

Summary
The study of the immune system has shifted from a purely dichotomous separation between the innate and adaptive arms to one that is now highly complex and reshaping our ideas of how steady‐state health is assured. It is now clear that immune cells do not neatly fit into these two streams and immune homeostasis depends on continual dialogue between multiple lineages of the innate (including dendritic cells, innate lymphoid cells, and unconventional lymphocytes) and adaptive (T and B lymphocytes) arms together with a finely tuned synergy between the host and microbes which is essential to ensure immune homeostasis. Innate lymphoid cells are critical players in this new landscape. Here, we discuss recent studies that have elucidated in detail the development of ILCs from their earliest progenitors and examine factors that influence their identification and ability to drive immune homeostasis and long‐term immune protection.
Keywords: homeostasis, innate immunity, innate lymphoid cells, mucosal immunity, protection
1. INTRODUCTION
The world of innate immune cells has greatly expanded in recent years. Broadly it includes innate lymphoid cells (ILCs) together with an array of unconventional lymphocytes such as γδ T cells, CD1‐restricted NKT cells, and mucosal‐associated invariant T (MAIT) cells. ILCs are distinct from other newly described innate cells as they lack recombined antigen‐specific receptors characteristic of B and T lymphocytes and many of the phenotypic lineage markers that define other immune cell subsets. Indeed, ILCs are enriched when the two genes Rag1 and Rag2 that regulate recombination machinery, selection, and diversity in other lymphocytes, are deleted.1 Nevertheless, ILCs exhibit a number of features that are reminiscent of T cells implying that they may be the innate counterparts of adaptive lineages.2 ILCs have generally been regarded to be an almost exclusively tissue‐resident population found at the barrier surfaces such as the skin, lungs, and intestinal tract.3 New evidence now suggests that colonization of tissues, replenishment, and rapid dissemination of ILCs depends at least partly on the capacity of these cells to move around the body in response to pro‐inflammatory signals allowing them to fight infection and maintain immune homeostasis. Here, we discuss the specific transcriptional pathways that are essential to regulate the generation and maintenance of ILCs. We focus on how recent findings are reshaping our understanding of the complexity of homeostatic regulation at barrier surfaces forcing us to rebuild the rules by which we understand how the immune system operates.
2. INNATE LYMPHOID CELL SUBSETS
Innate lymphoid cells are a heterogeneous family of immune cells that have shed new light on the architecture of the immune response and our understanding of how immune protection is orchestrated. ILCs express germline‐encoded receptors that enables them to respond rapidly to stimuli. In many cases, precisely how these receptors work has been unclear as little is known about the ligands activating the receptors. Recent evidence, however, suggests that NKp46 can recognize the cognate ligand complement factor P,4 and NKp44 can recognize platelet‐derived growth factor (PDGF)‐DD produced by tumors,5 highlighting additional crucial roles in recognizing soluble tissue components, in addition to recognition of pathogen‐derived ligands6, 7, 8, 9 to protect against infections and to mediate tissue repair. This feature allows them to deliver front line defense against the continual assault on the body from both foreign and commensal organisms as well as antigens derived from food and environmental sources.
Although we have only recently been readily able to dissect the diversity of ILC populations due to their rarity, NK cells, and lymphoid tissue‐inducer (LTi) cells were discovered more than 30 years ago. This established their prototypical roles in tumor immunosurveillance (NK cells)5, 10 and in the formation of secondary lymphoid tissues (LTi cells)11, 12 during embryogenesis, respectively. Our understanding of this family has now greatly expanded with the discovery of new previously unrecognized members that have been classified into three main subsets: ILC1, ILC2, and ILC3s.13 These groupings are largely aligned with effector T cells and are based on their expression of transcription factors and cytokine profiles.
ILC1s predominantly produce IFN‐γ following stimulation. They are defined by the surface receptors NK1.1 and NKp46 (CD335) together with their lack of lineage specific markers (including CD3, CD4, CD8, CD19, CD11c, and transcription factor RORγt). This reveals a heterogeneous population that can be further separated into NK cells (which express CD49b, also known as DX5) and non‐NK ILC1s (which express CD49a or VLA‐1α). Both NK cells and ILC1 express the transcription factor T‐BET (encoded by Tbx21), but generally only NK cells express EOMES (encoded by Eomesodermin, also referred as T‐box brain protein 2). These factors are associated with IFN‐γ production and anti‐tumoral activities. NK cells and ILC1 also differ in their lifecycle as NK cells seem to continuously recirculate around the body while non‐NK ILC1s appear to reside mostly in tissues such as the liver. In addition, it is likely that the specific tissues inhabited by ILC1 significantly influence their phenotype and function. For example, it has been shown that salivary gland ILC1 are phenotypically distinct from liver ILC1 or from intraepithelial ILC1.14
ILC2s produce interleukin(IL)‐5, IL‐9 and IL‐13 together with tissue repair factors such as amphiregulin. They are defined by their expression of the surface markers ICOS (Inducible T cell costimulator), KLRG1 (Killer cell lectin‐like receptor subfamily G member 1), Sca1, ST2 (IL‐33R), CD25 (IL‐2Rα), and IL‐7R together with the transcription factors GATA3 (GATA binding protein 3) and nuclear receptor RORα (RAR‐related orphan receptor α).15 Some variability in the expression of ST2, KLRG1, and CD25 has been observed depending on the tissue location and stimulus,16 while in most tissues, ICOS is reliably expressed and indeed required for their survival and cytokine production. ILC2 are mainly involved in responses to allergic stimuli and parasites and are thus found at several sites throughout the body including the lungs, spleen, gut, liver, and skin.3, 17
ILC3 are characterized by the expression of RORγt and production of IL‐22 and/or IL‐17. They are found enriched in mucosal tissues such as the intestine and are demarcated into three distinct subpopulations by their expression of CD4 and the NK receptor, NKp46 (encoded by Ncr1, natural cytotoxicity triggering receptor 1). LTi cells, which orchestrate the generation of lymphoid tissues during fetal development, express the coreceptor CD4 together with the chemokine receptor CCR6, but lack NKp46 expression.11, 12 Two additional populations of ILC3 are defined by their lack of CD4 expression combined with their expression, or lack of, NKp46. Although the marker CCR6 has been used to divide ILC populations into “helper” and “cytotoxic” ILC populations,18 it does not always appear to show clearly definable subsets within the ILC3 population. NKp46+CCR6− ILC3 correspond to the effector population that depends on the upregulation of T‐BET for their formation.19, 20 CCR6+ ILC3 have been shown to express Major Histocompatibility Class II expression and exhibit some antigen processing capacity. This feature allows them to limit the expansion of commensal bacteria‐responsive CD4+ T cells through activation induced cell death thus preventing subsequent intestinal disease and dysregulation of the microbiome.21
2.1. Development of early innate lymphoid progenitors in bone marrow
ILCs are thought to arise from all‐lymphoid progenitors (ALPs) which contains the common lymphoid progenitor (CLP) and the IL‐7Rα+ multipotent ILC progenitors.18, 22, 23, 24, 25 The major progenitor potential lies within the α4β7 fraction of the CLP.26 Although all ILCs derive from an IL‐7Rα+ progenitor, an additional stage, termed the early innate lymphoid progenitor (EILP) has recently been defined and notably is marked by the expression of the transcription factor T cell factor‐1 (TCF‐1, encoded by the gene Tcf7).27 Tcf7 + progenitors expressed only low levels of IL‐7Rα, Zbtb16 (also known as Plzf), and Id2 (Inhibitor of DNA binding 2).27 What was distinct about this cell type was that it did not fit with the known linear progression of ILC differentiation that had been previously described. Distinct from other members of the progenitor network, the EILP did not express IL‐7Rα. This was perplexing but such a step in ILC differentiation could occur if EILPs did not arise from the ALP; or alternately, ILC progenitors could transition through a stage that depended on the downregulation and subsequent re‐expression of IL‐7Rα as normally occurs in developing thymocytes (Figure 1).28 Thus, the EILP would represent an intermediate developmental stage in which IL‐7Rα is transiently downregulated. Indeed, when the IL7rCre strain was crossed to a ROSA26‐YFP reporter strain and the Tcf7 EGFP reporter, the temporal expression of Tcf7 and IL‐7R amongst IL‐7R+ an IL‐7R− cells could be ascertained.29 Indeed, it was then clear that the IL‐7R− population carried the imprint of previous IL‐7R expression and that the EILP defines a critical step in ILC generation. Importantly, this work defined the link between the very early progenitor stages of the ALP and ILCP (ILC progenitor), and the EILP, and crucially pinpointed the requirement for differential regulation of receptor expression for this transition that may well have been normally overlooked (Figure 1).29 IL‐7R expression is therefore highly dynamic and tightly regulated by TCF‐130 resulting in early expression in development, but subsequently downregulated to allow the EILP to give rise to ILCP.
Figure 1.
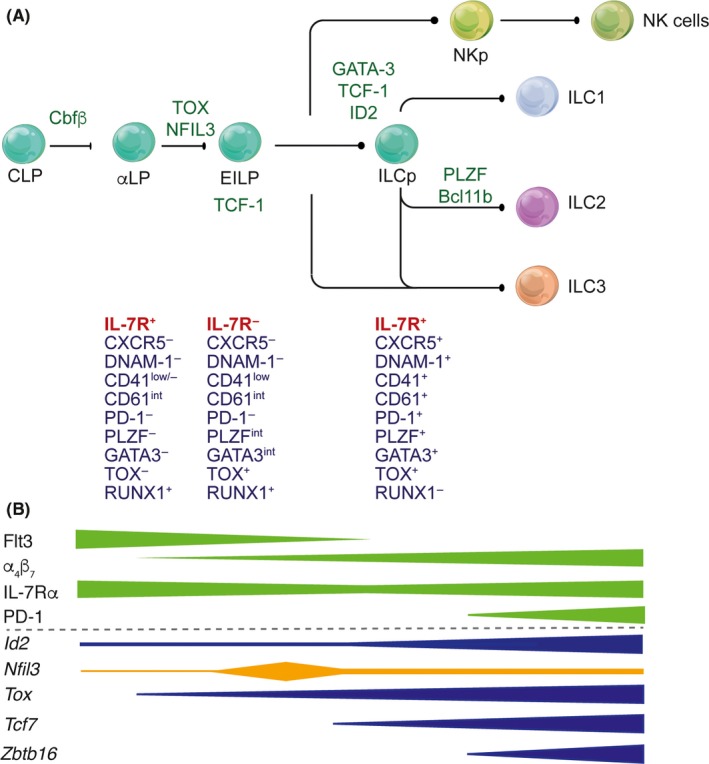
(A) Transcriptional regulation of ILC development from the common lymphoid progenitor (CLP) to mature ILC subsets 1, 2, and 3. It is now clear that the CLP transits through a series of intermediates including the early innate lymphoid progenitor (EILP) which in contrast with stages both preceding and following the EILP, downregulate the expression of IL‐7R. (B) Differential regulation of transcription factors and surface receptors is both dynamic and essential for diversification of ILC subsets
2.2. The thymic pathway
Although ILCs in the adult typically originate from the bone marrow, emerging data points to an additional network that regulates thymic progenitors that are normally destined to establish T cell identity to adopt an innate fate. This possibility challenges the current paradigm but is plausible as ILC express many transcription factors that are characteristic of the T cell lineage including TCF‐1, GATA‐3, and Bcl11b together and signaling molecules ICOS, PD‐1, LTA, LCK, and ITK.26, 30, 31, 32, 33, 34 This shared transcriptional network suggests that the fate decisions of T cells and ILCs are inextricably linked. Providing substance to the possibility of a thymic ILC origin, Miyazaki et al35 showed that the E proteins E2A and HEB, which interact with DNA binding proteins (Id), suppress the induction of the ILC gene program by promoting T cell specific genes such as Notch receptors. This effect is executed by interfering with the level of ID2 expression, a transcription factor necessary for the development of all ILC and the maintenance of several mature subsets.35, 36 Overexpression of ID3, which can function similarly to ID2, promotes the generation of NK cells from thymocytes,37 while overexpression of ID1 in transgenic mice enhances the development of ILC2 in multiple organs, most notably in the thymus.38, 39 ID1 itself is not generally found in immune cells but ectopic expression of Id proteins, or the removal of their E protein binding partners, serves to reciprocally enhance their expression and drive ILC development. The thymus is not essential for the formation of ILC2 but they can be generated from thymic progenitors when they are cultured with IL‐7 and IL‐3339, 40 suggesting that the balance of innate and adaptive immune cell fate outcomes depends on the combination of transcription factors together with external stimuli encountered by cells. This effect has also been shown in vivo.35, 41 Similarly, deletion of transcription factors that normally define T cell identify, such as BCL11b, can have the capacity to derepress the dominant T cell developmental pathway in the thymus resulting in NK cells development.42 In addition, however, BCL11b can fine tune the balance between ILC2 and ILC3 in the periphery to act as a sensitive rheostat for ILC subset development in response to stimuli.43, 44 Within the thymus, maintaining the delicate balance of ILC subsets appears to be crucial for the integrity of the thymus and the emergency generation of ILC. Indeed, while intrathymic ILC3 play a critical role in thymic regeneration where they produce abundant IL‐22 to drive thymic repair following graft‐versus‐host disease,45, 46 ILC2 have recently been shown to be the dominant ILC population within the thymus after birth.47 The accumulation of ILC2 in the thymus over time raises the possibility that type 2 cytokines produced by these cells play a role in supporting normal thymic function. However, the exact origin of both of these subsets is yet to be explored.48, 49
Collectively, these findings challenge the notion that the bone marrow is the only source of ILCs in the adult and instead raises the idea that an evolutionary mechanism has arisen providing multiple pathways to generate ILCs to protect the body against insults.
2.3. Key drivers of ILC subset differentiation
A core group of transcription factors are essential for the early development of ILCs. These include Inhibitor of DNA binding 2 (ID2, Idb2), T cell factor 1 (TCF‐1, encoded by Tcf7), Nuclear factor interleukin‐3 (NFIL3, E4 bp4), Thymocyte selection associated high mobility group box (TOX, Tox), and GATA binding protein 3 (GATA3, Gata3). These factors collaborate to orchestrate the sequential restriction of progenitors into individual ILC lineages and have been described in detail elsewhere.50, 51, 52, 53, 54
2.3.1. NK cells
Several NK cell progenitors have been identified to give rise to different peripheral NK cell subsets. NK cells are guided through progressive developmental stages by the expression of specific transcription factors necessary for NK lineage commitment and maturation. The pre‐pro NK cell precursor population is the earliest identified committed NK cell progenitor that shows a highly enriched capacity to generate NK cells.55 These cells express the IL‐2Rβ chain (CD122), Sca‐1, IL‐7Rα, and ID2 but lacked markers typically expressed on fully differentiated mature NK cells such as NK1.1, NKp46, and CD49b.55 In the bone marrow, this progenitor further committed into NK progenitor (NKP)55, 56 and subsequently into immature and mature NK cells under the influence of a core transcription program including but not restricted to Id2,55 Gata3,57 Nfil3,58 Klf2,59 Eomes,60 Tbx21,61 Tcf7,27 Tox, 62 and Ets‐1 (Figure 2).63 Interestingly, NFIL3 expression is essential for the development of all ILC subsets64 but appears to be only required early and transiently to implement the ILC program as Nfil3 deletion in ID2+ mature NK cells does not affect NK cell survival or function.26, 50, 65
Figure 2.
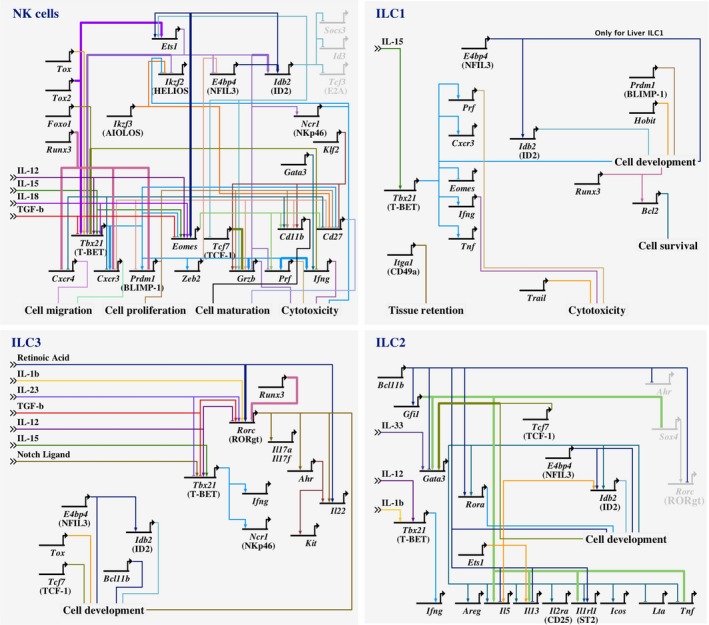
Gene regulatory network illustrating the involvement of key transcription factors in NK cells, ILC1, ILC2, and ILC3 development using BioTapestry software (Version 7.1.1m, biotapestry.org). Critical extracellular signals influencing ILC functions and mediating ILC plasticity are depicted. Networks were designed based on ChIP data or reporter assays61, 79, 91, 92, 102, 176, 177, 178, 179, 180 (thick lines) together with gene deletion or overexpression systems (regular lines).18, 19, 40, 43, 44, 57, 58, 59, 60, 61, 64, 72, 74, 75, 78, 79, 85, 86, 87, 89, 90, 91, 92, 93, 95, 98, 110, 118, 128, 169, 174, 176, 177, 178, 179, 180, 182, 183, 184, 185, 186, 187 Nonexpressed genes are depicted in gray. Linkages are color‐coded for clarity only. The individual genes are shown in schematic form only. The lines indicate the direct binding of the protein encoded by the indicated gene to the regulatory regions of the linked target genes, which leads to transcriptional activation or repression. Runx3, runt related transcription factor 3; Ets1, ETS proto‐oncogene 1; Ikzf2 (HELIOS), IKAROS family zinc finger 2; Ikzf3 (AILOS), IKAROS family zinc finger 3; Nfil3, nuclear factor, interleukin 3 regulated; Idb2, inhibitor of DNA binding 2; Socs3, suppressor of cytokine signaling 3; Id3, inhibitor of DNA binding 3; Tcf3 (E2A), transcription factor 3; Prf1, Perforin 1; Gzmb, granzyme B; Tcf7, transcription factor 7; Zeb2, zinc finger E‐box‐binding homeobox 2; Prdm1 (BLIMP1), PR domain zinc finger protein 1; Foxo1, forkhead box protein O1; Tox, thymocyte selection associated high mobility group box; Gata3, GATA binding protein 3; Itga1, integrin subunit alpha 1; Ifng, interferon‐gamma; Areg, amphiregulin; Il2ra, interleukin‐2 receptor subunit alpha; Icos, inducible T cell costimulator; Lta, lymphotoxin alpha; Tnf, tumor necrosis factor; Sox4, SRY‐Box 4; Rorc, RAR related orphan receptor C; Rora, RAR related orphan receptor A; Gfi1, growth factor independent 1; Bcl11b, B cell CLL/lymphoma 11B; Ahr, aryl hydrocarbon receptor. CD, cluster of differentiation
Immature and mature NK cells are present in peripheral tissues and blood and are characterized by the expression of NK1.1 and NKp46. CD11b and CD27 expression subdivides NK cells into immature (Imm, CD11b−/lowCD27+), mature 1 (M1, CD11b+CD27+) and mature 2 (M2, CD11b+CD27−) subsets.66 These mature subsets may express other surface molecules, such as KLRG1,67 CD62L,68 and DNAM‐169 that further characterize their specific phenotype and functions. In parallel with the progressive change in surface molecule expression during maturation, NK cell function is also affected. Most strikingly, mature NK cells become less proliferative and produce less cytokine but conversely, they gain cytotoxic function as they further mature from M1 into M2 populations.70
2.3.2. ILC1
ILC1 originate from the innate lymphoid cell progenitor (ILCP) which also gives rise to other subsets of ILCs such as ILC2 and some ILC3.71 ILC1 express NK1.1 and NKp46 but are distinct from NK cells. They generally lack CD49b or EOMES expression but depend strongly on T‐bet expression72 in contrast with splenic NK cells which only partly rely on this factor.18, 73, 74 In addition, while BLIMP1 is required to fully upregulate T‐BET expression in splenic NK cells, genetic deletion does not result in a marked defect in NK cell populations.75 Nevertheless, an unexpected synergy between BLIMP1 and HOBIT (homolog of BLIMP1 in T cells or ZNF683), a transcription factor normally controls tissue‐residency in CD8+ T cells, is essential for their development (Figure 2).12, 64, 65, 74, 76, 77, 78 ILC1 also preferentially express other surface molecules including IL‐7Rα, TRAIL (Tumor necrosis factor apoptosis‐inducing ligand), and CD49a (also known as integrin alpha‐1) and their survival is strongly regulated by the transcription factor RUNX3.79 TRAIL expression is dictated by signaling though the NKp46 receptor.80, 81, 82 In certain inflammatory conditions, NK cells and ILC3 are converted into ILC1‐like cells (sometimes referred to as ex‐NK cells and ex‐ILC3 respectively) and downregulate EOMES or RORγt expression in favor of T‐BET and TRAIL expression and exhibit enhanced IFN‐γ production.83, 84 ILC2 stimulated with IL‐1β potentiating IL‐12 responsiveness, or IL‐12 itself, also acts as a potent driver for this subset to acquire features of ILC1 cells.85, 86, 87, 88 Thus, in inflammatory settings each subset appears to be able to reprogram its capability to become IFN‐γ‐producing cells with potent effector functions.
2.3.3. ILC2
The development of ILC2 is guided by the core transcriptional regulators RORα,40 GATA3,22, 89 TCF‐1,90, 91 Gfi1,92 and Bcl11b (Figure 2).43, 93 ILC2 are typically characterized by their expression of ST2 (IL‐33R), ICOS, and GATA3. GATA3 is expressed by ILC subsets and their progenitors and is thus required for their development. ILC2 express GATA3 at high levels in mature cells while ILC3 depend on sustained GATA3 expression to maintain NKp46+ identity and IL‐22 production by repressing the ILC3 LTi cell program.94, 95 A number of different ILC2 subsets are now recognized, including (a) natural ILC2 (nILC2, ST2+Thy1highKLRG1intermediateIL‐17RBlow/−) and (b) inflammatory ILC2 (iILC2, ST2−Thy1lowKLRG1highIL‐17RB+) which are distinguished by their level of expression of the Killer Cell Lectin Like Receptor G1 (KLRG1).14 iILC2 are induced to undergo significant proliferation in response to IL‐25 while IL‐33 can drive some expansion of nILC214 and play an important role in the egress of ILC from the bone marrow through regulation of CXCR4.96 Programmed cell death program 1 (PD‐1, or CD279) signaling has recently been shown to negatively regulate KLRG1+ ILC2 limiting their capacity to inadvertently expand and induce pathology.97 Although ST2 has typically been used to identify ILC2, iILC2, in contrast with nILC2, do not express ST2 and thus this receptor cannot be universally used to identify ILC2 across different tissues or under different conditions of inflammation.
2.3.4. ILC3
RORγt is the cornerstone transcription factor identified as essential for the development of ILC398 and opened the door to the identification of the three major different subsets of ILC3 (Figure 2). The expression of the T cell co‐receptor CD4 distinguishes the prototypic CD4‐expressing ILC3, LTi cells, and CD4 negative populations. LTi act to orchestrate the generation of nascent lymphoid tissue initiated by the interaction between LTi cells and stromal organizer cells and that is dependent upon LTi‐expressed lymphotoxin.99, 100 CD4− ILC3 are distinguished by the expression of the natural cytotoxicity receptor (NCR), NKp46, resulting in NKp46+ and NKp46− ILC3 subsets. The transition between NKp46− and NKp46+ ILC3 depends on induction of T‐BET via NOTCH2 interactions19 and this could be regulated by the strength of the inflammatory stimulus in the environment.20 These two cornerstone studies opened the way to begin to tease apart the precise machinery that guides the development of ILC3, particularly NCR+ ILC3, revealing that a number of transcription factors are essential for the development of these cells. These include the aryl hydrocarbon receptor (AHR) which is regulated in ILC3 by RUNX379 and is sensitive to signals derived from the microbiota and dietary components that generate aryl hydrocarbons.101, 102, 103, 104
2.4. The complexity of the NK cell and ILC1 subsets and consequences of disruption of NKp46 signaling pathway
Our ability to ascertain the functions of NK cells has relied heavily on their identification as NK1.1+ cells. NK1.1+ cells have been attributed the important defense mechanism of immunesurveillance protecting from the emergence of cancer.105, 106, 107 With the discovery of ILC subsets and tracking of cells using the NKp46 receptor in combination with transcriptional regulators, it has become clear that the classical NK cell compartment contained not one, but two subsets of cells—ILC1 and NK cells. Thus, it could no longer be assumed that all the functions credited to NK cells were in fact due to NK cells alone but instead may reflect the outcome of a mixed population of cells that also contained ILC1. Further complicating the interpretation of the data attributable to individual subsets was the emergence of plasticity between a number of ILC subsets implying that alternate flexible programs that did not neatly fit into the subset classification could be identified. In an unexpected twist, recently three groups identified a point mutation in the Ncr1 gene in the congenic CD45.1+ mice used for many studies of lymphocyte tracking and function analysis (Figure 3A, Table 1).80, 81, 82 These lines, all derived from Jackson Laboratories, exhibited a single amino acid mutation from cysteine to arginine at amino acid 14 (C14R) which is localized in the region of the signal peptide of Ncr1. This mutation did not alter the overall expression of Ncr1 mRNA but significantly impaired the surface expression of NKp46 through failed trafficking within the cell.80, 81, 82 These findings have major implications for previous studies of NK cells and ILC1 where these mice have been used, even further complicating our interpretation of earlier data (Table 1).
Figure 3.
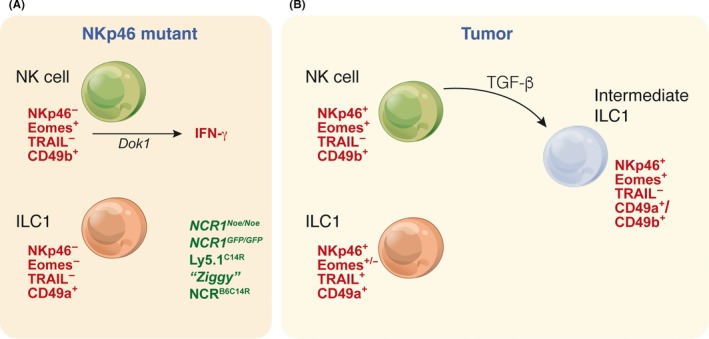
NK cells and ILC1 exhibit transitional phenotypes and functional alterations in response to tumors. (A) Multiple models have now been identified that highlight that deleted or unstable NKp46 results in ablation of TRAIL expression and impaired anti‐tumor activity. (B) Distinct NK cell and ILC1 subsets exist at steady‐state but under the influence of TGF‐β, tumor ILC1 acquire the expression of EOMES and surface markers such as CD49b normally expressed by NK cells
Table 1.
NKp46 regulation of ILC function
| Genetic modification | Phenotype | References | |
|---|---|---|---|
| Mutation | |||
| Ly5.1C14R (C14R) |
|
|
Jang et al82 |
| ‘Ziggy’ (C14R) |
|
Turchinovich et al80 | |
| Ly5.1C14R (C14R) (+ >300 additional genes) |
|
Almeida et al81 | |
| NCRB6C14R (C14R) |
|
Almeida et al81 | |
| Ncr1 Noé/Noé (W32R) |
|
|
Narni‐Mancinelli et al110 |
| Ncr1 Noé/Noé (W32R) |
|
|
Glasner et al111, 188 |
| Deletion | |||
| NCR gfp/gfp |
|
|
Gazit et al108 |
| NCR gfp/gfp | Wang et al112 | ||
| NCR gfp/gfp |
|
Glasner et al189 | |
| NCR1 iCre/iCre |
|
|
Narni‐Mancinelli et al109 |
The mutant mice, referred to as Ly5.1C14R, showed normal numbers of NK cells and ILC1 but a modest alteration to the immature and mature NK cell subsets.81 To date, several models affecting Ncr1 expression have been generated. These include the Ncr1 gfp/gfp , 108 Ncr1 iCre/iCre , 109 and Ncr1 Noé/Noé 110 mice. The Ncr1 gfp/gfp mice have a complete loss of NKp46 expression, while Ncr1 Noé/Noé mice exhibit a mutation at W32R110 which affects surface expression of NKp46.110, 111 A recent study also suggests that ILC1 development is intrinsically dependent on NKp46112 but this work contrasts with other studies that did not observe such effect.81, 113 Thus, the exact consequences of Ncr1 loss requires deeper investigation to determine precisely how it affects ILC1 and NK cell function. Nevertheless, the loss of stable expression of NKp46 on the surface of cells has been shown to be broadly important and crucial for protection from influenza virus82 and tumor control81 but paradoxically appears to confer higher resistance in MCMV infection.82 In some cases, such as influenza, the hemagglutinin and neurominidase viral proteins have been purported as endogenous ligands6, 108, 114 while B16F10 melanoma cells are known to be controlled by NK cells.106, 115 However, the resistance to MCMV infection identified in this study was a surprise and was correlated with enhanced IFN‐γ expression.82 Increased IFN‐γ production was attributed to the reduction in expression of the gene Dok1 which has previously been proposed to augment IFN‐γ.116 However, enhanced IFN‐γ expression was not observed in all studies of mice carrying the C14R mutation.81 Using exome sequencing, more than 300 genes were found to differ between Ly5.1C14R mice and their Ly5.2 counterparts.81 It is therefore likely that many associations between NKp46 expression and function will emerge and that the newly developed NCRB6C14R mouse, generated on an C57BL/6 background and carrying only the NCR mutation without disruption of other genes, will be an important tool in future studies.81 Despite this, the total loss of NKp46 expression, or unstable expression, was associated with the loss of TRAIL in both NK cells and ILC1.80, 81, 113 Similar to NKp46, TRAIL was transcribed but was unable to migrate to the cell surface in the absence of NKp46.113 This might occur if NKp46 and TRAIL comprise a single protein complex in cytoplasmic vesicles although the mechanism of release is not yet known. Collectively, these studies highlight the confounding nature of some earlier studies and the necessity to systematically ascertain the roles of NK cells and ILC1 in models where genes affecting function are not unknowingly disrupted.
2.5. ILC plasticity and the common default pathway
The broad subsets of ILCs have, through the development of elegant and novel tools and vigorous investigation, been relatively well‐elucidated. However, many questions remain around the programs that define each subpopulation, as well as the cellular and molecular triggers that allow so called “plasticity,” or the capacity to adopt a different phenotype. This attribute potentially enables the different ILC subsets unprecedented flexibility to respond to the encountered stimuli. Key transcription factors define the fundamental lineage fate program adopted by early progenitors. RORγt+ ILC3 were the first subset in which transformation into a cell type with characteristics mirroring those of ILC1 was identified, establishing this phenotype in response to pro‐inflammatory stimulation83. These cells, known as ex‐ILC3, carried the historical imprint of RORγt expression but become capable of producing IFN‐γ. Subsequently, it has been discovered that when ILC3 or ILC2 are exposed to a combination of stimuli, including IL‐1β, IL‐12, and IL‐33 for ILC2 (to generate ex‐ILC2) or IL‐12 in the case of ILC3, inflammatory pathways and T‐BET expression are activated, enabling the production of IFN‐γ.86, 87, 88 These stimuli reprogram both the phenotypic identity and function of ILC subsets such that they could now masquerade as ILC1‐like cells that potentially drive immune pathology. Transforming growth factor (TGF)‐β was shown to be a significant driver of the conversion of NK cells into intermediate NK‐ILC1 and ILC1 cells (Figure 3B).84, 117 TGF‐β accumulation often occurs in tumors in which it drives immunosuppression. Under TGF‐β stimulation, anti‐tumor NK cells are converted into intermediate NK‐ILC1 and ILC1 cells that are unable to control tumor growth and metastases formation.84, 117
Within ILC3 subsets, T‐BET also drives the development of NCR+ ILC319, 20 but this appears not to be a terminal end state for these cells as using fluorescent fate‐mapping (YFP), both YFP+ NCR+ and NCR− ILC3 have been detected suggesting that NCR+ ILC3 can revert to the NCR− phenotype.118 Similar to ILC1, TGF‐β is also a key driver of the formation of NCR+ ILC3.118 Why such an interconversion might exist is not yet completely clear but it could act as a brake on the formation of this highly reactive effector subset as an inflammatory stimulus subsides. This would limit immunopathology that may occur as a sequel to a prolonged or uncontrolled response.118 NCR− ILC3 are intriguing—they share many features with NCR+ ILC3 such as their capacity to constitutively produce IL‐22 but are even more closely related to LTi cells. Although NCR− and NCR+ ILC3 differ by several hundred genes, only a handful of genes are differentially expressed between LTi cells and NCR− ILC3 making them almost indistinguishable despite that LTi cells are derived from a fetal liver progenitors while NCR− ILC3 arise from a bone marrow progenitor.118, 119, 120 Similarly, NCR+ ILC3 have been mapped to express different levels of the transcription factor RORγt (dissecting high and intermediate expressing populations) and these populations differed by fewer than 100 genes. Again, these subpopulations appear to be phenotypically distinct, but why they would share such closely aligned gene signatures is not clear.118
3. IMMUNE HOMEOSTASIS AT MUCOSAL SURFACES: ILC NETWORKS IN THE GUT
The mucosa is colonized by the bulk of immune cells found in the body. These cells sense information from intestinal contents such as the trillions of microbes that inhabit the gut and food components. This landscape poses considerable challenges to maintain health. To that end, the immune system is charged with the task of balancing responses to maintain mucosal homeostasis. Fending off invading pathogens is clearly important, but maintaining immune homeostasis at these highly vulnerable surfaces is perhaps the single most important function that prevents succumbing to disease. In both the gut and the lung, the epithelium physically separates microbes from the immune cells but a constant dialogue between these compartments drives the integration of signals that guides homeostasis. For example, in addition to physical interactions between microbes and immune cells, it has been uncovered that metabolites generated by microbes provide essential signals to immune cells in the host‐microbiota homeostatic network.
3.1. Maintaining ILC at mucosal surfaces
If continuous protection is to be afforded by ILCs, then it is necessary for these cells to position themselves, and regenerate, at mucosal surfaces despite the pressures exerted by exposure to constant insults. The development and maintenance of this protective shield depends on two features. First, the provision of survival factors such as cytokines within the local tissue microenvironment. Second, the deposition of ILC at mucosal sites.
The cytokine IL‐7 is essential for the development of ILCs, particularly ILC3, which are severely reduced in IL‐7‐deficient mice.121 IL‐7 stimulation is the key to drive proliferation and survival of ILC3 but it also plays a role in preserving ILC2 and ILC3 numbers to maintain lymph node size.122 ILC3 also play a key role, distinct from the effects on stromal cell123 and dendritic cell124 number which also affect lymph node size, in gating transit of immune cells into lymph nodes via high endothelial venules. This contrasts markedly with ILC2 which have no apparent impact on this gating function.122 Although IL‐7 is integral to ILCs, IL‐7‐independent pathways that support ILC survival also exist. This includes IL‐15 which has well‐described roles in the development of NK cells125, 126 and ILC173 but can drive a separate program to at least partially support ILC2 and ILC3 in the intestine.127 Responsiveness, however, is modulated by sensitivity to IL‐15 which differs amongst the subsets in the gut. Intestinal NK1.1+ cells expand readily in response to IL‐15 but RORγt+ NKp46+ ILC3 are unaffected.121 In NK cells, sustained responsiveness to IL‐15 is maintained by the continuous expression of the transcription factor ID2.128 Whether ID2 plays a similar role in other ILC subsets in establishing differential responsiveness is not yet clear. In addition to IL‐7 and IL‐15, that regulate ILC maintenance at steady‐state, IL‐2 can also play a role in supporting ILC2, ILC3 and tuning NK cell sensitivity.129, 130 IL‐2 availability often depends on efficient local competition for supplies by ILC from regulatory CD4+ T cells, which strongly influence the outcome of local interactions.
It has become clear that cytokine maintenance of ILCs depends on complex interactions with specialized epithelial cells that are found in the gut and lungs. In the intestine, the number of ILC2 and ILC3 are delicately and reciprocally balanced. The gut epithelium is complex and composed of well‐known absorptive enterocytes, goblet cells, and Paneth cells found in the crypts, while other cells are rather less frequent and much more poorly characterized or understood. This includes the chemosensory epithelial Tuft (or “brush”) cells in the epithelial lining of the intestines. Their development is regulated by the transcription factor Pou domain, class 2, transcription factor 3, Pou2f3, and they play a central role in triggering the induction of type 2 immune responses following parasite infection. IL‐25 is essential to drive the amplification of ILC2131 but until recently, the exact cell type that produced this cytokine was unknown. Analyses of intestinal epithelial cells revealed that only a very small proportion of cells, the Tuft cells, produced IL‐25.132, 133, 134 Establishing this exclusivity was facilitated by generation of an IL‐25 reporter mouse line. This mouse also revealed that Tuft cells were not the source of other important epithelial cytokines such as IL‐33 and thymic stromal lymphopoietin (TSLP) that can also activate ILC2.35 Elucidation of this pathway is exciting and prompts us to ask whether other novel cell types found in the intestine, which as yet relatively poorly characterized, might also contribute to maintaining the ILC network and the elegant cooperation between epithelial and immune cells that drives homeostatic balance between ILC2 and ILC3.
3.2. Tissue residency and circulation
Except for NK cells which are mostly circulating,135, 136 ILCs have generally been thought to be largely restricted to the tissues in which they are found, having established their niche early in ontogeny.137, 138 This view of ILCs is predicated on several pieces of evidence including (a) ILCs are poorly replaced following transplantation,137, 139 (b) mature ILCs do not appear to exchange between mice in which the circulatory system is conjoined in models of parabiosis,137 and (c) few ILC that express a mature phenotype are found in the bone marrow.18, 71 In mice ILC replacement is extremely poor following exposure to lethal doses of γ‐irradiation,137 and in patients who display mutations in the common γ chain cytokine receptor subunit IL‐2Rγ, or the tyrosine kinase JAK3, tissue ILCs fail to be effectively reconstituted.139 In part, this has been attributed to the extremely slow turnover of ILCs.140 Taken together, these findings have implied that ILCs are fundamentally sessile and are not readily replaced by bone marrow‐derived cells. They are thus proposed to be replenished predominantly by local expansion within peripheral tissues in response to various stimuli.137 This model prevails if it is assumed that (a) bone marrow‐resident progenitors would generate mature ILC within the bone marrow itself, (b) circulating ILC exhibit the same phenotype, receptor and transcription factor expression as characterized for mature tissue‐resident ILC and (c) that homing receptor expression is static rather than dynamically regulated in response to the many cues to which ILCs are exposed. However, mounting evidence suggests that both local movements of ILCs can occur to “concentrate” them in an area (eg, ILC3 accumulate in perifollicular areas of Peyer's Patch and intestinal crypts141, 142), as well as more generalized movements required for effector cell distribution and immunosurveillance within the body (Figure 4). Thus, it remains to be determined whether ILCs actually exhibit life‐long tissue residency and fail to move in response to infection or insults.
Figure 4.
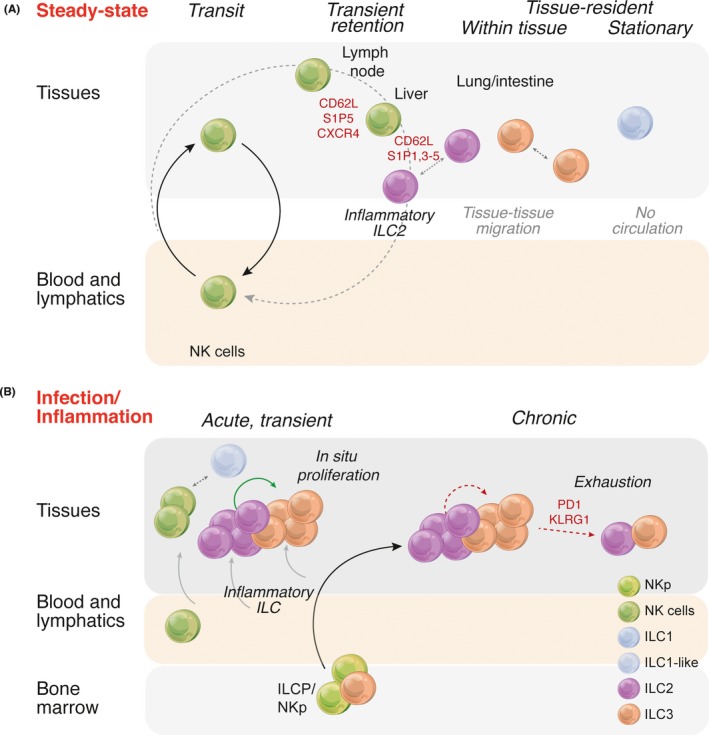
Regulation of ILCs at (A) steady‐state and during (B) infection and inflammation. (A) It is emerging that ILCs express a range of surface receptors that allow fine tuning of the positioning of different populations in tissues or between tissues dispelling the notion that ILCs are entirely sedentary within tissues. NK cells remain the most mobile with the capacity to move freely in the blood engaged in immunosurveillance, or to be recruited into tissues by modulating receptor expression. Only ILC1 appear to exhibit a truly tissue‐resident existence. (B) A proposed model for the replenishment of ILC in tissues. At steady‐state, slow proliferation of ILC within the tissues themselves allows the balance of subsets to be maintained. In an acute transient infection this may also be the case and that any temporary depletion would be rapidly replaced through enhanced local proliferation. If inflammation continues this might result in depletion that is not readily overcome by local proliferation and could lead to a state of “exhaustion” akin to that exhibited by T cells. Alternately, inflammation may drive differentiation of bone marrow progenitors and export in the blood to the affected tissues
ILCs are no doubt more radioresistant than a number of other immune cell lineages such T cells, B cells and most dendritic cells. This feature appears to be protective. In the gut, irradiation induces rapidly proliferating epithelial cells in the intestinal crypts to undergo apoptosis and then to be regenerated from Lgr5+ intestinal stem cells.143, 144 Radioresistant ILC3 protect the intestinal stem cell pool via their secretion of IL‐22.145 Complete loss of IL‐22 results in increased pathology in the gut, a severe loss of intestinal stem cells and significantly reduced survival.145 IL‐22 secreted by radioresistant ILC3 also drives thymic regeneration following irradiation which induces significant damage across thymocytes and thymic epithelial cells.45 Thus, radioresistance is an important attribute of the ILC family which has important implications in tissue regeneration and protection against graft‐versus‐host disease following bone marrow transplantation.
Recent studies indicate that circulating ILCs display their own unique molecular program. This appears to be distinct from ILC phenotypes that have been previously described or that might be predicted.146 Although CD117+ ILCP were shown by single cell sequencing to express transcripts for genes known to control ILC differentiation, they lacked a number of signature genes such as TBX21, GATA3, and RORC and cytokines including IL‐5, ‐13, ‐17, ‐22, and IFN‐γ found in mature cells. This study highlights that circulating ILC progenitor cells occur in the blood to enable seeding and establishment of ILCs in more distant tissues sites. Although it is purported that the major role of these cells is early in development, it provides a labile population responsive to triggering from inflammatory stimuli that could afford rapid repopulation of tissues. In the case of HIV, irreparable loss of ILCs, mainly ILC3, has been shown to occur but this arose from depletion of circulating ILC progenitors and under conditions of chronic stimulation, neither local or circulating ILC stores were capable of rebuilding the integrity of the tissues and mucosal protective barrier.147 Furthermore, other ILC subsets have been shown to express receptors that undoubtedly drive their entry into both the lymphatic and blood systems creating a pathway for their distribution to other tissue sites. For example, inflammatory ILC2 are sensitive to sphingosine 1‐phosphate(S1P)‐medicated chemotaxis during anti‐helminth immunity.148 Thus, evidence is beginning to emerge that suggests in some situations at least, ILCs can be mobilized and deployed to tissues to ensure mucosal protection (Figure 4).
NK cells have been typically regarded at undergoing continual recirculation in the blood, a feature essential to mediate immunosurveillance allowing rapid and potent responses to tumors. However, NK cells also express a variety of chemokine receptors and can be provoked to migrate in response to factors that do not belong to the chemokine superfamily. These include the proinflammatory protein chemerin149 and the G‐protein coupled receptor S1P5 150 molecule that can affect trafficking of NK cells both at steady‐state and during inflammation. S1P5, regulated by the expression of the transcription factor T‐BET, is critical for the egress of NK cells from the bone marrow and lymph nodes. NK cells can then return to these tissues via a mechanism that is dependent on CD62L expression.150, 151, 152 Differential gradients of S1P5 and the S1P transporter SPNS2 on tissues, particularly lymphatic endothelial cells, combined with CXCR4 expression provide the spatial cues for NK cell localization in tissues.151 Although NK cells are found at relatively high frequency in the peripheral blood, they are most frequent in the nonlymphoid organs lung and liver and most abundant in the spleen.153, 154 A number of factors are necessary for accumulation in tissue‐specific sites. For example, chemokine receptor 1 (CCR1) is necessary for the accumulation of NK cells in the liver.155 Thus, multiple organs harbor a significant reservoir of NK cells separate from those found in the blood and under certain physiological conditions such as pregnancy,156 or atopic or contact dermatitis,157, 158 these are massively expanded.
From an evolutionary perspective, the notion that ILCs might only be replenished from local sources would leave the body extremely vulnerable—ILCs would be exposed to depletion by a severe highly acute infection, or more damaging long‐term by a chronic infection without a mechanism to quickly deploy progenitors, or differentiated cells, to replace these cells. Local proliferation could provide some protection, but this is likely to be limited and an infection rapidly outstrip the capacity to generate new cells. Thus, the immune system would be quickly disabled, compromising the mucosal barrier in a life‐threatening manner, and negating the principal role of ILC in maintaining these barriers.
The mechanisms that supports the expansion and contraction of ILCs and their capacity to circulate either at steady‐state or during a response remain contentious and poorly characterized. ILC2 can expand significantly following exposure to an allergen (eg, papain or Aspergillus protease) or the alarmin IL‐33 and these cells produce large amounts of cytokines.159 Subsequent to the peak of this response, the number of ILC2 decline but the mechanisms that regulate this reduction are not clear. ILC2, similar to NK cells, express the inhibitory receptors PD‐1 and KLRG1 which perhaps act to regulate this arm of the response.26, 33, 67, 97 PD‐1 has been reported in T cells as a mark of immune exhaustion, but separately, this marker can also reflect immune activation. Nevertheless, it highlights an intriguing new regulatory circuitry that appears to be very finely tuned to maintain immune homeostasis requiring substantial more investigation to unravel all the molecular partners involved.
3.3. ILC3 are essential to maintain immune homeostasis
ILC3 are highly enriched in the gut mucosal tissues and rapidly respond to the cytokine milieu elicited by the colonization of microbes. Often, we view the role of these cells through the lens of driving immune protection. It is, however, the ability to maintain immune homeostasis that is one of the most fundamental aspects that ensures our health. This requires the capacity of the barrier tissues to continually adjust to unpredictable conditions at those surfaces and to integrate signals from the bacterial communities, epithelial cells, and immune cells. How then do ILCs, particularly ILC3s, participate in orchestrating this type of barrier defense is not well‐understood yet.
LTi cells in the embryo establish the sites at which lymph nodes and mucosal‐associated secondary tissues develop.16 These CD4+CD3− cells were first discovered in 1997 while a related population termed LTi‐like cells have been identified in the cryptopatches of mice.160 This population interacts with B cells to promote IgA production161 and the expression of lymphotoxin by ILC3s is critical for both IgA and lymphoid tissue development.161, 162 Subsequently, LTi and LTi‐like cells have both been identified in murine98, 163, 164, 165 and human tissues166, 167 demonstrating that they are highly conserved between species. In contrast, other ILC3 subsets are scattered along the intestine within the lamina propria where they can expand locally in response to microbial colonization.
Despite the prevalence of ILC3s in the gut, they are not uniformly distributed throughout the entire intestinal tract, being more frequent in the jejunum than the ileum.141 This distribution appears to be driven by the heterogeneity of microbes within the intestinal tract which generate metabolites such ligands for aryl hydrocarbon and short chain fatty acids that stimulate ILCs, drive regional specialization and differential distribution. For example, Lactobacillus, Streptococcus, and Enterococcus are localized mainly in the jejunum while segmented filamentous bacteria, Enterobacteriaceae, Bacteroides, and Clostridium are found lower in the intestinal tract, principally the distal ileum and colon.141 Whether microbial stimulation is required for the development of ILC3 still remains contentious. Some studies have demonstrated a paucity of NCR+ ILC3 in germ‐free mice163, 164 while other studies show these populations are preserved.140, 164, 168 However, the ILC populations are significantly amplified by stimulation from microbial communities and the administration of antibiotics eliminates this stimulus and allows the distribution of ILCs to normalize. Expansion of ILC3 appears to depend on the expression of AHR which is induced by tryptophan metabolism to generate indole ligands from the breakdown of glucosinolate glucobrassicin from cruciferous vegetables169, 170. Recently, the nuclear protein WASH (Wiskott‐Aldrich syndrome protein and SCAR homologue) has been implicated in the recruitment of Arid1a to the Ahr promoter to activate AHR expression.171 This expansion drives the production of IL‐22 by ILC3 which is essential for fucosylation of gut epithelial cells via the induction of the fucosyltransferase, Fut2.172 IL‐22 production also promotes the production of the epithelial derived antimicrobial peptide RegIIIγ which is essential for the control of enteric infections such as Citrobacter rodentium.120, 169, 173
In C. rodentium infection, loss of IL‐22 produced by the NKp46+ ILC3 subset does not in itself compromise the capacity to control bacterial colonization as IL‐22 production can be maintained through the NKp46− ILC3 subset.174 This raised the notion that innate and adaptive immune cells are highly redundant and challenged our understanding of how overlapping cell types might contribute to maintaining gut homeostasis. The loss of CD4+ T cell input, however, resulted in prolonged phosphorylation of STAT3, an activation step that is normally only induced transiently in response to microbial colonization.175 Thus, the absence of CD4+ T cells uncouples the functionality of NKp46+ ILC3 and this cannot be retrieved by sustained IL‐22 secreted by NKp46− ILC3. Instead, it results in impaired host lipid metabolism in the gut. Collectively, these studies demonstrate that sequential interactions between ILC3 and CD4+ T cells shape microbial populations allowing the establishment of commensal populations associated with a noninflammatory state and challenged our understanding of how the immune system established this landscape to maintain immune homeostasis.21, 173, 174
4. CONCLUSIONS
Elucidation of the key players in the ILC family add an entirely new dimension to how we view the complex interactome necessary for immune protection. ILCs are strategically positioned at all the peripheral and mucosal sites, pivotally positioning them to sense environmental changes and almost immediate responsiveness to any perceived challenges. We are gradually learning the signals that are capable of activating ILCs in autoimmune, allergic and pathogen‐driven responses but still know little about the mechanisms that retain tight control on such pathways to maintain the cells in a quiescent but “alert” state. With the discovery of potential new subsets, intermediate cell types and the gradual emergence of the pathways of ILC plasticity it will be important to understand the cues that allow ILC subsets to adapt to the changing landscape. Those features are drastically different at the beginning of a pathogen or allergen challenge compared with the established setting of an infection or tumor. Although in some cases the transition cell types, for example ILC1 in tumors, seems to disable the function of these cells it is not clear that this would also be true in a pathogen infection or whether pathogens can coopt ILCs to disable their immediate early functions and facilitate pathogen invasion. Mechanisms to replenish ILC in the face of tissue destruction are essential. The current models in which ILC are viewed as relatively static and undergo slow self‐renewal do not appear to fulfill the criteria to ensure that homeostasis would be maintained in the event of a crisis. At mucosal and cutaneous barriers, many insults could easily unravel into highly destructive sequel if multiple avenues are not available to repopulate ILCs. Indeed evidence suggests that ILCs may come from the bone marrow, circulation, local repositioning, or perhaps the thymus, which is emerging as a source of new ILC. We as yet know little about some of these sources, or even how to identify the cells that contribute to the repopulation. This will require a significant shift in our approach to thinking about what progenitors might look like and the circumstances and triggers that might mediate their rapid recruitment to the body's surfaces beyond the finite local tissue reservoirs.
AUTHOR CONTRIBUTIONS
All authors researched data for the article, made substantial contributions to discussions of the content, wrote the article, and reviewed and/or edited the manuscript before submission.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
ACKNOWLEDGEMENTS
This work was supported by grants and fellowships from the National Health and Medical Research Council of Australia (1054925, 1124907) and The Rebecca L. Cooper Foundation Medical Research Foundation (G. T. B). N. J. was supported by a fellowship from the Foundation ARC pour la recherche sur le cancer. This study was made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC Independent Research Institute Infrastructure Support Scheme (Walter and Eliza Hall Institute of Medical Research).
Almeida FF, Jacquelot N, Belz GT. Deconstructing deployment of the innate immune lymphocyte army for barrier homeostasis and protection. Immunol Rev. 2018;286:6‐22. 10.1111/imr.12709
This article is part of a series of reviews covering Innate Lymphoid Cells appearing in Volume 286 of Immunological Reviews.
REFERENCES
- 1. Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell. 2014;159:94‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vivier E, van de Pavert SA, Cooper MD, Belz GT. The evolution of innate lymphoid cells. Nat Immunol. 2016;17:790‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim CH, Hashimoto‐Hill S, Kim M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol. 2016;37:68‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Narni‐Mancinelli E, Gauthier L, Baratin M, et al. Complement factor P is a ligand for the natural killer cell‐activating receptor NKp46. Sci Immunol. 2017;2:eaam9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barrow AD, Edeling MA, Trifonov V, et al. Natural killer cells control tumor growth by sensing a growth factor. Cell. 2018;172:534‐548 e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mandelboim O, Lieberman N, Lev M, et al. Recognition of haemagglutinins on virus‐infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409:1055‐1060. [DOI] [PubMed] [Google Scholar]
- 7. Jarahian M, Watzl C, Fournier P, et al. Activation of natural killer cells by Newcastle disease virus hemagglutinin‐neuraminidase. J Virol. 2009;83:8108‐8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jarahian M, Fiedler M, Cohnen A, et al. Modulation of NKp30‐ and NKp46‐mediated natural killer cell responses by poxviral hemagglutinin. PLoS Pathog. 2011;7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arnon TI, Lev M, Katz G, Chernobrov Y, Porgador A, Mandelboim O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur J Immunol. 2001;31:2680‐2689. [DOI] [PubMed] [Google Scholar]
- 10. Dadi S, Chhangawala S, Whitlock BM, et al. Cancer immunosurveillance by tissue‐resident innate lymphoid cells and innate‐like T cells. Cell. 2016;164:365‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mebius RE, Rennert R, Weissman IL. Developing Lymph Nodes Collect CD4+ CD3− LTb+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity. 1997;7:493‐504. [DOI] [PubMed] [Google Scholar]
- 12. Adachi S, Yoshida H, Kataoka H, Nishikawa S. Three distinctive steps in Peyer's patch formation of murine embryo. Int Immunol. 1997;9:504‐514. [DOI] [PubMed] [Google Scholar]
- 13. Spits H, Artis D, Colonna M, et al. Innate lymphoid cells – A proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145‐149. [DOI] [PubMed] [Google Scholar]
- 14. Jiao Y, Huntington ND, Belz GT, Seillet C. Type 1 innate lymphoid cell biology: Lessons learnt from natural killer cells. Front Immunol. 2016;7:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells–how did we miss them? Nat Rev Immunol. 2013;13:75‐87. [DOI] [PubMed] [Google Scholar]
- 16. Huang Y, Guo L, Qiu J, et al. IL‐25‐responsive, lineage‐negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat Immunol. 2015;16:161‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walker JA, McKenzie AN. Development and function of group 2 innate lymphoid cells. Curr Opin Immunol. 2013;25:148‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klose CS, Flach M, Mohle L, et al. Differentiation of type 1 ILCs from a common progenitor to all helper‐like innate lymphoid cell lineages. Cell. 2014;157:340‐356. [DOI] [PubMed] [Google Scholar]
- 19. Rankin LC, Groom JR, Chopin M, et al. The transcription factor T‐bet is essential for the development of NKp46(+) innate lymphocytes via the Notch pathway. Nat Immunol. 2013;14:389‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klose CS, Kiss EA, Schwierzeck V, et al. A T‐bet gradient controls the fate and function of CCR6‐RORgammat+ innate lymphoid cells. Nature. 2013;494:261‐265. [DOI] [PubMed] [Google Scholar]
- 21. Hepworth MR, Fung TC, Masur SH, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria‐specific CD4(+) T cells. Science. 2015;348:1031‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moro K, Yamada T, Tanabe M, et al. Innate production of T(H)2 cytokines by adipose tissue‐associated c‐Kit(+)Sca‐1(+) lymphoid cells. Nature. 2010;463:540‐544. [DOI] [PubMed] [Google Scholar]
- 23. Possot C, Schmutz S, Chea S, et al. Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nat Immunol. 2011;12:949‐958. [DOI] [PubMed] [Google Scholar]
- 24. Cherrier M, Ohnmacht C, Cording S, Eberl G. Development and function of intestinal innate lymphoid cells. Curr Opin Immunol. 2012;24:277‐283. [DOI] [PubMed] [Google Scholar]
- 25. Yang Q, Saenz SA, Zlotoff DA, Artis D, Bhandoola A. Cutting edge: Natural helper cells derive from lymphoid progenitors. J Immunol. 2011;187:5505‐5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seillet C, Mielke LA, Amann‐Zalcenstein DB, et al. Deciphering the innate lymphoid cell transcriptional program. Cell Rep. 2016;17:436‐447. [DOI] [PubMed] [Google Scholar]
- 27. Yang Q, Li F, Harly C, et al. TCF‐1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat Immunol. 2015;16:1044‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu Q, Erman B, Park JH, Feigenbaum L, Singer A. IL‐7 receptor signals inhibit expression of transcription factors TCF‐1, LEF‐1, and RORgammat: Impact on thymocyte development. J Exp Med. 2004;200:797‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harly C, Cam M, Kaye J, Bhandoola A. Development and differentiation of early innate lymphoid progenitors. J Exp Med. 2018;215:249‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kamachi F, Isshiki T, Harada N, Akiba H, Miyake S. ICOS promotes group 2 innate lymphoid cell activation in lungs. Biochem Biophys Res Commun. 2015;463:739‐745. [DOI] [PubMed] [Google Scholar]
- 31. Paclik D, Stehle C, Lahmann A, Hutloff A, Romagnani C. ICOS regulates the pool of group 2 innate lymphoid cells under homeostatic and inflammatory conditions in mice. Eur J Immunol. 2015;45:2766‐2772. [DOI] [PubMed] [Google Scholar]
- 32. Maazi H, Patel N, Sankaranarayanan I, et al. ICOS:ICOS‐ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity. 2015;42:538‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu Y, Tsang JC, Wang C, et al. Single‐cell RNA‐seq identifies a PD‐1hi ILC progenitor and defines its development pathway. Nature. 2016;539:102‐106. [DOI] [PubMed] [Google Scholar]
- 34. Benson DM Jr, Bakan CE, Mishra A, et al. The PD‐1/PD‐L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT‐011, a novel monoclonal anti‐PD‐1 antibody. Blood. 2010;116:2286‐2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miyazaki M, Miyazaki K, Chen K, et al. The E‐Id protein axis specifies adaptive lymphoid cell identity and suppresses thymic innate lymphoid cell development. Immunity. 2017;46:818‐834 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yokota Y, Mansouri A, Mori S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix‐loop‐helix inhibitor Id2. Nature. 1999;397:702‐706. [DOI] [PubMed] [Google Scholar]
- 37. Heemskerk MH, Blom B, Nolan G, et al. Inhibition of T cell and promotion of natural killer cell development by the dominant negative helix loop helix factor Id3. J Exp Med. 1997;186:1597‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang HC, Qian L, Zhao Y, et al. Downregulation of E protein activity augments an ILC2 differentiation program in the thymus. J Immunol. 2017;198:3149‐3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gentek R, Munneke JM, Helbig C, et al. Modulation of signal strength switches notch from an inducer of T cells to an inducer of ILC2. Front Immunol. 2013;4:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wong SH, Walker JA, Jolin HE, et al. Transcription factor RORalpha is critical for nuocyte development. Nat Immunol. 2012;13:229‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang S, Miyazaki Y, Shinozaki Y, Yoshida H. Augmentation of antigen‐presenting and Th1‐promoting functions of dendritic cells by WSX‐1(IL‐27R) deficiency. J Immunol. 2007;179:6421‐6428. [DOI] [PubMed] [Google Scholar]
- 42. Li P, Burke S, Wang J, et al. Reprogramming of T cells to natural killer‐like cells upon Bcl11b deletion. Science. 2010;329:85‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walker JA, Oliphant CJ, Englezakis A, et al. Bcl11b is essential for group 2 innate lymphoid cell development. J Exp Med. 2015;212:875‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Califano D, Cho JJ, Uddin MN, et al. Transcription factor Bcl11b controls identity and function of mature type 2 innate lymphoid cells. Immunity. 2015;43:354‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dudakov JA, Hanash AM, Jenq RR, et al. Interleukin‐22 drives endogenous thymic regeneration in mice. Science. 2012;336:91‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dudakov JA, Mertelsmann AM, O'Connor MH, et al. Loss of thymic innate lymphoid cells leads to impaired thymopoiesis in experimental graft‐versus‐host disease. Blood. 2017;130:933‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jones R, Cosway EJ, Willis C, et al. Dynamic changes in intrathymic ILC populations during murine neonatal development. Eur J Immunol. 2018. 10.1002/eji.201847511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miller CN, Proekt I, von Moltke J, et al. Thymic tuft cells promote an IL‐4‐enriched medulla and shape thymocyte development. Nature. 2018;559(7715):627‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bornstein C, Nevo S, Giladi A, et al. Single‐cell mapping of the thymic stroma identifies IL‐25‐producing tuft epithelial cells. Nature. 2018;559(7715):622‐626. [DOI] [PubMed] [Google Scholar]
- 50. Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol. 2015;15:415‐428. [DOI] [PubMed] [Google Scholar]
- 51. Huang Q, Seillet C, Belz GT. Shaping innate lymphoid cell diversity. Front Immunol. 2017;8:1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lim AI, Verrier T, Vosshenrich CA, Di Santo JP. Developmental options and functional plasticity of innate lymphoid cells. Curr Opin Immunol. 2017;44:61‐68. [DOI] [PubMed] [Google Scholar]
- 53. De Obaldia ME, Bhandoola A. Transcriptional regulation of innate and adaptive lymphocyte lineages. Annu Rev Immunol. 2015;33:607‐642. [DOI] [PubMed] [Google Scholar]
- 54. Zhong C, Zhu J. Transcriptional regulators dictate innate lymphoid cell fates. Protein Cell. 2017;8:242‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carotta S, Pang SH, Nutt SL, Belz GT. Identification of the earliest NK‐cell precursor in the mouse BM. Blood. 2011;117:5449‐5452. [DOI] [PubMed] [Google Scholar]
- 56. Rosmaraki EE, Douagi I, Roth C, Colucci F, Cumano A, Di Santo JP. Identification of committed NK cell progenitors in adult murine bone marrow. Eur J Immunol. 2001;31:1900‐1909. [DOI] [PubMed] [Google Scholar]
- 57. Ali AK, Oh JS, Vivier E, Busslinger M, Lee SH. NK cell‐specific Gata3 ablation identifies the maturation program required for bone marrow exit and control of proliferation. J Immunol. 2016;196:1753‐1767. [DOI] [PubMed] [Google Scholar]
- 58. Seillet C, Huntington ND, Gangatirkar P, et al. Differential requirement for Nfil3 during NK cell development. J Immunol. 2014;192:2667‐2676. [DOI] [PubMed] [Google Scholar]
- 59. Rabacal W, Pabbisetty SK, Hoek KL, et al. Transcription factor KLF2 regulates homeostatic NK cell proliferation and survival. Proc Natl Acad Sci USA. 2016;113:5370‐5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gordon SM, Chaix J, Rupp LJ, et al. The transcription factors T‐bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Townsend MJ, Weinmann AS, Matsuda JL, et al. T‐bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20:477‐494. [DOI] [PubMed] [Google Scholar]
- 62. Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA‐binding factor TOX for the development of lymphoid tissue‐inducer cell and NK cell lineages. Nat Immunol. 2010;11:945‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barton K, Muthusamy N, Fischer C, et al. The Ets‐1 transcription factor is required for the development of natural killer cells in mice. Immunity. 1998;9:555‐563. [DOI] [PubMed] [Google Scholar]
- 64. Seillet C, Rankin LC, Groom JR, et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med. 2014;211:1733‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Firth MA, Madera S, Beaulieu AM, et al. Nfil3‐independent lineage maintenance and antiviral response of natural killer cells. J Exp Med. 2013;210:2981‐2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4‐stage developmental program. Blood. 2009;113:5488‐5496. [DOI] [PubMed] [Google Scholar]
- 67. Huntington ND, Tabarias H, Fairfax K, et al. NK cell maturation and peripheral homeostasis is associated with KLRG1 up‐regulation. J Immunol. 2007;178:4764‐4770. [DOI] [PubMed] [Google Scholar]
- 68. Peng H, Sun R, Tang L, Wei H, Tian Z. CD62L is critical for maturation and accumulation of murine hepatic NK cells in response to viral infection. J Immunol. 2013;190:4255‐4262. [DOI] [PubMed] [Google Scholar]
- 69. Martinet L, Ferrari De Andrade L, Guillerey C, et al. DNAM‐1 expression marks an alternative program of NK cell maturation. Cell Rep. 2015;11:85‐97. [DOI] [PubMed] [Google Scholar]
- 70. Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. 2006;176:1517‐1524. [DOI] [PubMed] [Google Scholar]
- 71. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508:397‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Daussy C, Faure F, Mayol K, et al. T‐bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. 2014;211:563‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. 2014;41:354‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fuchs A, Vermi W, Lee JS, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL‐12‐ and IL‐15‐responsive IFN‐gamma‐producing cells. Immunity. 2013;38:769‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Holmes ML, Huntington ND, Thong RP, et al. Peripheral natural killer cell maturation depends on the transcription factor Aiolos. EMBO J. 2014;33:2721‐2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bernink JH, Peters CP, Munneke M, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14:221‐229. [DOI] [PubMed] [Google Scholar]
- 77. Cortez VS, Fuchs A, Cella M, Gilfillan S, Colonna M. Cutting edge: Salivary gland NK cells develop independently of Nfil3 in steady‐state. J Immunol. 2014;192:4487‐4491. [DOI] [PubMed] [Google Scholar]
- 78. Mackay LK, Minnich M, Kragten NA, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459‐463. [DOI] [PubMed] [Google Scholar]
- 79. Ebihara T, Song C, Ryu SH, et al. Runx3 specifies lineage commitment of innate lymphoid cells. Nat Immunol. 2015;16:1124‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Turchinovich G, Ganter S, Barenwaldt A, Finke D. NKp46 calibrates tumoricidal potential of type 1 innate lymphocytes by regulating TRAIL expression. J Immunol. 2018;200:3762‐3768. [DOI] [PubMed] [Google Scholar]
- 81. Almeida FF, Tognarelli S, Marçais A, et al. A point mutation in the Ncr1 signal peptide impairs the development of innate lymphoid cell subsets. OncoImmunology. 2018. 10.1080/2162402x.2018.1475875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jang Y, Gerbec ZJ, Won T, et al. Cutting edge: Check your mice‐a point mutation in the Ncr1 locus identified in CD45.1 congenic mice with consequences in mouse susceptibility to infection. J Immunol. 2018;200:1982‐1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vonarbourg C, Mortha A, Bui VL, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor‐expressing RORgammat(+) innate lymphocytes. Immunity. 2010;33:736‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gao Y, Souza‐Fonseca‐Guimaraes F, Bald T, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18:1004‐1015. [DOI] [PubMed] [Google Scholar]
- 85. Lim AI, Menegatti S, Bustamante J, et al. IL‐12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med. 2016;213:569‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ohne Y, Silver JS, Thompson‐Snipes L, et al. IL‐1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol. 2016;17:646‐655. [DOI] [PubMed] [Google Scholar]
- 87. Silver JS, Kearley J, Copenhaver AM, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. 2016;17:626‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bal SM, Bernink JH, Nagasawa M, et al. IL‐1beta, IL‐4 and IL‐12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol. 2016;17:636‐645. [DOI] [PubMed] [Google Scholar]
- 89. Mjosberg J, Bernink J, Golebski K, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity. 2012;37:649‐659. [DOI] [PubMed] [Google Scholar]
- 90. Mielke LA, Groom JR, Rankin LC, et al. TCF‐1 controls ILC2 and NKp46+RORgammat+ innate lymphocyte differentiation and protection in intestinal inflammation. J Immunol. 2013;191:4383‐4391. [DOI] [PubMed] [Google Scholar]
- 91. Yang Q, Monticelli LA, Saenz SA, et al. T cell factor 1 is required for group 2 innate lymphoid cell generation. Immunity. 2013;38:694‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Spooner CJ, Lesch J, Yan D, et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol. 2013;14:1229‐1236. [DOI] [PubMed] [Google Scholar]
- 93. Yu Y, Wang C, Clare S, et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J Exp Med. 2015;212:865‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhong C, Cui K, Wilhelm C, et al. Group 3 innate lymphoid cells continuously require the transcription factor GATA‐3 after commitment. Nat Immunol. 2016;17:169‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yagi R, Zhong C, Northrup DL, et al. The transcription factor GATA3 is critical for the development of all IL‐7Rα‐expressing innate lymphoid cells. Immunity. 2014;40:378‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Stier MT, Zhang J, Goleniewska K, et al. IL‐33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J Exp Med. 2018;215:263‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Taylor S, Huang Y, Mallett G, et al. PD‐1 regulates KLRG1+ group 2 innate lymphoid cells. J Exp Med. 2017;214:1663‐1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Eberl G, Marmon S, Sunshine M‐J, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor Rorγt in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64‐72. [DOI] [PubMed] [Google Scholar]
- 99. Bar‐Ephraim YE, Mebius RE. Innate lymphoid cells in secondary lymphoid organs. Immunol Rev. 2016;271:185‐199. [DOI] [PubMed] [Google Scholar]
- 100. Mebius RE. Organogenesis of lymphoid tissues. Nat Rev Immunol. 2003;3:292‐303. [DOI] [PubMed] [Google Scholar]
- 101. Gomez de Aguero M, Ganal‐Vonarburg SC, Fuhrer T, et al. The maternal microbiota drives early postnatal innate immune development. Science. 2016;351:1296‐1302. [DOI] [PubMed] [Google Scholar]
- 102. van de Pavert SA, Ferreira M, Domingues RG, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. 2014;508:123‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mortha A, Chudnovskiy A, Hashimoto D, et al. Microbiota‐dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Spencer SP, Wilhelm C, Yang Q, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science (New York, NY). 2014;343:432‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM. In vivo natural killer cell activities revealed by natural killer cell‐deficient mice. Proc Natl Acad Sci USA. 2000;97:2731‐2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Smyth MJ, Thia KY, Street SE, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med. 2000;191:661‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235‐271. [DOI] [PubMed] [Google Scholar]
- 108. Gazit R, Gruda R, Elboim M, et al. Lethal influenza infection in the absence of the natural killer cell receptor gene Ncr1. Nat Immunol. 2006;7:517‐523. [DOI] [PubMed] [Google Scholar]
- 109. Narni‐Mancinelli E, Chaix J, Fenis A, et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci USA. 2011;108:18324‐18329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Narni‐Mancinelli E, Jaeger BN, Bernat C, et al. Tuning of natural killer cell reactivity by NKp46 and Helios calibrates T cell responses. Science. 2012;335:344‐348. [DOI] [PubMed] [Google Scholar]
- 111. Glasner A, Simic H, Miklic K, et al. Expression, function, and molecular properties of the killer receptor Ncr1‐Noe. J Immunol. 2015;195:3959‐3969. [DOI] [PubMed] [Google Scholar]
- 112. Wang Y, Dong W, Zhang Y, Caligiuri MA, Yu J. Dependence of innate lymphoid cell 1 development on NKp46. PLoS Biol. 2018;16:e2004867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Sheppard S, Schuster IS, Andoniou CE, et al. The murine natural cytotoxic receptor NKp46/NCR1 controls TRAIL protein expression in NK cells and ILC1s. Cell Rep. 2018;22:3385‐3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Draghi M, Pashine A, Sanjanwala B, et al. NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J Immunol. 2007;178:2688‐2698. [DOI] [PubMed] [Google Scholar]
- 115. Grundy MA, Zhang T, Sentman CL. NK cells rapidly remove B16F10 tumor cells in a perforin and interferon‐gamma independent manner in vivo. Cancer Immunol Immunother. 2007;56:1153‐1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Celis‐Gutierrez J, Boyron M, Walzer T, et al. Dok1 and Dok2 proteins regulate natural killer cell development and function. EMBO J. 2014;33:1928‐1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Cortez VS, Ulland TK, Cervantes‐Barragan L, et al. SMAD4 impedes the conversion of NK cells into ILC1‐like cells by curtailing non‐canonical TGF‐beta signaling. Nat Immunol. 2017;18:995‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Viant C, Rankin LC, Girard‐Madoux MJ, et al. Transforming growth factor‐beta and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 ILCs. Sci Signal. 2016;9:ra46. [DOI] [PubMed] [Google Scholar]
- 119. Robinette ML, Fuchs A, Cortez VS, et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol. 2015;16:306‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lee JS, Cella M, McDonald KG, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2012;13:144‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Satoh‐Takayama N, Lesjean‐Pottier S, Vieira P, et al. IL‐7 and IL‐15 independently program the differentiation of intestinal CD3‐NKp46+ cell subsets from Id2‐dependent precursors. J Exp Med. 2010;207:273‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Yang J, Cornelissen F, Papazian N, et al. IL‐7‐dependent maintenance of ILC3s is required for normal entry of lymphocytes into lymph nodes. J Exp Med. 2018;215:1069‐1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867‐878. [DOI] [PubMed] [Google Scholar]
- 124. Moussion C, Girard JP. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature. 2011;479:542‐546. [DOI] [PubMed] [Google Scholar]
- 125. Lodolce JP, Boone DL, Chai S, et al. IL‐15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669‐676. [DOI] [PubMed] [Google Scholar]
- 126. Kennedy MK, Glaccum M, Brown SN, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15‐deficient mice. J Exp Med. 2000;191:771‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Robinette ML, Bando JK, Song W, Ulland TK, Gilfillan S, Colonna M. IL‐15 sustains IL‐7R‐independent ILC2 and ILC3 development. Nat Commun. 2017;8:14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Delconte RB, Shi W, Sathe P, et al. The helix‐loop‐helix protein ID2 governs NK cell fate by tuning their sensitivity to interleukin‐15. Immunity. 2016;44:103‐115. [DOI] [PubMed] [Google Scholar]
- 129. Gasteiger G, Hemmers S, Bos PD, Sun JC, Rudensky AY. IL‐2‐dependent adaptive control of NK cell homeostasis. J Exp Med. 2013;210:1179‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Roediger B, Kyle R, Yip KH, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol. 2013;14:564‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type‐2 immunity. Nature. 2010;464:1367‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. von Moltke J, Ji M, Liang HE, Locksley RM. Tuft‐cell‐derived IL‐25 regulates an intestinal ILC2‐epithelial response circuit. Nature. 2016;529:221‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Gerbe F, Sidot E, Smyth DJ, et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature. 2016;529:226‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Howitt MR, Lavoie S, Michaud M, et al. Tuft cells, taste‐chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science. 2016;351:1329‐1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Sojka DK, Plougastel‐Douglas B, Yang L, et al. Tissue‐resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife. 2014;3:e01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Peng H, Jiang X, Chen Y, et al. Liver‐resident NK cells confer adaptive immunity in skin‐contact inflammation. J Clin Invest. 2013;123:1444‐1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (New York, NY). 2015;350:981‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. O'Sullivan TE, Rapp M, Fan X, et al. Adipose‐resident group 1 innate lymphoid cells promote obesity‐associated insulin resistance. Immunity. 2016;45:428‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Vely F, Barlogis V, Vallentin B, et al. Evidence of innate lymphoid cell redundancy in humans. Nat Immunol. 2016;17:1291‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sawa S, Cherrier M, Lochner M, et al. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. 2010;330:665‐669. [DOI] [PubMed] [Google Scholar]
- 141. Kim SH, Cho BH, Kiyono H, Jang YS. Microbiota‐derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer's patches. Sci Rep. 2017;7:3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Luci C, Reynders A, Ivanov II, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol. 2009;10:75‐82. [DOI] [PubMed] [Google Scholar]
- 143. Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003‐1007. [DOI] [PubMed] [Google Scholar]
- 144. Hua G, Thin TH, Feldman R, et al. Crypt base columnar stem cells in small intestines of mice are radioresistant. Gastroenterology. 2012;143:1266‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hanash AM, Dudakov JA, Hua G, et al. Interleukin‐22 protects intestinal stem cells from immune‐mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. 2012;37:339‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Lim AI, Li Y, Lopez‐Lastra S, et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell. 2017;168:1086‐1100 e1010. [DOI] [PubMed] [Google Scholar]
- 147. Kloverpris HN, Kazer SW, Mjosberg J, et al. Innate lymphoid cells are depleted irreversibly during acute HIV‐1 infection in the absence of viral suppression. Immunity. 2016;44:391‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Huang Y, Mao K, Chen X, et al. S1P‐dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science (New York, NY). 2018;359:114‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Parolini S, Santoro A, Marcenaro E, et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood. 2007;109:3625‐3632. [DOI] [PubMed] [Google Scholar]
- 150. Walzer T, Chiossone L, Chaix J, et al. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1‐phosphate receptor. Nat Immunol. 2007;8:1337‐1344. [DOI] [PubMed] [Google Scholar]
- 151. Fang V, Chaluvadi VS, Ramos‐Perez WD, et al. Gradients of the signaling lipid S1P in lymph nodes position natural killer cells and regulate their interferon‐gamma response. Nat Immunol. 2017;18:15‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Jenne CN, Enders A, Rivera R, et al. T‐bet‐dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206:2469‐2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Gregoire C, Chasson L, Luci C, et al. The trafficking of natural killer cells. Immunol Rev. 2007;220:169‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Tomasello E, Yessaad N, Gregoire E, et al. Mapping of NKp46(+) cells in healthy human lymphoid and non‐lymphoid tissues. Front Immunol. 2012;3:344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Wald O, Weiss ID, Wald H, et al. IFN‐gamma acts on T cells to induce NK cell mobilization and accumulation in target organs. J Immunol. 2006;176:4716‐4729. [DOI] [PubMed] [Google Scholar]
- 156. Moffett‐King A. Natural killer cells and pregnancy. Nat Rev Immunol. 2002;2:656‐663. [DOI] [PubMed] [Google Scholar]
- 157. Buentke E, Heffler LC, Wilson JL, et al. Natural killer and dendritic cell contact in lesional atopic dermatitis skin–Malassezia‐influenced cell interaction. J Invest Dermatol. 2002;119:850‐857. [DOI] [PubMed] [Google Scholar]
- 158. O'Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell‐ and B cell‐independent adaptive immunity mediated by natural killer cells. Nat Immunol. 2006;7:507‐516. [DOI] [PubMed] [Google Scholar]
- 159. Martinez‐Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen‐experienced group 2 innate lymphoid cells acquire memory‐like properties and enhance allergic lung inflammation. Immunity. 2016;45:198‐208. [DOI] [PubMed] [Google Scholar]
- 160. Kanamori Y, Ishimaru K, Nanno M, et al. Identification of novel lymphoid tissues in murine intestinal mucosa where clusters of c‐kit+ IL‐7R+ Thy1+ lympho‐hemopoietic progenitors develop. J Exp Med. 1996;184:1449‐1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Tsuji M, Suzuki K, Kitamura H, et al. Requirement for lymphoid tissue‐inducer cells in isolated follicle formation and T cell‐independent immunoglobulin A generation in the gut. Immunity. 2008;29:261‐271. [DOI] [PubMed] [Google Scholar]
- 162. Kruglov AA, Grivennikov SI, Kuprash DV, et al. Nonredundant function of soluble LTα3 produced by innate lymphoid cells in intestinal homeostasis. Science (New York, NY). 2013;342:1243‐1246. [DOI] [PubMed] [Google Scholar]
- 163. Satoh‐Takayama N, Vosshenrich CA, Lesjean‐Pottier S, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958‐970. [DOI] [PubMed] [Google Scholar]
- 164. Sanos SL, Bui VL, Mortha A, et al. RORγt and commensal microflora are required for the differentiation of mucosal interleukin IL22‐producion NKp46+ cells. Nat Immunol. 2009;10:83‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Eberl G. Development and evolution of RORgammat+ cells in a microbe's world. Immunol Rev. 2012;245:177‐188. [DOI] [PubMed] [Google Scholar]
- 166. Montaldo E, Teixeira‐Alves LG, Glatzer T, et al. Human RORgammat(+)CD34(+) cells are lineage‐specified progenitors of group 3 RORgammat(+) innate lymphoid cells. Immunity. 2014;41:988‐1000. [DOI] [PubMed] [Google Scholar]
- 167. Cupedo T, Crellin NK, Papazian N, et al. Human fetal lymphoid tissue‐inducer cells are interleukin 17‐producing precursors to RORC+ CD127+ natural killer‐like cells. Nat Immunol. 2009;10:66‐74. [DOI] [PubMed] [Google Scholar]
- 168. Reynders A, Yessaad N, Vu Manh TP, et al. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat‐ lymphoid cells. EMBO J. 2011;30:2934‐2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kiss EA, Vonarbourg C, Kopfmann S, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (New York, NY). 2011;334:1561‐1565. [DOI] [PubMed] [Google Scholar]
- 170. Li Y, Innocentin S, Withers DR, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629‐640. [DOI] [PubMed] [Google Scholar]
- 171. Xia P, Liu J, Wang S, et al. WASH maintains NKp46(+) ILC3 cells by promoting AHR expression. Nat Commun. 2017;8:15685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu Rev Immunol. 2014;32:403‐432. [DOI] [PubMed] [Google Scholar]
- 173. Qiu J, Guo X, Chen Z‐ME, et al. Group 3 innate lymphoid cells inhibit T‐cell‐mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Rankin LC, Girard‐Madoux MJ, Seillet C, et al. Complementarity and redundancy of IL‐22‐producing innate lymphoid cells. Nat Immunol. 2016;17:179‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Mao K, Baptista AP, Tamoutounour S, et al. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature. 2018;554:255‐259. [DOI] [PubMed] [Google Scholar]
- 176. Levanon D, Negreanu V, Lotem J, et al. Transcription factor Runx3 regulates interleukin‐15‐dependent natural killer cell activation. Mol Cell Biol. 2014;34:1158‐1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Ramirez K, Chandler KJ, Spaulding C, et al. Gene deregulation and chronic activation in natural killer cells deficient in the transcription factor ETS1. Immunity. 2012;36:921‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Vong QP, Leung WH, Houston J, et al. TOX2 regulates human natural killer cell development by controlling T‐BET expression. Blood. 2014;124:3905‐3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Male V, Nisoli I, Kostrzewski T, et al. The transcription factor E4 bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med. 2014;211:635‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Deng Y, Kerdiles Y, Chu J, et al. Transcription factor Foxo1 is a negative regulator of natural killer cell maturation and function. Immunity. 2015;42:457‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Jeevan‐Raj B, Gehrig J, Charmoy M, et al. The transcription factor Tcf1 contributes to normal NK cell development and function by limiting the expression of granzymes. Cell Rep. 2017;20:613‐626. [DOI] [PubMed] [Google Scholar]
- 182. van Helden MJ, Goossens S, Daussy C, et al. Terminal NK cell maturation is controlled by concerted actions of T‐bet and Zeb2 and is essential for melanoma rejection. J Exp Med. 2015;212:2015‐2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Yun S, Lee SH, Yoon SR, et al. TOX regulates the differentiation of human natural killer cells from hematopoietic stem cells in vitro. Immunol Lett. 2011;136:29‐36. [DOI] [PubMed] [Google Scholar]
- 184. Kallies A, Carotta S, Huntington ND, et al. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood. 2011;117:1869‐1879. [DOI] [PubMed] [Google Scholar]
- 185. Boos MD, Yokota Y, Eberl G, Kee BL. Mature natural killer cell and lymphoid tissue‐inducing cell development requires Id2‐mediated suppression of E protein activity. J Exp Med. 2007;204:1119‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Mielke LA, Jones SA, Raverdeau M, et al. Retinoic acid expression associates with enhanced IL‐22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. 2013;210:1117‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Zook EC, Ramirez K, Guo X, et al. The ETS1 transcription factor is required for the development and cytokine‐induced expansion of ILC2. J Exp Med. 2016;213:687‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Glasner A, Isaacson B, Mandelboim O. Expression and function of NKp46 W32R: The human homologous protein of mouse NKp46 W32R (Noe). Sci Rep. 2017;7:40944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Glasner A, Levi A, Enk J, et al. NKp46 receptor‐mediated interferon‐g production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity. 2018;48:1‐13. [DOI] [PubMed] [Google Scholar]