

Abstract
The reaction intermediates of reduced bovine Cytochrome c Oxidase (CcO) were trapped following its reaction with oxygen at 50 μs to 6 msec by innovative freeze-quenching methods and studied by EPR. When the enzyme was reduced with either ascorbate or dithionite, distinct radicals were generated; X-band (9 GHz) and D-band (130 GHz) CWEPR measurements support the assignments of these radicals to ascorbyl and sulfur dioxide anion radical (SO 2−•) respectively. The X-band spectra show a linewidth of 12 G for the ascorbyl radical and 11 G for the SO 2−• radical and an isotropic g-value of 2.005 for both species. The D-band spectra reveal clear distinctions in the g-tensors and powder patterns of the two species. The ascorbyl radical spectrum displays approximate axial symmetry with g-values of gx = 2.0068, gy = 2.0066, and gz = 2.0023. The SO2−• radical has rhombic symmetry with g-values of gx = 2.0089, gy = 2.0052, and gz = 2.0017. When the contributions from the ascorbyl and SO2−• radicals were removed, no protein-based radical on CcO could be identified in the EPR spectra.
1. INTRODUCTION
Radical-based catalysis is integral to biological processes in organisms ranging from archaebacteria [1] to mammals [2]. Protein-based radicals are essential to a variety of enzymatic systems, [3], such as ribonucleotide reductases [4, 5] and heme peroxidases [6–12]. Cytochrome c Oxidase (CcO), which catalyzes the reduction of oxygen to water, has been proposed to involve a protein-based radical [13–15] in its catalytic chemistry. Such a radical would explain how CcO sustains turnover without releasing reactive oxygen species even under conditions of limited electron availability. Despite impressive efforts and a variety of experimental approaches, the identity and role of radical species formed during the reaction of oxygen with CcO remain elusive.
Mammalian CcO is a 204 kDa membrane protein, which functions as Complex IV in the electron transport chain. Electrons derived from catabolism of fats, amino acids, and sugars enter the chain at Complex I and arrive via Cytochome c (Cyt c) at CcO, where a bound oxygen molecule is the terminal electron acceptor. CcO catalyzes this exergonic reduction of oxygen to water, harnessing the released energy to pump four net protons across the inner mitochondrial membrane. The reaction of CcO with oxygen has been heavily studied using small molecule reductants such as ascorbate or dithionite as the external electron source for Cyt c and ultimately CcO. From these studies, several intermediates along the catalytic pathway have been identified. One such intermediate, termed “P,” is believed to contain a protein-based radical, although its identity is highly debated [14, 16–18].
Wilson et al. detected a radical during the reaction of oxygen with fully reduced bovine CcO; the authors concluded that it was an artifact from the reductant, although they could not rule out a protein-based radical [18]. In a radioactive iodide study, Babcock and coworkers reported a radical on the modified tyrosine, Y244, which is believed to donate a proton and an electron during catalysis [13]. In the reaction of oxygen with the fully reduced forms of two bacterial oxidases, P. denitrificans aa3 and E. coli bo3, de Vries and coworkers suggested that as many as three radicals, ascribed to tryptophanyl and an unknown “6 msec” species, form during turnover [14, 17]. However, neither the locations of the radicals nor their role in catalysis could be determined; furthermore, their formation could not be correlated with the expected steps in the catalytic cycle. In addition, L-ascorbic acid was used as the primary reductant, but its effect on the EPR spectra was not addressed.
L-ascorbic acid (AH2) is an endogenous water-soluble antioxidant present in up to millimolar concentrations in the human body [19]. It has pKa values of 4.2 and 11.6 [20] and exists predominantly as the anion, AH−, at neutral pH. Ascorbate oxidation involves the loss of a proton and an electron, which may occur via several possible mechanisms: (1) AH− may lose an electron forming the neutral free radical, AH•, which deprotonates to form the semidehydroascorbate radical anion, A−•; (2) AH− deprotonates to form the dianion, A2-, which then releases an electron to form A−•; (3) AH− forms A−• directly by a concerted proton-electron transfer. The first mechanism can be ruled out by its unfavorable midpoint potential (E0 = +766 mV) [21]. The second mechanism is suggested to occur in the reduction of Cyt c [22]. The third mechanism has been demonstrated in cytochrome b561 [23]. In addition, recent energy calculations propose that a di-radical forms, although the mechanism is unclear [24, 25].
Although the mechanism may vary, the end product is conventionally accepted to be A−•, which has been used as a spectroscopic indicator of oxidative stress due to its half-life of ~50 sec [26, 27]. A−• has an unpaired electron in a highly delocalized π-system, conferring stability as the “terminal small-molecule antioxidant” [28]. A second one-electron oxidation produces dehydroascorbate, which is reduced and recycled in mitochondria to ascorbate [29–31]. Thus, the redox chemistry of ascorbate has immense physiological relevance as well as value as a laboratory reagent.
Sodium dithionite, which is another common reductant, has a midpoint potential of E0 = −420 mV at pH 7 [32]. The dithionite ion, S2O42- can be considered a dimer of SO2−• radicals, as the S-S bond is exceptionally long [33]; the reduction reactions of dithionite were suggested to occur via the SO2−• radical, whose EPR spectra have been studied [34, 35]. SO2−• may arise from thermal decomposition prior to reduction or in the commercial preparation of sodium dithionite [35].
In biological systems, multiple radical species often occur in mechanisms involving radical migration between tryptophan, tyrosine, glycine, and cysteine residues as well as from a porphyrin pi-cation radical in heme systems [3]. These systems give rise to complex EPR signatures. In addition, radicals originating from reductants used for reducing biological systems may further complicate EPR spectra. Thus, it is essential to identify these reductant-based radical components in order to make assignments of biologically relevant protein-based radicals.
To determine how the radicals formed from dithionite and ascorbate can affect EPR spectra obtained during the oxygen chemistry of CcO, we systematically investigated the reactions of these reductants with CcO. We found that when ascorbate/Cyt c or dithionite are used to reduce bovine CcO prior to its reaction with oxygen, ascorbyl or SO2−• radicals are generated and may be trapped under conditions typically used to detect reaction intermediates. We report the first multi-frequency EPR characterization of these radicals.
2. MATERIALS AND METHODS
CcO was purified from bovine heart tissue via the method described by Yoshikawa [36]. Enzyme concentrations were determined by taking the optical absorption difference between fully-reduced CcO at 604 nm and the oxidized enzyme at 630 nm, using an extinction coefficient of 23.3 mM−1 cm−1 [37].
Natural abundance L-ascorbic acid was obtained from Fisher Scientific. Isotopically labeled L-ascorbic acid was obtained from Omicron Biochemicals. A 1 M stock solution was prepared in degassed 0.2 M sodium phosphate buffer with the pH adjusted to 7.4 with NaOH. A final concentration of 10 mM ascorbate was added to degassed 160 μM CcO and incubated with 0.5 μM Cyt c as the electron carrier.
Sodium dithionite was obtained from Sigma. A 1 M stock solution was prepared in degassed 0.2 M sodium phosphate buffer at pH 7.4. A final concentration of 6 mM dithionite was added to degassed 600 μM CcO without Cyt c.
Rapid freeze-quench (RFQ) is a novel technology used to trap transient radicals formed at room temperature for spectroscopic characterization at low temperature. RFQ samples were prepared using a custom-built device described by Lin et al. [38] and Egawa et al. [39]. Resting CcO, reduced with either ascorbate or dithionite in a gas-tight syringe, was mixed with oxygenated buffer in the RFQ device at room temperature. Samples were freeze-quenched on a timescale of 50 μsec to 6 msec. The frozen powder was packed into 4-mm O.D. (X-band) and 0.55 mm O.D. (D-band) precision-bore quartz EPR tubes immersed in liquid nitrogen. Hand-quenched samples were prepared in similar EPR tubes and frozen in liquid nitrogen on the timescale of minutes.
X-band (9 GHz) measurements were made on a Varian E-line spectrometer. A finger dewar was filled with liquid nitrogen and inserted into the EPR cavity preserved the sample at 77 K. Experimental conditions were: modulation amplitude, 3.2 G; microwave power, 1 mW; receiver gain, 2.5 × 104; microwave frequency, 9.107 GHz.
D-band (130 GHz) two pulse echo-detected spectra were obtained on a spectrometer described elsewhere [40, 41] using the following parameters: temperature, 7 K; repetition rate, 30 Hz; 30 averages per point; 90 degree pulse, 50 ns; time τ between pulses, 130 ns. For both X- and D-band spectra, the field was calibrated using Mn2+ doped in MgO [42].
Low temperature optical absorption was measured at 77 K on a modified Linkam THMS600 microscope stage system with a halogen light source.
3. RESULTS AND DISCUSSION
3.1. Single-turnover reaction of O2 with ascorbate-reduced CcO
Fig. 1A shows optical absorption measurements of the reaction intermediates freeze-quenched at 50 to 6000 μs. The visible band exhibits a red-shift from 603 nm (50 μs) to 604 nm (150 μs), where it remains for ~1000 μs until shifting to 602 nm and finally to 599 nm (6000 μs). These shifts suggest progression from the fully reduced form, R, to the pulsed oxidized form of the enzyme, OH. We find that the visible α-band of the pulsed oxidized form at 599 nm is red-shifted relative to the resting oxidized form at 597 nm, consistent with that reported in the literature [43–45]. The Soret band is saturated at the high enzyme concentration needed for EPR measurements and hence, is not shown.
Fig. 1.
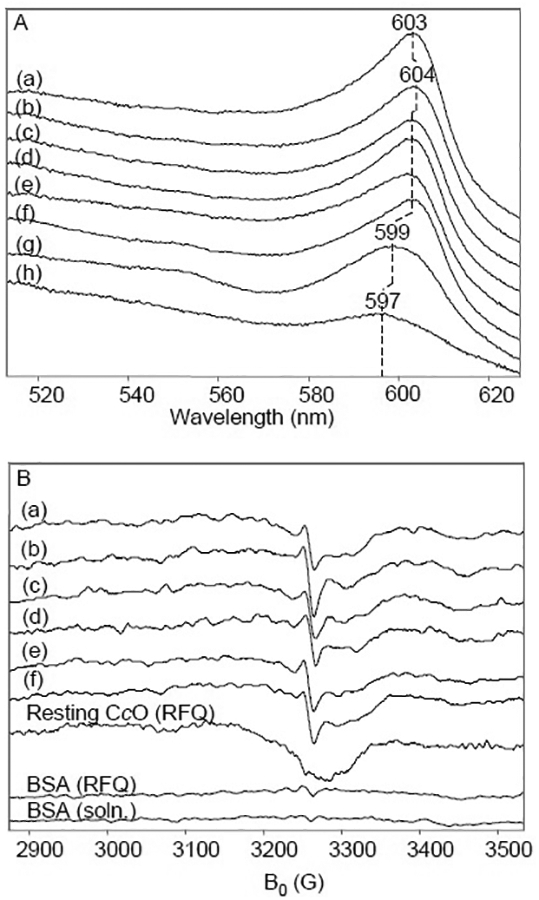
Low-temperature optical absorption and X-band CW-EPR measurements of bovine CcO. A. 80 μM CcO completely reduced by 10 mM ascorbate and catalytic amounts of Cyt c under Ar then mixed with O2-saturated buffer and rapid freeze-quenched at time points (a) 50 μs, (b) 150 μs, (c) 300 μs, (d) 400 μs, (e) 540 μs, (f) 1 ms, (g) 6 ms. (h) is a sample of the resting enzyme. Optical absorption measurements were made at 83 K. Solutions were prepared in 200 mM NaPi buffer, pH 7.4, containing 0.2% w/v n-decyl-β-D-maltoside (DM). B. X-band EPR under the same conditions with quench times of (a) 50 μs, (b) 150 μs, (c) 300 μs, (d) 540 μs, (e) 1 ms, (f) 6 ms. (h) is a sample made by running resting enzyme through the RFQ device. The bottom two spectra are control samples of BSA run through the RFQ device (upper) and simply frozen in solution (lower). The conditions of EPR spectroscopy were: microwave power, 1 mW; microwave frequency, 9.1 GHz; modulation amplitude, 3.2 G; temperature 77 K.
In the corresponding X-band CW-EPR spectra for the RFQ samples, a narrow radical with a linewidth of 12 G overlies features of cupric CuA in all samples containing oxygen-containing intermediates (Fig. 1B). The isolated CuA2+ signal is seen clearly in the RFQ resting CcO sample and can be simulated with the parameters, gx = 1.99, gy = 2.02, and gz = 2.18, consistent with reported values [46–48]. To demonstrate that the narrow radical was not formed due to passing a non-specific protein solution through the RFQ device, resting CcO and BSA were mixed with buffer and freeze-quenched in a similar fashion. As shown in Fig. 1B, the narrow radical was not generated in these controls.
High frequency EPR was used to determine the g-tensor of the radical. The radical has a powder pattern consistent with near axial symmetry and can be simulated with gx = 2.0068, gy = 2.0066, and gz = 2.0023 (Fig. 2a). The isotropic g-value calculated from the trace of the g-tensor matches values reported for the ascorbyl radical at room temperature, g = 2.0052 [49].
Fig. 2.
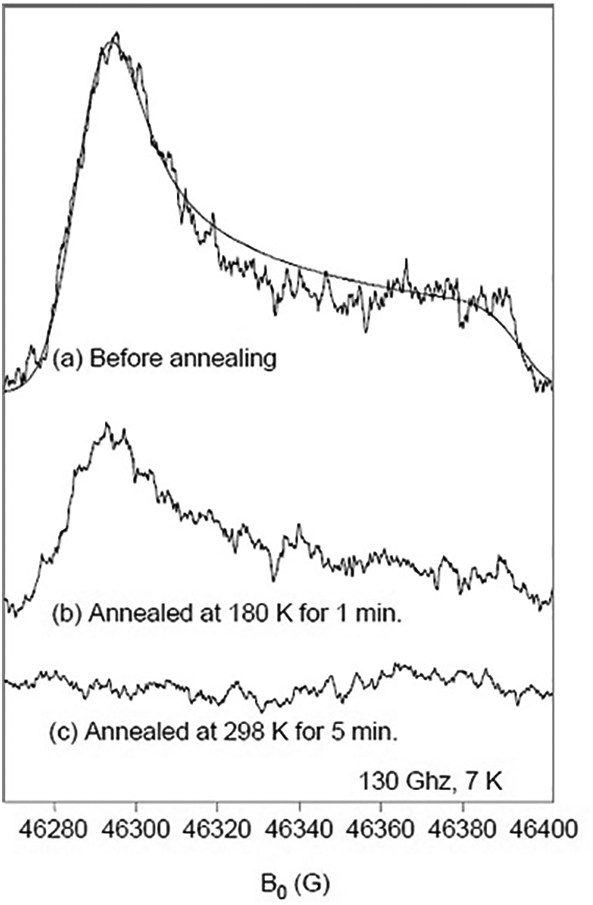
D-band EPR spectra of RFQ CcO prepared with ascorbate. (a) D-band EPR measurements of 80 μM CcO, completely reduced by 10 mM ascorbate and catalytic amounts of Cyt c under Ar, then mixed with O2-saturated buffer and rapid freeze-quenched at 150 μs. A simulation with gx = 2.0068, gy = 2.0066, and gz= 2.0023 in the dotted line. Sample from (a) annealed at the conditions: (b) 180 K for 1 min and (c) 298 K for 5 min. The conditions for the D-band Hahn echo-detected spectra were: microwave frequency, 129.998 GHz; repetition rate, 50 Hz; averages per point, 100; 90 degree pulse, 50 ns; time between pulses, 130 ns; temperature, 7K.
Annealing experiments were performed, in which the 150 μs sample was warmed to either 180 K or 298 K then cooled back to 7 K for EPR measurements. The paramagnetic species diminishes in the D-band spectra after annealing (Fig. 2b and c). The stability of this radical species is consistent with its assignment as the ascorbyl radical. The same radical species could be observed by hand-quenching the CcO reaction during multiple-turnover conditions as shown in Fig. 3.
Fig. 3.
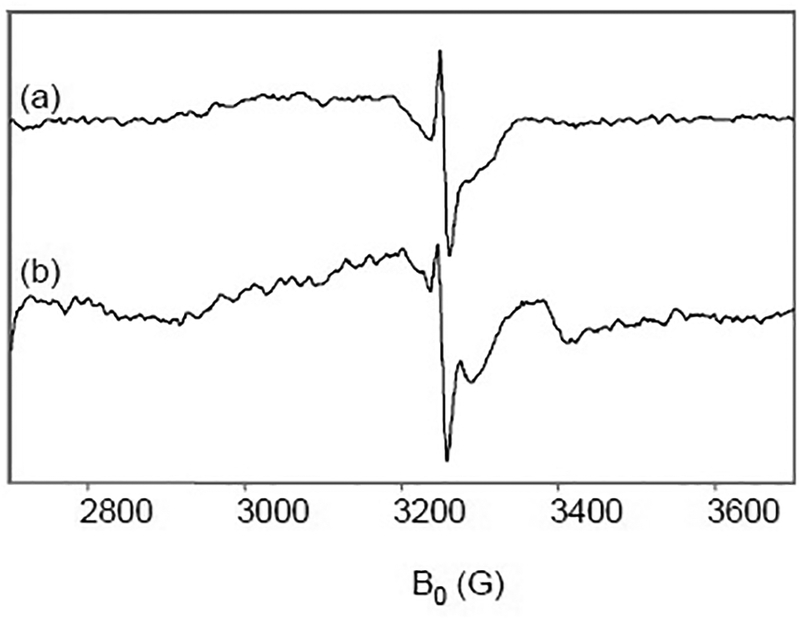
X-band EPR spectra of CcO under multiple-turnover and single-turnover. (a) 150 μM CcO completely reduced by 10 mM ascorbate and catalytic amounts of Cyt c under Ar then mixed with O2-saturated buffer and hand-quenched at 5 min. (b) 150 μM CcO completely reduced by 10 mM. ascorbate and catalytic amounts of Cyt c under Ar then mixed with O2-saturated buffer and rapid freeze-quenched at 150 μs. EPR conditions as in Fig. 1.
3.2. Multiple-turnover reaction of O2 with ascorbate-reduced CcO
The stability of the radical observed in the single-turnover reaction suggested that large amounts of radical could be trapped in the multiple-turnover reaction. In particular, an increase in radical would be seen if its production depended on the CcO reaction—either in the reduction of CcO to produce R or in the oxygen chemistry to produce P, F, or OH. To test this, we prepared hand-quenched samples in which the ascorbate-reduced CcO was mixed with O2-saturated buffer and allowed to turn over for several minutes. An accumulation of the relatively stable radical species was detected by X-band CWEPR. Fig. 3 shows the amount of unpaired spin in the multiple-turnover compared to the single-turnover samples. The signal-to-noise is higher in the multiple-turnover samples, which are frozen liquid, compared to the RFQ samples, which are frozen powders. The origin of the feature at ~3360 G in the RFQ sample spectrum is unknown at present but it does not interfere with our analysis of the signal at g~2.005.
The principal g-values and stability of the radical species at room temperature were consistent with the ascorbyl radical rather than a protein radical arising from CcO. To confirm the assignment, experiments were carried out with isotopically-labeled ascorbate. L-[1-13C]-ascorbic acid was used to repeat the hand-quenched and RFQ experiments. A characteristic splitting of the radical signal due to its coupling to the 13C nucleus was observed in both samples (Fig. 4). Lowering the power to 0.3 mW and narrowing the field sweep to 50 G allowed detection of the radical species with minimal contributions from CuA2+. The linewidth of the natural abundance ascorbyl radical is remarkably narrow, measuring 9 G, yet subtle hyperfine coupling broaden the edges of the signal (arrows). The hyperfine structure can be fit by adding 4 protons, expected from the hydrogen-atoms attached at the C4, C5, and C6 positions [49]. Adding the 13C nucleus with an isotropic hyperfine coupling of 6.54 in the C1 position simulates the splitting seen in the isotopic data, similar to that reported for the ascorbyl radical (5.74 G) in the literature [49, 50]. The hyperfine structure was well-fit using isotropic values in both the frozen liquid and RFQ samples, suggesting that the hyperfine anisotropy is too small to significantly influence the quality of the simulations. The simulation parameters are shown in Table 1.
Fig. 4.
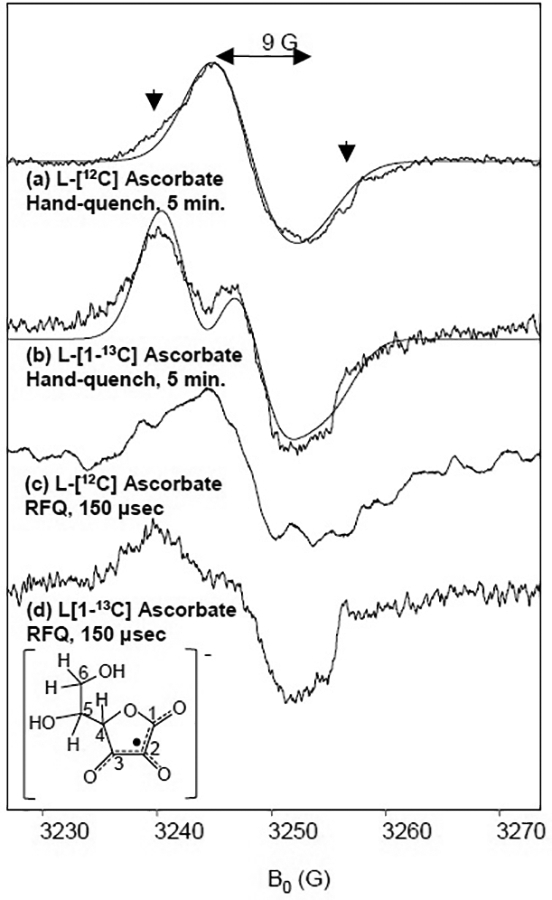
X-band EPR spectra of CcO prepared with isotopically-labeled ascorbate. 150 μM CcO completely reduced by (a) 10 mM natural abundance ascorbate or (b) 10 mM L-[1-13C] ascorbate and catalytic amounts of Cyt c under Ar then mixed with O2-saturated buffer and hand-quenched at 5 min. 150 μM CcO completely reduced by (c) 10 mM natural abundance ascorbate or (d) 10 mM L-[1-13C] ascorbate and catalytic amounts of Cyt c under Ar then mixed with O2-saturated buffer and rapid freeze-quenched at 150 μs. Simulations are shown in the dotted lines with parameters shown in Table 1. EPR conditions were: frequency, 9.1 GHz; power, 0.3 mW; temperature, 77 K.
Table 1.
Simulation parameters for the ascorbyl radical.
| L-[12C] Ascorbic Acid |
L-[1-13C] Ascorbic Acid |
|
|---|---|---|
| gx | 2.0068 | 2.0068 |
| gy | 2.0066 | 2.0066 |
| gz | 2.0023 | 2.0023 |
| aH4 | 1.76a | 1.76a |
| aH5 | 0.07a | 0.07a |
| aH6,1 | 0.19a | 0.19a |
| aH6,2 | 0.19a | 0.19a |
| ac1 | - | 6.54a |
Hyperfine couplings are given in units of Gauss.
To determine if the ascorbyl radical originated from the one-electron reduction reaction of Cyt c, Cyt c and ascorbate were mixed in various ratios and frozen within 5 minutes. One study on the reduction of single-site metalloenzymes suggested that the dianion acts as the major reductant, forming the anionic ascorbate radical, which decays by a slow disproportionation reaction [22]. However, unlike the situation in the presence of CcO, we found that no radicals accumulate at 5 minutes in preparations of 1:1 or 20:1 Cyt c to reductant (Fig. 5a and b). Thus, upon the reduction of Cyt c alone, the ascorbyl radical initially formed must undergo further reactions which render it EPR silent. One possibility is that the disproportionation reaction is somehow accelerated under our experimental conditions. Another possibility is that the ascorbyl radical also acts as a reductant for Cyt c, producing the diamagnetic product dehydroascorbate in a reaction that is somehow prevented in the presence of CcO. Although the chemistry of the process is unclear at this stage, the end result is that ascorbate reduces Cyt c alone without the generation of an EPR-detectable concentration of radicals, while reduction in the presence of CcO produces ascorbyl radical signals as early as 50 μs which are stable on the timescale of minutes.
Fig. 5.
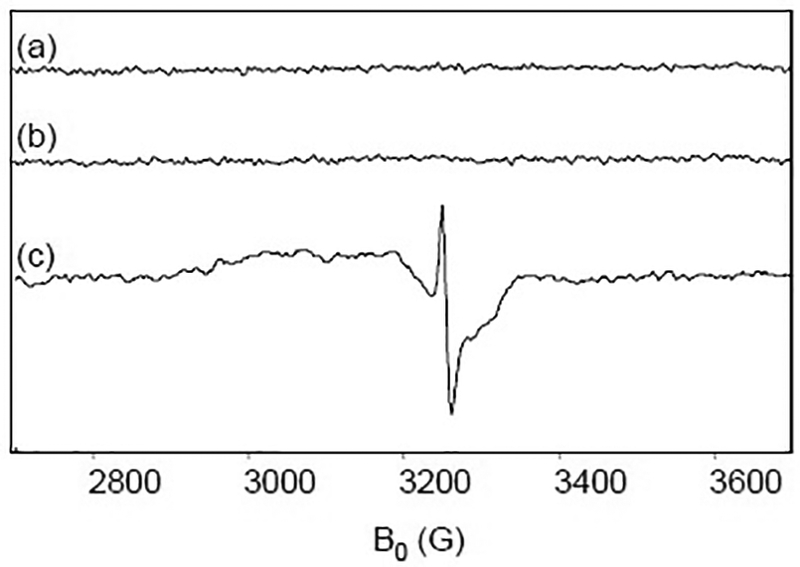
X-band EPR spectra of Cyt c and ascorbate. (a) 5 mM Cyt c is reduced with 5 mM ascorbate and frozen at 77 K at 5 min. (b) 0.5 mM Cyt c is reduced with 10 mM ascorbate and frozen at 77 K at 5 min. (c) 150 μM CcO and 0.5 μM Cyt c are reduced with 10 mM ascorbate and frozen at 77 K at 5 min. The pH of all samples is 7.4. EPR conditions as in Fig. 1.
A major physiological function of cellular ascorbate is to quench cytotoxic oxoferryl species and protein-based radicals produced on hemoglobin during oxidative stress [51]. Similar reactions have been demonstrated in leghaemoglobin [52] and Mb [53]. Thus, a possible mechanism in CcO is that ascorbate reacts with an oxoferryl heme or a protein-based radical to generate the observed radical species. We stress that detection of the ascorbyl free radical neither proves nor disproves that CcO produces a protein-based radical in the oxygen reaction.
3.3. Previously reported radicals in the reaction of O2 with ascorbate-reduced CcO
Wilson et al. studied bovine CcO using ascorbate as the reductant to create multiple turnover conditions and reported a g ~ 2 radical with a linewidth of 8 G that amounted to as much as 10% of the enzyme concentration [18]. They noted that when the enzyme is reduced with only ferrocytochrome c, which was passed on a column to remove excess ascorbate, the magnitude of the radical signal decreased to ~1% of the enzyme concentration; however, they were unable to determine if the signal resulted from a residual ascorbyl radical or a protein-based radical.
In 2007, Wiertz et al. reported the presence of two radicals in the reaction of ascorbate-reduced Paracoccus denitrificans aa3 with oxygen [14] The first species, present from 83 μs to ~1 ms, was assigned as a tryptophanyl radical derivative of W272 (P. denitrificans CcO numbering, W236 in bovine) based on simulations of Q-band data. A radical with similar features in X-band spectra was reported in the bo3 oxidase from E. coli [17]. A second unidentified, narrow radical existed from 83 μs through 6 msec. The authors suggested the source to be a “main-chain radical” but concluded that its identity and functionality could not be determined. This second “6 msec” species accounted for ~0.5% of the enzyme concentration and had g-values reported as 2.0022, 1.9965, 1.9994, which differ from those reported here for the ascorbyl radical.
For the bovine enzyme, when we subtract the ascorbyl radical and oxidized CuA from our data, we are unable to identify a protein-based radical. In addition, no protein-based radical was detected when CcO was reduced with dithionite, which was subsequently removed by a gel-filtration column. The data indicate that protein-based are not observed in bovine CcO when the fully-reduced enzyme is reacted with oxygen; in addition, ascorbate produces an ascorbyl radical in the oxygen reaction of bovine CcO. These data contrast the bacterial system, in which a tryptophanyl radical arises and no ascorbyl radical is reported even when the same concentration (10 mM) is used as a reductant [14].
Either the mechanism of the bovine enzyme or the reagents—specifically, the reductants and mediators—may cause these differences between the bovine and the bacterial systems. The observations of Wilson et al. along with ours are most consistent with differing mechanisms between mammalian and bacterial enzymes. This would suggest that electron donation from an amino acid is unnecessary in the robust mammalian chemistry but occurs in bacterial enzymes. This difference is interesting because in both systems, the metal centers of the fully-reduced enzyme are capable of donating the four electrons necessary to complete the reduction. If radicals were to form in the reaction of bovine CcO, they could be quenched by ascorbate, obscuring the detection of any protein-based radicals. However, it should be noted that in the studies of the bacterial enzymes by de Vries and coworkers, phenazine ethosulfate (PES) was used as the redox mediator in lieu of the physiological reductant, Cyt c, used by us and Wilson et al.
3.4. Reaction of O2 with dithionite-reduced CcO
To avoid the complication from the ascorbyl radical, we examined the CcO reaction by using dithionite as a reductant instead of ascorbate/Cyt c. In this series of experiments, the enzyme was exposed to a ten-fold excess of sodium dithionite prior to the reaction with O2. RFQ samples were prepared as in the ascorbate experiments. X-band CW-EPR measurements show a narrow radical signal with a linewidth of 11 G in intermediates trapped from 50 μs through 6000 μs (Fig. 6a–e). When the narrow radical signal was subtracted from the data, no additional protein-based radicals were detected. The narrow radical disappeared upon annealing for 1 minute, leaving only the CuA2+ signal (Fig. 6f). In addition, hand-quench samples, prepared by mixing CcO reduced by dithionite and oxygen and quenched at one minute, showed only cupric CuA with no evidence of the narrow radical (data not shown).
Fig. 6.
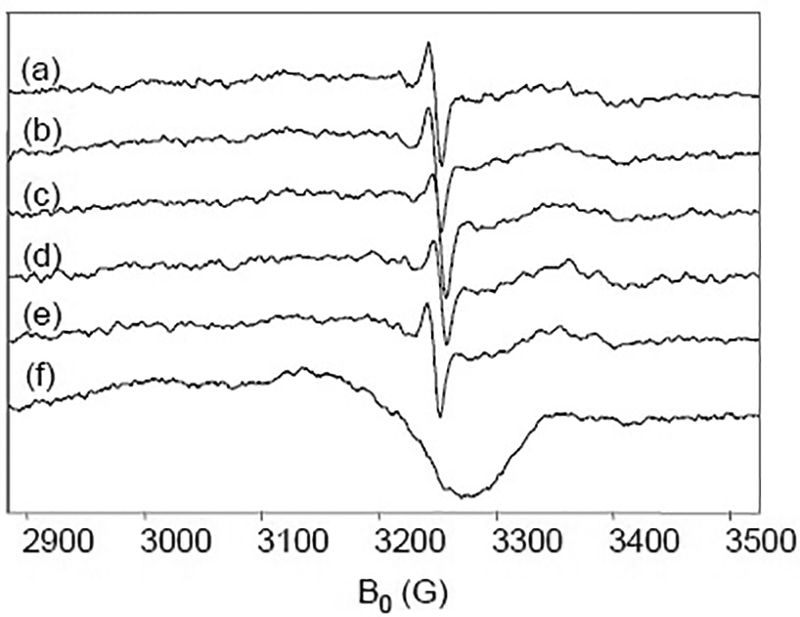
X-band EPR spectra of CcO prepared with dithionite. 150 μM CcO completely reduced by 1.5 mM dithionite under Ar then mixed with O2-saturated buffer and rapid freeze-quenched at (a) 50, (b)150, (c) 350, (d) 450, and (e) 6000 μs. (f) 350 μs sample annealed for 1 min. at 298 K. EPR conditions as in Fig. 1.
Because previous studies suggested that dithionite spontaneously dissociates into breakdown products such as SO2−• [34, 35], the reductant was tested with both redox and non-redox active proteins. 115 μM CcO, BSA, or Mb was mixed with 50 mM dithionite and freeze-quenched at 50 μs. An intense signal with a linewidth of 11 G was seen in all three enzyme samples (Fig. 7a–c). These observations suggest that the radical arises spontaneously from dithionite and not from interactions with proteins.
Fig. 7.
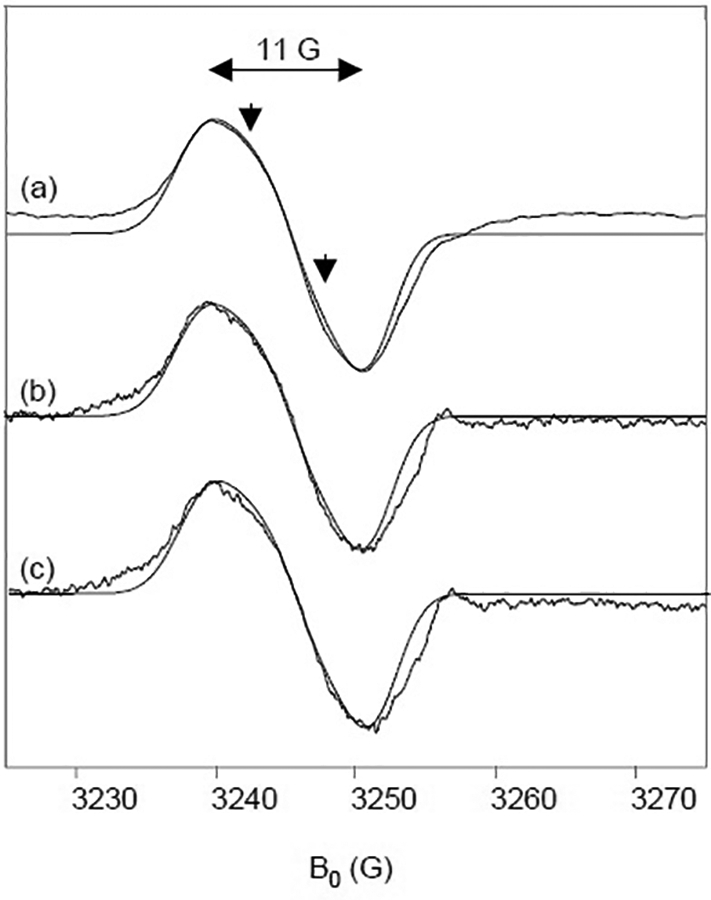
X-band EPR spectra of various proteins prepared with dithionite. (a) 115 μM CcO and 50 mM Na2S2O4 rapid freeze-quenched at 50 μs. (b) 115 μM bovine serum albumin and 50 mM Na2S2O4 rapid freeze-quenched at 50 μs. X-band EPR conditions as in Fig. 1.
To further characterize the radical species, 50 mM dithionite solutions were freeze-quenched with oxygenated or deoxygenated buffer and the samples were measured by D-band and X-band EPR. High-frequency spectra show perturbations of the gx and gz features in the oxygenated sample (Fig. 8b) compared to the deoxygenated sample (Fig. 8a), the origin of which are unknown at this time. However, both of these high-frequency oxygenated and deoxygenated spectra are well-fit by simulations with g-tensor values at 2.0089, 2.0052, and 2.0017 (Fig. 8–b, dotted lines). The isotropic g-value is 2.0053, which is in good agreement with the literature value of SO2−• [54–56]. This study is the first report of the g-tensor components of SO2−•. The radical in the samples of 50 mM dithionite with (Fig. 8e) and without oxygen (Fig. 8d) is identical to the radical produced when reacting CcO with oxygen (Fig. 8f). Using the well-defined g-values, the X-band data can be simulated using parameters in Table 2. The simulations, which are optimized to fit the slight asymmetry of the lineshape (Fig. 7, arrows), include two protons. These protons are likely H-bonding water molecules in the vicinity of SO2−•. In addition, the radical nearly disappears when annealed for 1 min. at 180 K (Fig. 8c), which is consistent with our annealing experiments performed at X-band. Unlike the ascorbyl radical, the SO2−• radical is unstable at room temperature under these experimental conditions.
Fig. 8.
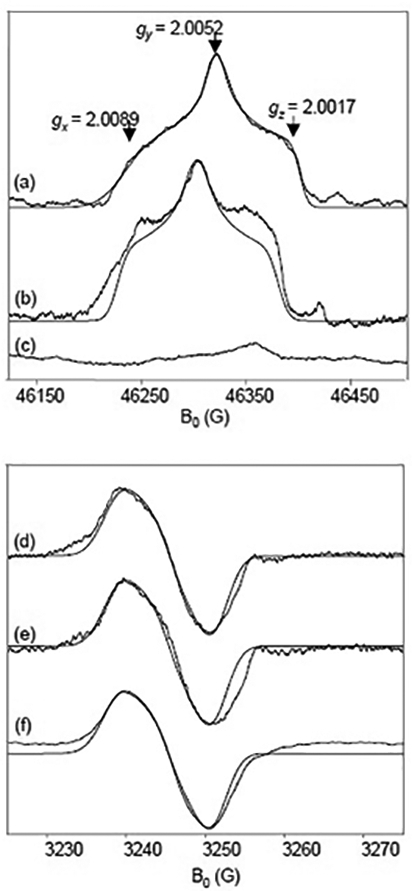
D-band (top) and X-band (bottom) EPR spectra of dithionite with and without oxygen. (a) and (d) 50 mM Na2S2O4 in degassed buffer rapid freeze-quenched at 50 μs. (b) and (e) 50 mM Na2S2O4 with oxygenated buffer and 115 μM CcO rapid freeze-quenched at 50 μs. (c) Sample from (b) annealed at 180 K for 1 min. (f) 50 mM Na2S2O4 in oxygenated buffer rapid freeze-quenched at 50 μs. D-band and X-band EPR conditions as in Figs. 2 and 1, respectively.
Table 2.
Simulation parameters for the sulfur dioxide anion radical.
Hyperfine couplings are given in units of Gauss.
4. CONCLUSIONS
This study reports g-tensors for the ascorbyl and SO2−• radicals and establishes that these radicals are formed and can be trapped in the course of CcO reacting with oxygen when ascorbate/Cyt c or dithionite is used as the reductant. In the frozen solution X-band spectra, the ascorbyl and SO2−• radicals both appear as relatively featureless single-line spectra with zero crossings at g ~ 2.005. The high-frequency spectra provide the resolution of peaks associated with the principal g-values and reveal that the ascorbyl radical has near-axial symmetry while the SO2−• is much more rhombic. This study demonstrates the utility of high-frequency EPR to distinguish between radicals that appear similar at X-band, thereby allowing for unequivocal identification.
Protein-based free radicals play an integral role in the chemistry of life processes and are often studied by EPR spectroscopy. In assessing protein-based radical mechanisms, byproducts from reductants must not be neglected as they may significantly impact the analyses. In bacterial CcO, catalytically relevant radicals are formed, but no such radical has yet been unequivocally identified in the mammalian enzyme. Identifying the origin of reducing equivalents used in catalysis is crucial to understanding the link between redox chemistry and proton translocation. However, the question of whether a radical arises in the catalytic reaction of mammalian CcO with O2 remains unclear. Our studies show that ascorbate and dithionite—two universally used reductants in studies of redox proteins—can contribute to EPR spectra. Hence, great care must be taken in the interpretation of data to understand their roles in the reactions studied.
5. ACKNOWLEDGEMENTS
This work was funded by the National Institutes of Health (NIH) grants GM074982 to D.L.R. and GM075920 to G.J.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Buckel W and Golding BT, Radical enzymes in anaerobes, Annu Rev Microbiol 60 (2006) 27–49. [DOI] [PubMed] [Google Scholar]
- [2].Marsh EN, A radical approach to enzyme catalysis, Bioessays 17 (1995) 431–41. [DOI] [PubMed] [Google Scholar]
- [3].Stubbe J and van Der Donk WA, Protein Radicals in Enzyme Catalysis, Chem Rev 98 (1998) 705–762. [DOI] [PubMed] [Google Scholar]
- [4].Sjoberg BM, Reichard P, Graslund A and Ehrenberg A, The tyrosine free radical in ribonucleotide reductase from Escherichia coli, J Biol Chem 253 (1978) 6863–5. [PubMed] [Google Scholar]
- [5].Licht S, Gerfen GJ and Stubbe J, Thiyl radicals in ribonucleotide reductases, Science 271 (1996) 477–81. [DOI] [PubMed] [Google Scholar]
- [6].Yonetani T, Schleyer H and Ehrenberg A, Studies on Cytochrome c Peroxidase: Electron Paramagnetic Resonance Absorptions of the Enzyme and Complex ES in Dissolved and Crystalline Forms, J. Biol. Chem 241 (1966) 3240–43. [PubMed] [Google Scholar]
- [7].Wittenberg BA, Kampa L, Wittenberg JB, Blumberg WE and Peisach J, The electronic structure of protoheme proteins. II. An electron paramagnetic resonance and optical study of cytochrome c peroxidase and its derivatives, J Biol Chem 243 (1968) 1863–70. [PubMed] [Google Scholar]
- [8].Goodin DB, Mauk AG and Smith M, Studies of the radical species in compound ES of cytochrome c peroxidase altered by site-directed mutagenesis, Proc Natl Acad Sci U S A 83 (1986) 1295–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hiner AN, Martinez JI, Arnao MB, Acosta M, Turner DD, Lloyd Raven E and Rodriguez-Lopez JN, Detection of a tryptophan radical in the reaction of ascorbate peroxidase with hydrogen peroxide, Eur J Biochem 268 (2001) 3091–8. [DOI] [PubMed] [Google Scholar]
- [10].Choinowski T, Blodig W, Winterhalter KH and Piontek K, The crystal structure of lignin peroxidase at 1.70 A resolution reveals a hydroxy group on the cbeta of tryptophan 171: a novel radical site formed during the redox cycle, J Mol Biol 286 (1999) 809–27. [DOI] [PubMed] [Google Scholar]
- [11].Pogni R, Baratto MC, Giansanti S, Teutloff C, Verdin J, Valderrama B, Lendzian F, Lubitz W, Vazquez-Duhalt R and Basosi R, Tryptophan-based radical in the catalytic mechanism of versatile peroxidase from Bjerkandera adusta, Biochemistry 44 (2005) 4267–74. [DOI] [PubMed] [Google Scholar]
- [12].Pogni R, Baratto MC, Teutloff C, Giansanti S, Ruiz-Duenas FJ, Choinowski T, Piontek K, Martinez AT, Lendzian F and Basosi R, A tryptophan neutral radical in the oxidized state of versatile peroxidase from Pleurotus eryngii: a combined multifrequency EPR and density functional theory study, J Biol Chem 281 (2006) 9517–26. [DOI] [PubMed] [Google Scholar]
- [13].Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT, Oxygen activation and reduction in respiration: involvement of redox-active Tyrosine 244, Science 290 (2000) 1588–90. [DOI] [PubMed] [Google Scholar]
- [14].Wiertz FG, Richter OM, Ludwig B and de Vries S, Kinetic resolution of a tryptophan-radical intermediate in the reaction cycle of Paracoccus denitrificans cytochrome c oxidase, J Biol Chem 282 (2007) 31580–91. [DOI] [PubMed] [Google Scholar]
- [15].Michel H, The mechanism of proton pumping by cytochrome c oxidase, Proc Natl Acad Sci U S A 95 (1998) 12819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Svistunenko DA, Wilson MT and Cooper CE, Tryptophan or tyrosine? On the nature of the amino acid radical formed following hydrogen peroxide treatment of cytochrome c oxidase, Biochim Biophys Acta 1655 (2004) 372–80. [DOI] [PubMed] [Google Scholar]
- [17].Wiertz FG, Richter OM, Cherepanov AV, MacMillan F, Ludwig B and de Vries S, An oxo-ferryl tryptophan radical catalytic intermediate in cytochrome c and quinol oxidases trapped by microsecond freeze-hyperquenching (MHQ). FEBS Lett. 575 (2004) 127–30. [DOI] [PubMed] [Google Scholar]
- [18].Wilson MT, Jensen P, Aasa R, Malmstrom BG and Vanngard T, An investigation by e.p.r. and optical spectroscopy of cytochrome oxidase during turnover, Biochem J 203 (1982) 483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bergsten P, Amitai G, Kehrl J, Dhariwal KR, Klein HG and Levine M, Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation, J Biol Chem 265 (1990) 2584–7. [PubMed] [Google Scholar]
- [20].Shaskus J and Haake P, AScorbic Acid. 2. Nucleophilic Reactivity of Ascorbate Anion towards Acyl Carbon and Phosphorus, J. Org. Chem 49 (1984) 197–199. [Google Scholar]
- [21].Pelizzetti E, Metnasti E and Pramauro E, Kinetics and mechanism of the oxidation of ascorbic acid by Tris(1,10-phenanthroline)iron(III) and its derivatives in aqueous acidic perchlorate media, Inorganic Chemistry 15 (1976) 2898–900. [Google Scholar]
- [22].Al-Ayash AI and Wilson MT, The mechanism of reduction of single-site redox proteins by ascorbic acid, Biochem J 177 (1979) 641–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Njus D, Jalukar V, Zu JA and Kelley PM, Concerted proton-electron transfer between ascorbic acid and cytochrome b561, Am J Clin Nutr 54 (1991) 1179S–1183S. [DOI] [PubMed] [Google Scholar]
- [24].Bailey DM, George WO and Gutowski M, Theoretical studies of L-ascorbic acid (vitamin C) and selected oxised, anionic and free-radical forms, J. of Molecular Structure: THEOCHEM 910 (2009) 61–68. [Google Scholar]
- [25].Allen RN, Shukla MK, Reed D and Leszczynski J, Ab Initio Study of the Structural Properties of Ascorbic Acid (Vitamin C), International Journal of Quantum Chemistry 106 (2006) 2934–2943. [Google Scholar]
- [26].Buettner GR and Chamulitrat W, The catalytic activity of iron in synovial fluid as monitored by the ascorbate free radical, Free Radic Biol Med 8 (1990) 55–6. [DOI] [PubMed] [Google Scholar]
- [27].Buettner GR and Jurkiewicz BA, Ascorbate free radical as a marker of oxidative stress: an EPR study, Free Radic Biol Med 14 (1993) 49–55. [DOI] [PubMed] [Google Scholar]
- [28].Buettner GR, The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate, Arch Biochem Biophys 300 (1993) 535–43. [DOI] [PubMed] [Google Scholar]
- [29].May JM, Li L, Qu ZC and Cobb CE, Mitochondrial recycling of ascorbic acid as a mechanism for regenerating cellular ascorbate, Biofactors 30 (2007) 35–48. [DOI] [PubMed] [Google Scholar]
- [30].Li X, Cobb CE and May JM, Mitochondrial recycling of ascorbic acid from dehydroascorbic acid: dependence on the electron transport chain, Arch Biochem Biophys 403 (2002) 103–10. [DOI] [PubMed] [Google Scholar]
- [31].Li X, Cobb CE, Hill KE, Burk RF and May JM, Mitochondrial uptake and recycling of ascorbic acid, Arch Biochem Biophys 387 (2001) 143–53. [DOI] [PubMed] [Google Scholar]
- [32].Mayhew SG, The redox potential of dithionite and SO-2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase, Eur J Biochem 85 (1978) 535–47. [DOI] [PubMed] [Google Scholar]
- [33].Dunitz JD, Structure of Dithionite, J. Amer. Chem. Soc 78 (1956) 878–9. [Google Scholar]
- [34].Hodgson WG, Neaves A and Parker CA, Detection of Free Radicals in Sodium Dithionite by Paramagnetic Resonance, Nature 178 (1956) 489. [Google Scholar]
- [35].Janzen EG, Electron spin resonance study of the SO2-. formation in the thermal deocmposition of sodium dithionite, sodium and potassium metabisulfite, and sodium hydrogen sulfite., J. of Physical Chemistry 76 (1972) 157–162. [Google Scholar]
- [36].Yoshikawa S, Choc MG, O’Toole MC and Caughey WS, An infrared study of CO binding to heart cytochrome c oxidase and hemoglobin A. Implications re O2 reactions, J Biol Chem 252 (1977) 5498–508. [PubMed] [Google Scholar]
- [37].Tsubaki M, Shinzawa K and Yoshikawa S, Effects of crystallization on the heme-carbon monoxide moiety of bovine heart cytochrome c oxidase carbonyl, Biophys J 63 (1992) 1564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lin Y, Gerfen GJ, Rousseau DL and Yeh SR, Ultrafast microfluidic mixer and freeze-quenching device, Anal Chem 75 (2003) 5381–6. [DOI] [PubMed] [Google Scholar]
- [39].Egawa T, Durand JL, Hayden EY, Rousseau DL and Yeh SR, Design and evaluation of a passive alcove-based microfluidic mixer, Anal Chem 81 (2009) 1622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Krymov V and Gerfen GJ, Analysis of the tuning and operation of reflection resonator EPR spectrometers, J Magn Reson 162 (2003) 466–78. [DOI] [PubMed] [Google Scholar]
- [41].Ranguelova K, Girotto S, Gerfen GJ, Yu S, Suarez J, Metlitsky L and Magliozzo RS, Radical sites in Mycobacterium tuberculosis KatG identified using electron paramagnetic resonance spectroscopy, the three-dimensional crystal structure, and electron transfer couplings, J Biol Chem 282 (2007) 6255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Burghaus O, Rohrer M, Plato M and Mobius K, A novel high-field/high-frequency EPR and ENDOR spectrometer operating at 3 mm wavelength, Meas Sci Tech 3 (1992) 765–774. [Google Scholar]
- [43].Antonini E, Brunori M, Greenwood C, Colosimo A, Wilson MT, Oxygen “pulsed” cytochrome c oxidase: functional properties and catalytic relevance, PNAS 74 (1977) 3128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Young LJ and Palmer G, Redox-cycled oxidase. One of the reaction products of reduced cytochrome c, cytochrome c oxidase, and dioxygen, J Biol Chem 261 (1986) 13031–3. [PubMed] [Google Scholar]
- [45].Okunuki K, Hagihara B, Sekuzu I and Horio T, Studies on the cytochrome system, Proceedings of the International Symposium on Enzyme Chemistry (1958) 264–272. [Google Scholar]
- [46].Hoffman BM, Roberts JE, Swanson M, Speck SH and Margoliash E, Copper electron-nuclear double resonance of cytochrome c oxidase, Proc Natl Acad Sci U S A 77 (1980) 1452–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Beinert H, Griffiths DE, Wharton DC, Sands RH, Properties of the copper associated with cytochrome oxidase as studied by paramagnetic resonance spectroscopy, J. Biol. Chem 237 (1962) 2337–46. [PubMed] [Google Scholar]
- [48].Zhen Y, Schmidt B, Kang UG, Antholine W and Ferguson-Miller S, Mutants of the CuA site in cytochrome c oxidase of Rhodobacter sphaeroides: I. Spectral and functional properties, Biochemistry 41 (2002) 2288–97. [DOI] [PubMed] [Google Scholar]
- [49].Laroff GP, Fessenden RW and Schuler RH, The electron spin resonance spectra of radical intermediates in the oxidation of ascorbic acid and related substances, J Am Chem Soc 94 (1972) 9062–73. [DOI] [PubMed] [Google Scholar]
- [50].O’Malley PJ, Density Functional Calculations Modelling the Spin Density Distribution, Hyperfine Couplings, and Hydrogen Bonding Environment of the Ascorbate (Vitamin C) Free Radical, J. Phys. Chem. B 105 (2001) 11290–11293. [Google Scholar]
- [51].Dunne J, Caron A, Menu P, Alayash AI, Buehler PW, Wilson MT, Silaghi-Dumitrescu R, Faivre B and Cooper CE, Ascorbate removes key precursors to oxidative damage by cell-free haemoglobin in vitro and in vivo, Biochem J 399 (2006) 513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Moreau S, Puppo A and Davies MJ, The reactivity of ascorbate with different redox states of leghaemoglobin, Phytochemistry 39 (1995) 1281–6. [DOI] [PubMed] [Google Scholar]
- [53].Giulivi C and Cadenas E, The reaction of ascorbic acid with different heme iron redox states of myoglobin. Antioxidant and prooxidant aspects, FEBS Lett 332 (1993) 287–90. [DOI] [PubMed] [Google Scholar]
- [54].Streeter I, Wain AJ, Davis J and Compton RG, Cathodic reduction of bisulfite and sulfur dioxide in aqueous solutions on copper electrodes: an electrochemical ESR study, J Phys Chem B 109 (2005) 18500–6. [DOI] [PubMed] [Google Scholar]
- [55].Zhu J, Petit K, Colson O, DeBolt S and Sevilla MD, Reactlons of HS and S* wlth Molecular Oxygen, H2S, HS-, and S2’: Formatlon of SO2’, HSSH’, HSS2’, and HSS’, Journal of Physical Chemistry 95 (1991) 3676–81. [Google Scholar]
- [56].Ozawa T, Setaka M and Kwan T, ESR studies of the Sulfite Racial Anion, Bull. Chem. Soc. Japan 44 (1971) 3473–3474. [Google Scholar]