

Actin assembly and type I myosins are both required for clathrin-mediated endocytosis. Here Pedersen and Drubin show that type I myosins anchor actin assembly factors to the plasma membrane at sites of clathrin-mediated endocytosis, facilitating force generation by actin assembly.
Abstract
The actin cytoskeleton generates forces on membranes for a wide range of cellular and subcellular morphogenic events, from cell migration to cytokinesis and membrane trafficking. For each of these processes, filamentous actin (F-actin) interacts with membranes and exerts force through its assembly, its associated myosin motors, or both. These two modes of force generation are well studied in isolation, but how they are coordinated in cells is mysterious. During clathrin-mediated endocytosis, F-actin assembly initiated by the Arp2/3 complex and several proteins that compose the WASP/myosin complex generates the force necessary to deform the plasma membrane into a pit. Here we present evidence that type I myosin is the key membrane anchor for endocytic actin assembly factors in budding yeast. By mooring actin assembly factors to the plasma membrane, this myosin organizes endocytic actin networks and couples actin-generated forces to the plasma membrane to drive invagination and scission. Through this unexpected mechanism, myosin facilitates force generation independent of its motor activity.
Introduction
Clathrin-mediated endocytosis (CME) is a highly conserved cellular process for internalizing soluble and membrane-associated cargos into nascent vesicles derived from the plasma membrane (PM). During the final stages of CME, the PM is bent into a deep pit that constricts at its neck and then undergoes scission. Clathrin and associated adaptor proteins are able to deform the PM when it is under low tension; however, force from the actin cytoskeleton is needed to bend the PM when it is under high tension in mammalian cells (Batchelder and Yarar, 2010; Boulant et al., 2011) or pressed against a cell wall by turgor pressure in fungal cells (Aghamohammadzadeh and Ayscough, 2009). Growing evidence indicates that actin assembly usually occurs during the final stages of CME, even in situations where F-actin is not required (Grassart et al., 2014).
Assembly of an Arp2/3 complex–derived actin network at endocytic sites generates force for PM deformation during CME, but experiments in live cells indicate that type I myosins are also required for force generation (Sun et al., 2006; Lewellyn et al., 2015). In the actin assembly–based force generation model, new monomers join endocytic actin networks near the PM, pushing F-actin in the network deeper into the cytoplasm (Kaksonen et al., 2003, 2005; Picco et al., 2015). F-actin is attached to the apex of endocytic pits by adaptor proteins, so this inward movement of the actin network drags the tip of the growing membrane invagination inward and facilitates transition from a U-shaped pit to an omega-shaped pit (Skruzny et al., 2012; Hassinger et al., 2017). Reconstitution experiments indicate that growth of endocytic actin networks is sufficient to generate force: beads coated with the endocytic Arp2/3 complex activator from Saccharomyces cerevisiae, the Wiscott–Aldrich syndrome protein (WASP) homologue Las17, assemble actin tails and are motile in budding yeast cytoplasmic lysates or in the presence of actin, Arp2/3 complex, and capping protein (Michelot et al., 2010). However, budding yeast cells harboring mutations in the genes encoding type I myosins assemble robust endocytic actin networks but fail to internalize vesicles by CME (Sun et al., 2006). These data demonstrate that both actin assembly and type I myosin activity are required for CME in vivo.
The Arp2/3 complex is activated at the base of endocytic sites (near the PM) by nucleation-promoting factors (NPFs). In budding yeast, the principal NPFs, Las17 and the type I myosins Myo5 and Myo3, exist in a complex termed the WASP/myosin complex that stays at the base of the endocytic site while the coat internalizes (Kaksonen et al., 2005). This complex also includes the homologues of WASP interacting protein (Vrp1) and the F-BAR protein Toca-1 (Bzz1) along with the NPF regulator Bbc1 (Soulard et al., 2002). Previously, we distilled this complex down to a single engineered Myo5-Las17 fusion protein with nonessential domains deleted to reveal the minimal activities of the WASP/myosin complex required to build an actin cytoskeleton capable of generating force for CME. Our fusion protein revealed that the following domains are sufficient to support CME even in the absence of all other members of the WASP/myosin complex: (a) a domain for recruitment to endocytic sites, (b) an NPF domain to activate the Arp2/3 complex, (c) a functional myosin motor domain, and (d) a membrane binding domain from Myo5 (Lewellyn et al., 2015). However, the mechanistic contributions of the activities provided by Myo5 in our minimal fusion protein were unclear.
To determine the mechanistic contributions of membrane binding by Myo5 during CME, we engineered budding yeast strains lacking membrane-bound Myo5. Our results reveal that Myo5 serves as the key membrane anchor for the actin assembly factors of the WASP/myosin complex.
Results and discussion
To investigate how type I myosin membrane binding contributes to CME, we made S. cerevisiae strains with MYO5 genes encoding mutant proteins linked to a C-terminal 13Myc tag for immunoblotting to ensure that the mutant alleles were expressed (Fig. S1 A). We examined CME phenotypes using live-cell imaging of Sla1-GFP as a marker for the endocytic coat and Abp1-mRFP as a marker for endocytic actin networks. Because Myo3 and Myo5 are redundant under laboratory conditions (Goodson et al., 1996), we used a background with the MYO3 gene deleted throughout our study. When we deleted MYO5 completely or replaced it with a mutant lacking the membrane-binding Tail homology 1 (TH1) domain (Feeser et al., 2010; Fernández-Golbano et al., 2014), CME was severely defective. While F-actin still assembled at endocytic sites, the sites turned over slowly and became depolarized (Fig. 1 A), puncta of Sla1-GFP failed to move off of the PM (Fig. 1, B and C; and Video 1), and lifetimes of Sla1-GFP and Abp1-mRFP at endocytic sites were extended in comparison to wild-type MYO5 cells (Fig. S1 B). Surprisingly, rather than being restricted to endocytic sites, endocytic actin networks marked by Abp1-mRFP frequently formed motile comets deep in the cytoplasm (Fig. 1, A and D; and Video 1; Lewellyn et al., 2015). These observations suggest that actin assembly can become uncoupled from endocytic sites in mutants lacking membrane-bound Myo5.
Figure 1.
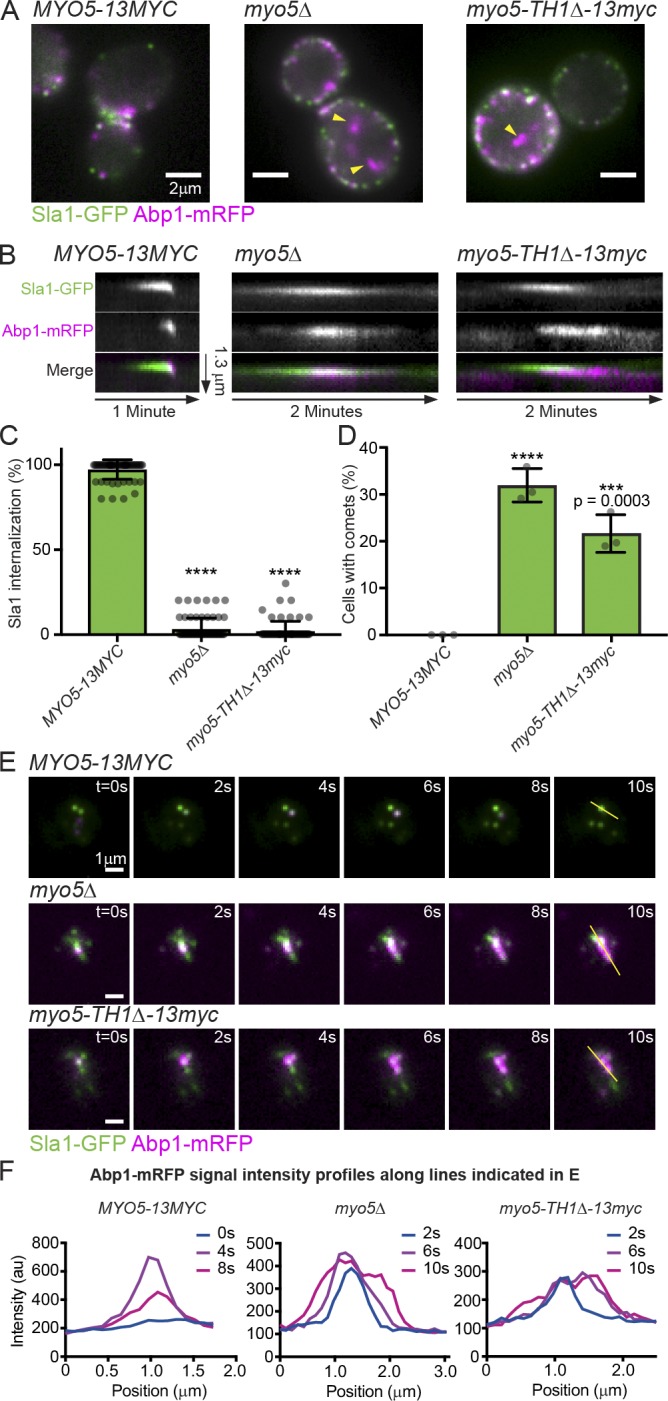
Membrane-bound Myo5 restricts actin assembly to endocytic sites. (A) Stills from epifluorescence videos of cells endogenously expressing Sla1-GFP (green) to label endocytic coats and Abp1-mRFP (magenta) to label endocytic actin networks. Yellow arrowheads mark cytoplasmic actin comets in mutants. See Video 1. (B) Kymographs of individual attempted endocytic events. Extracellular space is up and the cytoplasm down. (C) Quantification of Sla1-GFP patch internalization during a 2-min video. Each data point represents the proportion of patches internalized in one cell. n = 61 (MYO5), 64 (myo5Δ), and 65 (myo5-TH1Δ) cells observed. Asterisks denote statistical significance compared with MYO5 by one-way ANOVA (F = 5168) followed by Dunnett’s test. Error bars are SD. (D) Quantification of the proportion of cells manifesting cytoplasmic actin comets in epifluorescence videos. Each data point represents a separate experiment during which at least 110 cells were observed. Asterisks denote statistical significance compared with MYO5 as in C (F = 82.29). Error bars are SD. (E) Montages from TIRFM videos of cells endogenously expressing Sla1-GFP (green) and Abp1-mRFP (magenta). Yellow lines indicate selections used for intensity profiles in F. Montages are representative of observations from 80 (MYO5), 156 (myo5Δ), and 71 (myo5-TH1Δ) cells. See Video 2. (F) Abp1-mRFP fluorescence intensity profiles from the indicated time points along the line indicated in E. ****, P < 0.0001. All cells are myo3Δ.
All observed mutant cells lacking membrane-bound Myo5 had negligible rates of endocytic vesicle internalization (Fig. 1 C), but not all cells had cytoplasmic actin comets in epifluorescence videos (Fig. 1 D). To resolve this discrepancy, we imaged our mutants lacking membrane-binding competent Myo5 using total internal reflection fluorescence microscopy (TIRFM), reasoning that actin comets could be an exaggerated manifestation of a more penetrant actin assembly phenotype occurring close to the PM. In wild-type MYO5 cells, TIRFM videos revealed discrete actin assembly events at each Sla1-GFP–marked endocytic site after 16 ± 4 s (n = 60 sites, mean ± SD), even in situations where multiple sites were clustered together. In myo5Δ and myo5-TH1Δ cells, however, actin assembly initiated at individual endocytic sites after 20 ± 7 s (n = 85 and 50 sites, P = 0.0001 and 0.0022, respectively, compared with MYO5 by one-way ANOVA [F = 9.509] followed by Dunnett’s test) and then spread as a wave in the plane of the PM, frequently engulfing neighboring endocytic sites (Fig. 1, E and F; Fig. S1, C and D; and Video 2). This phenomenon was evident in every cell examined; thus, delocalized actin assembly is a fully penetrant phenotype. Together, these data indicate that Myo5 restricts actin assembly to endocytic sites.
The Arp2/3 complex nucleates actin assembly during CME, so aberrant actin assembly in myo5 mutants suggests that membrane binding by Myo5 may restrict actin assembly to endocytic sites through localizing Arp2/3 complex activity. To determine whether the Arp2/3 complex becomes mislocalized in myo5 mutants deficient in membrane-binding, we visualized the complex by creating an in-frame GFP fusion of ARC15, which encodes the S. cerevisiae homologue of the Arp2/3 complex subunit ARPC5, at its endogenous genomic locus. In wild-type MYO5 cells, the Arp2/3 complex localized exclusively to Abp1-mRFP patches at the cell cortex, but in myo5Δ cells or cells expressing the myo5-TH1Δ membrane-binding mutant, 99 ± 2% (myo5Δ) and 100% (myo5-TH1Δ) of cytoplasmic Abp1-mRFP-labeled actin comets colocalized with the Arp2/3 complex (Fig. 2 A and Video 3). To test whether the mislocalized Arp2/3 complex was active, we treated cells with CK-666, a reversible small molecule inhibitor of the Arp2/3 complex (Nolen et al., 2009), or with an equivalent control concentration of DMSO (Fig. S1, E and F). Addition of CK-666 to cells containing cytoplasmic actin comets caused rapid dissolution of the cytoplasmic actin structures (Fig. 2 B) and drastically reduced the percentage of cells with visible comets (Fig. 2 C). These results show that Myo5 constrains Arp2/3 complex activity to endocytic sites.
Figure 2.

Membrane-bound Myo5 restricts the activity of the Arp2/3 complex and its regulators to endocytic sites. (A) Stills from epifluorescence videos of cells endogenously expressing Arc15-GFP (green) to label the Arp2/3 complex and Abp1-mRFP (magenta). Yellow arrowheads mark cytoplasmic actin comets in mutants. Percentages are proportions of Abp1-mRFP comets with associated Arc15-GFP signal ± SD; n = 192 (myo5Δ) and 207 (myo5-TH1Δ) total comets observed. See Video 3. (B) Montages from epifluorescence videos during which 250 mM CK-666 or 0.5% DMSO was added to myo5-TH1Δ cells endogenously expressing Abp1-mRFP. Yellow arrowheads indicate cytoplasmic actin comets that dissolve upon CK-666 addition and persist upon DMSO addition. Times are minutes:seconds. Montages are representative of seven experiments. (C) Quantification of the proportion of cells displaying cytoplasmic actin comets in epifluorescence videos in the presence of 0.5% DMSO or 250 µM CK-666. Each data point represents a separate experiment during which at least 50 cells were observed. Asterisks denote statistical significance according to Student’s t tests corrected for multiple comparisons using the two-stage setup method of Benjamani, Kreiger, and Yekutieli with a false discovery rate of 5%. Error bars are SD. (D) Stills from epifluorescence videos of MYO5 and myo5-TH1Δ cells endogenously expressing Abp1-mRFP (magenta) and GFP-tagged Arp2/3 complex regulators (green). Yellow boxes indicate Abp1-mRFP comets with the corresponding GFP-tagged protein at the tip or, in the case of Pan1, absent from the comet. These selections are enlarged twofold and shown in grayscale below. Percentages are proportions of Abp1-mRFP comets with associated GFP signal ± SD; n = 143, 343, 328, 188, and 179 total comets observed for Las17, Vrp1, Bbc1, Bzz1, and Pan1, respectively. See Videos 4, 5, 6, 7, and 8. All cells are myo3Δ.
The observed mislocalization of active Arp2/3 complex suggested that membrane binding by Myo5 retains Arp2/3 complex activators (NPFs) at endocytic sites. The S. cerevisiae genome encodes four NPFs. The primary NPF activities are contained within the WASP/myosin complex and comprise (a) Las17 and (b) Vrp1 working in cooperation with Myo3 or Myo5. These factors remain at the base of CME sites during membrane invagination (Kaksonen et al., 2005; Sun et al., 2006). Abp1 and the budding yeast intersectin homologue Pan1 have also been reported to have NPF activity, but these activities are not essential for CME (Duncan et al., 2001; Goode et al., 2001; Sun et al., 2006). To test whether the WASP/myosin complex is restricted to endocytic sites by Myo5, we localized each member of the complex by epifluorescence microscopy. While all members of the WASP/myosin complex (Las17, Vrp1, Bbc1, and Bzz1) localized to cortical CME sites in wild-type MYO5 cells, they were found at the tips of the cytoplasmic actin comets in myo5-TH1Δ cells. Conversely, Pan1 localized exclusively to the cell cortex in both wild-type MYO5 and myo5-TH1Δ cells (Fig. 2 D and Videos 4, 5, 6, 7, and 8). The myo5-TH1Δ protein itself similarly localized to the tips of the cytoplasmic actin comets (Fig. S1 G and Video 9; Lewellyn et al., 2015). The relocalization of the WASP/myosin complex in the absence of membrane binding by Myo5 indicates that Myo5 restricts actin assembly to endocytic sites by constraining the location of Arp2/3 complex activation.
Membrane binding by Myo5 could be important for restricting Arp2/3 complex activation to sites of CME by physically linking the WASP/myosin complex to the PM at CME sites, by preventing continued WASP/myosin complex activity after vesicle internalization, or by preventing aggregation of WASP/myosin complex proteins in the cytoplasm. Knowing the origin of actin comets in myo5 mutants would help to distinguish between these models. We suspected that actin comets represented actin internalized from endocytic sites without coincident vesicle internalization for several reasons. First, we repeatedly observed comets that appeared to pull away from the PM in epifluorescence videos (Fig. 3 A and Video 3). Second, actin waves originated at CME sites in TIRFM videos (Figs. 1 E and S1 C and Video 2). Finally, Sla1-GFP–labeled patches failed to move off the PM in Myo5 mutants (Fig. 1 C), making it unlikely that the actin comets represented internalized vesicles that failed to disassemble the WASP/myosin complex. Pulse-chase experiments with the membrane dye FM4-64 supported this conclusion by revealing that myo5 mutants internalized very small amounts of dye (Fig. 3, B and C). While the trace amount of dye internalized in myo5 mutants is similar to the amount internalized in CME-defective sla2Δ cells (Peng et al., 2015), we cannot exclude the possibility that small amounts of membrane internalize in these mutants. Because actin comets appear to originate at the PM in myo5 mutants, and these same mutants fail to internalize membrane by CME, we conclude that Myo5 plays the role of anchoring the WASP/myosin complex to the PM at endocytic sites.
Figure 3.

Myo5 couples the assembling actin network to the PM during CME. (A) Montages from the same epifluorescence videos shown in Fig. 2 A. Yellow arrowheads indicate actin comets pulling away from the PM. Similar behavior was observed in at least 50 cells across many different experiments. See Video 3. (B) Spinning disc confocal images of cells subjected to a 5-min pulse of FM4-64 staining followed by a 5-min chase with imaging media. (C) Quantification of percentage of FM4-64 signal internalized during a 5-min chase. n = 20 (MYO5), 19 (myo5Δ), and 22 (myo5-TH1Δ) cells. Asterisks denote statistical significance compared with MYO5 by one-way ANOVA (F = 767.1) followed by Dunnett’s test. Error bars are SD. ****, P < 0.0001. (D) Kymographs of individual endocytic events following washout of CK-666 or no washout in wild-type MYO5 and myo5Δ cells endogenously expressing Sla1-GFP (green) and Abp1-mRFP (magenta). See Fig. S2 for montages. Kymographs are representative of 13 (MYO5) and 14 (myo5Δ) separate experiments. See Video 10. (E) Kymographs as in D from videos of cells endogenously expressing Vrp1-GFP (green) and Abp1-mRFP (magenta). Kymographs are representative of 8 (MYO5) and 16 (myo5Δ) separate experiments. See Video 11. All cells are myo3Δ.
To directly visualize the role of Myo5 as a membrane anchor for the WASP/myosin complex, we performed CK-666 washout experiments. We pretreated cells with CK-666 to synchronize CME events, stalling them at the stage before actin assembly. We then added media to dilute the CK-666 below its effective concentration and observed growth of endocytic actin networks by epifluorescence microscopy. In wild-type MYO5 cells, actin assembly after CK-666 washout coincided with internalization of Sla1-GFP patches (Fig. 3 D; Fig. S2, A and B; and Video 10). In mutant cells deficient for membrane-bound Myo5, actin still assembled at endocytic sites after CK-666 washout, but Sla1-GFP patches failed to move off the PM. Sla1-GFP signal decreased coincident with actin assembly, likely reflecting recruitment of endocytic disassembly factors by F-actin (Toret et al., 2008). Strikingly, flares of Abp1-mRFP signal were frequently observed to separate from endocytic sites and internalize (Fig. 3 D; Fig. S2, A and B; and Video 10). These flares moved off the PM with an average velocity of 240 ± 80 nm/s, similar to the reported speed of Abp1 patches after vesicle scission in wild-type cells of 230 nm/s (Fig. S2 C; Kaksonen et al., 2003). A one-sample t test comparing the measured actin flare velocities to the known value of 230 nm/s yielded a P value of 0.55, supporting the hypothesis that these two actin structures move similarly. These data strongly suggest that the observed flares represent actin internalization uncoupled from membrane internalization. Neither Abp1-mRFP nor Sla1-GFP signal internalized in control or mutant cells without CK-666 washout (Fig. 3 D). When we performed CK-666 washout experiments on cells expressing the WASP/myosin complex protein Vrp1-GFP, we once again observed actin flares peeling off the PM (Fig. 3 E and Video 11). However, unlike Sla1-GFP, which remained on the PM (Fig. 3 D), Vrp1-GFP puncta occasionally splintered perceptibly, with a subset of the protein internalizing with the Abp1-mRFP–labeled actin flare (Fig. 3 E). This behavior contrasts starkly with the behavior of Vrp1 puncta in wild-type MYO5 cells, which remain on the PM during CME (Fig. 3 E; Kaksonen et al., 2005). Thus Myo5 membrane binding is crucial for coupling actin assembly to vesicle internalization through anchorage of the WASP/myosin complex to the PM at endocytic sites.
Thus far, we have revealed a novel function for Myo5 in anchoring actin assembly to the PM during CME. While we have observed uncoupling of actin assembly from endocytic sites upon deletion of the membrane-binding domain of Myo5, we have not identified the other end of this crucial linkage: how Myo5 contacts actin assembly factors to anchor them. To elucidate this end of the Myo5 membrane-actin linkage, we scored cytoplasmic actin comet formation in strains harboring a series of myo5 alleles encoding domain-deleted versions of the protein, each linked to a C-terminal 13Myc tag for immunoblotting to ensure that the mutant alleles were expressed (Figs. 4 A and S3 A). While deletion of the membrane-binding TH1 domain caused formation of actin comets, no other individual domain truncation elicited this phenotype (Fig. 1 D; Fig. 4, B and C; and Video 12; Lewellyn et al., 2015). Because Myo5 motor activity itself is crucial for CME (Sun et al., 2006; Idrissi et al., 2012; Lewellyn et al., 2015), we reasoned that Myo5 may be redundantly linked to the actin cytoskeleton through binding of its motor domain to F-actin and one of its other domains to actin assembly factors in the WASP/myosin complex. To identify these domains, we made myo5 alleles encoding proteins with pairwise deletions, combining motor domain deletion with deletion of the F-actin–binding TH2 domain (Geli et al., 2000), the poly-proline domain-binding Src homology 3 (SH3) domain, and the Arp2/3 complex-binding Connector/acidic (CA) domain. Both myo5-motorΔ TH2Δ and myo5-motorΔ SH3Δ cells contained cytoplasmic actin comets (Fig. 4, B and D; and Video 13), indicating that Myo5 is linked to the endocytic actin cytoskeleton through both its motor domain and domains in the nonmotor tail of the protein.
Figure 4.

Molecular determinants of myosin-mediated coupling of actin assembly to endocytic sites. (A) Schematic diagram of Myo5 domains and deletion mutants analyzed. Domain boundaries are marked with the corresponding amino acid number. All mutants were linked to a C-terminal 13Myc tag. (B) Quantification of the proportion of cells harboring the indicated MYO5 truncation mutant displaying cytoplasmic actin comets in epifluorescence videos. Each data point represents a separate experiment during which at least 70 cells were observed. Data points for MYO5 and myo5-TH1Δ are repeated from Fig. 1 C. Asterisks denote statistical significance compared with MYO5 or between bracketed variants after one-way ANOVA (F = 18.88) followed by Sidak’s multiple comparisons test. Error bars are SD. (C–E) Stills from epifluorescence videos of cells endogenously expressing Sla1-GFP (green) and Abp1-mRFP (magenta). Yellow arrowheads indicate cytoplasmic actin comets in mutants. See Videos 12, 13, and 14. (F) Growth of cells harboring truncation mutations in MYO5 on YPD plates. Photos are representative of results from three separate experiments. (G) Schematic diagram illustrating membrane anchoring of actin assembly factors by Myo5. All cells are myo3Δ.
Myo5 is normally recruited to endocytic sites and bound into the WASP/myosin complex by its SH3 domain, with possible contributions from the TH2 domain (Sun et al., 2006; MacQuarrie et al., 2018 Preprint). Because the entire WASP/myosin complex becomes uncoupled from endocytic sites in myo5 membrane-binding mutants (Fig. 2 D) and myo5 motor mutants unable to bind the other WASP/myosin proteins, we concluded that Myo5 exerts its actin-membrane bridging function by directly anchoring the WASP/myosin complex to the PM. To test whether Myo5 is the membrane anchor for the WASP/myosin complex, we expressed versions of each myo5 truncation that formed comets fused via a flexible peptide linker to Vrp1, a WASP/myosin complex protein that normally binds to Myo5. If Myo5 is the membrane anchor for the WASP/myosin complex, we reasoned that this linkage might eliminate actin comet formation in myo5 truncation mutants that fail to bind to the WASP/myosin proteins but not in mutants that fail to bind to the PM. Vrp1 linkage to myo5-TH1Δ failed to rescue the cytoplasmic actin comet phenotype, but the linkage did rescue comet formation for myo5-motorΔ TH2Δ and the myo5-motorΔ SH3Δ mutant proteins (Fig. 4 E and Video 14), supporting the conclusion that Myo5 binds to and anchors other WASP/myosin complex components to the PM. Internalization of Sla1-GFP patches and lifetimes of Sla1-GFP and Abp1-mRFP patches were not rescued in these mutants, confirming the essential role of the Myo5 motor domain in CME (Fig. S3, B and C). A detailed characterization of the contribution of Myo5 motor activity to CME will be the subject of a future study. Nevertheless, myo5 mutants that did not exhibit comets were healthier than mutants that did exhibit comets, as illustrated by restored growth of the cells at high temperatures, indicating that cytoplasmic actin comets represent a bona fide pathology on the cellular level (Fig. 4 F). Our data demonstrate that Myo5 anchors assembling actin networks to the PM at endocytic sites redundantly through its motor and tail domains (Fig. 4 G).
A recent study demonstrated that the WASP/myosin complex is recruited to endocytic sites as a ring around the presumptive invagination site in the plane of the PM and that this symmetrical organization of Arp2/3 complex activators results in optimal force generation by actin assembly (Mund et al., 2018). Our results indicate that Myo5 anchors Arp2/3 complex activators to CME sites, most likely through direct membrane binding, and that this linkage is crucial for maintaining their ordered localization. This organization restricts actin nucleation to discrete areas on the PM, resulting in an ordered actin network that can generate force (Fig. 5 A; Mund et al., 2018). Absent membrane anchoring, Arp2/3 complex activation and subsequent actin nucleation can occur throughout the actin network, leading to disordered networks that fail to generate force to bend the membrane and, in the most catastrophic circumstances, pull away from the membrane completely (Fig. 5 B). This model differs from a previous interpretation where we proposed that Myo5 links actin filaments themselves to the PM at endocytic sites (Lewellyn et al., 2015). Interestingly, a requirement for a membrane anchor for NPFs has also been reported in engineered motile systems, and absence of the linkage causes a qualitatively similar phenotype to what we observed, with actin tails separating from their initial substrate (Co et al., 2007).
Figure 5.

Model for Myo5 function in anchoring actin assembly to the PM at endocytic sites. (A) Myo5 (yellow bananas) restricts activation of the Arp2/3 complex (gray avocados) by the WASP complex (blue widgets) to a discrete location, generating an actin array that grows predominantly in the same direction to generate force. (B) Absent this critical linkage, Arp2/3 activators splinter off of the PM, leading to Arp2/3 complex activation throughout the actin network. Delocalized Arp2/3 complex activation results in disordered actin arrays that fail to produce force. In the most catastrophic cases, the Arp2/3 complex and its activators pull away from the PM completely to form cytoplasmic actin comets (lower left of zoom).
PM-anchoring of actin assembly factors by Myo5 is a novel, conserved membrane-actin linkage at endocytic sites, adding to our understanding of the CME actin machinery. Previous studies have described how endocytic coat components recruit actin assembly factors to endocytic sites (Sun et al., 2015) and how movement of F-actin into the cytoplasm is coupled to inward movement of the endocytic coat (Skruzny et al., 2012). Our experiments reveal the mechanism by which actin assembly factors are anchored to the PM during actin assembly. Interestingly, the human homologue of Myo5, Myosin 1e, also interacts with a variety of endocytic proteins through its tail domains (Krendel et al., 2007), and mislocalization of Myosin 1e in human cells also leads to mislocalization of Arp2/3 complex activators (Cheng et al., 2012). These observations suggest that membrane anchoring of WASP family proteins is a conserved function of type I myosins.
It is particularly intriguing that unmooring of the actin assembly machinery from endocytic sites results in waves of actin assembly that spread across the PM (Fig. 1, E and F; Fig. S1 C; and Video 2). Similar waves of assembling F-actin manifest in diverse systems ranging from immune cells to oocytes and embryos (Weiner et al., 2007; Bement et al., 2015; Inagaki and Katsuno, 2017) and are usually generated by activator-inhibitor–coupled systems, where actin assembly leads to the delayed recruitment of an inhibitor of actin assembly (Allard and Mogilner, 2013). Indeed, F-actin itself indirectly recruits disassembly factors to endocytic sites (Toret et al., 2008), and negative feedback of F-actin on Arp2/3 regulators partially accounts for the transient assembly and disassembly of the actin machinery at endocytic sites (Wang et al., 2016). Our data substantiate the existence of such feedback relationships in CME by demonstrating that spatially uncoupling the actin assembly machinery (the WASP/myosin complex) from the endocytic disassembly factors (recruited by F-actin) results in traveling waves characteristic of activator-inhibitor–coupled systems.
Type I myosins are pervasive components of actin machinery involved in membrane trafficking events (Sokac et al., 2006; Almeida et al., 2011). Here we have shown that anchoring the actin assembly machinery to the PM is a principal role of type I myosin during CME. It is now important to determine whether type I myosins serve a similar mechanistic role during other membrane trafficking processes.
Materials and methods
Strains and plasmids
All budding yeast strains were derived from the wild-type diploid DDY1102 and propagated using standard techniques (Amberg et al., 2005). Mutant strains were constructed using plasmid DNA encoding URA3-marked MYO5 variants flanked by the MYO5 5′ and 3′ untranslated regions as templates for genomic integration in diploids that were MYO5/myo5Δ::HygMX6. Integration of the full-length mutant allele was ensured by cross selection for acquisition of uracil prototrophy and loss of hygromycin resistance. C-terminal GFP fusions, C-terminal 13Myc fusions, and truncations were constructed as described previously (Longtine et al., 1998). All strains were verified by PCR and immunoblotting. For growth comparisons, mid-log phase starter cultures were diluted to an OD600 of 0.1, 0.02, and 0.004, spotted on plates, and grown for 48 h at 25°C and 37°C. The strains used in this study are listed in Table S1. Plasmids used for genomic integration are listed in Table S2.
Live cell imaging
Cells were grown to mid-log phase in imaging media (synthetic minimal media supplemented with adenine, l-histidine, l-leucine, l-lysine, l-methionine, uracil, and 2% glucose), and then adhered to coverslips coated with 0.2 mg/ml concanavalin A.
Epifluorescence imaging was performed on a Nikon Eclipse Ti inverted microscope with a Nikon 100× 1.4-NA Plan Apo VC oil-immersion objective and an Andor Neo 5.5 sCMOS camera. GFP and mRFP fluorescence were excited using a Lumencore Spectra X LED light source with 550/515-nm and 470/422-nm excitation filters. For two-color imaging, channels were acquired sequentially using an FF-493/574-Di01 dual-pass dichroic mirror and FF01-512/630-25 dual-pass emission filters (Semrock). The system was controlled with either Metamorph or Nikon Elements software and maintained at 25°C by an environmental chamber (In Vivo Scientific). Frames were separated by either 2 or 1.15 s depending on the software used.
TIRFM imaging was performed on a Nikon Eclipse Ti2 inverted microscope with a Nikon CFI60 60× 1.49-NA Apo oil-immersion TIRF objective and a Hamamatsu Orca-Flash 4.0 V2 sCMOS camera. GFP and mRFP fluorescence were excited using 488- and 561-nm lasers and detected using a Chroma HC TIRF Quad Dichroic (C-FL TIRF Ultra Hi S/N 405/488/561/638) and Chroma HC Quad emission filters BP 525/550 and BP600/650, respectively. Channels were acquired sequentially. The system was controlled with Nikon Elements software and maintained at 25°C by an OkoLab environmental chamber. Frames were separated by 1 s.
All image analysis was performed using Fiji software (National Institutes of Health). For quantification of Sla1-GFP internalization and Sla1-GFP/Abp1-mRFP patch lifetimes, 10 radial kymographs were generated at random for each cell, measured, and judged as “internalized” if the Sla1-GFP patch moved at least 3 pixels (194 nm) toward the cell interior. The Manual Tracking Fiji plugin was used for particle tracking in Fig. S2 C. For figure panels and videos, individual cells were cropped, background fluorescence was uniformly subtracted from the image stack, and photo bleaching was corrected using a custom Fiji macro. Lookup tables used for display are linear and cover the full range of data. Figures were assembled in Adobe Illustrator CS6.
CK-666 experiments
CK-666 (≥98% pure) was purchased from Sigma Life Sciences, resuspended in DMSO, and used at 250 µM (Fig. S1 E). All CK-666 experiments were conducted in epifluorescence. For wash-in experiments, cells were adhered to a coverslip as described above and submerged in 500 µl imaging medium. After 10–20 frames of imaging, 500 µl of 500 µM CK-666 was added to the cells. To score actin comets across a population of cells treated with CK-666 (Fig. 2 C), cells were pretreated with 250 µM CK-666 for 10 min before imaging. For CK-666 washout experiments, cells were adhered to coverslips and submerged in 250 µl imaging medium with 250–500 µM CK-666. After 10–20 frames of imaging, 4 ml of fresh imaging medium was added to the cells, diluting CK-666 to ≤29 µM, well below the effective concentration (Fig. S1 E).
FM4-64 pulse-chase experiments
Cells were immobilized on coverslips, quickly equilibrated with a single wash of imaging medium with 10 µg/ml FM4-64 (Molecular Probes), and then incubated in imaging medium with 10 µg/ml FM4-64 for 5 min at room temperature. Coverslips were then washed vigorously with fresh imaging medium to eliminate excess FM4-64. Coverslips were photographed immediately to identify cells that stained brightly with FM4-64 at a zero time point; these cells were interpreted to be dead or to have compromised PM permeability and were eliminated from analysis. Coverslips were then photographed a second time after a 5-min chase.
Images were collected on a Nikon Eclipse Ti inverted microscope with a Nikon 100× 1.45-NA Plan Apo λ oil immersion objective, an Andor IXon X3 EM-CCD camera, and an Andor CSU-X spinning disc confocal equipment. FM4-64 fluorescence was excited using a 488-nm laser and detected with a Chroma 605/52-nm emission filter. The system was controlled with Nikon Elements software. Imaging was conducted at room temperature (∼23°C). The fraction of FM4-64 internalized was determined by dividing the total integrated signal of FM4-64 signal inside of the obvious PM by the total integrated FM4-64 signal of the cell.
Western blotting
Cells were grown to mid-log phase in rich medium (20 g/l bacto-peptone, 10 g/l bacto-yeast extract, and 2% glucose), harvested by centrifugation, washed with water, and lysed by bead beating in a lysis buffer with 5 mM Tris-HCl, pH 8, 5 mM EDTA, pH 8, and 15% TCA. The TCA precipitate was then washed with ice-cold acetone and resuspended in Tris/urea sample buffer (62.5 mM Tris, pH 6.8, 3 M urea, 1% sodium dodecyl sulfate, 5% β mercaptoethanol, and 0.05% bromophenol blue). Extracts were separated electrophoretically on 8% polyacrylamide gels and transferred to nitrocellulose. Membranes were blocked with 5% milk, probed with mouse monoclonal antibody clone 9E10 against a 13-myc tag (prepared from hybridoma supernatant; Lewellyn et al., 2015) and mouse monoclonal anti-Pgk1 (459250; Novex), and then incubated with secondary antibodies conjugated to infrared fluorophores (Li-Cor Biosciences). Bands were visualized using an Odyssey infrared imaging system (Li-Cor Biosciences).
Statistics and reproducibility of experiments
All experimental results presented were replicated in at least three distinct experiments to ensure reproducibility. For imaging experiments, multiple cells from each replicate were analyzed. As data from different days were indistinguishable, they were pooled for statistical analysis. The specific number of cells analyzed is indicated in each figure legend.
Statistical analysis was conducted in Prism 7.0 (GraphPad Software).
Online supplemental material
Fig. S1 shows a Western blot of the strains analyzed in Figs. 1 and 2, along with measurements of lifetimes of individual CME events in those cells. The figure also contains additional examples of the data shown in Fig. 1, E and F, results from experiments used to determine the appropriate concentration and treatment time for CK-666 experiments, and localization of Myo5-TH1Δ-GFP. Fig. S2 shows montages, additional examples, and quantification of the data shown in Fig. 3. Fig. S3 shows Western blots of the strains analyzed in Fig. 4, along with measurements of CME success and lifetimes of individual CME events in those cells. Tables S1 and S2 detail the strains and plasmids used in this study, respectively. Videos 1 and 2 are the sources of the stills shown in Fig. 1, A and E, respectively. Video 3 is the source of the stills shown in Fig. 2 A and the montage shown in Fig. 3 A. Video 4, 5, 6, 7, and 8 are the sources of the stills shown in Fig. 2 D. Video 9 is the source of the stills shown in Fig. S1 G. Videos 10 and 11 are the sources of the kymographs and montages shown in Fig. 3, D and E, and Fig. S2. Videos 12, 13, and 14 are the sources of the stills shown in Fig. 4, C, D, and E, respectively.
Supplementary Material
Acknowledgments
We thank J. Hassinger, D. Serwas, and M. Welch for critically evaluating the manuscript along with M. Akamatsu and members of the Drubin/Barnes laboratory for constant informal input. We thank H. Aaron and F. Ives for their microscopy training and assistance.
Spinning disc confocal microscopy was conducted at the University of California, Berkeley, Cancer Research Laboratory Molecular Imaging center, supported by the Gordon and Betty Moore Foundation. This work was supported by the National Institutes of Health grant R35GM118149 to D.G. Drubin.
The authors declare no competing financial interests.
Author contributions: R.T.A. Pedersen and D.G. Drubin conceived of the experiments. R.T.A. Pedersen generated the reagents, performed the experiments, and analyzed the data. R.T.A. Pedersen and D.G. Drubin wrote the manuscript. D.G. Drubin secured funding.
References
- Aghamohammadzadeh S., and Ayscough K.R.. 2009. Differential requirements for actin during yeast and mammalian endocytosis. Nat. Cell Biol. 11:1039–1042. 10.1038/ncb1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard J., and Mogilner A.. 2013. Traveling waves in actin dynamics and cell motility. Curr. Opin. Cell Biol. 25:107–115. 10.1016/j.ceb.2012.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida C.G., Yamada A., Tenza D., Louvard D., Raposo G., and Coudrier E.. 2011. Myosin 1b promotes the formation of post-Golgi carriers by regulating actin assembly and membrane remodelling at the trans-Golgi network. Nat. Cell Biol. 13:779–789. 10.1038/ncb2262 [DOI] [PubMed] [Google Scholar]
- Amberg D.C., Burke D.J., and Strathern J.N.. 2005. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual, 2005 Edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY; 205 pp. [Google Scholar]
- Batchelder E.M., and Yarar D.. 2010. Differential requirements for clathrin-dependent endocytosis at sites of cell-substrate adhesion. Mol. Biol. Cell. 21:3070–3079. 10.1091/mbc.e09-12-1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement W.M., Leda M., Moe A.M., Kita A.M., Larson M.E., Golding A.E., Pfeuti C., Su K.-C., Miller A.L., Goryachev A.B., and von Dassow G.. 2015. Activator-inhibitor coupling between Rho signalling and actin assembly makes the cell cortex an excitable medium. Nat. Cell Biol. 17:1471–1483. 10.1038/ncb3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S., Kural C., Zeeh J.-C., Ubelmann F., and Kirchhausen T.. 2011. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat. Cell Biol. 13:1124–1131. 10.1038/ncb2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Grassart A., and Drubin D.G.. 2012. Myosin 1E coordinates actin assembly and cargo trafficking during clathrin-mediated endocytosis. Mol. Biol. Cell. 23:2891–2904. 10.1091/mbc.e11-04-0383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Co C., Wong D.T., Gierke S., Chang V., and Taunton J.. 2007. Mechanism of actin network attachment to moving membranes: barbed end capture by N-WASP WH2 domains. Cell. 128:901–913. 10.1016/j.cell.2006.12.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan M.C., Cope M.J., Goode B.L., Wendland B., and Drubin D.G.. 2001. Yeast Eps15-like endocytic protein, Pan1p, activates the Arp2/3 complex. Nat. Cell Biol. 3:687–690. 10.1038/35083087 [DOI] [PubMed] [Google Scholar]
- Feeser E.A., Ignacio C.M.G., Krendel M., and Ostap E.M.. 2010. Myo1e binds anionic phospholipids with high affinity. Biochemistry. 49:9353–9360. 10.1021/bi1012657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Golbano I.M., Idrissi F.Z., Giblin J.P., Grosshans B.L., Robles V., Grötsch H., Borrás M.M., and Geli M.I.. 2014. Crosstalk between PI(4,5)P2and CK2 modulates actin polymerization during endocytic uptake. Dev. Cell. 30:746–758. 10.1016/j.devcel.2014.07.020 [DOI] [PubMed] [Google Scholar]
- Geli M.I., Lombardi R., Schmelzl B., and Riezman H.. 2000. An intact SH3 domain is required for myosin I-induced actin polymerization. EMBO J. 19:4281–4291. 10.1093/emboj/19.16.4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode B.L., Rodal A.A., Barnes G., and Drubin D.G.. 2001. Activation of the Arp2/3 complex by the actin filament binding protein Abp1p. J. Cell Biol. 153:627–634. 10.1083/jcb.153.3.627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson H.V., Anderson B.L., Warrick H.M., Pon L.A., and Spudich J.A.. 1996. Synthetic lethality screen identifies a novel yeast myosin I gene (MYO5): myosin I proteins are required for polarization of the actin cytoskeleton. J. Cell Biol. 133:1277–1291. 10.1083/jcb.133.6.1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassart A., Cheng A.T., Hong S.H., Zhang F., Zenzer N., Feng Y., Briner D.M., Davis G.D., Malkov D., and Drubin D.G.. 2014. Actin and dynamin2 dynamics and interplay during clathrin-mediated endocytosis. J. Cell Biol. 205:721–735. 10.1083/jcb.201403041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassinger J.E., Oster G., Drubin D.G., and Rangamani P.. 2017. Design principles for robust vesiculation in clathrin-mediated endocytosis. Proc. Natl. Acad. Sci. USA. 114:E1118–E1127. 10.1073/pnas.1617705114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idrissi F.-Z., Blasco A., Espinal A., and Geli M.I.. 2012. Ultrastructural dynamics of proteins involved in endocytic budding. Proc. Natl. Acad. Sci. USA. 109:E2587–E2594. 10.1073/pnas.1202789109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N., and Katsuno H.. 2017. Actin Waves: Origin of Cell Polarization and Migration? Trends Cell Biol. 27:515–526. 10.1016/j.tcb.2017.02.003 [DOI] [PubMed] [Google Scholar]
- Kaksonen M., Sun Y., and Drubin D.G.. 2003. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell. 115:475–487. 10.1016/S0092-8674(03)00883-3 [DOI] [PubMed] [Google Scholar]
- Kaksonen M., Toret C.P., and Drubin D.G.. 2005. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell. 123:305–320. 10.1016/j.cell.2005.09.024 [DOI] [PubMed] [Google Scholar]
- Krendel M., Osterweil E.K., and Mooseker M.S.. 2007. Myosin 1E interacts with synaptojanin-1 and dynamin and is involved in endocytosis. FEBS Lett. 581:644–650. 10.1016/j.febslet.2007.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewellyn E.B., Pedersen R.T.A., Hong J., Lu R., Morrison H.M., and Drubin D.G.. 2015. An Engineered Minimal WASP-Myosin Fusion Protein Reveals Essential Functions for Endocytosis. Dev. Cell. 35:281–294. 10.1016/j.devcel.2015.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M.S., McKenzie A., Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., and Pringle J.R.. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 14:953–961. [DOI] [PubMed] [Google Scholar]
- MacQuarrie C.D., Mangione M., Carroll R., James M., Gould K.L., and Sirotkin V.. 2018. Adaptor protein Bbc1 regulates localization of Wsp1 and Vrp1 during endocytic actin patch assembly. bioRxiv. 10.1101/389015 (Preprint posted August 9, 2018). [DOI] [PMC free article] [PubMed]
- Michelot A., Costanzo M., Sarkeshik A., Boone C., Yates J.R. III, and Drubin D.G.. 2010. Reconstitution and protein composition analysis of endocytic actin patches. Curr. Biol. 20:1890–1899. 10.1016/j.cub.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mund M., van der Beek J.A., Deschamps J., Dmitrieff S., Hoess P., Monster J.L., Picco A., Nédélec F., Kaksonen M., and Ries J.. 2018. Systematic Nanoscale Analysis of Endocytosis Links Efficient Vesicle Formation to Patterned Actin Nucleation. Cell. 174:884–896.e17. 10.1016/j.cell.2018.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B.J., Tomasevic N., Russell A., Pierce D.W., Jia Z., McCormick C.D., Hartman J., Sakowicz R., and Pollard T.D.. 2009. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 460:1031–1034. 10.1038/nature08231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Grassart A., Lu R., Wong C.C.L., Yates J. III, Barnes G., and Drubin D.G.. 2015. Casein kinase 1 promotes initiation of clathrin-mediated endocytosis. Dev. Cell. 32:231–240. 10.1016/j.devcel.2014.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picco A., Mund M., Ries J., Nédélec F., and Kaksonen M.. 2015. Visualizing the functional architecture of the endocytic machinery. eLife. 4:1–29. 10.7554/eLife.04535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skruzny M., Brach T., Ciuffa R., Rybina S., Wachsmuth M., and Kaksonen M.. 2012. Molecular basis for coupling the plasma membrane to the actin cytoskeleton during clathrin-mediated endocytosis. Proc. Natl. Acad. Sci. USA. 109:E2533–E2542. 10.1073/pnas.1207011109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokac A.M., Schietroma C., Gundersen C.B., and Bement W.M.. 2006. Myosin-1c couples assembling actin to membranes to drive compensatory endocytosis. Dev. Cell. 11:629–640. 10.1016/j.devcel.2006.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulard A., Lechler T., Spiridonov V., Shevchenko A., Shevchenko A., Li R., and Winsor B.. 2002. Saccharomyces cerevisiae Bzz1p is implicated with type I myosins in actin patch polarization and is able to recruit actin-polymerizing machinery in vitro. Mol. Cell. Biol. 22:7889–7906. 10.1128/MCB.22.22.7889-7906.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Martin A.C., and Drubin D.G.. 2006. Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Dev. Cell. 11:33–46. 10.1016/j.devcel.2006.05.008 [DOI] [PubMed] [Google Scholar]
- Sun Y., Leong N.T., Wong T., and Drubin D.G.. 2015. A Pan1/End3/Sla1 complex links Arp2/3-mediated actin assembly to sites of clathrin-mediated endocytosis. Mol. Biol. Cell. 26:3841–3856. 10.1091/mbc.E15-04-0252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toret C.P., Lee L., Sekiya-Kawasaki M., and Drubin D.G.. 2008. Multiple pathways regulate endocytic coat disassembly in Saccharomyces cerevisiae for optimal downstream trafficking. Traffic. 9:848–859. 10.1111/j.1600-0854.2008.00726.x [DOI] [PubMed] [Google Scholar]
- Wang X., Galletta B.J., Cooper J.A., and Carlsson A.E.. 2016. Actin-Regulator Feedback Interactions during Endocytosis. Biophys. J. 110:1430–1443. 10.1016/j.bpj.2016.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner O.D., Marganski W.A., Wu L.F., Altschuler S.J., and Kirschner M.W.. 2007. An actin-based wave generator organizes cell motility. PLoS Biol. 5:e221 10.1371/journal.pbio.0050221 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.