

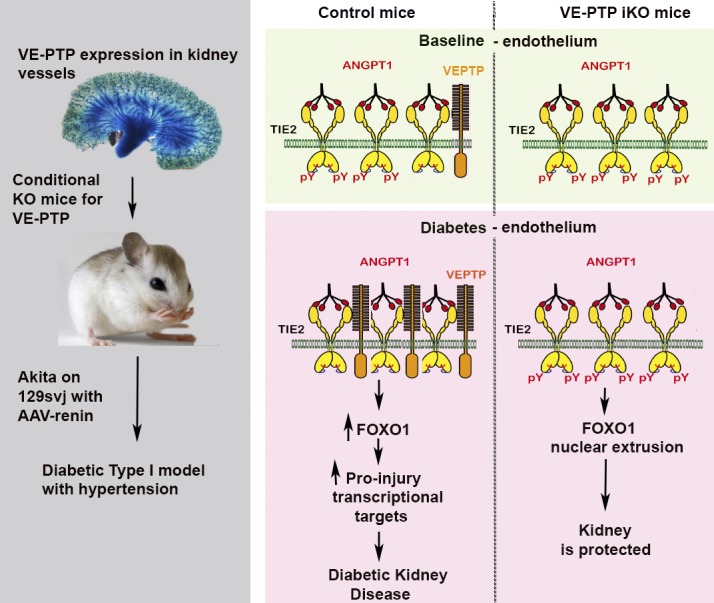
Diabetic nephropathy is a leading cause of kidney failure. VE-PTP phosphatase expression is increased in the endothelium of rodents with diabetes and hypertension. Genetic deletion of VE-PTP reduces kidney injury in diabetic mice, suggesting it may be a therapeutic target.
Abstract
Diabetic nephropathy is a leading cause of end-stage kidney failure. Reduced angiopoietin-TIE2 receptor tyrosine kinase signaling in the vasculature leads to increased vascular permeability, inflammation, and endothelial cell loss and is associated with the development of diabetic complications. Here, we identified a mechanism to explain how TIE2 signaling is attenuated in diabetic animals. Expression of vascular endothelial protein tyrosine phosphatase VE-PTP (also known as PTPRB), which dephosphorylates TIE2, is robustly up-regulated in the renal microvasculature of diabetic rodents, thereby reducing TIE2 activity. Increased VE-PTP expression was dependent on hypoxia-inducible factor transcriptional activity in vivo. Genetic deletion of VE-PTP restored TIE2 activity independent of ligand availability and protected kidney structure and function in a mouse model of severe diabetic nephropathy. Mechanistically, inhibition of VE-PTP activated endothelial nitric oxide synthase and led to nuclear exclusion of the FOXO1 transcription factor, reducing expression of pro-inflammatory and pro-fibrotic gene targets. In sum, we identify inhibition of VE-PTP as a promising therapeutic target to protect the kidney from diabetic injury.
Graphical Abstract
Introduction
Damage to the microvasculature in diabetes results in kidney failure, blindness, and nerve damage (Andersen et al., 1983; Krolewski et al., 1985; Frank, 2004; Pambianco et al., 2006). It is estimated that by the year 2040 there will be 650 million people suffering from diabetes worldwide, and ∼30–40% will develop diabetic kidney disease (DKD; Ogurtsova et al., 2017). These staggering numbers and associated burden on society underscore the urgent need to identify and develop new and effective therapies, which can prevent or slow the progression of DKD.
While the pathogenesis of microvascular complications of diabetes including DKD is complex, a large body of evidence exists to support a central role for dysregulation of major angiogenic signaling pathways (Nyengaard and Rasch, 1993; Cooper et al., 1999; Østerby et al., 2002; Guo et al., 2005; Kanesaki et al., 2005; Nakagawa et al., 2007; Kosugi et al., 2009; Sivaskandarajah et al., 2012; Saharinen et al., 2017). Altered expression of the angiopoietin ligands has been reported in rodents and patients with diabetic microvascular disease and is proposed to lead to reduced activity of the vascular receptor tyrosine kinase TIE2 (tyrosine kinase with Ig and EGF homology domains 2, also known as TEK in humans). Loss of the major TIE2 agonist/ligand ANGPT1 results in more aggressive renal disease in diabetic mice (Lee et al., 2007; Jeansson et al., 2011; Battiprolu et al., 2012; Dessapt-Baradez et al., 2014), while an increased circulating level of the TIE2 context-dependent antagonist, ANGPT2, has been linked to adverse cardiovascular, retinal, and renal outcomes in patients (Hackett et al., 2000, 2002; Chong et al., 2004; Augustin et al., 2009; David et al., 2009, 2010; Chang et al., 2013; Shroff et al., 2013; Antai et al., 2016). In a variety of vascular diseases, including malignant tumors, sepsis, obesity, and diabetes, administration of recombinant ANGPT1 or ANGPT1-mimetics has had beneficial effects on the vasculature by reducing vascular permeability and inflammation—phenotypes also observed in DKD (Joussen et al., 2002; Cho et al., 2006; Lee et al., 2007; Bitto et al., 2008; Pellegrinelli et al., 2014; Han et al., 2016). Indeed, TIE2 receptor activation has been described as the “gatekeeper” of vascular quiescence, promoting endothelial survival, junctional stability, and reduced responsiveness to TNFα (Fiedler et al., 2006).
In vivo, TIE2 activity is regulated by its ligands but also by the vascular endothelial protein tyrosine phosphatase (VE-PTP; also known as protein tyrosine phosphatase, receptor type B [PTPRB]), which potently dephosphorylates the receptor (Winderlich et al., 2009; Souma et al., 2018). Here, we report that VE-PTP transcript and protein levels are strongly up-regulated in the diabetic kidney in mice in vivo and that inhibition of VE-PTP in a mouse model of severe DKD (Harlan et al., 2018) potently protects against kidney damage. Furthermore, we demonstrate that VE-PTP inhibition affects two major molecular pathways that have been linked to DKD and are downstream of TIE2 phosphorylation in vivo: (1) increased endothelial NO synthase (eNOS) phosphorylation and (2) reduced nuclear expression of the transcription factor forkhead box protein O1 (FOXO1) and its transcriptional targets including EDN1, CTGF, PDGFB, and ANGPT2. In sum, we provide evidence to support a model whereby VE-PTP is a driver of DKD and show that VE-PTP inhibition is a promising vasculoprotective strategy for the kidney in diabetes.
Results
VE-PTP is highly expressed in the renal endothelium and is up-regulated in diabetes
VE-PTP is a vascular phosphatase with an endothelial-specific expression pattern in embryos (Fachinger et al., 1999; Carra et al., 2012; Takahashi et al., 2017). However, the details of tissue-specific VE-PTP expression in diabetes have not been reported. To characterize VE-PTP expression throughout different endothelial beds, we first analyzed mRNA expression levels of Veptp in different tissues of C57Bl/6 adult mice. The highest expression of Veptp was found in the kidney, lung, liver, and heart (Fig. S1 A). In a previously reported VeptplacZ/wt mouse reporter strain (Bäumer et al., 2006), a β-galactosidase (glb1) cassette replaces the second and third exons and is under control of the endogenous promoter. This reporter mouse line was used to examine the expression pattern of the phosphatase within the kidney. Strong Veptp expression was observed in glomerular capillaries, afferent and efferent arterioles, and interlobular vessels (Fig. 1, A and B). In peritubular capillaries of adult kidneys, weak expression of VE-PTP was observed, and no VE-PTP–positive cells were found outside of the endothelium.
Figure 1.

VE-PTP expression in adult kidneys in health and diabetes. (A) Kidney expression of Veptp in nondiabetic (VeptpLacZ/wt) and diabetic (Akita, VeptpLacZ/wt) mice is shown in 400-µm-thick vibratome sections stained with X-Gal. Higher-power views of boxed regions in the upper panels are shown in the lower panels. Bars, 1 mm (upper panel); 125 µm (lower panel). (B) Immunofluorescent staining shows colocalization of β-galactosidase (red) with an endothelial marker, endomucin (green), in a glomerulus from a kidney section of a nondiabetic (VeptpLacZ/wt) mouse. The presence of renin (magenta) allows the distinction between the afferent arteriole (white dotted line) and the efferent arteriole (yellow dotted line). Nuclei are stained with DAPI. Bars, 25 µm. (C and D) VE-PTP abundance in kidneys of diabetic and hypertensive mice. Whole-kidney lysates from the Ins2Akita/wt (Akita), db/db, and hypertensive mice after RenAAV (Ren) injection were analyzed by Western blot. (E) 24-h osmotic stimulation by high d-glucose, l-glucose or mannitol increased VE-PTP protein levels in hGECs. (F) 24-h 1% oxygen incubation resulted in elevated VE-PTP levels in hGECs. (G) mRNA analysis of kidney tissue from Ins2Akita/wt mice showed an elevation of Veptp expression that is reversed by administration of acriflavine (ACF) to reduce HIF activity. Western blot quantification from n = 4 per group; representative blots are shown; Values are means ± SEMs. *, P < 0.05; **, P < 0.01. Experiments were repeated at least once. Ctrl., control; rel., relative.
Next, we examined renal VE-PTP expression in mouse models of type 1 diabetes (Ins2Akita/wt; Akita) and type 2 diabetes (db/db; Wang et al., 1999). Similar to nondiabetic controls, VE-PTP expression remained restricted to the endothelium in diabetic mice as observed in Akita, VeptplacZ/wt mice (Fig. 1 A). However, expression levels of VE-PTP were increased in the renal cortex of both Akita and db/db mice (Fig. 1, C and D). As patients with DKD often have coexisting hypertension due to activation of the renin-angiotensin system, we tested the influence of blood pressure (BP) on VE-PTP expression in a renin-expressing, adeno-associated virus (RenAAV; Ren)–treated hypertensive mouse model (Fig. 1 C; Harlan et al., 2015). Notably, VE-PTP expression was also increased in the hypertensive mouse model. Outside the kidney, elevated VE-PTP expression was also observed in whole-lung lysate from diabetic Akita and renin-overexpressing mice (Fig. S1, B and C), suggesting extensive effects on the vasculature.
VE-PTP expression is increased by hypoxic and hyperosmolar conditions
To uncover the mechanism leading to increased expression of VE-PTP in kidneys from diabetic animals, VE-PTP protein expression was measured in primary human glomerular endothelial cells (hGECs) exposed to a variety of stimuli. We found that hyperglycemia simulated by 30 mM d-glucose as well as osmotic stress induced by 30 mM l-glucose or mannitol resulted in up-regulation of VE-PTP in hGECs (d-glucose: 1.76-fold; l-glucose: 1.61-fold; mannitol: 2.45-fold increase compared with control samples; Fig. 1 E). Similar induction of VE-PTP protein levels was observed under 1% hypoxia (2.28-fold increase; Fig. 1 F), as reported in lung blood endothelium with hypoxic stimuli (Gong et al., 2015). As renal hypoxia is a well-described component of DKD (Nangaku, 2006; Norman and Fine, 2006; Nangaku and Eckardt, 2007), we hypothesized that hypoxia-induced VE-PTP expression might explain the up-regulation of VE-PTP seen in mouse models of diabetes. To determine whether enhanced levels of VE-PTP in diabetic mice were transcriptionally regulated by hypoxia-inducible factor (HIF), we analyzed mRNA expression levels of Veptp in diabetic Akita mice following five once-daily injections of acriflavine, an inhibitor of HIFα subunit dimerization with HIF1β (Lee et al., 2009). While Veptp mRNA levels were elevated in untreated Akita mice, expression was reduced to control levels following acriflavine treatment (Fig. 1 G).
Generation of an inducible Veptp KO mouse model with enhanced TIE2 activity
The dramatic VE-PTP up-regulation in diabetic mice, combined with the increased susceptibility to renal disease observed in diabetic Angpt1 KO mice, suggested that VE-PTP deletion or inhibition as a means to activate TIE2 might be an effective therapeutic strategy for treatment of DKD (Jeansson et al., 2011). To examine this possibility and to bypass the embryonic lethality observed in conventional Veptp KO mice, we generated a VE-PTP–inducible KO mouse model (VE-PTP iKO mice) using the Cre-LoxP system paired with whole-body doxycycline-inducible deletion driven by the ubiquitously expressed Rosa26 promotor (Veptpflox/flox; Rosa26rtTA/rtTA; tetO-CrTg/wt, hereafter referred to as VE-PTP iKO mice; Fig. 2 A; Belteki et al., 2005). Using this system, 98.3 ± 0.5% deletion efficiency was achieved following doxycycline induction at postnatal day 0 (P0) as shown by Western blot analysis of kidney lysate harvested from 8-wk-old VE-PTP iKO mice (Fig. 2 B). Although deletion of Veptp any time before embryonic day 10.5 (E10.5) resulted in embryonic lethality, postnatal deletion was compatible with normal lifespan, fertility, and growth of mice (Fig. S2). As predicted, deletion of the VE-PTP phosphatase resulted in robust TIE2 activation; we observed a 3.5 ± 0.5–fold elevation of TIE2 tyrosine phosphorylation levels in kidney protein lysates as well as in isolated glomeruli from VE-PTP iKO mice (Fig. 2, B and C).
Figure 2.

Loss of VE-PTP decreases BP and increases GFR through elevated eNOS activity. (A) Targeting strategy to generate VE-PTP iKO mice; mice with a floxed Veptp exon 2 were bred to mice carrying rtTA/TetO-Cre transgenes under control of the ubiquitously expressed Rosa26 promoter. Upon induction with doxycycline, the second exon of Veptp is excised resulting in a null allele. (B) Postnatal deletion of Veptp in VE-PTP iKO mice leads to 98.3 ± 0.5% reduction of VE-PTP protein in total kidney tissue; TIE2 tyrosine phosphorylation is increased 3.5-fold in kidneys from KO mice (quantification from n = 6 per group, representative blot). (C) Elevated TIE2 tyrosine phosphorylation is observed in glomeruli isolated and pooled from kidneys of two VE-PTP iKO mice. (D) Mean systolic BP of VE-PTP iKO mice on a mixed genetic background at 42 wk of age is 28.37 ± 5 mm Hg lower than control (Ctrl.) littermates (n = 9 per group). NOS inhibition (L-NAME treatment) resulted in normalization of BP in VE-PTP iKO mice to control values (n = 6 per group). (E) Genetic deletion of Veptp increases GFR by 25%. This effect is reversed by NOS blockade (L-NAME treatment; n = 6 per group). (F) No difference in urinary ACR was observed at 12 mo of age between WT control and VE-PTP iKO mice (n = 6–7 per group). (G) eNOS phosphorylation was increased by 91.8% in kidneys from VE-PTP iKO mice compared with controls (n = 6 per group, representative blot). (H) Knock-down of Veptp by siRNA in EA.Hy926 endothelial cells (ECs) increased eNOS phosphorylation by 65.2% (n = 4 per group, representative blot). Values are means ± SEMs. n.s., not significant, *, P < 0.05; **, P < 0.01; ***, P < 0.001. Experiments were repeated at least once for each Western blot. IB, immunoblotting; IP, immunoprecipitation; rel. relative.
VE-PTP iKO mice on a mixed genetic background have a higher glomerular filtration rate (GFR) and lower BP at baseline due to enhanced eNOS signaling
To understand the role of VE-PTP in renal physiology, we measured baseline levels of GFR, BP, and urinary albumin/creatinine ratios (ACRs) in male and female VE-PTP iKO mice at 42 wk of age. At baseline, systolic BP was significantly lower by an average of 28.37 ± 5 mm Hg in VE-PTP iKO mice compared with control littermates; BP was measured by tail-cuff plethysmography for VE-PTP iKO mice compared with control littermates (Fig. 2 D). The genetic make-up of VE-PTP iKO and littermate controls was on a mixed background, including the C57Bl/6 strain. Additionally, we found that VE-PTP–deficient mice had higher GFRs at 393.3 ± 29.12 µl/min compared with 316.4 ± 15.7 µl/min in control animals (Fig. 2 E). We detected no difference in urinary ACR between control and VE-PTP–deficient mice (WT: 43.5 ± 11 µg/mg, VE-PTP iKO: 67.4 ± 30 µg/mg; Fig. 2 F), suggesting that increased GFR was not the result of increased glomerular capillary pressure. Instead, VE-PTP iKO mice demonstrated increased cortical renal blood flow as measured by multimodality magnetic resonance imaging (MRI) as a possible explanation for the increased GFR (Fig. S3).
We hypothesized that the observed GFR and BP phenotypes at baseline might be due to elevations in eNOS phosphorylation, a known downstream target of TIE2 receptor activation, which should lead to a reduction in vascular resistance and/or vasodilation; therefore, we analyzed levels of eNOS phosphorylation, which revealed a 91.8 ± 17% increase in eNOS-phosphorylation (Ser1177) in kidney tissues from VE-PTP iKO mice compared with control kidneys (Fig. 2 G). Furthermore, we showed that knock-down of PTPRB, which encodes human VE-PTP, resulted in a 65.2 ± 20% increase in eNOS Ser1177 phosphorylation levels (Fig. 2 H) in human endothelial cells. To verify enhanced eNOS signaling as the mechanism underlying differences in GFR and BP in VE-PTP iKO mice, we treated mice with the NOS inhibitor NG-nitro–l-arginine methyl ester (L-NAME) via drinking water. Blockade of NOS signaling resulted in an increase of BP in VE-PTP iKO mice (VE-PTP iKO: 35.64 ± 14 mm Hg) that was similar to BP in control animals (Fig. 2 D). Additionally, L-NAME treatment resulted in a decline of GFR in both groups (control: −73.0 ± 8 µl/min; VE-PTP iKO: −63.4 ± 7 µl/min; Fig. 2 E), demonstrating the functional dependence of elevated GFR and reduced BP upon NO signaling in VE-PTP–deficient mice.
VE-PTP deletion preserves the microvasculature and kidney function in diabetes
To study the effect of VE-PTP inhibition on the development of DKD, VE-PTP iKO mice were bred to the Akita mouse on a 129S6.SvEvTac background. Mice were backcrossed for five generations as the genetic 129S6.SvEvTac background has been shown to be more susceptible to the development of chronic kidney disease compared with other genetic backgrounds (Yang et al., 2005; Gurley et al., 2010). Veptp was deleted at P0, and male mice were aged for 12 wk before we administered RenAAV to induce hypertension through activation of renin–angiotensin system signaling (Fig. S4; Harlan et al., 2015, 2018). At 11 wk of age, diabetic Akita mice exhibited elevated systolic BP, which further increased after RenAAV injections to levels of 183.59 mm Hg in Akita and 190.72 mm Hg in Akita/VE-PTP iKO mice (Table 1). Blood glucose levels were increased to equal levels in VE-PTP WT and deficient mice under diabetic conditions. In contrast to baseline conditions on a mixed genetic background strain, we did not detect any difference in systolic BP levels between nondiabetic RenAAV-treated or control male VE-PTP WT and iKO mice on the 129S6.SvEvTac background when measured by tail-cuff plethysmography (Fig. S5, A and B). However, unexpectedly, VE-PTP deficiency led to significantly decreased levels of triglycerides and slowed the loss of body weight in diabetic mice (Table 1).
Table 1. Analysis of renal function, BP, blood glucose, plasma insulin, plasma triglycerides, and body weight of diabetic and nondiabetic WT and VE-PTP iKO mice.
| WT | VE-PTP iKO | Akita/Ren | Akita/Ren/VE-PTP iKO | P value Akita/Ren vs. Akita/ Ren/VE-PTP iKO | |
|---|---|---|---|---|---|
| GFR, 11 wk (μl/min) | 288.55 ± 13 | 290.73 ± 15 | 368.50 ± 27 | 478.24 ± 22 | ** |
| GFR, 24 wk (μl/min) | 357.63 ± 39.13 | 391.39 ± 22 | 242.63 ± 37 | 480.32 ± 79 | * |
| ACR, 24 wk | 142.61 ± 27 | 171.36 ± 38 | 1,549.40 ± 255 | 741.18 ± 160 | *** |
| Systolic BP, 11 wk (mm Hg) | 92.77 ± 7 | 90.04 ± 9 | 154.86 ± 7 | 148.70 ± 5 | n.s. |
| Systolic BP, 24 wk (mm Hg) | 107.43 ± 3 | 100.91 ± 6 | 183.59 ± 6 | 190.72 ± 9 | n.s. |
| Blood glucose (mg/dl) | 219.27 ± 9 | 205.39 ± 8 | 836.49 ± 34 | 938.36 ± 105 | n.s. |
| Plasma insulin (ng/ml) | 1.07 ± 0.1 | 0.7 ± 0.11 | 0.26 ± 0.04 | 0.23 ± 0.05 | n.s. |
| Plasma triglycerides (mg/dl) | 63.18 ± 5 | 57.05 ± 12 | 184.6 ± 24 | 100.8 ± 17 | ** |
| Body weight (g) | 33.84 ± 0.8 | 32.02 ± 0.8 | 24.52 ± 1.1 | 28.66 ± 0.9 | * |
n = 10–12/group. Values are means ± SEMs. n.s., P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To understand the molecular basis of protective effects observed in VE-PTP–deficient mice, we analyzed TIE2 signaling in these mice. Western blot analysis of lung tissue from normoglycemic control and VE-PTP–deficient mice confirmed a loss of VE-PTP and a corresponding increase of TIE2 tyrosine phosphorylation (+147.5 ± 9% compared with control tissue; Fig. 3, A and B). In diabetic hypertensive mice, VE-PTP levels in protein lysates harvested from the lung or kidney were markedly increased by 178.2 ± 24% (Fig. 3 A) and 151.2 ± 36%, respectively, (Fig. 3 C) compared with normoglycemic controls. Accordingly, TIE2 tyrosine phosphorylation levels were reduced (−45.9 ± 14% compared with controls) in these diabetic control mice (Fig. 3 B). Genetic deletion of VE-PTP in Ins2Akita/wt, RenAAV+ (Akita/Ren) mice resulted in an elevation of TIE2 phospho-tyrosine levels to a similar degree as observed in tissue harvested from normoglycemic VE-PTP iKO mice (239.4 ± 53% compared with controls; Fig. 3 B).
Figure 3.

TIE2 is activated, and kidney function is preserved in VE-PTP iKO in mice with hypertension and diabetes. (A and B) Western blot and immunoprecipitation (IP) analysis of lung tissue from Ins2Akita/wt;RenAAV+ mice showed an elevation of VE-PTP protein and reduced TIE2 phosphorylation compared with WT control (Ctrl.) samples. Deletion of VE-PTP restored TIE2 phosphorylation in lungs from diabetic, hypertensive mice (quantification from n = 6 per group, representative blot). (C) Western blot analysis of lysates from kidney tissue from Ins2Akita/wt;RenAAV+ mice also showed an increase in VE-PTP compared with control. (D) Assessment of renal function in diabetic mice before and after RenAAV administration. GFR was increased in all 12-wk-old diabetic mice compared with nondiabetic controls; GFRs were higher in VE-PTP iKO diabetic mice compared with control diabetic mice. 12 wk after RenAAV injection, diabetic control mice showed a decline in GFR whereas diabetic mice lacking VE-PTP had stable GFR (n = 10–12 per group). (E) Measurement of urinary ACRs showed lower ACRs in diabetic VE-PTP iKO mice at 10 wk following RenAAV administration compared with diabetic controls (n = 10–12 per group). Values are means ± SEMs. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Experiments were repeated at least once.
To further determine renal function in the absence and presence of VE-PTP, we measured GFRs at 11 wk of age, before RenAAV administration, as well as at 12 wk after renin overexpression. At baseline, diabetic control and VE-PTP iKO mice both exhibited elevated GFRs. At 12 wk after RenAAV injection Ins2Akita/wt, RenAAV+ mice showed a marked decline in GFR whereas VE-PTP–deficient Akita/Ren mice showed a preserved GFR (Fig. 3 D and Table 1). Assessment of urinary ACRs was performed biweekly as a surrogate in vivo marker for the degree of glomerular injury. Diabetic control mice showed a significantly higher increase in ACR over time than diabetic VE-PTP iKO mice (Fig. 3 E and Table 1).
Histopathological study of kidney sections from diabetic, hypertensive males showed severe glomerular injury with matrix expansion, mild-to-severe glomerulosclerosis, tubulo-interstitial fibrosis, as well as proteinaceous casts in the tubules. By contrast, diabetic, hypertensive VE-PTP iKO mice showed reduced glomerular matrix accumulation and glomerulosclerosis (Fig. 4, A and B). VE-PTP deficiency led to reduced deposition of fibrin and collagen in glomeruli as visualized by Martius Scarlet Blue (MSB) staining (Fig. S5, D and E). Furthermore, we observed fewer glomeruli with de novo αSMA-expressing cells in glomeruli from diabetic, hypertensive VE-PTP iKO mice compared with diabetic, hypertensive VE-PTP control littermates (Fig. 4, C and D; Young et al., 1995). No αSMA-positive glomeruli were detected in nondiabetic control or VE-PTP iKO mice. Additionally, we quantitatively analyzed the expression of glomerular endothelial and podocyte markers, including endomucin and nephrin (Fig. 4 E). Immunofluorescence analysis revealed a significant loss of glomerular endothelial cells and podocytes in Akita/Ren mice. By contrast, we found no significant reduction in endomucin or nephrin expression in VE-PTP–deficient diabetic, hypertensive mice compared with nondiabetic controls. Electron microscopic studies showed preservation of glomerular slit diaphragms and podocyte foot processes in kidneys from VE-PTP iKO mice compared with kidneys from Akita/Ren mice, which showed flattened podocyte foot processes (Fig. S5 F). Additionally, thickening of the glomerular basement membrane observed in Akita/Ren mice was also prevented in mice lacking VE-PTP (Fig. 4 F).
Figure 4.

Diabetic kidney injury is reduced in VE-PTP iKO mice. (A and B) Histological analysis of kidney sections from diabetic, hypertensive mice showed mesangial expansion, glomerulosclerosis, and interstitial changes, which were less severe in VE-PTP iKO diabetic mice compared with controls (periodic acid Schiff [PAS] staining; n = 10–12 per group). (C and D) Immunofluorescent staining showed a reduction of αSMA-positive glomeruli in diabetic VE-PTP iKO mice compared with control Akita+/RenAAV mice (n = 10–12 per group). (E) Immunofluorescent staining for nephrin and endomucin in kidney tissue showed decreased expression in control (Ctrl.) diabetic mice compared with VE-PTP iKO diabetic, hypertensive mice (n = 10–12 per group). (F) Analysis of electron microscopy images showed a reduction of glomerular basement membrane (GBM) thickening in samples harvested from diabetic VE-PTP iKO kidneys compared with diabetic VE-PTP WT mice. Bars, 100 µm (A); 5 µm (B); 50 µm (C); 20 µm (D and E); 4 µm (F). n = 1–2 mice per group; 38–52 width measurements/genotype. Values are means ± SEMs. n.s., not significant, *, P < 0.05; **, P < 0.01; ***, P < 0.001; rel. relative.
VE-PTP inhibition leads to reduced levels of the transcription factor FOXO1 and its downstream targets
To understand how TIE2 activation by VE-PTP inhibition results in vascular protection, we determined its effect on the regulation of FOXO1, a transcription factor that is increased in diabetic tissues. We found elevated AKT protein levels and decreased FOXO1 in hGECs following VE-PTP/PTPRB siRNA knock-down compared with control cells (Fig. 5, A and B). The reduction of FOXO1 also led to a decrease in genes known to be regulated by FOXO1, including PDGFB, ANGPT2, CTGF, and EDN1 (Fig. 5 C). Further analysis by immunofluorescent staining showed a reduction of FOXO1 in nuclei of hGECs 24 h after VE-PTP knock-down (Fig. 5 D). Consistent with these in vitro data, Foxo1 mRNA expression levels were reduced in renal cortical tissue from the kidneys of VE-PTP iKO mice in vivo (Fig. 5 E). Expression of FOXO1 target genes including Angpt2, Pdgfb, Ctgf, and Edn1 were also decreased in the renal cortex of VE-PTP iKO mice (Angpt2: −62.3 ± 5%, Pdgfb: −44.8 ± 3% Ctgf: −64.4 ± 4%, Edn1: −60.4 ± 4%; Fig. 5 F), suggesting a model whereby disrupted TIE2-FOXO1 signaling is involved in the pathogenesis of DKD, which is corrected through VE-PTP inhibition (Fig. 6).
Figure 5.

FOXO1 is decreased following VE-PTP inhibition. (A) siRNA knock-down of VE-PTP/PTPRB in hGECs led to elevated AKT protein levels and reduced FOXO1 protein expression shown by Western blot analysis (n = 4 per group, representative blot shown). (B and C) qPCR analysis showed reduced expression of FOXO1 and FOXO1 target genes ANGPT2, PDGFB, CTGF, and EDN1 (n = 4 per group). (D) Immunofluorescent staining showed decreased nuclear FOXO1 expression (n = 3 per group) in hGECs following VE-PTP/PTPRB knock-down. Bars, 25 µm. (E) Down-regulation of Foxo1 in mRNA isolated from renal cortex of VE-PTP iKO mice (n = 6 per group). (F) Reduced expression of the FOXO1 targets Angpt2, Pdgfb, Ctgf, and Edn1 was seen in kidneys from VE-PTP iKO diabetic mice compared with controls (Ctrl.). n = 6 per group. Experiments were repeated at least once. siNC, si negative control.
Figure 6.

Schematic model of TIE2 signaling in diabetic tissues with and without VE-PTP inhibition. Balanced TIE2 signaling in healthy endothelium stabilizes the vasculature. Under diabetic conditions, TIE2 signaling is reduced due to increased VE-PTP expression; this results in decreased eNOS phosphorylation, as well as increased Foxo1 levels and its downstream pro-fibrotic and pro-inflammatory targets Angpt2, Pdgfb, Ctgf, and Edn1. Inhibition of VE-PTP restores TIE2 phosphorylation and downstream signaling pathways resetting a “healthy” endothelial phenotype.
Discussion
Microvascular injury in diabetes results in kidney failure, blindness, and nerve damage and is strongly associated with dysregulation of major vascular signaling pathways. While anti–vascular endothelial growth factor agents have been successful in the treatment of diabetic eye complications (Cheung et al., 2010; Cheung and Wong, 2012; Do et al., 2013; Elman et al., 2015), the continued requirement for vascular endothelial growth factor signaling in the renal glomerulus throughout life precludes their use for treating DKD in patients (Eremina et al., 2003, 2008). Here, we identify a new vascular-specific therapeutic target that may protect kidney function in patients with diabetes: TIE2 activation through inhibition of the phosphatase VE-PTP.
In diabetes, an association between dysregulation of the angiopoietin–TIE2 pathway and worse outcomes has been reported in both rodents and humans (Jeansson et al., 2011; Chang et al., 2013; Dessapt-Baradez et al., 2014; Antai et al., 2016). Previous models suggest suppression of TIE2 activation may occur due to increased circulating levels of the context-dependent antagonistic ligand, ANGPT2 (Rizkalla et al., 2005; Jeansson et al., 2011; Dessapt-Baradez et al., 2014). In keeping with these models, an increased ANGPT2:ANGPT1 ratio has been linked to increased risk of both micro- and macrovascular complications in patients (Augustin et al., 2009; Koh, 2013). In our study, we were surprised to discover a dramatic up-regulation of the TIE2-inhibiting phosphatase VE-PTP in diabetic kidney tissue, identifying an alternative mechanism to explain suppression of the TIE2 vasculoprotective pathway in disease. We found that VE-PTP expression in cultured glomerular endothelial cells is potently stimulated by both hypoxia and increased osmolarity. In vivo, acriflavine, an inhibitor of HIF subunit dimerization, reduced Veptp transcript levels in a mouse model of type I diabetes, which is consistent with prior studies that have shown that HIF2α up-regulates VE-PTP in endothelial cells (Gong et al., 2015). As both hypoxia and hyperosmolarity occur in diabetic tissues and increased endothelial HIF2α levels have been reported in diabetes (Chakravarthy et al., 1998; Kowluru et al., 2001; Akita et al., 2003; El-Remessy et al., 2003; Gunton et al., 2005; Xiao et al., 2013; HoWangYin et al., 2014; Gong et al., 2015), we propose a model whereby the diabetic milieu itself down-regulates angiopoietin–TIE2 signaling through increased endothelial VE-PTP expression leading to dephosphorylation of the TIE2 receptor.
To determine VE-PTP’s potential as a novel diabetic kidney therapy, we generated a conditional VE-PTP transgenic mouse model. We found Veptp is essential for early embryogenesis, but postnatal global deletion was compatible with normal lifespan, fertility, and growth of mice. Importantly, phase I and IIa trials using a small-molecule inhibitor of VE-PTP, AKB-9778, have been completed in patients with diabetic macular edema, demonstrating the safety of VE-PTP inhibition in humans (Campochiaro et al., 2015). In mice, postnatal VE-PTP deletion led to ligand-independent constitutive activation of TIE2 and was not harmful to the vasculature or kidney function. In fact, VE-PTP iKO mice of mixed genetic background exhibited lower systolic BP and enhanced renal function (GFR was 25% higher) compared with control littermates in nondiabetic conditions. The BP effect appears to translate to humans as systolic BP was reduced in patients in the phase 1B trial after AKB-9778 treatment (Campochiaro et al., 2015). In mice, we showed these physiological effects were mediated by enhanced eNOS signaling as differences were abrogated by the administration of the NOS inhibitor L-NAME. While glomerular hyperfiltration is believed to be the first step in the development of DKD and, hence, is harmful, no albuminuria or glomerular hypertrophy was observed in VE-PTP iKO mice, suggesting the enhanced GFR due to VE-PTP inhibition is not the result of glomerular capillary hypertension. Instead, MRI studies showed enhanced renal blood flow in VE-PTP iKO mice, providing a plausible explanation for the observed increase in GFR, although other factors, such as an increased ultrafiltration coefficient, cannot be excluded.
In the setting of diabetes and severe hypertension, VE-PTP iKO mice were protected from the development of proteinuria, loss of GFR, and glomerular histopathological changes. Despite the beneficial effects on BP in control and VE-PTP nondiabetic mice, we did not observe lower BPs in the Ins2Akita;RenAAV (Akita/Ren) model, likely due to the severity of hypertension from RenAAV expression and/or the difference in genetic background strains (129S6.SvEvTac strain for diabetic studies vs. mixed strain for baseline studies). Furthermore, insulin and glucose levels were not different, and both KO and control diabetic mice exhibited hyperfiltration, suggesting the kidney-protective effects of VE-PTP inhibition are independent of glucose levels, BP, and effects on hyperfiltration. However, we did find that triglyceride levels were lower in VE-PTP iKO mice, suggesting other possible protective effects, which were not fully explored.
The elevation of VE-PTP identified in endothelial cells of diabetic mice and in glomeruli isolated from biopsy samples of patients (He et al., 2008) suggests that increased expression of the phosphatase may be pathogenic and an important driver of kidney disease progression in diabetes. Intriguingly, preliminary results from diabetic patients treated with the VE-PTP inhibitor AKB 9778 showed a reduction in albuminuria (Peters, K., M. Brigell, and S. Pakola. 2018. American Society of Nephrology Kidney Week). We posit that inhibition of this negative regulator can reset the endothelial phenotype, restoring it toward a nondiabetic state. TIE2 activation initiates a cascade of downstream signaling events, including activation of PI3K and AKT, which increases eNOS phosphorylation and local NO production, a molecular pathway previously shown to be renoprotective in diabetic mouse models (Zhao et al., 2006; Cheng and Harris, 2014; Takahashi and Harris, 2014). The BP results and L-NAME studies discussed above confirm VE-PTP inhibition increases eNOS phosphorylation in vivo. In addition to effects on eNOS, activation of the ANGPT-TIE2 pathway results in phosphorylation of the FOXO1 transcription factor, resulting in its exclusion from the nucleus. It has previously been reported that activation of FOXO1 in the endothelium occurs in the setting of diabetes (Battiprolu et al., 2012; Fiorentino et al., 2013) and leads to up-regulation of a number of genes promoting fibrogenesis and endothelial cell apoptosis such as Pdgfb, Ctgf, Edn1, and Angpt2 (Baelde et al., 2007; Dharaneeswaran et al., 2014; Dang et al., 2017). Hence, reducing FOXO1-mediated events through TIE2 activation is predicted to be vasculoprotective. In this study, we confirmed that inhibition of VE-PTP leads to decreased FOXO1 nuclear accumulation in hGECs and shows for the first time that long-term activation of TIE2 leads to decreased expression of Foxo1 mRNA in kidney tissue and reduction of its transcriptional targets.
In sum, we have shown that the endothelial-specific phosphatase VE-PTP is strongly up-regulated in the setting of diabetes, suppressing the vasculoprotective actions of the angiopoietin–TIE2 signaling cascade. Inhibition of VE-PTP using an inducible KO mouse model restored TIE2 activation and protected the kidney from diabetic injury, resulting in favorable downstream molecular signatures, including decreased expression of Foxo1. Taken altogether, these data provide a compelling rationale to consider VE-PTP inhibition as a candidate therapy to prevent and/or slow progression of DKD.
Materials and methods
Generation of mouse lines and husbandry
The Veptp lacZ reporter mouse line was used to analyze VE-PTP expression in adult kidney tissue (Bäumer et al., 2006; Winderlich et al., 2009). We generated a mouse line carrying a conditional Veptp floxed allele on the genetic C57Bl/6 background (Fig. 2 A). Conditionally ready Ptprbtm2a(EUCOMM)Hmgu JM8A3.N1.C2 ES embryonic stem cells (C57/BL6N background) were obtained from EUCOMM and injected into blastocysts in The Northwestern University Transgenic and Targeted Mutagenesis Laboratory core transgenic facility. Chimeric mice were then mated with C57BL6/N mice from Charles River Laboratories. Founder heterozygotes were identified by PCR and Southern blot genotyping. Mice were subsequently maintained on a C57BL6/N background by intercrossing or backcrossing to C57BL6/N. Mice were bred under standard husbandry conditions in standard cages with environmental enrichment. KO mice were genotyped by PCR by using the following primers: VeptpFlox, forward 5′-GCGTCTATCCAGTGGAGGACT TTC-3′, reverse 5′-CCAGGTGCCGTTCATTCAGC-3′ (WT: 489-bp product; VeptpFlox: 546-bp); VeptpDelete, forward 5′-GACAGCAAGGCGCATAACGATA-3′, reverse 5′-CCAGGTGCCGTTCATTCAGC-3′ (234-bp product). The Veptp floxed mice were further bred on strains carrying Rosa26rtTA/rtTA (Gt[ROSA]26Sortm1(rtTA,EGFP)Nagy) and tetO-Cre (Tg(tetO-cre)1Jaw) transgenes (Belteki et al., 2005) on mixed genetic backgrounds. Deletion of the floxed Veptp alleles was induced postnatally by 0.5% wt/vol doxycycline via drinking water for 21 d. VE-PTP iKO (Veptpflox/flox, Rosa26-rtTA+/+, tetO-CreTg/+) mice were then bred for five generations on the Ins2Akita 129/SvEv background for diabetic studies. Male littermates were aged to 12 wk before a single dose of 1010 GC RenAAV was administered intravenously (Harlan et al., 2015, 2018). Diabetic mice were then further observed until 24 wk of age. The mouse colony was housed, maintained, and phenotyped at the Center for Comparative Medicine of Northwestern University following ethical procedures approved by the Institutional Animal Care and Use Committee.
RNA isolation and quantitative PCR (qPCR)
RNA was isolated from various organs by using the standard trizol (Life Technologies) extraction protocol. cDNA was reverse-transcribed by using the iSCRIPT synthesis kit (Bio-Rad). Murine mRNA levels were determined by SYBR green qPCR on a 7500 real-time cycler (Applied Biosystems). The following primers were used (m, mouse; h, human): m/hGAPDH-F: 5′-AAGGTCATCCCAGAGCTGAA-3′, m/hGAPDH-R: 5′-CTGCTTCACCACCTTCTTGA-3′; mVeptp-F: 5′-TCTATCCAAGGAATATGAGG-3′, mVeptp-R: 5′-ACCCCATGGTCTGGCCACACC-3′; hPDGFB-F: 5′-GATCCGCTCCTTTGATGATC-3′, hPDGFB-R: 5′-GTCTCACACTTGCATGCCAG-3′; mPecam1-F: 5′-CGGTGTTCAGCGAGATCC-3′, mPecam1-R: 5′-ACTCGACAGGATGGAAATCAC-3′; mFoxo1-F: 5′-ACATTTCGTCCTCGAACCAGCTCA-3′, mFoxo1-R: 5′-ATTTCAGACAGACTGGGCAGCGTA-3′; hFOXO1-F:3′-GCCATGTAAGTCCCATCAGGA-5′, hFOXO1-R: 5′-ATCGGAACAAGAACGTGGAATC-3′; mAngpt2-F: 5′-GATCTTCCTCCAGCCCCTAC-3′, mAngpt2-R: 5′-TTTGTGCTGCTGTCTGGTTC-3′;, m/hEDN1-F: 5′-TCTCTGCTGTTTGTGGCTTG 3′, m/hEDN1-R: 5′-GACTGGGAGTGGGTTTCTCC-3′; mCtgf-F: 5′-GTGCCAGAACGCACACTG-3′, mCtgf-R: 5′-CCCCGGTTACACTCCAAA-3′; and hCtgf-F: 5′-TTGGCCCAGACC CAACTA-3′, hCtgf-R: 5′-GCAGGAGGCGTTGTCATT-3′.
Histology and immunostaining
Kidney tissues were fixed overnight in 4% paraformaldehyde in PBS and embedded in paraffin blocks followed by 5-µm-thick sectioning for immunofluorescent staining. Paraffin sections were deparaffinized and stained for periodic acid Schiff or MSB or processed for immunofluorescent staining following standard protocols for antigen retrieval, blocking in 5% BSA/3% donkey serum in PBS, and antibody incubation. The following primary antibodies were used: rabbit–VE-PTP-c (University of Muenster, Germany; Nawroth et al., 2002), endomucin (Abcam; Ab106100), pecam1 (CD31; BD Bioscience; 55337), β-galactosidase (Life Technologies; A11132), nephrin (R&D Systems; AF3159), α-SMA (Sigma-Aldrich; F3777), and renin (Aviva Systems Biology; OACA02177). Immunofluorescent images were obtained using a Nikon A1 confocal microscope from the Center of Advanced Microscopy at Northwestern University. For X-Gal staining, VeptpLacZ/wt and Akita, VeptpLacZ/wt animals were anesthetized using ketamine/xylazine and perfused through the left ventricle with heparinized saline (10 U/ml heparin in PBS) until the exudate was free of blood. Animals were then perfused with 20 ml 0.2% paraformaldehyde to fix the tissues. Both kidneys of each animal were then collected and 400-µm sections were obtained using a vibratome set with frequency 40 Hz, amplitude 1.2 mm, and speed 30 mm/s (Microm HM650V; Thermo Scientific). Sections were permeabilized with three washes of rinse solution (2 mM MgCl2, 0.01% Na deoxycholate, 0.1% IGEPAL CA-630, and 100 mM Tris buffer, pH 8) before incubation in the LacZ staining solution (2 mM MgCl2, 0.01% Na deoxycholate, 0.1% IGEPAL CA-630, 5 mM K4Fe(CN)6, 5 mM K3Fe(CN)6, and 1 mg/ml X-Gal in PBS, pH 7.5) at 37°C for 2 h. Chloroquine (0.3 mM final) was added at each washing and staining step to inactivate endogenous β-galactosidase. After staining, the reaction was quenched by replacing the staining solution with 10% phosphate-buffered formalin overnight. Sections were cleared in 1% KOH solution up to 24 h and mounted between coverslips for imaging.
Ultrastructure analysis by electron microscopy
Dissected kidneys were fixed in 0.1 M cacodylate buffer (pH 7.5) containing 2% glutaraldehyde and 4% formaldehyde and postfixed in 1% OsO4. Fixed specimens were then dehydrated and embedded in epoxy resin for sectioning. Ultrathin resin sections were stained with a solution containing uranyl acetate and lead acetate and viewed in an FEI Tecnai G2 transmission electron microscope.
Protein analysis by Western blot
Protein samples from kidney tissue or cell culture were lysed in radioimmunoprecipitation assay buffer with protease inhibitor and phosphatase inhibitor cocktails (Sigma-Aldrich). Lysates were then separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes by the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked in 5% BSA/TBS-0.5% Tween-20 followed by incubation with primary antibodies overnight at 4°C. For immunoprecipitation, tissue lysates were incubated with anti-TIE2 antibody and subsequently with Dynabeads against Protein A (Invitrogen). The samples were eluted with Laemmli sample buffer with a reducing agent containing 200 mM dithiothreitol. The following antibodies were used for Western blot analysis: TIE2 antibody (Santa Cruz; C-20; SC-324), 4G-10 platinum anti-phosphotyrosine (EMD Millipore; 05–1050), α-tubulin antibody (Genscript; A01410), eNOS (Cell Signaling Technology; 49G3), phospho-eNOS (Cell Signaling Technology; C9C3), and FOXO1 (Cell Signaling Technology; C29H4). HRP-tagged secondary antibodies (Jackson ImmunoResearch) were used in a dilution of 1:10,000 in TBS-0.5% Tween. Quantification of protein expression was performed on multiple replicate experiments using the software ImageJ.
Glomerular protein isolation
Renal cortices were isolated from VE-PTP iKO mice and Cre-negative controls at 10 wk of age, and glomeruli were isolated by using a sieving protocol as previously described (Thongboonkerd, 2008). To increase yield, cortices from two littermate mice with matching genotype (four kidneys) were pooled into a single sample before sieving. After pooling, minced tissue was passed through 150- and 75-µm sieves before glomeruli were captured by using a 53-µm stainless steel sieve and resuspended in ice-cold sterile PBS (Gibco). Glomeruli were then collected by centrifugation (1,000 g for 5 min at 4°C) and frozen in liquid N2. For analysis of TIE2 phosphorylation, frozen glomeruli were thawed and lysed in radioimmunoprecipitation assay buffer (50 mM Tris, 150 mM NaCl, 1% IGEPAL CA-630, 0.5% Na deoxycholate, and 0.1% SDS, pH 7.5) containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich). Lysates were centrifuged (14,000 g for 15 min at 4°C), and 100 µg of the resulting supernatant was used for immunoprecipitation using anti-TIE2 antibodies as described above.
Urine and plasma analysis
Urine ACRs were determined by ELISA (Albuwell M kit; Exocell) and the microtiter-format colorimetric Jaffe reaction assay by using alkaline picrate. Blood glucose was measured by a Contour blood glucose meter (Bayer). Plasma insulin was determined by ELISA (Thermo Scientific) with the help of the Northwestern University Comprehensive Metabolic Core. Plasma triglycerides were measured by autoanalyzer at Eli Lilly & Company.
GFR and BP measurement
GFRs were measured by FITC-labeled sinistrin clearance obtaining seven consecutive blood draws over the time course of 75 min after retro-orbital 1% FITC sinistrin injection (56.1 μg sinistrin/g body weight; Fresenius Kabi). Plasma clearance of FITC-sinistrin was measured fluorometrically (λex = 488 nm/λex = 538 nm) from blood samples obtained via the saphenous vein at seven postinjection time points from 3 to 75 min. Systolic BP measurements were obtained by tail-cuff plethysmography (Kent Scientific). Mice were trained in the system for 3 consecutive days during the same time of day before BP measurements were recorded.
Measurement of renal blood flow by MRI
Multiple MRI techniques were used for the imaging of kidney anatomy and function by using a subset of mice (two to three) from each group (WT controls and VE-PTP iKO). All scans were conducted on a dedicated high-field small-animal MRI (7T Bruker Clinscan) by using a four-channel phased array coil placed on the mouse kidney region. Scans included a series of dynamic contrast-enhanced MRI (DCE-MRI) acquisitions following injection of gadolinium contrast and a set of T1/T2 mapping sequences for anatomical and quantitative functional assessment of the kidneys. From the DCE-MRI data, semiquantitative kidney maps were obtained from the sequential images 30 s after injection. This parameter was selected to qualitatively describe different regional kidney perfusion.
Inhibition of NOS signaling
1 g/ml L-NAME was administered to VE-PTP iKO mice and control littermates via the drinking water for 5 d before GFRs and BP were measured (Mülsch and Busse, 1990; Rees et al., 1990).
Inhibition of HIF signaling
To inhibit HIF subunit dimerization in diabetic Ins2Akita mice, acriflavine (Sigma-Aldrich) was administered intraperitoneally at 2 mg/kg daily for 5 d before kidney tissue was harvested for further analysis (Lee et al., 2009).
Cell culture
Primary human glomerular microvascular endothelial cells (Applied Cell Biology Research) and EA.Hy926 human endothelial cells were cultured in EBM-2 medium including EGM-BulletKit factors (Lonza) and DMEM high glucose, respectively, by using standard cell culture methods. For osmotic stimulation, cells were treated 24 h with 30 mM d-glucose, l-glucose or mannitol. Hypoxia was induced by incubation at 1% O2 in a custom-made oxygen chamber. For siRNA-mediated gene knock-down, Lipofectamine RNAi max transfection reagent (Thermo Fisher, 13778075) was used as recommended by the manufacturer. Briefly, 30 pmol of siRNA targeting human VE-PTP/PRPTB (Ambion Silence Select; s11536) or control scramble siRNA was transfected into the cultured cells on six-well plates, and the cells were harvested at 24 h and 48 h after transfection. (Souma et al., 2018).
Statistics
Quantitative phenotyping data were expressed as means and their corresponding SEs. Means were compared for statistical significance by two-tailed Student’s t test, one-way ANOVA with Bonferroni comparison, or two-way ANOVA accordingly. P < 0.05 was considered statistically significant.
Online supplemental material
Fig. S1 shows VE-PTP expression in tissues of WT and diabetic and hypertensive mice. Fig. S2 shows the Kaplan Meier Survival plot of VE-PTP–deficient mice versus control littermates. Fig. S3 shows renal perfusion images obtained by MRI following gadolinium contrast agent injection. Fig. S4 shows a schematic diagram of the generation of diabetic hypertensive VE-PTP iKO mice. Fig. S5 shows BP, insulin, and glucose levels and additional histological images from diabetic hypertensive mice versus control mice.
Acknowledgments
We thank Venus Onay, Megan Kelly, Ronnie Anderson, and Anna Woo for their technical support. We thank Edward Thorp and Matthew DeBerge for providing their oxygen chamber. We thank Sandra Acosta for her help with the vibratome sections.
This study was funded by National Institute of Health grants R01HL124120, T32DK108738, R01EY025799, and P30DK114857 (S.E. Quaggin). I.A. Carota was supported by the Lilly Innovation Fellowship Award program from Eli Lilly. Imaging was performed at the Northwestern University Center for Advanced Microscopy and supported by National Cancer Institute CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. The genetically engineered mice were generated with the assistance of Northwestern University Transgenic and Targeted Mutagenesis Laboratory (National Institutes of Health grant CA060553). The metabolic analysis was supported by the Comprehensive Metabolic Core at Northwestern University.
S.E. Quaggin holds patents related to therapeutic targeting of the ANGPT–TEK pathway in ocular hypertension and glaucoma and receives research support, owns stock in, and is a director of Mannin Research. S.E. Quaggin is an external advisory board member of Astra Zeneca. During the time of study completion and manuscript preparation, M.D. Breyer, S.S. Oladipupo, and I.A. Carota were employed by Eli Lilly & Company, and S. Yamaguchi was employed by Daiichi Sankyo. D. Vestweber is a scientific advisory board member of Aerpio Pharmaceuticals. The other authors declare no competing financial interests.
Author contributions: S.E. Quaggin, I.A. Carota, and M.D. Breyer designed the study; I.A. Carota, Y. Kenig-Kozlovsky, T. Onay, R. Scott, B.R. Thomson, T. Souma, C.S. Bartlett, Y. Li, D. Procissi, V. Ramirez, S. Yamaguchi, A. Tarjus, C.E. Tanna, C. Li, V. Eeremina, and S.S. Oladipupo carried out the experiments; S.E. Quaggin, I.A. Carota, R. Scott, B.R. Thomson, T. Souma, Y. Li, D. Procissi, A. Tarjus, D. Vestweber, S.S. Oladipupo, and M.D. Breyer analyzed the data; I.A. Carota, B.R. Thomson, T. Souma, R. Scott, A. Tarjus, and D. Procissi made the figures; D. Vestweber, S.S. Oladipupo, and M.D. Breyer provided reagents; S.E. Quaggin, I.A. Carota, and B.R. Thomson drafted and revised the paper; and all authors approved the final version of the manuscript.
References
- Akita T., Murohara T., Ikeda H., Sasaki K., Shimada T., Egami K., and Imaizumi T.. 2003. Hypoxic preconditioning augments efficacy of human endothelial progenitor cells for therapeutic neovascularization. Lab. Invest. 83:65–73. 10.1097/01.LAB.0000050761.67879.E4 [DOI] [PubMed] [Google Scholar]
- Andersen A.R., Christiansen J.S., Andersen J.K., Kreiner S., and Deckert T.. 1983. Diabetic nephropathy in Type 1 (insulin-dependent) diabetes: an epidemiological study. Diabetologia. 25:496–501. 10.1007/BF00284458 [DOI] [PubMed] [Google Scholar]
- Antai Z., Shvetsov M., Kozlovskaya L., Serova A., and Mukhin N.. 2016. Urinary excretion of angiogenesis factors in chronic glomerulonephritis patients: Association with clinical activity and urinary biomarkers of kidney injury. Nephrol. Dial. Transplant. 31(suppl_1):i429–i430. 10.1093/ndt/gfw188.25 [DOI] [Google Scholar]
- Augustin H.G., Koh G.Y., Thurston G., and Alitalo K.. 2009. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 10:165–177. 10.1038/nrm2639 [DOI] [PubMed] [Google Scholar]
- Baelde H.J., Eikmans M., Lappin D.W., Doran P.P., Hohenadel D., Brinkkoetter P.T., van der Woude F.J., Waldherr R., Rabelink T.J., de Heer E., and Bruijn J.A.. 2007. Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int. 71:637–645. 10.1038/sj.ki.5002101 [DOI] [PubMed] [Google Scholar]
- Battiprolu P.K., Hojayev B., Jiang N., Wang Z.V., Luo X., Iglewski M., Shelton J.M., Gerard R.D., Rothermel B.A., Gillette T.G., et al. 2012. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Invest. 122:1109–1118. 10.1172/JCI60329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer S., Keller L., Holtmann A., Funke R., August B., Gamp A., Wolburg H., Wolburg-Buchholz K., Deutsch U., and Vestweber D.. 2006. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood. 107:4754–4762. 10.1182/blood-2006-01-0141 [DOI] [PubMed] [Google Scholar]
- Belteki G., Haigh J., Kabacs N., Haigh K., Sison K., Costantini F., Whitsett J., Quaggin S.E., and Nagy A.. 2005. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 33:e51 10.1093/nar/gni051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto A., Minutoli L., Galeano M.R., Altavilla D., Polito F., Fiumara T., Calò M., Lo Cascio P., Zentilin L., Giacca M., and Squadrito F.. 2008. Angiopoietin-1 gene transfer improves impaired wound healing in genetically diabetic mice without increasing VEGF expression. Clin. Sci. (Lond.). 114:707–718. 10.1042/CS20070250 [DOI] [PubMed] [Google Scholar]
- Campochiaro P.A., Sophie R., Tolentino M., Miller D.M., Browning D., Boyer D.S., Heier J.S., Gambino L., Withers B., Brigell M., and Peters K.. 2015. Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates Tie2. Ophthalmology. 122:545–554. 10.1016/j.ophtha.2014.09.023 [DOI] [PubMed] [Google Scholar]
- Carra S., Foglia E., Cermenati S., Bresciani E., Giampietro C., Lora Lamia C., Dejana E., Beltrame M., and Cotelli F.. 2012. Ve-ptp modulates vascular integrity by promoting adherens junction maturation. PLoS One. 7:e51245 10.1371/journal.pone.0051245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy U., Hayes R.G., Stitt A.W., McAuley E., and Archer D.B.. 1998. Constitutive nitric oxide synthase expression in retinal vascular endothelial cells is suppressed by high glucose and advanced glycation end products. Diabetes. 47:945–952. 10.2337/diabetes.47.6.945 [DOI] [PubMed] [Google Scholar]
- Chang F.C., Lai T.S., Chiang C.K., Chen Y.M., Wu M.S., Chu T.S., Wu K.D., and Lin S.L.. 2013. Angiopoietin-2 is associated with albuminuria and microinflammation in chronic kidney disease. PLoS One. 8:e54668 10.1371/journal.pone.0054668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H., and Harris R.C.. 2014. Renal endothelial dysfunction in diabetic nephropathy. Cardiovasc. Hematol. Disord. Drug Targets. 14:22–33. 10.2174/1871529X14666140401110841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N., and Wong T.Y.. 2012. Changing trends of blindness: the initial harvest from translational public health and clinical research in ophthalmology. Am. J. Ophthalmol. 153:193–195. 10.1016/j.ajo.2011.11.022 [DOI] [PubMed] [Google Scholar]
- Cheung N., Mitchell P., and Wong T.Y.. 2010. Diabetic retinopathy. Lancet. 376:124–136. 10.1016/S0140-6736(09)62124-3 [DOI] [PubMed] [Google Scholar]
- Cho C.H., Sung H.K., Kim K.T., Cheon H.G., Oh G.T., Hong H.J., Yoo O.J., and Koh G.Y.. 2006. COMP-angiopoietin-1 promotes wound healing through enhanced angiogenesis, lymphangiogenesis, and blood flow in a diabetic mouse model. Proc. Natl. Acad. Sci. USA. 103:4946–4951. 10.1073/pnas.0506352103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong A.Y., Caine G.J., Freestone B., Blann A.D., and Lip G.Y.H.. 2004. Plasma angiopoietin-1, angiopoietin-2, and angiopoietin receptor tie-2 levels in congestive heart failure. J. Am. Coll. Cardiol. 43:423–428. 10.1016/j.jacc.2003.08.042 [DOI] [PubMed] [Google Scholar]
- Cooper M.E., Vranes D., Youssef S., Stacker S.A., Cox A.J., Rizkalla B., Casley D.J., Bach L.A., Kelly D.J., and Gilbert R.E.. 1999. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 48:2229–2239. 10.2337/diabetes.48.11.2229 [DOI] [PubMed] [Google Scholar]
- Dang L.T.H., Aburatani T., Marsh G.A., Johnson B.G., Alimperti S., Yoon C.J., Huang A., Szak S., Nakagawa N., Gomez I., et al. 2017. Hyperactive FOXO1 results in lack of tip stalk identity and deficient microvascular regeneration during kidney injury. Biomaterials. 141:314–329. 10.1016/j.biomaterials.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S., Kümpers P., Hellpap J., Horn R., Leitolf H., Haller H., and Kielstein J.T.. 2009. Angiopoietin 2 and cardiovascular disease in dialysis and kidney transplantation. Am. J. Kidney Dis. 53:770–778. 10.1053/j.ajkd.2008.11.030 [DOI] [PubMed] [Google Scholar]
- David S., Kümpers P., Lukasz A., Fliser D., Martens-Lobenhoffer J., Bode-Böger S.M., Kliem V., Haller H., and Kielstein J.T.. 2010. Circulating angiopoietin-2 levels increase with progress of chronic kidney disease. Nephrol. Dial. Transplant. 25:2571–2576. 10.1093/ndt/gfq060 [DOI] [PubMed] [Google Scholar]
- Dessapt-Baradez C., Woolf A.S., White K.E., Pan J., Huang J.L., Hayward A.A., Price K.L., Kolatsi-Joannou M., Locatelli M., Diennet M., et al. 2014. Targeted glomerular angiopoietin-1 therapy for early diabetic kidney disease. J. Am. Soc. Nephrol. 25:33–42. 10.1681/ASN.2012121218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharaneeswaran H., Abid M.R., Yuan L., Dupuis D., Beeler D., Spokes K.C., Janes L., Sciuto T., Kang P.M., Jaminet S.S., et al. 2014. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ. Res. 115:238–251. 10.1161/CIRCRESAHA.115.303227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do D.V., Nguyen Q.D., Khwaja A.A., Channa R., Sepah Y.J., Sophie R., Hafiz G., and Campochiaro P.A.. READ-2 Study Group . 2013. Ranibizumab for edema of the macula in diabetes study: 3-year outcomes and the need for prolonged frequent treatment. JAMA Ophthalmol. 131:139–145. 10.1001/2013.jamaophthalmol.91 [DOI] [PubMed] [Google Scholar]
- Elman M.J., Ayala A., Bressler N.M., Browning D., Flaxel C.J., Glassman A.R., Jampol L.M., and Stone T.W.. Diabetic Retinopathy Clinical Research Network . 2015. Intravitreal Ranibizumab for diabetic macular edema with prompt versus deferred laser treatment: 5-year randomized trial results. Ophthalmology. 122:375–381. 10.1016/j.ophtha.2014.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Remessy A.B., Abou-Mohamed G., Caldwell R.W., and Caldwell R.B.. 2003. High glucose-induced tyrosine nitration in endothelial cells: role of eNOS uncoupling and aldose reductase activation. Invest. Ophthalmol. Vis. Sci. 44:3135–3143. 10.1167/iovs.02-1022 [DOI] [PubMed] [Google Scholar]
- Eremina V., Sood M., Haigh J., Nagy A., Lajoie G., Ferrara N., Gerber H.P., Kikkawa Y., Miner J.H., and Quaggin S.E.. 2003. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Invest. 111:707–716. 10.1172/JCI17423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eremina V., Jefferson J.A., Kowalewska J., Hochster H., Haas M., Weisstuch J., Richardson C., Kopp J.B., Kabir M.G., Backx P.H., et al. 2008. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 358:1129–1136. 10.1056/NEJMoa0707330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinger G., Deutsch U., and Risau W.. 1999. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene. 18:5948–5953. 10.1038/sj.onc.1202992 [DOI] [PubMed] [Google Scholar]
- Fiedler U., Reiss Y., Scharpfenecker M., Grunow V., Koidl S., Thurston G., Gale N.W., Witzenrath M., Rosseau S., Suttorp N., et al. 2006. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat. Med. 12:235–239. 10.1038/nm1351 [DOI] [PubMed] [Google Scholar]
- Fiorentino L., Cavalera M., Menini S., Marchetti V., Mavilio M., Fabrizi M., Conserva F., Casagrande V., Menghini R., Pontrelli P., et al. 2013. Loss of TIMP3 underlies diabetic nephropathy via FoxO1/STAT1 interplay. EMBO Mol. Med. 5:441–455. 10.1002/emmm.201201475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank R.N. 2004. Diabetic retinopathy. N. Engl. J. Med. 350:48–58. 10.1056/NEJMra021678 [DOI] [PubMed] [Google Scholar]
- Gong H., Rehman J., Tang H., Wary K., Mittal M., Chaturvedi P., Zhao Y.Y., Komarova Y.A., Vogel S.M., and Malik A.B.. 2015. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Invest. 125:652–664. 10.1172/JCI77701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunton J.E., Kulkarni R.N., Yim S., Okada T., Hawthorne W.J., Tseng Y.H., Roberson R.S., Ricordi C., O’Connell P.J., Gonzalez F.J., and Kahn C.R.. 2005. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell. 122:337–349. 10.1016/j.cell.2005.05.027 [DOI] [PubMed] [Google Scholar]
- Guo M., Ricardo S.D., Deane J.A., Shi M., Cullen-McEwen L., and Bertram J.F.. 2005. A stereological study of the renal glomerular vasculature in the db/db mouse model of diabetic nephropathy. J. Anat. 207:813–821. 10.1111/j.1469-7580.2005.00492.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley S.B., Mach C.L., Stegbauer J., Yang J., Snow K.P., Hu A., Meyer T.W., and Coffman T.M.. 2010. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am. J. Physiol. Renal Physiol. 298:F788–F795. 10.1152/ajprenal.90515.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett S.F., Ozaki H., Strauss R.W., Wahlin K., Suri C., Maisonpierre P., Yancopoulos G., and Campochiaro P.A.. 2000. Angiopoietin 2 expression in the retina: upregulation during physiologic and pathologic neovascularization. J. Cell. Physiol. 184:275–284. 10.1002/1097-4652(200009)184:3<275::AID-JCP1>3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- Hackett S.F., Wiegand S., Yancopoulos G., and Campochiaro P.A.. 2002. Angiopoietin-2 plays an important role in retinal angiogenesis. J. Cell. Physiol. 192:182–187. 10.1002/jcp.10128 [DOI] [PubMed] [Google Scholar]
- Han S., Lee S.J., Kim K.E., Lee H.S., Oh N., Park I., Ko E., Oh S.J., Lee Y.S., Kim D., et al. 2016. Amelioration of sepsis by TIE2 activation-induced vascular protection. Sci. Transl. Med. 8:335ra55 10.1126/scitranslmed.aad9260 [DOI] [PubMed] [Google Scholar]
- Harlan S.M., Ostroski R.A., Coskun T., Yantis L.D., Breyer M.D., and Heuer J.G.. 2015. Viral transduction of renin rapidly establishes persistent hypertension in diverse murine strains. Am. J. Physiol. Regul. Integr. Comp. Physiol. 309:R467–R474. 10.1152/ajpregu.00106.2015 [DOI] [PubMed] [Google Scholar]
- Harlan S.M., Heinz-Taheny K.M., Sullivan J.M., Wei T., Baker H.E., Jaqua D.L., Qi Z., Cramer M.S., Shiyanova T.L., Breyer M.D., and Heuer J.G.. 2018. Progressive Renal Disease Established by Renin-Coding Adeno-Associated Virus-Driven Hypertension in Diverse Diabetic Models. J. Am. Soc. Nephrol. 29:477–491. 10.1681/ASN.2017040385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Sun Y., Takemoto M., Norlin J., Tryggvason K., Samuelsson T., and Betsholtz C.. 2008. The glomerular transcriptome and a predicted protein-protein interaction network. J. Am. Soc. Nephrol. 19:260–268. 10.1681/ASN.2007050588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HoWangYin K.Y., Loinard C., Bakker W., Guérin C.L., Vilar J., d’Audigier C., Mauge L., Bruneval P., Emmerich J., Lévy B.I., et al. 2014. HIF-prolyl hydroxylase 2 inhibition enhances the efficiency of mesenchymal stem cell-based therapies for the treatment of critical limb ischemia. Stem Cells. 32:231–243. 10.1002/stem.1540 [DOI] [PubMed] [Google Scholar]
- Jeansson M., Gawlik A., Anderson G., Li C., Kerjaschki D., Henkelman M., and Quaggin S.E.. 2011. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Invest. 121:2278–2289. 10.1172/JCI46322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joussen A.M., Poulaki V., Tsujikawa A., Qin W., Qaum T., Xu Q., Moromizato Y., Bursell S.E., Wiegand S.J., Rudge J., et al. 2002. Suppression of diabetic retinopathy with angiopoietin-1. Am. J. Pathol. 160:1683–1693. 10.1016/S0002-9440(10)61115-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanesaki Y., Suzuki D., Uehara G., Toyoda M., Katoh T., Sakai H., and Watanabe T.. 2005. Vascular endothelial growth factor gene expression is correlated with glomerular neovascularization in human diabetic nephropathy. Am. J. Kidney Dis. 45:288–294. 10.1053/j.ajkd.2004.09.020 [DOI] [PubMed] [Google Scholar]
- Koh G.Y. 2013. Orchestral actions of angiopoietin-1 in vascular regeneration. Trends Mol. Med. 19:31–39. 10.1016/j.molmed.2012.10.010 [DOI] [PubMed] [Google Scholar]
- Kosugi T., Heinig M., Nakayama T., Connor T., Yuzawa Y., Li Q., Hauswirth W.W., Grant M.B., Croker B.P., Campbell-Thompson M., et al. 2009. Lowering blood pressure blocks mesangiolysis and mesangial nodules, but not tubulointerstitial injury, in diabetic eNOS knockout mice. Am. J. Pathol. 174:1221–1229. 10.2353/ajpath.2009.080605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru R.A., Tang J., and Kern T.S.. 2001. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 50:1938–1942. 10.2337/diabetes.50.8.1938 [DOI] [PubMed] [Google Scholar]
- Krolewski A.S., Warram J.H., Christlieb A.R., Busick E.J., and Kahn C.R.. 1985. The changing natural history of nephropathy in type I diabetes. Am. J. Med. 78:785–794. 10.1016/0002-9343(85)90284-0 [DOI] [PubMed] [Google Scholar]
- Lee K., Zhang H., Qian D.Z., Rey S., Liu J.O., and Semenza G.L.. 2009. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA. 106:17910–17915. 10.1073/pnas.0909353106 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lee S., Kim W., Moon S.O., Sung M.J., Kim D.H., Kang K.P., Jang K.Y., Lee S.Y., Park B.H., Koh G.Y., and Park S.K.. 2007. Renoprotective effect of COMP-angiopoietin-1 in db/db mice with type 2 diabetes. Nephrol. Dial. Transplant. 22:396–408. 10.1093/ndt/gfl598 [DOI] [PubMed] [Google Scholar]
- Mülsch A., and Busse R.. 1990. NG-nitro-L-arginine (N5-[imino(nitroamino)methyl]-L-ornithine) impairs endothelium-dependent dilations by inhibiting cytosolic nitric oxide synthesis from L-arginine. Naunyn Schmiedebergs Arch. Pharmacol. 341:143–147. [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Sato W., Glushakova O., Heinig M., Clarke T., Campbell-Thompson M., Yuzawa Y., Atkinson M.A., Johnson R.J., and Croker B.. 2007. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J. Am. Soc. Nephrol. 18:539–550. 10.1681/ASN.2006050459 [DOI] [PubMed] [Google Scholar]
- Nangaku M. 2006. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 17:17–25. 10.1681/ASN.2005070757 [DOI] [PubMed] [Google Scholar]
- Nangaku M., and Eckardt K.U.. 2007. Hypoxia and the HIF system in kidney disease. J. Mol. Med. (Berl.). 85:1325–1330. 10.1007/s00109-007-0278-y [DOI] [PubMed] [Google Scholar]
- Nawroth R., Poell G., Ranft A., Kloep S., Samulowitz U., Fachinger G., Golding M., Shima D.T., Deutsch U., and Vestweber D.. 2002. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. 21:4885–4895. 10.1093/emboj/cdf497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman J.T., and Fine L.G.. 2006. Intrarenal oxygenation in chronic renal failure. Clin. Exp. Pharmacol. Physiol. 33:989–996. 10.1111/j.1440-1681.2006.04476.x [DOI] [PubMed] [Google Scholar]
- Nyengaard J.R., and Rasch R.. 1993. The impact of experimental diabetes mellitus in rats on glomerular capillary number and sizes. Diabetologia. 36:189–194. 10.1007/BF00399948 [DOI] [PubMed] [Google Scholar]
- Ogurtsova K., da Rocha Fernandes J.D., Huang Y., Linnenkamp U., Guariguata L., Cho N.H., Cavan D., Shaw J.E., and Makarof L.E.. 2017. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 128:40–50. 10.1016/j.diabres.2017.03.024 [DOI] [PubMed] [Google Scholar]
- Østerby R., Hartmann A., and Bangstad H.J.. 2002. Structural changes in renal arterioles in Type I diabetic patients. Diabetologia. 45:542–549. 10.1007/s00125-002-0780-2 [DOI] [PubMed] [Google Scholar]
- Pambianco G., Costacou T., Ellis D., Becker D.J., Klein R., and Orchard T.J.. 2006. The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes. 55:1463–1469. 10.2337/db05-1423 [DOI] [PubMed] [Google Scholar]
- Pellegrinelli V., Rouault C., Veyrie N., Clément K., and Lacasa D.. 2014. Endothelial cells from visceral adipose tissue disrupt adipocyte functions in a three-dimensional setting: partial rescue by angiopoietin-1. Diabetes. 63:535–549. 10.2337/db13-0537 [DOI] [PubMed] [Google Scholar]
- Rees D.D., Palmer R.M., Schulz R., Hodson H.F., and Moncada S.. 1990. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol. 101:746–752. 10.1111/j.1476-5381.1990.tb14151.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizkalla B., Forbes J.M., Cao Z., Boner G., and Cooper M.E.. 2005. Temporal renal expression of angiogenic growth factors and their receptors in experimental diabetes: role of the renin-angiotensin system. J. Hypertens. 23:153–164. 10.1097/00004872-200501000-00026 [DOI] [PubMed] [Google Scholar]
- Saharinen P., Eklund L., and Alitalo K.. 2017. Therapeutic targeting of the angiopoietin-TIE pathway. Nat. Rev. Drug Discov. 16:635–661. 10.1038/nrd.2016.278 [DOI] [PubMed] [Google Scholar]
- Shroff R.C., Price K.L., Kolatsi-Joannou M., Todd A.F., Wells D., Deanfield J., Johnson R.J., Rees L., Woolf A.S., and Long D.A.. 2013. Circulating angiopoietin-2 is a marker for early cardiovascular disease in children on chronic dialysis. PLoS One. 8:e56273 10.1371/journal.pone.0056273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaskandarajah G.A., Jeansson M., Maezawa Y., Eremina V., Baelde H.J., and Quaggin S.E.. 2012. Vegfa protects the glomerular microvasculature in diabetes. Diabetes. 61:2958–2966. 10.2337/DB11-1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souma T., Thomson B.R., Heinen S., Carota I.A., Yamaguchi S., Onay T., Liu P., Ghosh A.K., Li C., Eremina V., et al. 2018. Context-dependent functions of angiopoietin 2 are determined by the endothelial phosphatase VEPTP. Proc. Natl. Acad. Sci. USA. 115:1298–1303. 10.1073/pnas.1714446115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., and Harris R.C.. 2014. Role of endothelial nitric oxide synthase in diabetic nephropathy: lessons from diabetic eNOS knockout mice. J. Diabetes Res. 2014:590541 10.1155/2014/590541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Kim R., Lauhan C., Park Y., Nguyen N.G., Vestweber D., Dominguez M.G., Valenzuela D.M., Murphy A.J., Yancopoulos G.D., et al. 2017. Expression of receptor-type protein tyrosine phosphatase in developing and adult renal vasculature. PLoS One. 12:e0177192–e0177192. 10.1371/journal.pone.0177192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongboonkerd V. 2008. Renal and urinary proteomics. Proteomics Clin. Appl. 2:947–949. 10.1002/prca.200890024 [DOI] [PubMed] [Google Scholar]
- Wang J., Takeuchi T., Tanaka S., Kubo S.K., Kayo T., Lu D., Takata K., Koizumi A., and Izumi T.. 1999. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Invest. 103:27–37. 10.1172/JCI4431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winderlich M., Keller L., Cagna G., Broermann A., Kamenyeva O., Kiefer F., Deutsch U., Nottebaum A.F., and Vestweber D.. 2009. VE-PTP controls blood vessel development by balancing Tie-2 activity. J. Cell Biol. 185:657–671. 10.1083/jcb.200811159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H., Gu Z., Wang G., and Zhao T.. 2013. The possible mechanisms underlying the impairment of HIF-1α pathway signaling in hyperglycemia and the beneficial effects of certain therapies. Int. J. Med. Sci. 10:1412–1421. 10.7150/ijms.5630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T., Huang Y.G., Ye W., Hansen P., Schnermann J.B., and Briggs J.P.. 2005. Influence of genetic background and gender on hypertension and renal failure in COX-2-deficient mice. Am. J. Physiol. Renal Physiol. 288:F1125–F1132. 10.1152/ajprenal.00219.2004 [DOI] [PubMed] [Google Scholar]
- Young B.A., Johnson R.J., Alpers C.E., Eng E., Gordon K., Floege J., Couser W.G., and Seidel K.. 1995. Cellular events in the evolution of experimental diabetic nephropathy. Kidney Int. 47:935–944. 10.1038/ki.1995.139 [DOI] [PubMed] [Google Scholar]
- Zhao H.J., Wang S., Cheng H., Zhang M.-Z., Takahashi T., Fogo A.B., Breyer M.D., and Harris R.C.. 2006. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J. Am. Soc. Nephrol. 17:2664–2669. 10.1681/ASN.2006070798 [DOI] [PMC free article] [PubMed] [Google Scholar]