

The transcriptomic memory in poplar trees after recovery from drought-heat stress changes with stress frequency and intensity and involves a complex interplay of common and tissue-specific factors.
Abstract
Throughout the temperate zones, plants face combined drought and heat spells in increasing frequency and intensity. Here, we compared periodic (intermittent, i.e., high-frequency) versus chronic (continuous, i.e., high-intensity) drought-heat stress scenarios in gray poplar (Populus× canescens) plants for phenotypic and transcriptomic effects during stress and after recovery. Photosynthetic productivity after stress recovery exceeded the performance of poplar trees without stress experience. We analyzed the molecular basis of this stress-related memory phenotype and investigated gene expression responses across five major tree compartments including organs and wood tissues. For each of these tissue samples, transcriptomic changes induced by the two stress scenarios were highly similar during the stress phase but strikingly divergent after recovery. Characteristic molecular response patterns were found across tissues but involved different genes in each tissue. Only a small fraction of genes showed similar stress and recovery expression profiles across all tissues, including type 2C protein phosphatases, the LATE EMBRYOGENESIS ABUNDANT PROTEIN4-5 genes, and homologs of the Arabidopsis (Arabidopsis thaliana) transcription factor HOMEOBOX7. Analysis of the predicted transcription factor regulatory networks for these genes suggested that a complex interplay of common and tissue-specific components contributes to the coordination of post-recovery responses to stress in woody plants.
INTRODUCTION
Climate change increases the frequency and intensity of extreme events such as heat waves and drought (IPCC, 2014). Plants, particularly long-living trees, have evolved flexible mechanisms to cope with environmental stresses (Harfouche et al., 2014). Poplar (Populus spp) is a widely used model in tree research that combines moderate genome size, a complete genome reference, fast growth, rapid maturation, and wide geographic distribution with economic relevance for wood and biomass production (Taylor, 2002; Tuskan et al., 2006). Poplar is also suitable for transcriptome studies across a variety of tissues. For instance, co-expression patterns underlying cambial growth and wood formation have been investigated by sampling multiple sections across tree trunks (Sundell et al., 2017). Various physiological changes have been observed in plants in response to abiotic stresses. Drought limits water uptake by roots and results in reduced transpiration and photosynthesis, which can have severe effects on growth and yield (Aroca et al., 2012; Osakabe et al., 2014). These processes are mediated by well-known molecular responses of cells to drought, frequently triggered by the plant hormone abscisic acid (ABA; Shinozaki and Yamaguchi-Shinozaki, 2007; Osakabe et al., 2014). In addition to ABA-responsive elements binding factors (ABF), members of the NOAPICAL MERISTEM, ARABIDOPSIS ACTIVATING FACTOR and CUP-SHAPED COTYLEDON (NAC) and of the dehydration-responsive element binding (DREB) transcription factor families orchestrate pronounced gene expression changes upon drought stress, as demonstrated in Arabidopsis (Arabidopsis thaliana) and crop species (Nakashima et al., 2014).

Stress exposure also alters gene expression beyond the duration of the stress phase, forming a molecular “memory” (Crisp et al., 2016; Fleta-Soriano and Munné-Bosch, 2016). A well-studied effect of stress-related memory is enhanced tolerance toward subsequent stress events, as reflected by response differences between the first and subsequent stress challenges (Ding et al., 2012, 2013; Liu et al., 2016). This “primed response” is characterized by gene expression changes that induce damage protection, growth regulation, osmotic readjustment, and the coordination of hormonal crosstalk (Ding et al., 2013). Such an expression memory can also involve chromatin remodeling through histone modifications (Sani et al., 2013; Lämke et al., 2016). Phenotypically, plants primed by drought stress have shown a higher photosynthesis rate during subsequent stress periods than nonprimed plants (Wang et al., 2014). Even in the absence of a further stress challenge, plant performance can have signs of a stress-related memory after successful stress recovery, significantly differing from untreated control plants (Xu et al., 2010; Hagedorn et al., 2016). The molecular basis of this post-recovery phenotype is still largely unexplored.
In the present study, we focused on gene expression changes during stress and after recovery in a woody plant species. In particular, we investigated expression characteristics of stress-related memory, which we define in the context of this work as a post-recovery steady state in stress-treated trees that is distinct from that of nontreated trees. Our analysis not only contrasts stress scenarios that differ in frequency and intensity but also compares responses in different tissues of poplar trees. We simulated predicted regional climate conditions (IPCC, 2014) by applying simultaneous drought and heat spells at the elevated atmospheric carbon dioxide (CO2) concentrations expected in 2050 to explore how gray poplars (Populus × canescens) that have recovered from drought-heat stress differ from nontreated plants. We characterized the stress response and the stress-related post-recovery memory regarding leaf photosynthesis phenotypes and transcriptional responses of young (“sink”) and mature (“source”) leaves (Vanzo et al., 2015), phloem-bark, developing xylem and roots. Two stress scenarios of equal total duration were compared, contrasting periodic, intermittent stress (PS) with chronic, continuous stress (CS).
The post-recovery effects of abiotic stress we observed at the transcriptome level extend the established concept of a molecular memory after stress exposure. While previous work has analyzed gene expression changes in response to recurrent versus initial stress challenges, our analysis also investigates stress-induced shifts in steady state after recovery, i.e., before a new stress challenge. Still, the aspect of recurrent versus one-time stress is covered by the two stress scenarios, which are compared with control scenarios not only at the end of the stress phase but also after recovery. The multifactorial study sheds light on the regulatory architecture of memory-related gene expression networks after different climatic challenges and across multiple tree organs and tissues.
RESULTS
Impact of Drought-Heat Stress Periods on Post-Recovery Photosynthetic Performance
To obtain a systems-level view of the stress response and recovery in a woody plant species, we subjected groups of gray poplar (Populus × canescens) trees to one of four climate scenarios and collected transcriptome samples from five tree compartments (organs and wood tissues) at two subsequent time points (Figure 1). In addition, we took phenotypic measurements of photosynthesis in attached leaves (Figure 1A). The experiment was performed in climate chambers under highly controlled conditions, including a chronic drought and heat stress scenario at elevated CO2 levels (CS scenario), a periodic drought and heat stress scenario at elevated CO2 levels with two intermediate recovery periods (PS scenario), a control scenario at elevated CO2 levels (EC scenario) and a control scenario at ambient CO2 levels (AC scenario) (Vanzo et al., 2015). AC represents the current temperate climate as a reference point, which allowed us to estimate the effects of predicted future climate scenarios (EC, CS, and PS). The stress phase of 22 d was followed by a recovery period of 1 week under irrigation and temperature conditions equal to those for control plants (see Methods). Phenotypic photosynthetic performance of mature leaves was assessed using gas exchange measurements (Vanzo et al., 2015; Jud et al., 2016). During the stress phase, the net CO2 assimilation rate of leaves was significantly reduced for CS-treated poplar trees compared with the corresponding EC control trees (p.adj = 0.0347). PS-treated trees showed intermediate levels (Figure 1B). The same response pattern was found for the transpiration rate (Figure 1C) and stomatal conductance (Figure 1D). For all three physiological parameters, AC and EC controls were not significantly different. At the end of the recovery phase, the leaf transpiration rate and stomatal conductance of stress-treated trees reached similar levels to those of the control trees, suggesting that the trees indeed had recovered from the combined drought and heat spells (Figures 1C and 1D). The recovery of the physiological phenotype is also confirmed by the clear separation between stress phase and recovery measurements for each stress treatment and physiological parameter (Figures 1B to 1D). Remarkably, the leaf net CO2 assimilation rates of PS- and CS-treated trees not only recovered but were significantly higher than that of AC trees (p.adj = 0.0089 and p.adj = 0.0388, respectively), with intermediate levels for EC trees (Figure 1B). Evaluation of continuous net ecosystem exchange measurements throughout the entire experiment (Vanzo et al., 2015) showed that both PS- and CS-treatments led to a significant increase in the daily rates of canopy level C gain from photosynthesis during the second half of the recovery phase (d 26 to 29) compared with the control scenarios (Figure 1E).
Figure 1.

Effect of Climatic Stress on the Post-Recovery Photosynthetic Performance of Poplar Trees.
(A) The 3D experimental design to investigate the climate response of P. × canescens trees regarding memory aspects and systemic effects. Plants from four environmental conditions including the ambient CO2 control (AC), enhanced CO2 control (EC), periodic drought-heat stress (PS), and chronic drought-heat stress (CS) were examined both at the end of a 22-d stress phase (S; d22) and after one week of recovery (R; d29). At the start of the stress treatment (d0), the plants were 8.5 weeks old and already under AC and EC control climates for 25 d. For fully developed leaves, both phenotypic and transcriptomic measurements are available; for the four other tissues, only transcriptomic data are available.
(B) to (D) Comparison of leaf-level gas exchange rates (leaf No. 9 from the apex) across environmental conditions (center line, median; box limits, top and bottom quartiles; whiskers, maximum and minimum). For each of the two phases (separated by the vertical line), groups that do not share identical lowercase letters are significantly different (Kruskal-Wallis test with post hoc Dunn’s test, Benjamini-Hochberg adjustment, p.adj < 0.05).
(E) Carbon gain determined by online gas exchange analysis for the gas-tight sub-chamber of each environmental condition. The slope (shown by circles) was estimated from the last four measurements (d 26 to d 29). Different lowercase letters indicate a significant difference of slopes (Kruskal-Wallis test with post hoc Dunn’s test, Benjamini-Hochberg adjustment, p.adj < 0.05).
(F) Projection on the top two components from canonical correlation analysis between gas exchange data and log2(TPM+1)-transformed per-gene RNA-seq data of the 100 most varying genes in mature leaves across the four conditions and two treatment phases. Each data point represents the mean of biological replicates for the given group; because of destructive harvesting, stress phase RNA-seq measurements were obtained from biological samples that differed from those used for the continuous gas exchange measurements. Ellipses mark 0.75 confidence levels estimated from the replicates.
Shared Effects between Transcriptomic and Phenotypic Data
We integrated the photosynthetic gas exchange data with RNA sequencing (RNA-seq) data from mature leaves by regularized canonical correlation analysis (see Methods; Lê Cao et al., 2009). RNA-seq reads were mapped to the Populus trichocarpa reference genome (Tuskan et al., 2006; Sundell et al., 2017). Both data types shared major stress and recovery effects, as reflected by the first and second correlated component, respectively (Figure 1F). For component one, the most representative phenotypic variable is the ratio between net CO2 assimilation rate and stomatal conductance (Pearson correlation −0.96). The leaf transpiration rate showed a correlation of 0.95 with component one, which is consistent with stomatal closure upon drought stress (Osakabe et al., 2014). The dominating genes for component one also have known stress response functions. Among the top ten genes upregulated during stress (correlation < −0.95), six genes were annotated as encoding heat shock proteins (Potri.012G022400, Potri.010G195700, Potri.013G089200, Potri.017G130700, Potri.010G088600, Potri.010G053400), potentially acting as chaperones in protein folding. This could indicate a response to elevated leaf temperature caused by heat and a lack of transpirational cooling (Kotak et al., 2007). Indeed, the mean temperature of mature leaves in the experiment increased to more than 35°C during PS and CS, whereas it ranged from 27°C to 30°C after recovery and for the controls (see Supplemental Figure 4 of Vanzo et al., 2015). Considering all 589 genes that were upregulated under PS and CS in mature leaves (log2[fold change] > 1, p.adj < 0.05; Supplemental Data Set 1), protein folding is also the top enriched Gene Ontology (GO) category (p.adj = 5.64e-9; Supplemental Data Set 2). At the same time, the top downregulated variables associated with component one included an MYB (myeloblastosis) transcription factor (Potri.002G260000), a peroxidase (Potri.016G132666), and a glutaredoxin gene (Potri.014G134300), indicating changes in transcriptional regulation and stress signaling. Oxidoreductase, peroxidase, and transcription factor activity functions were also significantly enriched among the genes downregulated in mature leaves by both stresses, along with many other processes including protein phosphorylation, cell wall modification, proteolysis, transmembrane transport, cell division, and defense response (Supplemental Data Set 3). The recovery phase mature leaf samples were indistinguishable from control samples with respect to component one, suggesting the disappearance of major stress characteristics and thus successful recovery.
The second component linking phenotypic and gene expression data reveals differences between recovery phase samples and untreated samples (Figure 1F). Component two is characterized by an increased mean net CO2 assimilation rate in the recovery samples (Pearson correlation 0.77), which is consistent with the results of phenotypic data analysis (Figure 1B). Individual genes did not correlate significantly with component two, and the upregulated genes shared by both stress treatments after recovery were not enriched for specific functions (Supplemental Data Set 4). Nucleotide binding and ATPase activity for transmembrane movement were enriched among the downregulated genes of both stress treatments but much more pronounced for CS (Supplemental Data Set 5). PS-specific upregulation was enriched for stress response genes (e.g., the heat shock protein Potri.004G073600, Supplemental Data Set 4). Many genes were upregulated only for one stress treatment type; for instance, the putatively photosynthesis-associated plastocyanin-like domain gene Potri.001G332200 was only upregulated for PS (Figure 2; Supplemental Data Set 1). These results indicate that the post-recovery transcriptomes of PS and CS in mature leaves share more subtle, multivariate effects.
Figure 2.

System-Wide Comparison of Poplar Gene Expression Under Stress, Recovery, and Control Conditions.
(A) Projection on top two components from principal component analysis of log2(TPM+1)-transformed per-gene RNA-seq data from all samples across four climate conditions, two treatment phases, five different tissues, and three biological replicates per group. The biological replicates are tissue samples from different trees subjected to the same environmental condition and harvested at the same time. Data points are colored by tissue (LE1: young leaves, LE2: mature leaves, PHL: phloem, XYL: xylem, ROO: root).
(B) Principal component analysis of poplar trees with complete RNA-seq measurements from all five tissues, concatenating all tissue measurements from the same tree (see Methods). Ellipses mark the 0.75 confidence contour for stressed trees and all other trees (S: stress phase, R: recovery phase).
(C) Number of differentially expressed genes overlapping between PS and CS treatment or unique to each stress type. Differential expression was determined relative to untreated EC controls for each tissue and treatment phase separately (fold change > 2, p.adj < 0.05). The dashed box shows a zoom-in for the three bottommost groups.
(D) Comparison of antioxidant levels in mature leaves across environmental conditions. Groups that do not share identical lowercase letters are significantly different (Kruskal-Wallis test with post hoc Dunn’s test, Benjamini-Hochberg adjustment, p.adj < 0.05). The y axis gives the percentage of functional, reduced ascorbate relative to total ascorbate (oxidized and reduced forms).
(E) Stress-recovery overlap of upregulated genes. For each tissue, the percentage of stress phase down- or upregulation of genes upregulated in the recovery phase relative to EC control plants is given (fold change > 2, p.adj < 0.05).
Systemic and Tissue-Specific Stress Responses
In addition to the mature leaf data described so far, we also obtained RNA-seq measurements from young leaves, phloem-bark, developing xylem, and fine roots. To simplify the figure keys and descriptions, the term “tissues” hereafter refers to exactly this set of organs and tissues. The analysis across tissues provided a comprehensive systems-level view on the transcriptional responses occurring during stress and after recovery (Figure 2). The predominant gene expression variation across biological samples was attributable to distinct tissue characteristics (Figure 2A). Whole-tree gene expression profiles concatenating profiles of tissue samples from the same tree clearly separate PS and CS stress phase trees from controls and recovery phase trees (Figure 2B). In all tissues, PS and CS led to very similar transcriptomic responses relative to EC. For both up- and downregulated genes (abs[log2 fold change] > 1, p.adj < 0.05), the observed overlap between the stress types was always larger than one or both of the stress type-specific fractions, suggesting that both scenarios evoke similar molecular stress responses in the tree (Figure 2C, top). Among all tissues, the largest overlap between the two stress types was found in the developing xylem, indicating pronounced changes in the upward transport system of the plant. Significantly enriched GO functions (p.adj < 0.05) among the upregulated genes that overlap between PS and CS xylem samples include oxidoreductase activity, transcription factor activity, transporter activity, and response to stress. In the root, genes encoding recognition proteins (e.g., lectins, glycoproteins) and ATPases were activated by both stress types, whereas common stress responses in phloem-bark and leaves were dominated by protein folding processes (Supplemental Data Set 2). In total three genes were found to be upregulated for each stress scenario in each tissue: Potri.T044100 (one of two co-orthologs of the TCP [TEOSINTE BRANCHED1, CYCLOIDEA, PROLIFERATING CELL FACTORS] family transcription factors AT5G41030 and AT3G27010), Potri.008G133200 (one of two co-orthologs of the O-glycosyl hydrolase AT2G01630), and Potri.001G293000 (not annotated). The annotation of genes was taken from the Phytozome portal (Tuskan et al., 2006; Goodstein et al., 2012) throughout this work (see Methods).
Regarding the genes downregulated under both type of stress, the developing xylem showed many enriched processes, such as translation, microtubule-based movement, DNA replication, carbohydrate metabolic process, cell wall, electron transfer activity, and transmembrane transport, suggesting a downregulation of cell division and growth (Supplemental Data Set 3). Similarly, roots showed significant downregulation of nucleosome, cell wall, electron transfer activity, and carbohydrate metabolic process genes. For phloem-bark, we also observed a significant transcriptional decrease in genes involved in cell wall modification, microtubule-based movement, and carbohydrate metabolic process. In addition, a strong reduction in the expression of genes involved in proteolysis and response to oxidative stress was found in this tissue. The same was observed for downregulated genes in young leaves, with an additional enrichment in fatty acid biosynthetic process, transmembrane transport, DNA replication, and response to auxin. These results, along with the observations in mature leaves (see section "Shared Effects between Transcriptomic and Phenotypic Data"), indicate that both stress treatments (CS and PS) had similar effects, leading to the downregulation of growth-related processes across all tissues.
However, we also found differences between PS and CS. In leaf and root tissues, PS induced more gene expression changes than CS, whereas the CS response was more pronounced than the PS response in xylem and phloem-bark tissues. The PS-specific upregulated genes detected in root are enriched for ATPases acting as transporters. This upregulation might be at least partially attributable to priming effects, because PS plants had experienced their third stress phase, whereas CS plants were still in their first stress challenge at d 22 (d22; Figure 1A). CS-specific upregulated genes in developing xylem are enriched for ATPase activity and photosystem II functions. This pattern of upregulated gene expression is consistent with the previous observation that stem photosynthesis involving the use of internal CO2 from respiration may play a role in young poplar plants, especially during drought stress (Bloemen et al., 2016), although light penetration through the bark is limited (Pfanz et al., 2002). The CS-specific upregulation of photosystem genes may reflect the slightly more severe water deficiency during CS (shoot midday water potential [ψmd] −1.52 ± 0.10 MPa) relative to PS (ψmd −1.27 ± 0.05 MPa) and the controls (EC: ψmd −0.97 ± 0.07 MPa, AC: −0.97 ± 0.04 MPa), increasing the need for C assimilation via a pathway that does not lead to further dehydration promoted by open stomata. CS-specific downregulated genes in developing xylem are enriched for endoplasmatic reticulum and intracellular protein transport (Supplemental Data Set 3). The gene regulation patterns in the developing xylem illustrate the tissue specificity of stress responses, with a high similarity between periodic and chronic stress as well as stress-specific enhancement of various processes.
Post-Recovery Characteristics of Stress-Treated Trees
After 1 week of recovery from periodic or chronic stress, respectively, the total number of differentially regulated genes relative to the EC control plants was lower than that at the end of the stress phase across all poplar tissues (Figure 2C). This suggests that the transcriptomes had left the stress state and again approached the state of control plants. The stress recovery was physiologically confirmed by gas exchange (Figures 1C and 1D) and shoot water potential measurements, which had recovered to −0.72 ± 0.10 MPa and −0.93 ± 0.07 MPa in PS and CS, respectively (compared with the stress phase levels given in the previous paragraph; see Methods). In contrast to the stress phase observations, fewer differentially expressed genes were shared between PS and CS than were specifically up- or downregulated in one of the two stress scenarios (Figure 2C, bottom). This divergence between stress types during the recovery phase indicates that most stress-activated genes are no longer induced and that the recurrence or duration of drought-heat stress alters the post-recovery processes of plants. In all tissues except mature leaves, PS induced more upregulated genes during the recovery phase than CS.
The largest number of upregulated genes after recovery from PS occurred in young (sink) leaves (Vanzo et al., 2015), followed by mature (source) leaves. In young leaves, the upregulated genes were dominated by oxidation-reduction, coenzyme binding, hexosyl transferase, and carbohydrate metabolic process GO terms (Supplemental Data Set 4). The re-induction of carbohydrate metabolism gene expression after its decrease during stress (see section "Systemic and Tissue-Specific Stress Responses") indicates that growth processes were reactivated in young leaves. CS-specific expression patterns were characterized by the downregulation of genes involved in unfolded protein binding, protein folding, and response to stress for young leaves and the downregulation of oxidoreductase activity for phloem, which are indicative of stress recovery (Supplemental Data Set 5). By contrast, for mature leaves of post-recovery trees, PS-specific enrichment indicated the continued activity of several stress response genes, e.g., with functions as heat shock protein (Potri.004G073600) or drought-related late embryogenesis abundant protein (Potri.010G012100). Also, transcription factor genes showed PS-specific transcriptional upregulation in post-recovery mature leaves, e.g., Potri.006G221500, one of six poplar co-orthologs of Arabidopsis MYB123 involved in anthocyanin and pro-anthocyanidin biosynthesis. In agreement with that finding, anthocyanin levels of poplar leaves at the post-recovery time point were higher for PS than for EC and CS (Vanzo et al., 2015). Transporter activity tended to be enriched (p.adj = 0.06) among the genes that were upregulated in young leaves , including many aquaporin genes (Potri.001G235300, Potri.009G005400, Potri.009G013900, Potri.009G027200), some of which were also upregulated in other post-recovery tissues (phloem, mature leaves) of PS or CS trees, which could be an indication of drought decline (Supplemental Data Set 1).
Biochemical data that monitored the antioxidative system in mature leaves also confirm the recovery from the stressed state (Supplemental Data Set 6), matching the gene expression response profiles (Figure 2C). Leaves of PS-treated trees exhibited a significant decrease in relative reduced ascorbate content during the stress phase (p.adj = 0.0347), indicating increased scavenging of reactive oxygen species (AbdElgawad et al., 2016). By contrast, all stress-treated and control trees displayed similar leaf levels of relative reduced ascorbate at the end of the recovery phase (Figure 2D). In addition, we compared the treatment-related expression responses between post-recovery and stress-phase tissue samples to assess how much the molecular processes in each tissue differ between the two phases. Interestingly, the fraction of post-recovery upregulated genes that already showed upregulation during the stress phase varied widely among tissues, ranging from 58% in xylem to 7% in young leaves for PS and from 69% in xylem to 6% in mature leaves for CS (Figure 2E). This suggests that for some tissues, molecular processes after recovery resemble molecular processes during stress, whereas for other tissues, post-recovery and stress responses are largely different. For instance, ATPases and transport functions played a major role in developing xylem during both phases, whereas for young leaves, genes involved in carbohydrate metabolism were downregulated during stress (both PS and CS) and upregulated after recovery.
Among different tissues, the gene expression response patterns showed only a slight overlap (Figure 3). During the recovery phase, we did not find any differentially expressed gene that responded across all tissues in PS or CS. Nevertheless, the five tissues shared similar characteristic stress and post-recovery expression profiles involving distinct coexpression modules in each tissue (see Methods: Figure 4; Supplemental Data Set 7). Interestingly, more than half of these characteristic profiles exhibited a pronounced difference between stress-exposed plants and nontreated plants at the end of the recovery phase, which is indicative of stress-related memory (Figure 4, bottom). For example, young and mature leaf modules in the memory community C12 included the glutathione-S-transferase gene Potri.019G130566, which protects plants against oxidative damage, and the esterase gene Potri.017G062300, with highest similarity to an Arabidopsis gene involved in maintaining the integrity of photosynthetic membranes during abiotic stress (Lippold et al., 2012). For only a small fraction of genes, PS and CS showed similar cross-tissue memory response patterns (Figures 2C and 5A). Among the different tissues, the most pronounced agreement was found for young and mature leaves. The observed divergence between PS and CS was not caused by the fold change threshold (abs[log2 fold change] > 1); there were very few genes that satisfied the significance threshold (p.adj < 0.05) but not the fold change threshold (Supplemental Figure 1). Differences between PS and CS expression levels were consistent with the results of control-based comparisons (Figure 5B).
Figure 3.

Tissue overlap of differentially expressed poplar genes during the stress and recovery phases. Each Venn diagram gives the number of up- or downregulated genes for a specific treatment type and a specific phase in comparison with EC controls (LE1: young leaves, LE2: mature leaves, PHL: phloem, XYL: xylem, ROO: root; S: stress phase, R: recovery phase).
Figure 4.

Characteristic poplar gene expression profiles across stress, recovery, and control conditions shared by all tissues. Each network node represents a co-expression module of a specific tissue (see Methods), as indicated by the respective node color (identical code to Figure 2A) and the prefix of the node label (LE1: young leaves, LE2: mature leaves, PHL: phloem, XYL: xylem, ROO: root). Subsequent numbers in the node label identify the module within each tissue in decreasing order of module size, which is indicated by node size. Each module is represented by its eigengene profile, which is the first principal component oriented according to average expression. The correlation of module eigengenes was used to cluster the modules into communities (see Methods). The figure shows all communities that contain modules from all five tissues together with heatmaps of the corresponding eigengenes. Communities are marked by gray polygons and C identifiers (decreasing shades of gray with increasing identifier numbers). For correlation values > 0.7, edges are depicted between module nodes and the edge width represents the correlation strength. The heatmaps with background shading exhibit a pronounced difference between stress-exposed plants and nontreated plants at the end of the recovery phase for at least one stress type, indicative of stress-related memory (S: stress phase, R: recovery phase). Community C9 putatively represents age-related changes that only occur in nonstressed plants.
Figure 5.

Gene Expression Memory After Recovery from Periodic vs. Chronic Stress.
(A) Poplar genes with cross-tissue memory responses, i.e., transcriptional up- or downregulation in post-recovery stress relative to EC control samples. The heatmap shows PS and CS expression patterns of all genes with PS memory responses in at least two tissues (LE1: young leaves, LE2: mature leaves, PHL: phloem, XYL: xylem, ROO: root, R: recovery phase, S: stress phase).
(B) Direct PS (R) vs. CS (R) comparison of differentially expressed recovery genes determined relative to the EC control (see [A] and Figure 2C). For each tissue, volcano plots show the distribution of overlapping and stress type-specific differential genes (left: downregulation, right: upregulation), taking adjusted p-values and fold changes from the direct comparison. Volcano plots for the respective PS vs. EC and CS vs. EC comparisons are available in Supplemental Figure 1.
The co-analysis of spatially separate tissues provided insights into the complexity of coordinated whole-plant, long-term responses to periodically occurring stress. Strikingly, the stress and recovery profiles of individual genes along the different trees were not conserved across tissues (Figure 6). The expression patterns of the same gene correlated well across all tissues for only 0.2% of the genes (Figure 6A). The largest number of self-correlated genes was found between young and mature leaves, reflecting the functional similarity of these compartments (Figure 6B). Furthermore, 995 genes were self-correlated between phloem and xylem. Among these, functions in oxidation-reduction processes, carbohydrate, and protein metabolic processes as well as transmembrane transport and microtubule-based processes were abundant. The genes with the strongest self-correlation across all tissues included a large proportion of genes that exhibited a significant post-recovery memory pattern in PS (Figure 6A; Supplemental Data Set 1). The top five of these genes were the transcription factor genes HOMEOBOX7 (HB7) co-ortholog 1 (of 4; Potri.014G103000) and HB7 co-ortholog 3 (of 4; Potri.001G083700), as well as GLUTAREDOXIN C1 co-ortholog 2 (of 2; Potri.012G082800) and two clade A protein phosphatases of the 2C family (PP2Cs), the HIGHLY ABA-INDUCED1 (HAI1) ortholog (Potri.009G037300) and Potri.001G092100. In Arabidopsis, HB7 is transcriptionally induced by ABA and positively regulates PP2C gene expression (Valdés et al., 2012).
Figure 6.

Expression Self-correlation of Genes Across Tissues Based on Fully Sampled Individual Trees.
(A) The genes with the strongest self-correlation across all tissues. Among them are many genes with periodic stress memory expression patterns in at least one tissue, marked in black and annotated from top to bottom (letters a-q).
(B) Heatmap showing the number of genes (see scale at the bottom) with self-correlation > 0.8 between individual tissues (LE1: young leaves, LE2: mature leaves, PHL: phloem, XYL: xylem, ROO: root).
Transcription Factors Associated with Stress-Related Memory
Transcription factors (TFs) are key regulators at the top level of the molecular hierarchy. Because several memory genes showed similar stress and post-recovery responses across all tissues (Figure 6A), we were interested in whether common regulatory mechanisms exist among different tissues that may play a role in stress-related memory. We used our gene expression data from each tissue to infer regulatory relationships between known TFs (Jin et al., 2014; Berardini et al., 2015) and these 17 self-correlated memory genes, resulting in a gene regulatory network for each tissue (see Methods; Figure 7). Each of these tissue-specific networks has one main connected component or forms a single connected component, indicating that the self-correlated memory genes (Figure 7A, gray nodes) share common top candidates of regulatory TFs (which were computationally inferred for each gene by choosing the top five expression predictors; see Methods). The majority of candidate TFs were tissue-specific (Figure 7A, white nodes), but a considerable fraction co-occurred across two to four tissues. In particular, young and mature leaves shared ten candidate TFs. Edges were also shared across tissues, meaning that a specific TF was found in several tissues among the top five candidate regulators for a specific memory gene (Figure 7A, colored edges; Figure 7B). A relationship between HB7 co-ortholog 1 (of 4; Potri.014G103000) and HB7 co-ortholog 3 (of 4; Potri.001G083700) was predicted in all tissues except mature leaves. In the co-expression analysis, both genes fell into the tissue-specific co-expression modules of community C2, which was characterized by pronounced stress responses during PS and CS (Figure 4; Supplemental Data Set 7). After recovery, a significant (20-fold) PS upregulation of HB7 co-ortholog 3 (of 4) gene expression was observed for both xylem and mature leaves as well as a 200-fold PS upregulation of HB7 co-ortholog 1 (of 4) for xylem. By contrast, CS trees did not show significant changes in the expression of these genes relative to control trees (Supplemental Data Set 1). The two HB7-related TF genes were also central in the sense that together they covered all putative targets (non-TF memory genes) of their subnetwork and their removal would disconnect the network into several parts (Figure 7B). The HB7 TFs are members of the homeodomain Leu zipper family. In Arabidopsis, HB7 has been associated with drought stress responses as well as reduced cell elongation in leaves and inflorescence stems (Söderman et al., 1996; Hjellström et al., 2003). HB7 has also been identified as a drought stress memory gene that showed a stronger upregulation at the third stress experience than after a single incidence (Ding et al., 2013). Under nonstress conditions, HB7 overexpression has been related to an increase in chlorophyll content and photosynthesis rate (Ré et al., 2014), which is consistent with our physiological observations at the recovery phase.
Figure 7.

Gene Regulatory Networks of Stress-Related Multi-Tissue Memory Genes.
(A) Tissue-specific transcription factor networks around self-correlated genes (gray nodes, labeled by letter code from Figure 6A). For each tissue network, colored nodes and edges indicate their co-occurrence across several tissue networks (see color key). If nodes or edges occur only in one additional tissue (except the currently considered tissue indicated in the box at the top left of each network), they have the characteristic color of that additional tissue. For example, ten transcription factors occur only in the networks of both young and mature leaves (dark green and light green nodes in the first and second network, respectively). Likewise, nodes q and o are connected to the same transcription factor in these networks (dark green and light green edges in the first and second network, respectively).
(B) Regulatory relationships co-occurring across tissues. The edge width is proportional to the number of tissues where a specific regulatory relationship was found. Transcription factor nodes are colored according to their transcription factor family. Homeodomain Leu zipper, HD-ZIP. Basic domain/leucine zipper, bZIP. APETALA2, AP2. Ethylene response factor, ERF.
The expression of the HB7 co-ortholog 1 (of 4; Potri.014G103000) gene itself is putatively related to the expression of the TF genes Potri.006G138900, which was found to be a co-predictor with Potri.001G083700 for several putative target genes, and Potri.002G125400 (Figure 7B). Potri.002G125400 is annotated as ABSCISIC ACID RESPONSIVE ELEMENTS BINDING FACTOR2 (ABF2) co-ortholog 1 (of 2). Arabidopsis ABF2 is known to enhance drought tolerance (Nakashima et al., 2014). Potri.006G138900 is a member of the ethylene response factor/APETALA2 TF family. The closest Arabidopsis ortholog in its evolutionary family, PTHR31985:SF77 (Mi et al., 2017), is AT5G21960, which belongs to the DREB subfamily A-5, with established functions in drought stress response (Singh and Laxmi, 2015). In poplar, the Potri.006G138900 gene has been reported to be induced by four different types of osmotic stresses (Yao et al., 2017). In our data, significant upregulation of this gene was only observed for PS and not for CS. In Arabidopsis, RELATED TO AP2 1 (RAP2.1), a prominent member of the DREB gene subfamily A-5, is also more strongly induced after the repeated application of dehydration stress (Ding et al., 2013). RAP2.1 is a negative regulator of RD/COR (RESPONSIVE TO DESICCATION/ COLD-REGULATED) genes (Dong and Liu, 2010). Poplar RAP2.1 Potri.014G025200 and the other poplar DREB TF that most closely matches Arabidopsis RAP2.1 were significantly upregulated in xylem during both PS and CS. Consistently, the COR413 gene Potri.007G033801 was significantly downregulated under these conditions.
A relationship between two TCP family TFs was detected in young and mature leaves (Figure 7B; Supplemental Data Set 8). Both TFs, Potri.013G119400 and Potri.019G091300, are most similar to the Arabidopsis TF TCP4. The expression of Potri.013G119400 was significantly downregulated after recovery from PS in both young and mature leaves compared with the untreated controls. During PS and CS, both TFs were transcriptionally downregulated in young and mature leaves (Supplemental Data Set 1). In the remaining tissues, the expression patterns of the two TFs diverged from each other. For the developing xylem, Potri.013G119400 was downregulated but Potri.019G091300 was upregulated. The putative leaf target, Potri.010G230366, does not have a known function, but its expression was also strongly upregulated in developing xylem under both PS and CS, and after recovery from PS, upregulation was more than 50-fold. Potri.013G119400 expression was also downregulated in phloem and roots, whereas there was no change in expression for Potri.019G091300. TCP4 has been associated with cell elongation in hypocotyls and leaf morphogenesis (Challa et al., 2016). The differential regulation of these genes across tissues during stress and recovery in gray poplar may reflect different cell growth dynamics. Although water deficiency generally suppresses growth in aboveground poplar tissues, xylem structure and secondary cell wall formation play a central role in avoiding drought damage and are highly regulated (Paul et al., 2017; Sun et al., 2017).
Common and Tissue-Specific Processes Involved in Stress-Related Memory
To further elucidate and compare stress-related memory processes that take place in individual poplar tissues, we investigated the regulatory networks in mature leaves and developing xylem (Figure 8). These were the two tissues where the HB7 TFs, the top correlated genes within and across tissues (Figures 6A and 7B,), showed the strongest post-recovery memory response (Supplemental Data Set 1). For each tissue network, we specifically focused on TFs that were computationally associated with more than one putative target as predictors of gene expression (Figure 8A). Among the targets that were included in the core networks of both mature leaves and developing xylem, we found two PP2Cs (the HAI1 ortholog [Potri.009G037300] and an HAI3-related PP2C, Potri.001G092100), the two LATE EMBRYOGENESIS ABUNDANT PROTEIN4-5 (LEA4-5) co-orthologs, and a gene of unknown function with almost 100-fold upregulation in PS and more than 150-fold upregulation after PS recovery in mature leaves (Potri.004G044300), whose closest Arabidopsis match has been reported to be induced by ABA in guard cells (Leonhardt et al., 2004). The two PP2Cs and the LEA4-5 homologs were also upregulated during PS and after PS recovery in mature leaves (Figure 8A). LEA4-5 protein levels strongly increase in poplar under drought stress conditions for osmoprotection (Abraham et al., 2018). In Arabidopsis, LEA4-5 transcript and protein levels showed the largest response to ABA and salt stress within the LEA4 group (Olvera-Carrillo et al., 2010).
Figure 8.

Predicted Regulatory Stress-Related Memory Processes in Mature Leaves (LE2) and Developing Xylem (XYL).
(A) Core regulatory networks obtained by iteratively removing single-edge nodes from expression-based regulatory network predictions (Figure 7A). Node label colors refer to periodic stress-related expression patterns. Function annotation is shown for selected nodes discussed in the main text. Potri.010G193000 is a co-ortholog of the Arabidopsis thaliana MYB transcription factor AT5G05790, here abbreviated as MYB.
(B) Model of possible transcription factor complex formation in stress-related memory derived from protein-protein interaction data in Arabidopsis. Gene names are taken from the orthology information in Phytozome. For some ZHD ortholog groups, different Arabidopsis genes (marked by an Arabidopsis ZHD identifier after the slash) constitute the best BLASTP matches of poplar genes.
(C) Model suggesting physiological roles of PP2Cs and regulatory transcription factors in mature leaves during and after periodic stress.
PP2Cs are negative regulators of ABA signaling and hamper stomatal closure, as demonstrated by the protein interactions of the PP2C HYPERSENSITIVE TO ABA1 in Populus euphratica and the PP2C ABA-INSENSITIVE1 in P. trichocarpa, as well as the analysis of transgenic Arabidopsis plants overexpressing these genes (Chen et al., 2015; Yu et al., 2016). In Arabidopsis, the PP2C HAI1 (SENESCENCE ASSOCIATED GENE113; AT5G59220) prevents stomatal closure during leaf senescence, and its promoter is directly targeted by a NAC TF (NAC029, AtNAP; AT1G69490; Zhang and Gan, 2012). In the mature leaf regulatory network inferred from our data, the expression of the HAI1 ortholog (Potri.009G037300) was not only associated with the expression of the HB7-related TF genes (Potri.014G103000, Potri.001G083700) but also with the expression of the NAC TF gene Potri.011G123300 and the MYB family TF genes Potri.010G193000 and Potri.003G100100, which belong to different ortholog groups (Figure 8A). The association between the latter three genes and the HAI1 ortholog was not detected in developing xylem. In fact, the Pearson correlation coefficients in developing xylem were 0.61, 0.30, and 0.26, respectively, in contrast with the highly significant values in mature leaves (0.94, 0.91, and 0.93). The gene Potri.011G123300 belongs to the NAC TF family because of its NAM (no apical meristem) domain; it is a member of the NAC019-related subfamily of orthologs, PTHR31719:SF82 (Mi et al., 2017). NAC TFs, particularly the three Arabidopsis members of that subfamily, NAC019, NAC055, and NAC072, are of central importance in drought signal transduction via the ABA-dependent pathway (Tran et al., 2004; Singh and Laxmi, 2015). Gene expression of the MYB TF Potri.010G193000 is negatively correlated with the wood saccharification potential in poplar, which decreases under drought conditions (Wildhagen et al., 2018). Consistent with this observation, our expression data showed that Potri.010G193000 was upregulated under stress. The same pattern was observed for mature leaves and in the case of PS, even persisted after recovery. Furthermore, Potri.010G193000 is in general co-expressed with Potri.007G085700, the TF gene TGACG SEQUENCE-SPECIFIC BINDING PROTEIN1 (TGA1) (Wildhagen et al., 2018). Interestingly, several Arabidopsis orthologs of inferred regulators of HAI1 in mature poplar leaves were connected via experimental and literature-curated protein-protein interaction data (Arabidopsis Interactome Mapping Consortium, 2011; Berardini et al., 2015; Yazaki et al., 2016), including TGA1 (Figure 8B). The TCP4 orthologs discussed above (Potri.019G091300, Potri.013G119400) were strongly transcriptionally anti-correlated with the HB7 co-ortholog Potri.001G083700, which was found to be a central predictor for the two PP2Cs and both LEA4-5 orthologs in mature leaves.
In developing xylem, the HB7 co-ortholog Potri.001G083700 was also associated with these PP2Cs and LEA4-5 orthologs, which have known physiological functions related to drought responses. In both tissues, Potri.001G083700 continued to be upregulated after PS recovery. In addition, the core networks of xylem and mature leaves shared the putative target GLUTAREDOXIN C1 (GRXC1) co-ortholog 2 (of 2; Potri.012G082800), showing post-recovery PS upregulation in both tissues. With respect to abiotic stress, GRXC1 plays roles in signaling and oxidative stress tolerance (Li, 2014). Another shared putative target was Potri.002G117800, one of two NADH DEHYDROGENASE (UBIQUINONE) FE-S PROTEIN4 genes involved in the mitochondrial electron transfer chain that have been associated with thermotolerance in Arabidopsis (Kim et al., 2012). With respect to additional TFs, the xylem network showed several differences from the network from mature leaves (Figure 8A). The ethylene response factor/APETALA2 DREB TF Potri.006G138900 (also see section "Transcription Factors Associated with Stress-Related Memory") was a large hub, in addition to the HB7 TF Potri.001G083700, which formed a central hub in both tissues. The BASIC HELIX-LOOP-HELIX18-related TF Potri.009G081400 was unique to the xylem network, associated with GRX1, LEA4-5, and both PP2Cs, and still significantly upregulated after PS recovery. A xylem-specific, putative regulator of both HB7 TFs was the BASIC HELIX-LOOP-HELIX TF Potri.014G111400, one of three PHYTOCHROME-INTERACTING FACTOR3 genes. Arabidopsis PHYTOCHROME-INTERACTING FACTOR3 promotes hypocotyl elongation (Soy et al., 2012; Zhong et al., 2012). In summary, our data suggest that common TFs such as the HB7 homologs, particularly the central hub gene Potri.001G083700, work together with tissue-specific TFs to coordinate stress and post-recovery processes in different tissues.
DISCUSSION
Although drought stress is one of the major threats to plant growth, it is well-known that plants that have endured stress can show better photosynthetic performance than nonexposed plants under both subsequent stress (Wang et al., 2014) and well-watered conditions (Hagedorn et al., 2016), which may even lead to overcompensating plant growth (Xu et al., 2010). We investigated gray poplar trees that had experienced three weeks of drought-heat stress. After 1 week of recovery, we observed not only a complete reconstitution of transpiration and photosynthetic capacity along with a relaxation of water potentials but also an increased rate of carbon gain compared with nonstressed controls, for both a periodic and a chronic stress scenario. Transcriptomic analyses across five different organs and tissues revealed cellular processes occurring in response to combined drought and heat stress and after recovery. Post-recovery expression patterns showed significant differences from nontreated poplar trees and also between the two stress scenarios, although the PS and CS responses had been highly similar at the end of the stress phase. This observation substantiates the hypothesis that stress exposure influences the physiological state of a plant even after recovery and that this long-term response varies according to the frequency or duration of the stress intervals. This memory phenomenon in trees already occurs a few days after the stress has ceased, which hints at powerful molecular mechanisms that could potentially also make a difference in plant fitness across multiple successive years. Such a life-cycle investigation was outside the scope of this study. However, the results show that such a long-term investigation might be highly valuable when carefully designed.
Similar expression patterns in response to stress and after recovery were found throughout the tree. However, they were implemented by distinct genes in each tissue. Only a small number of genes showed a consistent response profile across all tissues. Apart from genes encoding signaling components and enzymes such as PP2Cs and GRXC1 or proteins with structural function like the LEA4-5 hydrophilins (Battaglia et al., 2008), this set of genes with putatively ubiquitous function contained several TFs, most prominently two HB7 homologs that also showed a PS-related post-recovery upregulation. HB7 contains a homeodomain and a Leu zipper motif. This protein architecture indicates that the TF forms dimers (Ariel et al., 2007). TF homo- or heterodimerization as well as multimerization allow for a high degree of regulatory fine-tuning of gene expression. We therefore speculate that TF complexes might play a role in shaping stress and post-recovery regulation of gene expression in the tissue-specific context (Figure 8B). Protein-protein interaction data from the model system Arabidopsis suggest that HB7 and some TFs from the NAC019 and MYB TF families with leaf-specific responses in poplar (best Arabidopsis matches AT4G27410 and AT5G05790, respectively) may all associate with TGA1 and a set of ZHD (zinc-finger homeodomain) TFs, which were expressed in all poplar tissues according to our data set. Another putative interactor of TGA1, the only HEAT STRESS TRANSCRIPTION FACTOR C-1 (HSFC1) TF in poplar (Potri.T137400), was predicted to be a regulator of the HB7 co-ortholog Potri.001G083700 and the two PP2Cs in phloem-bark (Supplemental Data Set 8). Members of the ZHD TF family form heterodimers that play a crucial role in floral development (Tan and Irish, 2006) as well as ABA responses (Wang et al., 2011) in Arabidopsis. Coexpression of the NAC019 family gene AT4G27410 and ZHD11 strongly induces the expression of EARLY RESPONSIVE TO DEHYDRATION STRESS1, which is upregulated by drought through the ABA-independent pathway (Tran et al., 2007).
Combined action of several TFs might regulate the expression of target genes. The upregulation of PP2Cs in mature poplar leaves observed in this study is consistent with the positive regulatory role of HB7 in PP2C expression reported for Arabidopsis (Valdés et al., 2012). Furthermore, NAC TFs regulate the PP2C HAI1 (Zhang and Gan, 2012) and other drought tolerance genes (Tran et al., 2004; Singh and Laxmi, 2015). PP2Cs inactivate type-2 SUCROSE NONFERMENTING1-related protein kinases (SnRK2s), which are positive regulators of ABA signaling, stomatal closure, and chlorophyll degradation (Nakashima et al., 2009; Fujii et al., 2011; Kulik et al., 2011; Valdés et al., 2012; Gao et al., 2016). Consistently, HB7 overexpression leads to an increased chlorophyll content and a higher rate of photosynthesis (Ré et al., 2014). Moreover, various PP2Cs interact with the photosynthetic machinery (Samol et al., 2012; Fuchs et al., 2013), and photosynthesis genes are upregulated in Arabidopsis snrk2 triple mutants (Nakashima et al., 2009), pointing to a relationship between PP2Cs and photosynthesis. Translating these findings to poplar, the model would explain the improved photosynthesis detected in recovered poplar trees after periodic stress (Figure 8C). Chlorophyll content estimates did not show differences from the controls (Vanzo et al., 2015) but were done noninvasively, in contrast with the Arabidopsis studies (Ré et al., 2014; Gao et al., 2016). The increase in ABA levels during drought stress inhibits the enzyme activity of clade A PP2Cs like HAI1 and HAI3 via interacting ABA receptors (Dupeux et al., 2011; Tischer et al., 2017). Because of this mechanism, leaf stomata can close during drought to prevent excessive water loss (Figure 8C). For poplar, stress-induced stomatal closure was confirmed by our measurements of transpiration and stomatal conductance. Protein-protein interactions of PP2Cs with a SnRK2 kinase and pyrabactin resistance-like ABA receptors and their effect on leaf stomatal closure in transgenic plants have been shown for several poplar species (Chen et al., 2015; Yu et al., 2016), providing evidence for the potential roles of PP2Cs in poplar leaves during and after stress. Apart from that, PP2Cs may also be involved in chromatin remodeling and the establishment of an epigenetic memory after stress (Asensi-Fabado et al., 2017).
The system-wide rearrangement of gene expression after stress recovery might also contribute to the improved tolerance against future stresses described previously (Wang et al., 2014; Crisp et al., 2016; Hilker et al., 2016). In Arabidopsis, HB7 and LEA4-5, prominent memory-related genes based on our study, are more strongly induced during repeated dehydration stress challenges than during the first stress challenge (Ding et al., 2013). Complementing such studies on recurrence-dependent changes in stress responses, our data provide a comprehensive view of stress-related molecular memory under nonstress, post-recovery conditions. The increased base levels of HB7 and LEA4-5 gene expression after stress recovery could potentially explain their higher expression levels during subsequent stress challenges. Consistent with such a model, the periodic stress response was greater than the chronic stress response in leaves, and the increase in post-recovery base level was only significant for periodic stress and not for chronic stress, suggesting a gradual base level increase along with several stress experiences. The same trend was observed in the photosynthesis data. A better understanding of the molecular changes, their timing, and their impact on the performance of plants is instrumental for providing guidelines for resource-efficient agroforestry water management and in the breeding of crop and tree cultivars that are genetically equipped for climate change scenarios. The present evaluation indicates that transcription factors function as central switches of molecular memory and may be important mediators of plant fitness during the persistent adaptation to recurrent abiotic stress. The biological hypotheses generated by our comprehensive data acquisition and integration pave the way for detailed mechanistic studies that will provide deeper insights into memory-related molecular processes in plants.
METHODS
Plant Material
The experiments were performed with wild-type gray poplar (Populus × canescens [INRA clone 7171-B4]; syn. Populus tremula × Populus alba) plants. Plantlets were amplified by micro-propagation under sterile conditions (Leplé et al., 1992) and raised for five weeks in 2.2-liter pots on a sandy soil (1:1 [v/v] silica sand and Fruhstorfer Einheitserde, initially mixed with slow-release fertilizers: Triabon [Compo] and Osmocote [Scotts Miracle-Gro], 1:1, 10 g L−1 soil; fertilized every 2 weeks with 0.1% [w/v] Hakaphos Grün [Compo]) in the greenhouse (16/8 h photoperiodicity with supplemental lighting [high-pressure sodium vapor lamp, Philips Son-T agro], 200-240 μmol photons m−2 s−1 at the canopy level, PAR; day/night temperature 22°C/18°C; and an ambient mean CO2 concentration of 380 μL L−1). To simulate specific climate scenarios, the plants were moved to phytotron chambers (see section "Simulated Climate Conditions and Harvesting Schedule"). Within each chamber, 12 plants were cultivated together in a gas-tight sub-chamber made of acrylic glass (∼1 m3), which enabled online analysis of canopy gas exchange (Vanzo et al., 2015).
Simulated Climate Conditions and Harvesting Schedule
The future climate scenarios simulated in our experiments comprised elevated CO2 (EC) and two abiotic stress scenarios under elevated CO2, periodic drought-heat stress (PS) and chronic drought-heat stress (CS). Before starting the abiotic stress scenarios, plants were cultivated for 25 d in the phytotron chambers under control conditions (daily maximum air temperature of 27°C, 50% relative air humidity) with either ambient (380 μL L−1) or elevated (500 μL L−1) CO2. The CO2 concentrations in all scenarios followed natural occurring diurnal variations. The elevated CO2 environment in the EC, PS, and CS scenarios was created by injection of pure CO2 (+ 120 μL L−1) into the air stream of the ambient CO2. At the top of the phytotron chambers, a combination of four lamp types (metal halide lamps: Osram Powerstar HQI- TS 400W/D [Osram], quartz halogen lamps: Osram Haloline 500W, blue fluorescent tubes: Philips TL-D 36W/BLUE, and UV-B fluorescent tubes: Philips TL 40W/12) was used to obtain a natural balance of simulated global radiation throughout the UV to infrared spectrum. The lamp types were arranged in several groups to get the natural diurnal variations of solar irradiance by switching appropriate groups of lamps on and off. The short-wave cutoff in the UV-B range of the spectrum was achieved by selected soda-lime and acrylic glass filters. A detailed description of the sun simulator facility is given by Thiel et al. (1996). The PAR at canopy level was 750-800 µmol photons m−2 s−1.
For chronic stress treatment, irrigation was gradually reduced for 22 d, down to 70% reduction compared with the controls. The periodic stress treatment included three cycles of reduced irrigation (50%, 60%, and 70% reduction compared with controls), each one lasting for 6 d; between the cycles, there were recovery periods lasting 2 d. In both stress scenarios, the daily maximum air temperature was set to 33°C during periods with reduced irrigation. Noninvasive gas exchange measurements were made continuously; destructive harvests of six plants per chamber were performed at the end of the stress phase and after 1 week of recovery (Vanzo et al., 2015). Mid-day shoot water potentials (ψmd) were determined at each sampling date (n = 6 plants per treatment, mean ± se) using a Scholander pressure chamber (Scholander et al., 1965). In chronically and periodically stress-treated plants, ψmd was more negative (−1.52 ± 0.10 and −1.27 ± 0.05 MPa, respectively) compared with a ψmd of −0.97 ± 0.04 MPa in AC and of −0.97 ± 0.07 MPa in EC shoots. At recovery, ψmd went back to −0.72 ± 0.10 and −0.93 ± 0.07 MPa in PS and CS, respectively, reaching comparable values to the untreated controls in AC (−0.77 ± 0.07 MPa) and EC (−0.90 ± 0.06 MPa).
Gas Exchange Measurements
Leaf-level gas exchange measurements were performed using two GFS-3000 instruments (Walz) with an 8 cm2 clip-on-type cuvette on attached leaves (No. 9 from the apex) of four biological replicates under standard conditions (30°C, 1000 μmol photons m−2 s−1, and air humidity of 10,000 μL L−1). The cuvette was flushed with synthetic air with the CO2 concentration of the respective growth condition. For each climate chamber, CO2 and water concentrations in the ambient air were measured every 20 min with two infrared gas analyzers (Rosemount 100/4P; Walz) from the outlet of the gas-tight sub-chamber containing the plants. Inlet air was also measured every 20 min. The whole plant (canopy) net CO2 assimilation and evapotranspiration rates (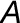 and
and 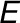 , respectively) were calculated based on the difference between the outlet and inlet concentrations of each sub-chamber (see Appendix 2 of von Caemmerer and Farquhar, 1981):
, respectively) were calculated based on the difference between the outlet and inlet concentrations of each sub-chamber (see Appendix 2 of von Caemmerer and Farquhar, 1981):
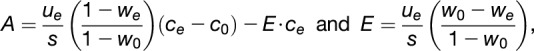 |
where 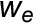 and
and 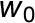 are the mole fractions of water vapor and
are the mole fractions of water vapor and  and
and 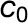 the mole fractions of CO2 in the incoming and outgoing airstreams, respectively;
the mole fractions of CO2 in the incoming and outgoing airstreams, respectively; 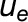 is the molar flow of air entering the chamber; and is the canopy leaf area, which was estimated for every day (Vanzo et al., 2015; Jud et al., 2016).
is the molar flow of air entering the chamber; and is the canopy leaf area, which was estimated for every day (Vanzo et al., 2015; Jud et al., 2016).
Organ and Tissue Sampling
Plants were harvested at noon on the last day of stress treatment and 7 d later at the end of the recovery period. Leaves (young leaves No. 4-6, mature leaves No. 9-12 counting from the apex, respectively) were immediately frozen in liquid N2. A stem segment was cut 10 cm above the stem base and immediately frozen in liquid N2. The roots were washed three times in water, carefully dabbed with filter paper, and frozen in liquid N2. All material was stored at −80°C until homogenization. Homogenization of plant materials was performed under liquid N2 with a mortar and pestle. The bark containing the phloem tissue was removed from the stem section with a scalpel. Young developing xylem tissue was obtained by scraping off the first 1–2 mm of the hardwood section. The homogenized mature leaf material was used for both biochemical analysis and RNA extraction, and all other materials were used only for RNA extraction. Although leaf and root samples were mixtures of several tissues, the different plant materials are referred to as tissues throughout this work.
Biochemical Measurements of the Antioxidative System
Enzyme activities and molecular antioxidant levels from four biological replicates were determined as previously described (AbdElgawad et al., 2016). Molecular antioxidants were quantified by HPLC after frozen plant materials were extracted in hexane (tocopherols) or ice-cold meta-phosphoric acid (ascorbate, glutathione). The enzyme activities of superoxide dismutase, peroxidase, catalase, ascorbate peroxidase, glutathione peroxidase, glutathione reductase, dehydroascorbate reductase, and monodehydroascorbate reductase were determined using a micro-plate reader after extracting frozen plant materials in potassium phosphate buffer supplemented with protease inhibitors.
RNA-Seq Analysis
RNA extraction was performed as described in Bi et al. (2015). Total RNA was extracted from 50 mg frozen tissue using an Aurum Total RNA Mini kit (Bio-Rad) following the manufacturer’s instructions. The RNA concentration was quantified using a NanoDrop 1000 spectrophotometer (NanoDrop, Peqlab GmbH). The 260/230 and 260/280 ratios were in the range of 1.90 to 2.67 (mean 2.21) and 1.94 to 2.45 (mean 2.12), respectively. RNA integrity was confirmed using an Agilent Bioanalyzer 2100 (Agilent Technologies). For each specific combination of environmental condition, time point, and tissue, RNA samples from three biological replicates were analyzed by Illumina sequencing (100 bp single reads, HiSeq 2500, Illumina, Inc.) of messenger RNA libraries (NEBNext Ultra directional RNA library prep Kit Illumina, New England Biolabs, Inc.), yielding RNA-seq reads for 120 samples in total. The biological replicates are samples from different individual trees grown under the same condition and harvested at the same time. For each tree, samples from all five tissues were sequenced, except for two cases where RNA extraction from the initial sample failed (260/230 ratio 0.42 and 1.4, respectively) and samples from additional trees had to be taken as replacement: AC recovery root sample replicate 1 and PS stress xylem sample replicate 3 (Supplemental Data Set 9). RNA-seq reads were aligned against the repeat-masked version of the Populus trichocarpa reference genome (assembly version v3.0; Tuskan et al., 2006) using TopHat2 (Kim et al., 2013). To account for the evolutionary distance between gray poplar and the reference genome, different alignment stringency levels were tested. For that purpose, three different sequencing libraries were randomly selected and RNA-seq reads mapped against the reference genome, allowing two to six mapping errors per read (Supplemental Figure 2). Approximately 70% of the RNA-seq reads were aligned when allowing a maximum of five errors in the read alignments, which is relatively similar to RNA-seq analysis in other plants (Mayer et al., 2012). Because of the relatively constant proportion of uniquely mapped reads for the considered error levels (Supplemental Figure 2B), we continued the analysis with the maximum threshold of five errors (Supplemental Figure 3).
Based on the read alignments and the P. trichocarpa annotation version v3.1 at the Phytozome platform (Tuskan et al., 2006; Goodstein et al., 2012), transcripts per million (TPM) gene expression levels were calculated using StringTie version 1.3.4 (Pertea et al., 2015). The biological replicates showed high Pearson correlation coefficients (computed by the cor function in R version 3.5.0 [R Core Team, 2018]) except for one single case (Supplemental Figure 4), which was excluded from further analysis. Differentially expressed genes between PS or CS and EC groups were identified using the R package DESeq2 version 1.20.0 (Love et al., 2014) using the script provided at http://ccb.jhu.edu/software/stringtie/dl/prepDE.py. Gene annotation including functional description, InParanoid orthology, and GO terms was retrieved from the P. trichocarpa reference annotation version v3.1 at the Phytozome platform (Tuskan et al., 2006; Goodstein et al., 2012). GO enrichment analysis for categories with at least 50 genes was performed in R version 3.5.0 (R Core Team, 2018) using fisher.test and multiple testing correction by p.adjust using the false discovery rate method.
Co-Expression Network Analysis
The co-expression network analysis focused on the environmental conditions with elevated CO2 levels, omitting the AC (ambient CO2) condition. For each tissue, log2(TPM+1)-transformed gene expression levels were averaged for each condition and time point and genes were filtered for a minimum coefficient of variation of 0.3 (Supplemental Figure 5). Individual co-expression modules for each tissue were determined using the R packages WGCNA version 1.64-1, flashClust version 1.01-2 and dynamicTreeCut version 1.63-1 (Langfelder and Horvath, 2008, 2012; Langfelder et al., 2008). The parameters were set to “hybrid signed” network, “average” agglomeration, split sensitivity 1, and a minimum cluster size of 50. The module eigengenes (Langfelder and Horvath, 2007), characteristic expression profiles of modules, were clustered across all tissues into communities according to their correlation. This step was performed using flashClust and dynamicTreeCut (“average” agglomeration, deep split set to true, and a minimum cluster size of 2). Communities that contained modules from all five tissues were visualized with the tkplot and plot functions in the R package igraph version 1.2.2 (Csardi and Nepusz, 2006). The corresponding heatmaps were plotted using the R packages pheatmap version 1.0.10, gridExtra version 2.3 and ggplot2 version 2.2.1 (Wickham, 2009; Auguie, 2017; Kolde, 2018).
Between-Tissue Correlations and Gene Regulatory Network Analysis
To investigate tissue-specific regulation of universally responding genes, we first determined individual genes that behaved similarly in all the tissues and then predicted their regulation by TFs using the RNA-seq data. In the first step, gene-gene correlations across individual trees from all treatment groups were computed using the cor function in R version 3.5.0 on the log2(TPM+1)-transformed gene expression data (R Core Team, 2018). In particular, correlation values of the same gene across all pairs of tissues were recorded, and the 17 genes with a median greater than 0.8 and significant post-recovery difference to controls in at least one tissue (abs[log2 fold change] > 1 and p.adj < 0.05 according to the DESeq2 analysis) were selected as query genes for further analysis. Because observations for these genes were quite complete (less than 20 values with expression level zero in the whole data set with 119 samples), we focused the regulatory network analysis on genes with at most 20 zero values. TF family annotation for P. trichocarpa and Arabidopsis (Arabidopsis thaliana) was downloaded from PlantTFDB (Jin et al., 2014) on March 9, 2018. Poplar genes were included as candidate TFs in the analysis if they themselves as well as their best Arabidopsis match according to the Phytozome annotation v3.1 (Tuskan et al., 2006; Goodstein et al., 2012) were both classified as TFs, resulting in 1346 candidates. For each query gene, the top regulatory candidates were determined from the gene expression data of each tissue separately using the R package GENIE3 version 1.2.1 (Huynh-Thu et al., 2010; Aibar et al., 2017). Networks were drawn with the R package igraph version 1.2.2 (Csardi and Nepusz, 2006). For visualization purposes, the top five candidates are shown for each query gene.
Protein-Protein Interaction Analysis
Experimental and literature-curated protein-protein interaction data for Arabidopsis were obtained from data sets of interactome publications and from the TairProteinInteraction file (time stamp: 2011-08-23) at The Arabidopsis Information Resource (Arabidopsis Interactome Mapping Consortium, 2011; Berardini et al., 2015; Yazaki et al., 2016) and compiled into a single network. The network was visualized with Graphviz version 2.36 (Gansner and North, 2000). Because of the prominent transcriptional stress-related memory response observed for the HB7 TF Potri.001G083700 and its predicted target, the HAI1 ortholog Potri.009G037300, combined with the known physiological role of HAI1 in Arabidopsis leaves and the dimerization motif of HB7, we investigated the interactomes of Arabidopsis orthologs given in Phytozome (Tuskan et al., 2006; Goodstein et al., 2012) for all TFs that were predicted to regulate Potri.009G037300 in mature leaves and showed a significant differential expression in PS vs. EC after recovery. The subnetwork connecting AT2G46680 (HB7) and AT4G27410 (closest match from the NAC019 orthology group) was evident from visual inspection of the network, and the connection to the MYB TF AT5G05790 was found computationally by neighborhood intersection. All three TFs did not have any interactions other than the ones shown in the subnetwork (Figure 8B).
Further Statistical Analysis
Treatment group comparisons for the gas exchange and antioxidant data were performed using the R package dunn.test with the false discovery rate method “bh” as a post hoc Dunn's test after application of the Kruskal-Wallis test using kruskal.test in R version 3.5.0 (R Core Team, 2018). Dimension reduction for data visualization was also done in R. To show common variation between the gas exchange data (eight parameters) and the log2(TPM+1)-transformed gene expression data in mature leaves, we selected the 100 most varying genes and applied regularized canonical correlation analysis using the rcc function from the mixOmics package version 6.3.2 (Lê Cao et al., 2009; Gonzalez et al., 2011) and an analytical estimate of the regularization parameter (Schäfer and Strimmer, 2005). Principal component analysis of the whole gene expression data set was performed with the prcomp function in R version 3.5.0 (R Core Team, 2018). Ellipses for 75% confidence levels were constructed from the expression data using the dataEllipse function of the R package car version 3.0-2 (Fox and Weisberg, 2011). Venn diagrams for differentially expressed genes (abs[log2 fold change] > 1 and p.adj < 0.05 according to the DESeq2 analysis) were created with the R package venn version 1.7 (Dusa, 2018), and the gene-wise expression heatmap was generated with the heatmap.2 function of the gplots R package version 3.0.1 (Warnes et al., 2016).
Accession Numbers
The RNA-seq data have been deposited in the ArrayExpress database at EMBL-EBI (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6121). R scripts for the data analysis are available at https://github.com/georgii-helmholtz/samm.
Supplemental Data
Supplemental Figure 1. Volcano plots for differential expression analysis of stress-treated trees relative to control trees in tissues harvested after recovery.
Supplemental Figure 2. Parameter selection for the alignment of RNA-seq reads.
Supplemental Figure 3. RNA-seq read mapping statistics.
Supplemental Figure 4. Gene expression correlation between biological replicates.
Supplemental Figure 5. Selection of genes used for co-expression network analysis.
Supplemental Data Set 1. Annotation and differential expression of stress (PS, CS) vs. control (EC) samples for all genes found in the RNA-seq analysis.
Supplemental Data Set 2. GO enrichment analysis of stress phase up-regulated genes (log2 fold change > 1, p.adj < 0.05) shared between PS and CS or specific to one of the stress scenarios.
Supplemental Data Set 3. GO enrichment analysis of stress phase downregulated genes.
Supplemental Data Set 4. GO enrichment analysis of recovery phase upregulated genes.
Supplemental Data Set 5. GO enrichment analysis of recovery phase downregulated genes.
Supplemental Data Set 6. Biochemical parameters of the anti-oxidative system (anti-oxidant levels and enzyme activities).
Supplemental Data Set 7. Co-expression modules identified from phase- and condition-dependent gene expression profiles for each tissue.
Supplemental Data Set 8. Tissue-specific gene regulatory networks for query genes that responded similarly across all tissues and showed a significant stress-related memory effect.
Supplemental Data Set 9. Tree origin and RNA-seq alignment statistics of tissue samples.
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
The work was financially supported by the European Science Foundation (ESF) Eurocores programme ‘EuroVOL’ within the joint research project ‘MOMEVIP’; the European Commission (EC), European Plant Phenotyping Network (EPPN) funded by the EU FP7 Research Infrastructures Programme (284443); the Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research) projects ‘PROBIOPA’ (0315412) and German Plant Phenotyping Network (DPPN) (031A053C); the Belgian National Fund for Scientific Research | Fonds pour la Formation à la Recherche dans l'Industrie et dans l'Agriculture (Training Fund for Research in Industry and Agriculture) (GA13511N); and by the Austrian Science Fund (FWF) (I655-B16). The authors thank Pascal Falter-Braun, Daniel Lang, and Georg Haberer for helpful comments.
AUTHOR CONTRIBUTIONS
H. Asard, A.H., K.P., K.F.X.M., and J.-P.S. designed the research; E.V., M.A.D., W.J., and R.R. performed the experiments; E.G., K.K., M.P., K.B., H.AbdElgawad, and M.S. analyzed the data; E.G. wrote the paper with contributions from K.K., M.S., A.R.S., K.P., K.F.X.M., and J.-P.S.; all authors checked and revised the manuscript.
Footnotes
Articles can be viewed without a subscription.
References
- AbdElgawad H., Zinta G., Hegab M.M., Pandey R., Asard H., Abuelsoud W. (2016). High salinity induces different oxidative stress and antioxidant responses in maize seedlings organs. Front. Plant Sci. 7: 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham P.E., Garcia B.J., Gunter L.E., Jawdy S.S., Engle N., Yang X., Jacobson D.A., Hettich R.L., Tuskan G.A., Tschaplinski T.J. (2018). Quantitative proteome profile of water deficit stress responses in eastern cottonwood (Populus deltoides) leaves. PLoS One 13: e0190019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aibar S., et al. (2017). SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14: 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Interactome Mapping Consortium (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333: 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariel F.D., Manavella P.A., Dezar C.A., Chan R.L. (2007). The true story of the HD-Zip family. Trends Plant Sci. 12: 419–426. [DOI] [PubMed] [Google Scholar]
- Aroca R., Porcel R., Ruiz-Lozano J.M. (2012). Regulation of root water uptake under abiotic stress conditions. J. Exp. Bot. 63: 43–57. [DOI] [PubMed] [Google Scholar]
- Asensi-Fabado M.A., Amtmann A., Perrella G. (2017). Plant responses to abiotic stress: The chromatin context of transcriptional regulation. Biochim. Biophys. Acta. Gene Regul. Mech. 1860: 106–122. [DOI] [PubMed] [Google Scholar]
- Auguie B. (2017). gridExtra: Miscellaneous functions for “grid” graphics. https://cran.r-project.org/web/packages/gridExtra/index.html (accessed 09/24/2018)
- Battaglia M., Olvera-Carrillo Y., Garciarrubio A., Campos F., Covarrubias A.A. (2008). The enigmatic LEA proteins and other hydrophilins. Plant Physiol. 148: 6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardini T.Z., Reiser L., Li D., Mezheritsky Y., Muller R., Strait E., Huala E. (2015). The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 53: 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Z., Merl-Pham J., Uehlein N., Zimmer I., Mühlhans S., Aichler M., Walch A.K., Kaldenhoff R., Palme K., Schnitzler J.P., Block K. (2015). RNAi-mediated downregulation of poplar plasma membrane intrinsic proteins (PIPs) changes plasma membrane proteome composition and affects leaf physiology. J. Proteomics 128: 321–332. [DOI] [PubMed] [Google Scholar]
- Bloemen J., Vergeynst L.L., Overlaet-Michiels L., Steppe K. (2016). How important is woody tissue photosynthesis in poplar during drought stress? Trees (Berl.) 30: 63–72. [Google Scholar]
- Challa K.R., Aggarwal P., Nath U. (2016). Activation of YUCCA5 by the transcription factor TCP4 integrates developmental and environmental signals to promote hypocotyl elongation in Arabidopsis. Plant Cell 28: 2117–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Zhang D., Zhang C., Xia X., Yin W., Tian Q. (2015). A putative PP2C-Encoding Gene Negatively Regulates ABA signaling in Populus euphratica. PLoS One 10: e0139466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp P.A., Ganguly D., Eichten S.R., Borevitz J.O., Pogson B.J. (2016). Reconsidering plant memory: Intersections between stress recovery, RNA turnover, and epigenetics. Sci. Adv. 2: e1501340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G., Nepusz T. (2006). The igraph software package for complex network research. InterJournal Complex Systems: 1695. http://igraph.org (accessed 09/18/2018)
- Ding Y., Fromm M., Avramova Z. (2012). Multiple exposures to drought ‘train’ transcriptional responses in Arabidopsis. Nat. Commun. 3: 740. [DOI] [PubMed] [Google Scholar]
- Ding Y., Liu N., Virlouvet L., Riethoven J.J., Fromm M., Avramova Z. (2013). Four distinct types of dehydration stress memory genes in Arabidopsis thaliana. BMC Plant Biol. 13: 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C.J., Liu J.Y. (2010). The Arabidopsis EAR-motif-containing protein RAP2.1 functions as an active transcriptional repressor to keep stress responses under tight control. BMC Plant Biol. 10: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupeux F., Antoni R., Betz K., Santiago J., Gonzalez-Guzman M., Rodriguez L., Rubio S., Park S.Y., Cutler S.R., Rodriguez P.L., Márquez J.A. (2011). Modulation of abscisic acid signaling in vivo by an engineered receptor-insensitive protein phosphatase type 2C allele. Plant Physiol. 156: 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusa A. (2018). venn: Draw Venn Diagrams. https://cran.r-project.org/web/packages/venn/index.html (accessed 08/28/2018)
- Fleta-Soriano E., Munné-Bosch S. (2016). Stress Memory and the inevitable effects of drought: A physiological perspective. Front. Plant Sci. 7: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J., Weisberg S. (2011). An R Companion to Applied Regression. (California: Sage; ). [Google Scholar]
- Fuchs S., Grill E., Meskiene I., Schweighofer A. (2013). Type 2C protein phosphatases in plants. FEBS J. 280: 681–693. [DOI] [PubMed] [Google Scholar]
- Fujii H., Verslues P.E., Zhu J.K. (2011). Arabidopsis decuple mutant reveals the importance of SnRK2 kinases in osmotic stress responses in vivo. Proc. Natl. Acad. Sci. USA 108: 1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansner E.R., North S.C. (2000). An open graph visualization system and its applications to software engineering. Softw. Pract. Exper. 30: 1203–1233. [Google Scholar]
- Gao S., Gao J., Zhu X., Song Y., Li Z., Ren G., Zhou X., Kuai B. (2016). ABF2, ABF3, and ABF4 promote ABA-mediated chlorophyll degradation and leaf senescence by transcriptional activation of chlorophyll catabolic genes and senescence-associated genes in Arabidopsis. Mol. Plant 9: 1272–1285. [DOI] [PubMed] [Google Scholar]
- Gonzalez I., Le Cao K.A., Dejean S. (2011). mixOmics: Omics data integration project. http://mixomics.org. (accessed 08/22/2018)
- Goodstein D.M., Shu S., Howson R., Neupane R., Hayes R.D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N., Rokhsar D.S. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40: D1178–D1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedorn F., et al. (2016). Recovery of trees from drought depends on belowground sink control. Nat. Plants 2: 16111. [DOI] [PubMed] [Google Scholar]
- Harfouche A., Meilan R., Altman A. (2014). Molecular and physiological responses to abiotic stress in forest trees and their relevance to tree improvement. Tree Physiol. 34: 1181–1198. [DOI] [PubMed] [Google Scholar]
- Hilker M., et al. (2016). Priming and memory of stress responses in organisms lacking a nervous system. Biol. Rev. Camb. Philos. Soc. 91: 1118–1133. [DOI] [PubMed] [Google Scholar]
- Hjellström M., Olsson A.S.B., Engström O., Söderman E.M. (2003). Constitutive expression of the water deficit-inducible homeobox gene ATHB7 in transgenic Arabidopsis causes a suppression of stem elongation growth. Plant Cell Environ. 26: 1127–1136. [Google Scholar]
- Huynh-Thu V.A., Irrthum A., Wehenkel L., Geurts P. (2010). Inferring regulatory networks from expression data using tree-based methods. PLoS One 5: e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IPCC (2014). Near-term Climate Change: Projections and Predictability. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, T.F. Stocker, D. Qin, G.K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y, Xia B.V. and Midgley P.M., eds (Cambridge: Cambridge University Press; ). [Google Scholar]
- Jin J., Zhang H., Kong L., Gao G., Luo J. (2014). PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42: D1182–D1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jud W., Vanzo E., Li Z., Ghirardo A., Zimmer I., Sharkey T.D., Hansel A., Schnitzler J.P. (2016). Effects of heat and drought stress on post-illumination bursts of volatile organic compounds in isoprene-emitting and non-emitting poplar. Plant Cell Environ. 39: 1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Lee U., Small I., des Francs-Small C.C., Vierling E. (2012). Mutations in an Arabidopsis mitochondrial transcription termination factor-related protein enhance thermotolerance in the absence of the major molecular chaperone HSP101. Plant Cell 24: 3349–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R. (2018). pheatmap: Pretty heatmaps. R package version 1.0.10. https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed 09/18/2018).
- Kotak S., Larkindale J., Lee U., von Koskull-Döring P., Vierling E., Scharf K.D. (2007). Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 10: 310–316. [DOI] [PubMed] [Google Scholar]
- Kulik A., Wawer I., Krzywińska E., Bucholc M., Dobrowolska G. (2011). SnRK2 protein kinases–key regulators of plant response to abiotic stresses. OMICS 15: 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämke J., Brzezinka K., Altmann S., Bäurle I. (2016). A hit-and-run heat shock factor governs sustained histone methylation and transcriptional stress memory. EMBO J. 35: 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. (2007). Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol. 1: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. (2012). Fast R functions for robust correlations and hierarchical clustering. J. Stat. Softw. 46: i11. [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Zhang B., Horvath S. (2008). Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 24: 719–720. [DOI] [PubMed] [Google Scholar]
- Lê Cao K.A., González I., Déjean S. (2009). integrOmics: An R package to unravel relationships between two omics datasets. Bioinformatics 25: 2855–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt N., Kwak J.M., Robert N., Waner D., Leonhardt G., Schroeder J.I. (2004). Microarray expression analyses of Arabidopsis guard cells and isolation of a recessive abscisic acid hypersensitive protein phosphatase 2C mutant. Plant Cell 16: 596–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplé J.C., Brasileiro A.C., Michel M.F., Delmotte F., Jouanin L. (1992). Transgenic poplars: Expression of chimeric genes using four different constructs. Plant Cell Rep. 11: 137–141. [DOI] [PubMed] [Google Scholar]
- Li S. (2014). Redox modulation matters: emerging functions for glutaredoxins in plant development and stress responses. Plants (Basel) 3: 559–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippold F., vom Dorp K., Abraham M., Hölzl G., Wewer V., Yilmaz J.L., Lager I., Montandon C., Besagni C., Kessler F., Stymne S., Dörmann P. (2012). Fatty acid phytyl ester synthesis in chloroplasts of Arabidopsis. Plant Cell 24: 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Staswick P.E., Avramova Z. (2016). Memory responses of jasmonic acid-associated Arabidopsis genes to a repeated dehydration stress. Plant Cell Environ. 39: 2515–2529. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer K.F., Waugh R., Brown J.W., Schulman A., Langridge P., Platzer M., Fincher G.B., Muehlbauer G.J., Sato K., Close T.J., Wise R.P., Stein N., et al. (2012). A physical, genetic and functional sequence assembly of the barley genome. Nature 491: 711–716. [DOI] [PubMed] [Google Scholar]
- Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., Thomas P.D. (2017). PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45: D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima K., Fujita Y., Kanamori N., Katagiri T., Umezawa T., Kidokoro S., Maruyama K., Yoshida T., Ishiyama K., Kobayashi M., Shinozaki K., Yamaguchi-Shinozaki K. (2009). Three Arabidopsis SnRK2 protein kinases, SRK2D/SnRK2.2, SRK2E/SnRK2.6/OST1 and SRK2I/SnRK2.3, involved in ABA signaling are essential for the control of seed development and dormancy. Plant Cell Physiol. 50: 1345–1363. [DOI] [PubMed] [Google Scholar]
- Nakashima K., Yamaguchi-Shinozaki K., Shinozaki K. (2014). The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 5: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olvera-Carrillo Y., Campos F., Reyes J.L., Garciarrubio A., Covarrubias A.A. (2010). Functional analysis of the group 4 late embryogenesis abundant proteins reveals their relevance in the adaptive response during water deficit in Arabidopsis. Plant Physiol. 154: 373–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakabe Y., Osakabe K., Shinozaki K., Tran L.S. (2014). Response of plants to water stress. Front. Plant Sci. 5: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S., Wildhagen H., Janz D., Polle A. (2017). Drought effects on the tissue- and cell-specific cytokinin activity in poplar. AoB Plants 10: plx067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanz H., Aschan G., Langenfeld-Heyser R., Wittmann C., Loose M. (2002). Ecology and ecophysiology of tree stems: Corticular and wood photosynthesis. Naturwissenschaften 89: 147–162. [DOI] [PubMed] [Google Scholar]
- R Core Team (2018). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. http://www.R-project.org (accessed 06/08/2018).
- Ré D.A., Capella M., Bonaventure G., Chan R.L. (2014). Arabidopsis AtHB7 and AtHB12 evolved divergently to fine tune processes associated with growth and responses to water stress. BMC Plant Biol. 14: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samol I., Shapiguzov A., Ingelsson B., Fucile G., Crèvecoeur M., Vener A.V., Rochaix J.D., Goldschmidt-Clermont M. (2012). Identification of a photosystem II phosphatase involved in light acclimation in Arabidopsis. Plant Cell 24: 2596–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sani E., Herzyk P., Perrella G., Colot V., Amtmann A. (2013). Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 14: R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer J., Strimmer K. (2005). A shrinkage approach to large-scale covariance matrix estimation and implications for functional genomics. Stat. Appl. Genet. Mol. Biol. 4: 32. [DOI] [PubMed] [Google Scholar]
- Scholander P.F., Bradstreet E.D., Hemmingsen E.A., Hammel H.T. (1965). Sap pressure in vascular plants: Negative hydrostatic pressure can be measured in plants. Science 148: 339–346. [DOI] [PubMed] [Google Scholar]
- Shinozaki K., Yamaguchi-Shinozaki K. (2007). Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 58: 221–227. [DOI] [PubMed] [Google Scholar]
- Singh D., Laxmi A. (2015). Transcriptional regulation of drought response: A tortuous network of transcriptional factors. Front. Plant Sci. 6: 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderman E., Mattsson J., Engström P. (1996). The Arabidopsis homeobox gene ATHB-7 is induced by water deficit and by abscisic acid. Plant J. 10: 375–381. [DOI] [PubMed] [Google Scholar]
- Soy J., Leivar P., González-Schain N., Sentandreu M., Prat S., Quail P.H., Monte E. (2012). Phytochrome-imposed oscillations in PIF3 protein abundance regulate hypocotyl growth under diurnal light/dark conditions in Arabidopsis. Plant J. 71: 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Wang C., Xiang N., Li X., Yang S., Du J., Yang Y., Yang Y. (2017). Activation of secondary cell wall biosynthesis by miR319-targeted TCP4 transcription factor. Plant Biotechnol. J. 15: 1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundell D., et al. (2017). AspWood: High-spatial-resolution transcriptome profiles reveal uncharacterized modularity of wood formation in Populus tremula. Plant Cell 29: 1585–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q.K., Irish V.F. (2006). The Arabidopsis zinc finger-homeodomain genes encode proteins with unique biochemical properties that are coordinately expressed during floral development. Plant Physiol. 140: 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor G. (2002). Populus: Arabidopsis for forestry. Do we need a model tree? Ann. Bot. 90: 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel S., Döhring T., Köfferlein M., Kosak A., Martin P., Seidlitz H.K. (1996). A phytotron for plant stress research: How far can artificial lighting compare to natural sunlight? J. Plant Physiol. 148: 456–463. [Google Scholar]
- Tischer S.V., Wunschel C., Papacek M., Kleigrewe K., Hofmann T., Christmann A., Grill E. (2017). Combinatorial interaction network of abscisic acid receptors and coreceptors from Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 114: 10280–10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran L.S., Nakashima K., Sakuma Y., Simpson S.D., Fujita Y., Maruyama K., Fujita M., Seki M., Shinozaki K., Yamaguchi-Shinozaki K. (2004). Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 16: 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran L.S., Nakashima K., Sakuma Y., Osakabe Y., Qin F., Simpson S.D., Maruyama K., Fujita Y., Shinozaki K., Yamaguchi-Shinozaki K. (2007). Co-expression of the stress-inducible zinc finger homeodomain ZFHD1 and NAC transcription factors enhances expression of the ERD1 gene in Arabidopsis. Plant J. 49: 46–63. [DOI] [PubMed] [Google Scholar]
- Tuskan G.A., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313: 1596–1604. [DOI] [PubMed] [Google Scholar]
- Valdés A.E., Overnäs E., Johansson H., Rada-Iglesias A., Engström P. (2012). The homeodomain-leucine zipper (HD-Zip) class I transcription factors ATHB7 and ATHB12 modulate abscisic acid signalling by regulating protein phosphatase 2C and abscisic acid receptor gene activities. Plant Mol. Biol. 80: 405–418. [DOI] [PubMed] [Google Scholar]
- Vanzo E., et al. (2015). Facing the future: Effects of short-term climate extremes on isoprene-emitting and non-emitting poplar. Plant Physiol. 169: 560–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Caemmerer S., Farquhar G.D. (1981). Some relationships between the biochemistry of photosynthesis and the gas exchange of leaves. Planta 153: 376–387. [DOI] [PubMed] [Google Scholar]
- Wang L., Hua D., He J., Duan Y., Chen Z., Hong X., Gong Z. (2011). Auxin Response Factor2 (ARF2) and its regulated homeodomain gene HB33 mediate abscisic acid response in Arabidopsis. PLoS Genet. 7: e1002172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Vignjevic M., Jiang D., Jacobsen S., Wollenweber B. (2014). Improved tolerance to drought stress after anthesis due to priming before anthesis in wheat (Triticum aestivum L.) var. Vinjett. J. Exp. Bot. 65: 6441–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes G.R., et al. (2016). gplots: Various R Programming Tools for Plotting Data. https://cran.r-project.org/web/packages/gplots/index.html (accessed 06/08/2018)
- Wickham H. (2009). ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag: New York. [Google Scholar]
- Wildhagen H., et al. (2018). Genes and gene clusters related to genotype and drought-induced variation in saccharification potential, lignin content and wood anatomical traits in Populus nigra. Tree Physiol. 38: 320–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Zhou G., Shimizu H. (2010). Plant responses to drought and rewatering. Plant Signal. Behav. 5: 649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W., Zhang X., Zhou B., Zhao K., Li R., Jiang T. (2017). Expression pattern of ERF gene family under multiple abiotic stresses in Populus simonii x P. nigra. Front. Plant Sci. 8: 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki J., et al. (2016). Mapping transcription factor interactome networks using HaloTag protein arrays. Proc. Natl. Acad. Sci. USA 113: E4238–E4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Yang L., Liu X., Tang R., Wang Y., Ge H., Wu M., Zhang J., Zhao F., Luan S., Lan W. (2016). Overexpression of poplar pyrabactin resistance-like abscisic acid receptors promotes abscisic acid sensitivity and drought resistance in transgenic Arabidopsis. PLoS One 11: e0168040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Gan S.S. (2012). An abscisic acid-AtNAP transcription factor-SAG113 protein phosphatase 2C regulatory chain for controlling dehydration in senescing Arabidopsis leaves. Plant Physiol. 158: 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S., Shi H., Xue C., Wang L., Xi Y., Li J., Quail P.H., Deng X.W., Guo H. (2012). A molecular framework of light-controlled phytohormone action in Arabidopsis. Curr. Biol. 22: 1530–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]