiovascular events (23). In control conditions, the levels of non-HDL cholesterol were predictors of cardiovascular complications—an association that is lost upon niacin treatment, suggesting the involvement of nonlipoprotein mechanisms. The results from the AIM-HIGH and HPS2-THRIVE studies have, however, resulted in marginalization of niacin in national guidelines for treatment of dyslipidemia and atherosclerotic CVD. To date, statin therapy remains the gold standard for treatment of these conditions due to its more potent effect on cholesterol levels (25). Niacin, however, is approved in many countries as therapy for patients with statin intolerance and as a broad lipid-lowering drug (26). Moreover, it has an off-target use in chronic kidney disease (CKD) and end-stage renal disease (ESRD), given its beneficial impact on factors affecting the glomerular filtration rate, including phosphate levels (27), oxidative stress (28, 29), inflammation (30–32), and HDL and TG levels (4, 33). Interestingly, HDL and TG levels have both been associated independently with CKD (34), indicating that niacin treatment may be relevant in these conditions, thus providing the rationale for future clinical trials to test the effects of niacin in CKD and ESRD patients.
NIACIN, ALSO A PRECURSOR FOR NAD+
Although less studied than other mechanisms of niacin, niacin is also an important precursor for NAD+ via the so-called Preiss-Handler pathway (Fig. 2). Hence, current literature on niacin’s mechanism of action needs to be revisited from an NAD+-centered point of view. Until very recently, NAD+ was, together with its reduced counterpart, NADH, perceived as a redox couple, whose function seemed to be fully established since the 1930s (35). In 2000, however, the discovery that NAD+ is a cosubstrate for the sirtuin family of deacylases, important longevity and metabolic regulators, heralded a whole new era in NAD+ research (36). Raising NAD+ levels was found to promote sirtuin 1 (SIRT1) activity, leading, among others, to the deacetylation and thus activation of the peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α), an important regulator of mitochondrial biogenesis (37, 38), thus putting sirtuins at the pinnacle of the control of mitochondrial homeostasis. In line with this, sirtuins were found to govern lipid and energy metabolism, acting as negative regulators of TG synthesis (39–41) and stimulating FA oxidation (42–44). The beneficial effects of niacin on serum TG and FFA levels might therefore at least in part be explained by raising NAD+ levels, which promotes sirtuin activity, mitochondrial biogenesis, and thus enhanced mitochondrial FA oxidation and reduced TG synthesis (Fig. 3).
Fig. 2.

Pathways modulating NAD+ levels in mammals. NAD+ can be synthesized either via salvage pathways from precursors such as niacin (nicotinic acid), nicotinamide (NAM), and nicotinamide riboside (NR) or de novo from tryptophan (Trp). In the first step of the Preiss-Handler pathway, niacin is converted into NA mononucleotide (NAMN) by nicotinate phosphoribosyltransferase (NAPRT). NAM mononucleotide adenylyltransferase (NMNAT) uses NAMN to generate NA adenine dinucleotide (NAAD), which gets converted into NAD+ by NAD synthetase (NADS). NAD+ synthesis from NAM and NR comprises their conversion into NAM mononucleotide (NMN) by NAM phosphoribosyltransferase (NAMPT) and NAM riboside kinase (NRK), respectively, and the subsequent conversion of NMN into NAD+ by NMNAT. NMN can also be recycled back into NR by CD73. The de novo NAD+ synthesis pathway from Trp consists of eight steps and merges with the Preiss-Handler pathway. Pathways that reduce NAD+ availability include the conversion of NAM into methylnicotinamide (MNA) by N-methyltransferase (NNMT) and NAD+ consumption by enzymes including the sirtuins, CD38, and PARP1.
Fig. 3.

Niacin’s mechanism of action explained by raising NAD+ levels? As a precursor for NAD+, niacin can activate the deacylase SIRT1, which might explain niacin’s beneficial effects on lipid homeostasis. First, SIRT1 activates PGC-1α, which leads to increased mitochondrial activity and FA oxidation. Moreover, SIRT1 activates LXR, which leads to increased expression of ABCA1, and thus increased circulating HDL particles. Furthermore, SIRT1 destabilizes SREBPs and thereby lowers SREBP-mediated FA, TG, and cholesterol synthesis and circulation.
Further supporting the hypothesis of NAD+-driven effects of niacin, SIRT1 was also shown to regulate the activity of important transcription factors that control lipid metabolism, including the LXR and SREBP families. More specifically, SIRT1-mediated deacetylation of LXR leads to its activation and subsequently to increased expression of its target genes, including Abca1 and Srebp-1c (45). Thus, increases of NAD+ levels after niacin administration might act upstream of the previously described effects of niacin on ABCA1-mediated cholesterol homeostasis (7) (Fig. 3). In line with this, Sirt1-KO mice show impaired cholesterol homeostasis and develop hepatic steatosis (45–47), whereas gain of SIRT1 function provides protection against hepatic steatosis (48). As LXR activation, however, also upregulates the expression of Srebp-1c (45), a key driver of FA and TG synthesis, the impact of SIRT1-LXR activation on lipid homeostasis is controversial. A possible explanation for this discrepancy could be the fact that SIRT1 not only deacetylates LXR, but also SREBP-1c itself and other members of the SREBP family, including SREBP-2, which governs cholesterol biosynthesis, and SREBP-1a, which is involved in FA, TG, and cholesterol biosynthesis (49). SIRT1-mediated deacetylation of these transcription factors leads to their destabilization and subsequently to decreased SREBP-mediated lipid synthesis (40, 50). Hence, activation of SIRT1 by NAD+ seems to promote LXR-mediated improvements in cholesterol homeostasis, while at the same time abrogating detrimental SREBP-mediated lipid synthesis (Fig. 3). Future work focused on the sirtuin family as putative mediators of niacin’s health benefits is therefore urgently needed.
Apart from niacin, NAD+ can be produced from two other precursor molecules, nicotinamide (NAM) and nicotinamide riboside (NR), or synthesized de novo from tryptophan (Fig. 2). Studies exploring these alternative NAD+-replenishing pathways provided further support for the hypothesis that niacin’s beneficial effects involve NAD+-mediated signaling.
For example, the expression of the rate-limiting enzyme for NAD+ synthesis from NAM, NAM phosphoribosyltransferase (NAMPT), is decreased in adipose tissue in obesity (51–53). In line, adipocyte-specific KO of Nampt leads to increased plasma FFA and TG levels and adipose tissue dysfunction (54). Moreover, inactivation of NAMPT in mature macrophages induces lipid accumulation in these cells (55), and the inhibition of NAMPT enhances hepatic steatosis in mice challenged with high-fat diet (56).
Interestingly, NAD+-consuming enzymes, including the poly(ADP-ribose)-polymerase (PARP) family, can be overactivated in some diseases by lipid overload and, hence, could be responsible for NAD+ depletion, as observed in steatotic livers (57). In line, several different approaches to increase NAD+ levels, including stimulation of NAD+ biosynthesis, e.g., by administration of NR, as well as inhibition of PARPs, were shown to protect from hepatic lipid accumulation in the case of nonalcoholic fatty liver disease (57–60) and alcoholic liver disease (61). Although administration of NAM failed to reproduce the benefits of niacin due to end-product inhibition of sirtuin activity (62), NR potently increases the use of lipids as energy substrates and ameliorates cholesterol profiles in mice (63), reminiscent to what is seen upon niacin treatment. Furthermore, also knockdown of nicotinamide N-methyltransferase (NNMT), which is responsible for the conversion of NAM into methylnicotinamide (MNA) (Fig. 2), leads to increased NAD+ levels and subsequently promotes SIRT1-target gene expression in adipose tissues and overall energy expenditure (64). Importantly, recent publications also highlighted that raising NAD+ levels provides protection against acute kidney injury in mice (59, 65), which might explain in part the beneficial impact of niacin on kidney function in patients with CKD and ESRD (27–34). Despite the wealth of preclinical studies studying the effects of NAD+ boosting on lipid metabolism, metabolic control, and longevity, large clinical trials focused on the consequences of NAD+ boosting on lipid homeostasis and CVDs in humans are still pending.
CONCLUSION AND PERSPECTIVES
Niacin, the oldest antidyslipidemic drug, has been intensively studied in the past, revealing several mechanisms that might explain its significant health benefits. Through its interaction with important players in lipid metabolism, such as β-chain ATP synthase, ABCA1, and DGAT2, niacin is capable of increasing the levels of circulating HDL particles while decreasing TG levels. However, niacin also has the ability to raise levels of NAD+ through the Preiss-Handler pathway. Interestingly, boosting NAD+ levels recapitulates several of the initially observed effects of niacin on lipid metabolism. These NAD+-driven effects are mainly mediated by SIRT1, which plays a key role in the modulation of important regulators of lipid metabolism such as PGC-1α, LXR, and SREBP. Although increasing evidence in literature points to the NAD+/sirtuin-axis as a mediator of niacin’s health benefits, future studies will be needed to tell whether niacin acts on its own or whether it should from now on be exclusively perceived as precursor for NAD+. These studies could also uncover important mechanisms of action for other NAD+ boosters, such as NR.
Footnotes
Abbreviations:
- CKD
- chronic kidney disease
- DGAT2
- diglyceride acyltransferase 2
- ESRD
- end-stage renal disease
- NAM
- nicotinamide
- NAMPT
- nicotinamide phosphoribosyltransferase
- NNMT
- nicotinamide N-methyltransferase
- NR
- nicotinamide riboside
- PARP
- poly(ADP-ribose)-polymerase
- PGC-1α
- peroxisome proliferator-activated receptor gamma coactivator 1-α
- SIRT1
- sirtuin 1
- TG
- triglyceride
This work was supported by European Research Council Grant ERC-2017-ADG-787702, Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung Grant SNSF 31003A_179435, and the École Polytechnique Fédérale de Lausanne. J. A. acts as a consultant to Mitobridge-Astellas and TES Pharma, which are developing NAD-boosting drugs.
REFERENCES
- 1.Elvehjem C. A., Madden R. J., Strong F. M., and Woolley D. W.. 1974. The isolation and identification of the anti-black tongue factor. Nutr. Rev. 32: 48–50. [DOI] [PubMed] [Google Scholar]
- 2.Council on Foods and Nutrition 1942. Niacin and niacin amide. J. Am. Med. Assoc. 118: 819. [Google Scholar]
- 3.Altschul R., and Hoffer A.. 1958. Effects of salts of nicotinic acid on serum cholesterol. BMJ. 2: 713–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shepherd J., Packard C. J., Patsch J. R., Gotto A. M., and Taunton O. D.. 1979. Effects of nicotinic acid therapy on plasma high density lipoprotein subfraction distribution and composition and on apolipoprotein A metabolism. J. Clin. Invest. 63: 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rader D. J., Alexander E. T., Weibel G. L., Billheimer J., and Rothblat G. H.. 2009. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 50(Suppl): S189–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boden W. E. 2000. High-density lipoprotein cholesterol as an independent risk factor in cardiovascular disease: assessing the data from Framingham to the Veterans Affairs High–Density Lipoprotein Intervention Trial. Am. J. Cardiol. 86(12A): 19L–22L. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L-H., Kamanna V. S., Ganji S. H., Xiong X-M., and Kashyap M. L.. 2012. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J. Lipid Res. 53: 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L-H., Kamanna V. S., Zhang M. C., and Kashyap M. L.. 2008. Niacin inhibits surface expression of ATP synthase beta chain in HepG2 cells: implications for raising HDL. J. Lipid Res. 49: 1195–1201. [DOI] [PubMed] [Google Scholar]
- 9.Martinez L. O., Jacquet S., Esteve J-P., Rolland C., Cabezón E., Champagne E., T. Pineau, V. Georgeaud, J. E. Walker, F. Tercé, et al. 2003. Ectopic beta-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature. 421: 75–79. [DOI] [PubMed] [Google Scholar]
- 10.Carlson L. A. 1963. Studies on the effect of nicotinic acid on catecholamine stimulated lipolysis in adipose tissue in vitro. Acta Med. Scand. 173: 719–722. [DOI] [PubMed] [Google Scholar]
- 11.Lauring B., Taggart A. K. P., Tata J. R., Dunbar R., Caro L., Cheng K., J. Chin, S. L. Colletti, J. Cote, S. Khalilieh, et al. 2012. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci. Transl. Med. 4: 148ra115. [DOI] [PubMed] [Google Scholar]
- 12.Ganji S. H., Tavintharan S., Zhu D., Xing Y., Kamanna V. S., and Kashyap M. L.. 2004. Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J. Lipid Res. 45: 1835–1845. [DOI] [PubMed] [Google Scholar]
- 13.Hu M., Chu W. C. W., Yamashita S., Yeung D. K., Shi L., Wang D., D. Masuda, Y. Yang, and B. Tomlinson. 2012. Liver fat reduction with niacin is influenced by DGAT-2 polymorphisms in hypertriglyceridemic patients. J. Lipid Res. 53: 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaren D. G., Han S., Murphy B. A., Wilsie L., Stout S. J., Zhou H., T. P. Roddy, J. N. Gorski, D. E. Metzger, M. K. Shin, et al. 2018. DGAT2 inhibition alters aspects of triglyceride metabolism in rodents but not in non-human primates. Cell Metab. 27: 1236–1248.e6. [DOI] [PubMed] [Google Scholar]
- 15.Berge K. G., and Canner P. L.. 1991. Coronary drug project: experience with niacin. Coronary Drug Project Research Group. Eur. J. Clin. Pharmacol. 40(Suppl 1): S49–S51. [PubMed] [Google Scholar]
- 16.Blankenhorn D. H., Johnson R. L., Nessim S. A., Azen S. P., Sanmarco M. E., and Selzer R. H.. 1987. The Cholesterol Lowering Atherosclerosis Study (CLAS): design, methods, and baseline results. Control. Clin. Trials. 8: 356–387. [DOI] [PubMed] [Google Scholar]
- 17.Brown G., Albers J. J., Fisher L. D., Schaefer S. M., Lin J. T., Kaplan C., X. Q. Zhao, B. D. Bisson, V. F. Fitzpatrick, and H. T. Dodge. 1990. Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N. Engl. J. Med. 323: 1289–1298. [DOI] [PubMed] [Google Scholar]
- 18.Brown B. G., Zhao X. Q., Chait A., Fisher L. D., Cheung M. C., Morse J. S., A. A. Dowdy, E. K. Marino, E. L. Bolson, P. Alaupovic, et al. 2001. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N. Engl. J. Med. 345: 1583–1592. [DOI] [PubMed] [Google Scholar]
- 19.Taylor A. J., Sullenberger L. E., Lee H. J., Lee J. K., and Grace K. A.. 2004. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 110: 3512–3517. [DOI] [PubMed] [Google Scholar]
- 20.Guyton J. R., Brown B. G., Fazio S., Polis A., Tomassini J. E., and Tershakovec A. M.. 2008. Lipid-altering efficacy and safety of ezetimibe/simvastatin coadministered with extended-release niacin in patients with type IIa or type IIb hyperlipidemia. J. Am. Coll. Cardiol. 51: 1564–1572. [DOI] [PubMed] [Google Scholar]
- 21.Sang Z. C., Wang F., Zhou Q., Li Y. H., Li Y. G., Wang H. P., and S. Y. Chen. 2009. Combined use of extended-release niacin and atorvastatin: safety and effects on lipid modification. Chin. Med. J. (Engl.). 122: 1615–1620. [PubMed] [Google Scholar]
- 22.Villines T. C., Stanek E. J., Devine P. J., Turco M., Miller M., Weissman N. J., L. Griffen, and A. J. Taylor. 2010. The ARBITER 6-HALTS Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6-HDL and LDL Treatment Strategies in Atherosclerosis): final results and the impact of medication adherence, dose, and treatment duration. J. Am. Coll. Cardiol. 55: 2721–2726. [DOI] [PubMed] [Google Scholar]
- 23.AIM-HIGH Investigators, W. E. Boden, J. L. Probstfield, T. Anderson, B. R. Chaitman, P. Desvignes-Nickens P, K. Koprowicz, R. McBride, K. Teo, and W. Weintraub. 2011. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 365: 2255–2267. [DOI] [PubMed] [Google Scholar]
- 24.HPS2-THRIVE Collaborative Group, M. J. Landray, R. Haynes, J. C. Hopewell, S. Parish, T. Aung, J. Tomson, K. Wallendszus, M. Craig, L. Jiang, et al. 2014. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 371: 203–212. [DOI] [PubMed] [Google Scholar]
- 25.Besseling J., van Capelleveen J., Kastelein J. J. P., and Hovingh G. K.. 2013. LDL cholesterol goals in high-risk patients: how low do we go and how do we get there? Drugs. 73: 293–301. [DOI] [PubMed] [Google Scholar]
- 26.Creider J. C., Hegele R. A., and Joy T. R.. 2012. Niacin: another look at an underutilized lipid-lowering medication. Nat. Rev. Endocrinol. 8: 517–528. [DOI] [PubMed] [Google Scholar]
- 27.Shimoda K., Akiba T., Matsushima T., Rai T., Abe K., and Hoshino M.. 1998. [Niceritrol decreases serum phosphate levels in chronic hemodialysis patients] Nippon Jinzo Gakkai Shi. 40: 1–7. Japanese. [PubMed] [Google Scholar]
- 28.Ganji S. H., Qin S., Zhang L., Kamanna V. S., and Kashyap M. L.. 2009. Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis. 202: 68–75. [DOI] [PubMed] [Google Scholar]
- 29.Hamoud S., Kaplan M., Meilin E., Hassan A., Torgovicky R., Cohen R., and T. Hayek. 2013. Niacin administration significantly reduces oxidative stress in patients with hypercholesterolemia and low levels of high-density lipoprotein cholesterol. Am. J. Med. Sci. 345: 195–199. [DOI] [PubMed] [Google Scholar]
- 30.Giri S. N. 2003. The combined treatment with taurine and niacin blocks the bleomycin-induced activation of nuclear factor-kappaB and lung fibrosis. Adv. Exp. Med. Biol. 526: 381–394. [DOI] [PubMed] [Google Scholar]
- 31.Westphal S., Borucki K., Taneva E., Makarova R., and Luley C.. 2006. Adipokines and treatment with niacin. Metabolism. 55: 1283–1285. [DOI] [PubMed] [Google Scholar]
- 32.Si Y., Zhang Y., Zhao J., Guo S., Zhai L., Yao S., H. Sang, N. Yang, G. Song, J. Gu, et al. 2014. Niacin inhibits vascular inflammation via downregulating nuclear transcription factor-κB signaling pathway. Mediators Inflammation 2014: 263786. [DOI] [PMC free article] [PubMed]
- 33.Grundy S. M., Mok H. Y., Zech L., and Berman M.. 1981. Influence of nicotinic acid on metabolism of cholesterol and triglycerides in man. J. Lipid Res. 22: 24–36. [PubMed] [Google Scholar]
- 34.Kurella M., Lo J. C., and Chertow G. M.. 2005. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. J. Am. Soc. Nephrol. 16: 2134–2140. [DOI] [PubMed] [Google Scholar]
- 35.Warburg O., and Christian W.. 1936. Pyridine, the hydrogen transfusing component of fermentative enzymes. Helv. Chim. Acta. 19: 79–88. [Google Scholar]
- 36.Imai S., Armstrong C. M., Kaeberlein M., and Guarente L.. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 403: 795–800. [DOI] [PubMed] [Google Scholar]
- 37.Rodgers J. T., Lerin C., Haas W., Gygi S. P., Spiegelman B. M., and Puigserver P.. 2005. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 434: 113–118. [DOI] [PubMed] [Google Scholar]
- 38.Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., P. J. Elliott, P. Puigserver, and J. Auwerx. 2009. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 458: 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim H-S., Xiao C., Wang R-H., Lahusen T., Xu X., Vassilopoulos A., G. Vazquez-Ortiz, W. I. Jeong, O. Park, S. H. Ki, et al. 2010. Hepatic specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 12: 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ponugoti B., Kim D. H., Xiao Z., Smith Z., Miao J., Zang M., S. Y. Wu, C. M. Chiang, T. D. Veenstra, and J. K. Kemper. 2010. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 285: 33959–33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker A. K., Yang F., Jiang K., Ji J. Y., Watts J. L., Purushotham A., O. Boss, M. L. Hirsch, S. Ribich, J. J. Smith, et al. 2010. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 24: 1403–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feige J. N., Lagouge M., Canto C., Strehle A., Houten S. M., Milne J. C., P. D. Lambert, C. Mataki, P. J. Elliott, and J. Auwerx. 2008. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 8: 347–358. [DOI] [PubMed] [Google Scholar]
- 43.Hirschey M. D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D. B., C. A. Grueter, C. Harris, S. Biddinger, O. R. Ilkayeva, et al. 2010. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 464: 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lagouge M., Argmann C., Gerhart-Hines Z., Meziane H., Lerin C., Daussin F., N. Messadeq, J. Milne, P. Lambert, P. Elliott, et al. 2006. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 127: 1109–1122. [DOI] [PubMed] [Google Scholar]
- 45.Li X., Zhang S., Blander G., Tse J. G., Krieger M., and Guarente L.. 2007. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell. 28: 91–106. [DOI] [PubMed] [Google Scholar]
- 46.Purushotham A., Schug T. T., Xu Q., Surapureddi S., Guo X., and Li X.. 2009. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9: 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang R-H., Li C., and Deng C-X.. 2010. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int. J. Biol. Sci. 6: 682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfluger P. T., Herranz D., Velasco-Miguel S., Serrano M., and Tschöp M. H.. 2008. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA. 105: 9793–9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eberlé D., Hegarty B., Bossard P., Ferré P., and Foufelle F.. 2004. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 86: 839–848. [DOI] [PubMed] [Google Scholar]
- 50.Shah S. A., Yoon G. H., Chung S. S., Abid M. N., Kim T. H., Lee H. Y., and M. O. Kim. 2017. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry. 22: 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caton P. W., Kieswich J., Yaqoob M. M., Holness M. J., and Sugden M. C.. 2011. Metformin opposes impaired AMPK and SIRT1 function and deleterious changes in core clock protein expression in white adipose tissue of genetically-obese db/db mice. Diabetes Obes. Metab. 13: 1097–1104. [DOI] [PubMed] [Google Scholar]
- 52.Mercader J., Granados N., Caimari A., Oliver P., Bonet M. L., and Palou A.. 2008. Retinol-binding protein 4 and nicotinamide phosphoribosyltransferase/visfatin in rat obesity models. Horm. Metab. Res. 40: 467–472. [DOI] [PubMed] [Google Scholar]
- 53.Yoshino J., Mills K. F., Yoon M. J., and Imai S.. 2011. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14: 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stromsdorfer K. L., Yamaguchi S., Yoon M. J., Moseley A. C., Franczyk M. P., Kelly S. C., N. Qi, S. Imai, and J. Yoshino. 2016. NAMPT-mediated NAD(+) biosynthesis in adipocytes regulates adipose tissue function and multi-organ insulin sensitivity in mice. Cell Reports. 16: 1851–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dahl T., Ranheim T., Holm S., Berge R., Aukrust P., and Halvorsen B.. 2011. Nicotinamide phosphoribosyltransferase and lipid accumulation in macrophages. Eur. J. Clin. Invest. 41: 1098–1104. [DOI] [PubMed] [Google Scholar]
- 56.Wang L-F., Wang X-N., Huang C-C., Hu L., Xiao Y-F., Guan X-H., Y. S. Qian, K. Y. Deng, and H. B. Xin. 2017. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKα/SREBP1 signaling pathway. Lipids Health Dis. 16: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gariani K., Ryu D., Menzies K. J., Yi H. S., Stein S., Zhang H., A. Perino, V. Lemos, E. Katsyuba, P. Jha, et al. 2017. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 66: 132–141. [DOI] [PubMed] [Google Scholar]
- 58.Gariani K., Menzies K. J., Ryu D., Wegner C. J., Wang X., Ropelle E. R., N. Moullan, H. Zhang, A. Perino, V. Lemos, et al. 2016. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology. 63: 1190–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Katsyuba E., Mottis A., Zietak M., Franco F. D., van der Velpen V., Gariani K., D. Ryu, L. Cialabrini, O. Matilainen, P. Liscio, et al. 2018. De novo NAD + synthesis enhances mitochondrial function and improves health. Nature. 563: 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trammell S. A., Weidemann B. J., Chadda A., Yorek M. S., Holmes A., Coppey L. J., A. Obrosov, R. H. Kardon, M. A. Yorek, and C. Brenner. 2016. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci. Rep. 6: 26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mukhopadhyay P., Horvath B., Rajesh M., Varga Z. V., Gariani K., Ryu D., Z. Cao, E. Holovac, O. Park, Z. Zhou, et al. 2017. PARP inhibition protects against alcoholic and non-alcoholic steatohepatitis. J. Hepatol. 66: 589–600. [DOI] [PubMed] [Google Scholar]
- 62.Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., and Sinclair D. A.. 2003. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 423: 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cantó C., Houtkooper R. H., Pirinen E., Youn D. Y., Oosterveer M. H., Cen Y., P. J. Fernandez-Marcos, H. Yamamoto, P. A. Andreux, P. Cettour-Rose, et al. 2012. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 15: 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kraus D., Yang Q., Kong D., Banks A. S., Zhang L., Rodgers J. T., E. Pirinen, T. C. Pulinilkunnil, F. Gong, Y. C. Wang, et al. 2014. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature. 508: 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tran M. T., Zsengeller Z. K., Berg A. H., Khankin E. V., Bhasin M. K., Kim W., C. B. Clish, I. E. Stillman, S. A. Karumanchi, E. P. Rhee, et al. 2016. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 531: 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]