

SUMMARY
Cytomegalovirus (CMV) has a high prevalence worldwide, is often fatal for immunocompromised patients, and causes bone marrow suppression. Deficiency of signal transducer and activator of transcription 1 (STAT1) results in severely impaired antiviral immunity. We have used cell-type restricted deletion of Stat1 to determine the importance of myeloid cell activity for the defense against murine CMV (MCMV). We show that myeloid STAT1 limits MCMV burden and infection-associated pathology in the spleen but does not affect ultimate clearance of infection. Unexpectedly, we found an essential role of myeloid STAT1 in the induction of extramedullary hematopoiesis (EMH). The EMH-promoting function of STAT1 was not restricted to MCMV infection but was also observed during CpG oligodeoxynucleotide-induced sterile inflammation. Collectively, we provide genetic evidence that signaling through STAT1 in myeloid cells is required to restrict MCMV at early time points post-infection and to induce compensatory hematopoiesis in the spleen.
Graphical Abstract
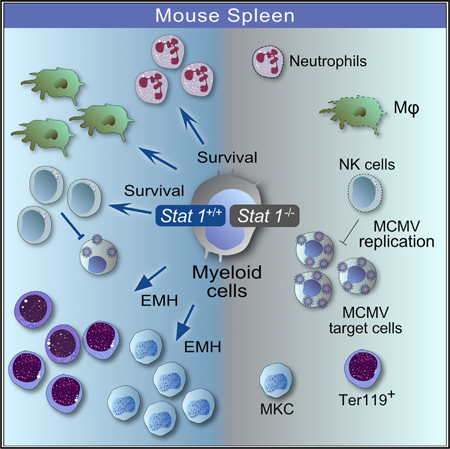
In Brief
Extramedullary hematopoiesis (EMH) is the formation of blood cells outside the bone marrow, usually in response to pathological conditions. Gawish et al. report here that STAT1 signaling in myeloid cells restricts early murine cytomegalovirus (MCMV) replication and promotes splenic EMH during acute infection and sterile inflammation.
INTRODUCTION
Cytomegalovirus (CMV), a member of the herpesvirus family, causes acute infection and establishes latency after resolution of the primary disease. Sero-positivity in the human population is up to 90%, with the potential of CMV to reactivate (Staras et al., 2006). Although the infection is usually asymptomatic in immunocompetent hosts, it can be fatal for immunocompromised patients, who develop severe immunopathology, including pneumonia, bone marrow failure (Almeida-Porada and Ascensăo, 1996; Griffiths et al., 2015; Sing and Ruscetti, 1995), splenomegaly, and splenic rupture (Alliot et al., 2001; Duarte et al., 2003). Despite the availability of antiviral drugs (Einsele et al., 2014), CMV is still a threat for the elderly (Pawelec et al., 2012), newborns, transplantation patients (Navarro, 2016), and intensive care unit patients (Frantzeskaki et al., 2015) and represents the most frequent cause of death among AIDS patients (Griffiths et al., 2015). Experimental infection of mice with murine CMV (MCMV) is established as a robust research model for human CMV (HCMV) infection (Brune et al., 2001). Similar to HCMV, MCMV has broad cellular and tissue tropism and can infect a wide range of immune and non-immune cells, such as epithelial cells, endothelial cells (Landolfo et al., 2003; Reddehase et al., 1985. 2002), monocytes, and macrophages (Henry et al., 2000; Hsu et al., 2009; Stoddart et al., 1994). In general, a fast and profound innate immune response is necessary to control CMV infection. It includes activation of dendritic cells (DCs) and natural killer (NK) cells and directs adaptive immune responses, which are crucial to clear the primary infection and to prevent reactivation of persistent CMV (Fodil-Cornu and Vidal, 2008; Loewendorf and Benedict, 2010). Although the protective functions of NK cells and DCs during CMV infection are well described (Alexandre et al., 2014; Brinkmann et al., 2015; Lisnić et al., 2015), only a few studies addressed the role of macrophages and monocytes. In the liver, resident macrophages and newly recruited monocytes modulate hepatitis (Borst et al., 2017) and facilitate recruitment and activation of NK cells and viral clearance (Hokeness et al., 2005; Salazar-Mather et al., 2002). Macrophage depletion with clodronate-loaded liposomes increases MCMV burden (Hanson et al., 1999), further supporting a protective role of these cells during infection. In contrast, monocytes and macrophages are target cells for MCMV (Hanson et al., 1999) and serve as dissemination vehicles to promote virus spread (Daley-Bauer et al., 2014). Although it is still a matter of debate, they may also be a latent reservoir for CMV (Koffron et al., 1998; Marquardt et al., 2011).
Signal transducer and activator of transcription 1 (STAT1) is a crucial component of the antiviral defense. The most prominent function of STAT1 is to mediate responses to all types of interferons (IFNs) (Boisson-Dupuis et al., 2012; Najjar and Fagard, 2010). Upon tyrosine phosphorylation by receptor-associated Janus kinases (JAKs), activated STAT1 translocates to the nucleus and induces several hundred genes whose products regulate a variety of cellular functions, such as antiviral activity, proliferation, and apoptosis. STAT1 not only induces cell-intrinsic antiviral activity but also has immune modulatory properties in cells of the innate and adaptive immune system (Najjar and Fagard, 2010). Mice deficient for STAT1 (Stat1−/−) are highly susceptible to bacterial (Decker et al., 2002; Meraz et al., 1996) and viral infections, including MCMV (Durbin et al., 1996; Gil et al., 2001). Consistently, patients carrying loss-of-function mutations in the STAT1 gene are severely immunocompromised and suffer from life-threatening bacterial and viral infections (Boisson-Dupuis et al., 2012).
Macrophages are widely used to study antiviral mechanisms, including those effective against CMV, and viral evasion strategies. Pre-treatment with IFNs makes them less susceptible for infections (Kropp et al., 2011; Presti et al., 2001), and the absence of IFN signaling components results in a failure to combat MCMV infection in vitro (Kropp et al., 2011; Presti et al., 2001; Strobl et al., 2005). However, it has remained elusive whether the antiviral activity of macrophages contributes to the immune defense against MCMV in vivo. We made use of mice that lack STAT1 in monocytes, macrophages (M), and polymorphonuclear neutrophils (PMNs; granulocytes), that is, Stat1flox/flox/LysMCre mice (herein referred to as Stat1ΔM/PMN mice), to study the role of STAT1-mediated functions of myeloid cells in the immune defense against MCMV.
Our study revealed that STAT1 in myeloid cells (i.e., monocytes, macrophages, and/or neutrophils) is crucial for the early control of MCMV infection and for induction of splenic extramedullary hematopoiesis (EMH) during viral infection and sterile inflammation. Intriguingly, the EMH phenotype is independent of the individual presence of the receptors for type I and II IFNs or IL-27.
RESULTS
Myeloid Cells Suppress Early MCMV Replication in the Spleen via STAT1
To investigate the importance of STAT1 signaling in myeloid cells in the anti-MCMV defense, we infected Stat1ΔM/PMN and Stat1fl/fl mice with salivary gland-derived MCMV (SG-MCMV) and determined viral titers in spleen and liver at 3 and 7 days post-infection (p.i.). In immunocompetent mice, MCMV replication is very efficiently controlled in the spleen (Sacher et al., 2012; Tay and Welsh, 1997), and viral load was close to the detection limit in Stat1fl/fl mice (Figure 1A). Viral titers were increased in Stat1ΔM/PMN mice compared with littermate controls and, as expected, even higher in mice that completely lack STAT1 (Stat1−/−) (Figure 1A, left). Viral load remained elevated in Stat1ΔM/PMN mice on day 7 p.i., although it considerably decreased compared with day 3 p.i. (Figure 1A, right). Thus, in contrast to Stat1−/− mice, which succumb to MCMV infection around day 5 p.i., Stat1ΔM/PMN mice are capable of controlling the infection. Viral load in the liver did not significantly differ between Stat1ΔM/PMN and Stat1fl/fl mice, although in Stat1ΔM/PMN animals, it appeared to be intermediate between Stat1fl/fl and Stat1−/− mice at day 3 p.i. (Figure 1B). MCMV was cleared from livers of Stat1fl/fl and Stat1ΔM/PMN mice at day 7 p.i. (data not shown). Consistently, MCMV genome copies in spleens, but not livers, were increased in Stat1ΔM/PMN compared with Stat1fl/fl mice at day 3 p.i. (Figures S1A and S1B). Mice that express only the Cre recombinase (LysM-Cre, Stat1+/+ Cre) did not differ from Stat1fl/fl mice with respect to the amount of viral genomes in spleen and liver (Figures S1A and S1B).
Figure 1. Stat1ΔM/PMN Mice Have Increased MCMV Load in Spleen and Salivary Glands Compared with Stat1fl/fl Mice but Are Capable of Clearing the Infection.

(A and B) Viral titers in (A) spleen and (B) liver at times indicated in Stat1fl/fl, Stat1ΔM/PMN, and Stat1−/− mice after intraperitoneal (i.p.) infection with 5 × 104 plaque-forming units (PFUs) of MCMV (n = 11–14, N = 2 or 3).
(C) Spleen and liver sections stained for the MCMV m123 protein at day 3 p.i. (left scale bar, 100 µm; right scale bar, 20 µm).
(D)Viral titers in salivary gland of MCMV-infected Stat1fl/fl and Stat1ΔM/PMN mice at times indicated (n = 15–20, N = 2).
DL, detection limit; horizontal lines and error bars indicate mean ± SEM; n, biological replicates; N, experimental repetitions. *p ≤ 0.05, **p ≤ 0.01, and ****p ≤ 0.0001.
See also Figure S1.
Immunohistological analysis for the immediate-early viral protein 1 (IE1, m123) revealed a profound increase in m123+ cells in spleens from Stat1ΔM/PMN compared with Stat1fl/fl mice (Figure 1C, top). Infected cells were localized in the red pulp and the marginal zone in Stat1ΔM/PMN mice, whereas the splenic architecture was completely lost in Stat1−/− mice, and m123+ cells were detectable throughout the whole organ (Figure 1C, top). In livers, the number of m123+ cells was similar in Stat1ΔM/PMN and Stat1fl/fl mice, whereas it was strongly increased in Stat1−/− mice (Figure 1C, bottom).
Although MCMV gets cleared from spleen and liver around day 7 p.i., viral titers start to increase in the salivary gland, the site where the virus persists and may get reactivated (Krmpotic et al., 2003). MCMV titers in the salivary glands of Stat1ΔM/PMN mice were higher than in those of control mice on day 14 p.i. (Figure 1D, left). The difference was less pronounced on day 21 p.i. (Figure 1D, middle) and infectious virus was cleared at 3 months p.i. in mice of both genotypes (Figure 1D, right). Furthermore, the abundance of MCMV genomes was similar in Stat1ΔM/PMN and Stat1fl/fl mice at 3 months p.i. (Figure S1C). Thus, STAT1 in myeloid cells contributes to the early control of MCMV in spleen and salivary glands but is dispensable for the ultimate control of the infection.
Myeloid STAT1 Protects from MCMV-Induced Spleen Pathology
To test whether the increased MCMV load in Stat1ΔM/PMN mice affects tissue damage, we performed histopathological analyses of H&E-stained spleen and liver sections at 3, 5, and 14 days p.i. (Figures 2A and 2B). Spleen pathology showed mainly cell death and marginal zone cell proliferation, peaked at 3 days p.i., and was higher in Stat1ΔM/PMN than in Stat1fl/fl mice (Figure 2A). Liver pathology was most pronounced at day 5 p.i. and remained unaffected by the absence of STAT1 in myeloid cells (Figure 2B).
Figure 2. Stat1ΔM/PMN Mice Have a More Severe Spleen Pathology and Larger Numbers of Apoptotic Splenocytes.

(A and B) Spleen (A) and (B) liver pathology of MCMV-infected Stat1fl/fl and Stat1ΔM/PMN mice at time points indicated and scored from H&E-stained sections (PBS: n = 18 or 19, N = 6; MCMV: n = 8, N = 2).
(C) Spleen sections of MCMV-infected mice (day 3 p.i.) stained for cleaved caspase-3. Representative pictures and quantifications (left scale bar, 100 µm; right scale bar, 20 µm; n = 5–8, N = 2).
(D–F) Flow cytometric analysis of apoptotic cells in spleens from MCMV-infected Stat1fl/fl and Stat1ΔM/PMN mice (day 3 p.i.). Percentage of viable, early apoptotic, and late apoptotic CD11b−/low F4/ 80+ cells (D), neutrophils (E), and NK cells (F) at day 3 p.i. and representative fluorescence-activated cell sorting (FACS) plots (n = 7 or 8, N = 2).
Mean percentages ± SEM are given. n, biological replicates; N, experimental repetitions. *p ≤ 0.05 (statistical significance between the genotypes); #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001, and ####p ≤ 0.0001 (statistical significance relative to the PBS control).
See also Figure S2.
Consistent with this finding, plasma levels of aspartate amino transaminase (AST) and alanine amino transaminase (ALT), two well-established markers for liver injury, were similar in both genotypes on days 3 and 5 p.i. (Figure S2A). Previous studies have shown that type I IFN-dependent production of CCL2 by liver-resident macrophages is required for the recruitment of inflammatory monocytes, which subsequently recruit antiviral NK cells through the secretion of CCL5 (Crane et al., 2009; Hokeness et al., 2005). We did not observe differences in the local expression of Ccl2 and Ccl5 mRNAs (Figure S2B), the recruitment of NK cells to the liver and the frequency of IFNγ+ liver NK cells (Figures S2C and S2D) between Stat1ΔM/PMN and Stat1fl/fl mice.
To confirm increased splenocyte death in Stat1ΔM/PMN mice, we performed immunohistochemical staining for cleaved caspase-3 (Casp3). The number of apoptotic splenocytes profoundly increased in Stat1ΔM/PMN mice upon infection, whereas it remained largely unchanged in Stat1fl/fl mice (Figure 2C). Flow cytometric analysis using annexin V and viability dye staining revealed an increased apoptosis of CD11b−/low F4/80+ macrophages (mainly red pulp macrophages), neutrophils, and NK cells in Stat1ΔM/PMN compared with Stat1fl/fl mice at day 3 p.i. (Figures 2D–2F). Apoptosis of CD11b+ F4/80+ cells containing macrophages and monocytes (also eosinophils and some DCs), CD11b+ F4/80− monocytes and DCs, and inflammatory monocytes (CD11b+ F4/80− Ly-6C+) did not significantly differ between genotypes (Figure S2F). Furthermore, T cell and B cell apoptosis was unaltered when comparing MCMV-infected Stat1ΔM/PMN and Stat1fl/fl mice (Figure S2E).
Taken together, these data indicate that STAT1 in myeloid cells has organ specific effects during the early immune response to MCMV: although it does not contribute to tissue damage in the liver, it inhibits viral replication and protects CD11b−/low F4/80+ macrophages, neutrophils, and NK cells from apoptotic death in the spleen.
Impaired Early Control of MCMV Infection in Stat1ΔM/PMN Mice Is Partially NK Cell Independent
NK cells are crucial for the early control of MCMV (Babić et al., 2011). We analyzed the production of IFNγ by splenic NK cells at 36 h p.i., the time point when IFNγ production by NK cells reaches peak levels (Loewendorf and Benedict, 2010). As reported previously, the frequency of NK cells in the spleen decreased upon MCMV infection (Daniels et al., 2001; Mitrović et al., 2012) but did not differ between Stat1ΔM/PMN and Stat1fl/fl mice (Figure 3A). IFNγ production by NK cells (Figure 3B) and serum levels of IFNγ (Figure 3C) were not affected by the absence of STAT1 in myeloid cells. Moreover, the frequency of granzyme B (GZMB)-expressing NK cells was similar in Stat1ΔM/PMN and control mice at 36 h, 3 days, and 5 days p.i. (Figures 3D and S3A). NK cell proliferation was analyzed by measuring cell cycle distribution using DAPI and Ki67 staining. MCMV infection resulted in a comparable shift of NK cells from G0 toward G1 and S/G2/M in Stat1ΔM/PMN and control mice, with a peak at day 3 p.i. (Figure 3E).
Figure 3. Increased Virus Titers in Stat1ΔM/PMN Mice Are Partially, but Not Completely, Dependent on NK Cells.

(A and B) Percentage of NK cells in controls (PBS) and MCMV-infected Stat1fl/fl and Stat1ΔM/PMN mice (A) and (B) percentage of IFNγ-producing NK cells and representative FACS plots (n = 3–6, N = 2).
(C) Plasma levels of IFNγ determined by ELISA (n = 9, N = 2).
(D) The percentage of GZMB-positive NK cells with representative FACS plots (n = 7–12, N = 2).
(E) NK cell proliferation of MCMV-infected and control (PBS) mice was evaluated at the times indicated by flow cytometry using DAPI/Ki67 double staining (n = 8, N = 2).
(F) Viral load in spleens from Stat1fl/fl and Stat1ΔM/PMN mice infected i.p. with 5 × 105 PFU of Δm157-MCMV (n = 4 or 5, N = 1).
(G) Viral load in spleens of MCMV-infected mice after NK cell depletion (αNK1.1) or control (PBS) treatment (n = 10, N = 2). Horizontal lines and error bars indicate mean ± SEM. **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 (statistical significance between the genotypes); ###p ≤ 0.001 and ####p ≤ 0.0001 (statistical significance relative to the PBS control). n, biological replicates; N, experimental repetitions.
See also Figure S3.
In C57BL/6 mice, MCMV-infected cells are predominantly recognized through interactions of the Ly49H receptor on NK cells with the m157 protein of MCMV (Arase et al., 2002). To test whether impaired virus control in Stat1ΔM/PMN mice still occurs in the absence of Ly49H/m157-dependent NK cell responses, we made use of a mutant MCMV that lacks m157 (Dm157-MCMV) (Bubić et al., 2004). We found higher Dm157-MCMV titers in spleens from Stat1ΔM/PMN mice compared with Stat1fl/fl mice (Figure 3F). Thus, the suppression of splenic MCMV replication by myeloid STAT1 is at least in part independent of the NK cell ability to control viral replication through Ly49H-dependent recognition. We next depleted NK cells and analyzed viral load in spleens from wild-type MCMV-infected mice. As expected, viral load increased in both genotypes upon NK cell depletion (Figure 3G). Stat1ΔM/PMN mice still exhibited a higher MCMV load than Stat1fl/fl mice after NK cell depletion, although the difference was less pronounced than in PBS controls (Figure 3G). Thus, the suppression of splenic MCMV replication by myeloid STAT1 is partly, but not completely, independent of NK cells.
STAT1 Signaling in Myeloid Cells Promotes Extramedullary Hematopoiesis during MCMV Infection
The spleen functions as a site for EMH, particularly under stress conditions (Chiu et al., 2015; Kim, 2010). Splenomegaly and spleen rupture caused by excessive EMH is a complication of CMV infection in humans (Alliot et al., 2001; Duarte et al., 2003). In support of previous studies (Jordan et al., 2013; Loh and Hudson, 1981; Lucia and Booss, 1981), Stat1fl/fl mice developed profound splenomegaly during MCMV infection, as indicated by a high spleen weight and elevated splenocyte numbers (Figures 4A and 4B). In contrast, spleen weight only modestly increased in Stat1ΔM/PMN mice (Figure 4A), and total cellularity did not change upon infection (Figure 4B). We next histologically analyzed the abundance of megakaryocytes, which is frequently used as an indicator of EMH (Lai et al., 1996; Villeval et al., 1997). Megakaryocyte numbers increased upon infection in Stat1fl/fl mice, while they remained unchanged in Stat1ΔM/PMN mice (Figure 4C), indicating that Stat1ΔM/PMN mice fail to induce infection-triggered EMH. Stat1ΔM/PMN and Stat1fl/fl mice showed comparable reductions of bone marrow cells and erythroblasts (total numbers and frequencies) upon MCMV infection (Figure S4A), thus excluding the possibility that the bone marrow is not suppressed in the absence of myeloid STAT1.
Figure 4. Stat1ΔM/PMN Mice Have Impaired Extramedullary Hematopoiesis during MCMV Infection.

(A and B) Spleen weight (A) and (B) cellularity in MCMV-infected Stat1fl/fl and Stat1ΔM/PMN mice.
(C) Number of megakaryocytes (MKC) per field counted on H&E-stained spleen sections.
(D) Total numbers of cell types indicated analyzed by flow cytometry.
(E) Erythrocyte precursor composition among erythroblasts in spleens according to size and CD44 expression.
(F) Number of hematopoietic progenitors determined by colony forming assays from spleen homogenates (day 5 p.i.). (G and H) Blood cell composition determined with a Vet ABC analyzer. Total numbers of platelets, RBC, and WBC (G) and percentage of lymphocytes, monocytes, and granulocytes (H).
In (A), (B), (D), and (E), n = 9–12, N = 3; in (C) and (F), n = 6–8, N = 2; and in (G) and (H), n = 10–14, N = 2 or 3. Mean values ± SEM are given. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 (statistical significance between the genotypes); #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001, and ####p ≤ 0.0001 (statistical significance relative to the PBS control). n, biological replicates; N, experimental repetitions.
See also Figure S4.
EMH upon MCMV infection results in increases of monocytes, macrophages, neutrophils, and erythroid cells, with a predominance of the erythroid lineage (Jordan et al., 2013). We found smaller numbers of erythroblasts, macrophages, and monocytes (CD11b−/low F4/80+, CD11b+ F4/80+, and CD11b+ F4/80− monocytes and/or DCs), NK cells, and B cells in infected Stat1ΔM/PMN mice than in littermate controls, whereas neutrophil (PMN) and T cell numbers did not differ between genotypes (Figures 4D and S4B).
Erythrocyte precursors (stages I, II, III, and IV) follow a 1:2:4:8 ratio, with stage I referring to proerythroblasts, stage II to basophilic erythroblasts, stage III to polychromatic erythroblasts, and stage IV to orthochromatic erythroblasts (Liu et al., 2013). MCMV infection did not grossly alter the relative abundance of erythroid precursors in Stat1fl/fl mice, whereas Stat1ΔM/PMN mice had a lower frequency of stage III precursors than Stat1fl/fl mice upon MCMV infection (Figure 4E). Thus, the absence of STAT1 in myeloid cells not only results in reduced numbers of erythroblasts but also alters the erythroid precursor cell composition during MCMV infection.
To functionally confirm the defect in EMH in Stat1ΔM/PMN mice, we analyzed the numbers of splenic hematopoietic progenitor cells using colony formation assays. Consistent with previous studies (Jordan et al., 2013), MCMV infection induced increases in BFU-E, CFU-M, CFU-G, and CFU-GEMM in Stat1fl/fl mice (Figure 4F). The number of BFU-E did not increase in Stat1ΔM/PMN mice, supporting a crucial role of myeloid STAT1 in MCMV-induced splenic erythropoiesis (Figure 4F). CFU-M and CFU-GEMM were unaltered, whereas the number of CFU-G was higher in Stat1ΔM/PMN than in Stat1fl/fl mice (Figure 4F).
To investigate whether differences in EMH correlate with differences in blood cell composition, we performed differential blood cell analysis on days 3, 5, and 9 p.i. (Figures 4G and 4H). MCMV infection caused severe cytopenia, as indicated by strong decreases in platelet and white blood cell (WBC) counts at day 3 p.i. (Figure 4G). Red blood cells (RBCs) modestly decreased upon MCMV infection at day 5 p.i. (Figure 4G). The decrease in RBC counts upon MCMV infection is comparable with those reported using other infection and inflammation models (Johns et al., 2009; Noyes et al., 2009; Schubert et al., 2008) and satisfies the definition of anemia (i.e., RBC counts lower than 2 SDs below the average of a population; Raabe et al., 2011). Stat1ΔM/PMN mice showed reduced platelet and RBC numbers compared with Stat1fl/fl mice at days 5 and 9, respectively (Figure 4G), indicating that blood cell recovery is delayed in the absence of myeloid STAT1. Moreover, lymphocyte abundance was lower and granulocyte abundance higher in Stat1ΔM/PMN than in Stat1fl/fl mice on day 5 p.i. (Figure 4H).
Taken together, these results suggest that myeloid STAT1 promotes extramedullary erythropoiesis and megakaryopoiesis and directly or indirectly restricts granulopoiesis during MCMV infection.
MCMV Infection Promotes EMH in the Absence of IFNAR1 or IFNGR2 in Myeloid Cells or the Complete Absence of IL-27Rα
Among cytokines that activate STAT1, type I and type II IFNs regulate stress-induced hematopoiesis in the bone marrow by multiple mechanisms, including direct effects on hematopoietic stem cells (HSCs) and various hematopoietic progenitors (Baldridge et al., 2010; de Bruin et al., 2014; Ehninger et al., 2014; Essers et al., 2009; Haas et al., 2015; Hirche et al., 2017; Sato et al., 2009). More recently, IL-27 has been shown to promote the expansion of HSCs and myeloid progenitors in the bone marrow (Furusawa et al., 2016). To test potential upstream signals, we made use of Ifnar1ΔM/PMN, Ifngr2ΔM/PMN, and IL-27Rα-deficient mice (Il27ra−/−/Wsx1−/−). As EMH correlates with splenomegaly and the abundance of erythroblasts (Jordan et al., 2013; Figure 4), we determined the extent of EMH on the basis of these parameters. Surprisingly, neither conditional loss of type I or type II IFN responsiveness in myeloid cells nor complete loss of IL-27 responsiveness was each individually sufficient to decrease splenomegaly and splenic erythropoiesis (Figures 5A–5C, 5F–5H, and 5K–5M). Ifnar1ΔM/PMN had an even modestly increased splenomegaly, although this did not translate into an increase in total splenocyte and erythroblast numbers (Figures 5A–5C). However, IL-27Rα deficiency resulted in a shift in the erythroblast composition from stage II and III toward stage I erythroblasts (Figure 5N), indicating that IL-27 signaling affects erythroblast differentiation during MCMV infection. Modest alterations in erythroblast composition were also observed in Ifngr2ΔM/PMN mice (Figure 5I). In contrast to Stat1ΔM/PMN mice, Ifnar1ΔM/PMN, Ifngr2ΔM/PMN, and Il27ra−/− mice did not have decreased platelet counts compared with control mice (Figures 5E, 5J, and 5O).
Figure 5. MCMV Infection Promotes EMH in the Absence of IFNAR1 or IFNGR2 in Myeloid Cells and in the Complete Absence of IL-27Rα.

(A–C, F–H, and K–M) Spleen weight (A, F, and K), (B, G, and L) cellularity, and (C, H, and M) total number of erythroblasts per spleen of Ifngr2ΔM/PMN, Ifnra1ΔM/PMN, and Il27ra−/− mice 5 days after MCMV infection.
(D, I, and N) Erythrocyte precursor composition among erythroblasts in spleens according to size and CD44 expression (day 5 p.i.).
(E, J, and O) Blood platelet counts determined with a Vet ABC analyzer (day 5 p.i.).
In (A)–(E) and (K)–(O), n = 8–14, N = 2; and in (F)–(J), n = 17–19, N = 3. Mean values ± SEM are given. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 (statistical significance between the genotypes); #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001, and ####p ≤ 0.0001 (statistical significance relative to the PBS control). n, biological replicates; N, experimental repetitions.
Taken together these data show that neither type I or type II IFN signaling in myeloid cells nor IL-27 activity is individually required for EMH in response to MCMV infection.
Inflammation-Associated EMH Crucially Depends on the Presence of STAT1 in Myeloid Cells
Having established that STAT1 drives splenic EMH during MCMV infection, we next asked the question whether this extends to inflammation-associated EMH. Hence, we induced sterile systemic inflammation by challenging mice with synthetic CpG-motif containing oligodeoxynucleotides (CpG-ODN), which induce; EMH with massive erythropoiesis at day 6 post-injection (Sparwasser et al., 1999). Stat1ΔM/PMN mice reacted with a comparable phenotype upon CpG-ODN challenge as during MCMV infection: spleen weight and cellularity remained almost un-changed in Stat1ΔM/PMN mice, whereas they strongly increased in Stat1fl/fl mice (Figures 6A and 6B). In addition, splenic erythroblast numbers were lower in Stat1ΔM/PMN than in Stat1fl/fl mice (Figure 6C), suggesting that CpG-ODN-induced EMH is impaired in these mice. Moreover, CpG-ODN-challenged Stat1ΔM/PMN mice had fewer monocytes and/or macrophages, B cells, and NK cells than control mice (Figures 6D and S5A), although the differences were less pronounced than during MCMV infection. Splenic erythroid precursor composition differed between Stat1ΔM/PMN and Stat1fl/fl mice upon CpG-ODN challenge (Figure 6E), although the stages affected were different compared with MCMV infection. As reduced EMH correlated with enhanced splenic cell death after MCMV infection, we performed immunohistochemical staining for cleaved Casp3. CpG-ODN treatment induced considerable splenocyte apoptosis in both Stat1fl/fl and Stat1ΔM/PMN mice. In contrast to MCMV infection, we did not detect differences between genotypes (Figures 6F and 6G), indicating that STAT1 promotes EMH independent of its anti-apoptotic activity.
Figure 6. Stat1ΔM/PMN Mice Have Impaired Extramedullary Hematopoiesis during Sterile Inflammation.

(A and B) Spleen weights (A) and (B) cellularity in Stat1fl/fl and Stat1ΔM/PMN mice 6 days after injection of 10 nM CpG-ODN.
(C and D) Total numbers of erythroblasts (C) and (D) cell types indicated were analyzed using flow cytometry.
(E) Erythrocyte precursor composition as in Figure 4E. Representative FACS plots and quantifications.
(F and G) Splenic sections of CpG-ODN-challenged mice (day 6 post-injection) stained for cleaved caspase-3. Representative pictures (F) and quantifications (G) of cells positive for active caspase-3 (left scale bar, 100 µm; right scale bar, 20 µm).
Mean values ± SEM are given (n = 6–8, N = 2). *p ≤ 0.05, **p ≤ 0.01, and ****p ≤ 0.0001 (statistical significance between the genotypes); #p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001, and ####p ≤ 0.0001 (statistical significance relative to the PBS control). n, biological replicates; N, experimental repetitions.
DISCUSSION
In this study we identified a crucial contribution of myeloid cells to the early antiviral defense against MCMV and discovered a novel function of STAT1 in stress-induced hematopoiesis in the spleen.
The role of monocytes and macrophages in the context of CMV infection is poorly understood. Conditional ablation of Stat1 allowed us to study the importance of STAT1 in myeloid cells in the immune response to MCMV. Monocytes and macrophages are target cells for MCMV in vivo and believed to promote dissemination of MCMV to distal organs (Daley-Bauer et al., 2014; Hanson et al., 1999; Henry et al., 2000; Hsu et al., 2009; Stoddart et al., 1994). We show that myeloid cells require STAT1 to restrict early MCMV infection and to prevent infection-associated tissue damage in the spleen. STAT1 may inhibit viral replication through its cell-intrinsic antiviral activity and by facilitating NK cell survival. Although it must be taken into consideration that LysM-Cre is expressed not only in monocytes and macrophages but also in neutrophils (Clausen et al., 1999), a recent study excluded a contribution of neutrophils to the defense against MCMV in the spleen (Stacey et al., 2014). It also seems unlikely that neutrophils disseminate MCMV to the salivary glands, as they are relatively short-lived cells and are not productively infected by MCMV (Stacey et al., 2014). In support of studies with clodronate-mediated depletion of macrophages (Hanson et al., 1999), we observed little impact of STAT1 deficiency in myeloid cells on MCMV infection in the liver. Organ specificity may reflect MCMV tropism, as macrophages are the major target cell in the spleen (Hanson et al., 1999), whereas the contribution of monocytes and macrophages to MCMV control in the liver may be masked by the high susceptibility of hepatocytes to MCMV infection (Sacher et al., 2012). Alternatively, functional heterogeneity of tissue resident macrophages (Gordon et al., 2014) may account for organ specificity. Our finding that in contrast to type I IFNs (Crane et al., 2009; Hokeness et al., 2005), STAT1 in myeloid cells is not involved in the induction of Ccl2 and the recruitment of NK cells supports previous studies indicating that Ccl2 is induced by type I IFN in a STAT1-independent manner (Zimmerer et al., 2007) and underscores the importance of STAT1-independent responses to IFNs to the immune response against MCMV (Gil et al., 2001).
In addition to the role of STAT1 in the antiviral defense, our study uncovered an unexpected function in the induction of splenic EMH, as evidenced by decreased splenomegaly, decreased megakaryocyte and erythroblast numbers, and a marked decrease in BFU-E in MCMV-infected Stat1ΔM/PMN mice compared with littermate controls. This correlated with a delayed recovery of Stat1ΔM/PMN mice from MCMV-induced peripheral blood thrombocytopenia and anemia and a more persistent MCMV-induced shift of WBCs from lymphocytes to granulocytes. As bone marrow failure frequently occurs during CMV infection after transplantation and is associated with pancytopenia and a poor prognosis, our findings are of clear clinical significance (Almeida-Porada and Ascensăo, 1996; Lin et al., 2011; Mutter et al., 1988; Sato et al., 2011; Sing and Ruscetti, 1995). EMH is a compensatory response to hematopoietic stress in humans and mice and is frequently induced by microbial infections (Chiu et al., 2015). During CMV infection, EMH causes splenomegaly, with splenic rupture being a rare but fatal complication (Alliot et al., 2001; Duarte et al., 2003). The crucial role of macrophages in homeostatic and stress-induced hematopoiesis in the bone marrow and in stress-induced EMH is just beginning to emerge (Chow et al., 2013; McCabe and MacNamara, 2016). We show that myeloid cells promote EMH through a process that requires STAT1. Importantly, STAT1 not only facilitates EMH during MCMV infection but also during sterile inflammation. This indicates that the EMH-promoting function of STAT1 may be separated from its antiviral activity and contributes to compensatory hematopoiesis under diverse inflammatory conditions. Further investigations will be required to delineate the exact mechanism for how myeloid STAT1 induces EMH. It is attractive to speculate that during MCMV infection, the main EMH-promoting function of STAT1 is to directly or indirectly protect macrophages from cell death. Our data indicate that the cell-protective activity of STAT1 is not the sole mechanism for how STAT1 drives EMH, as we did not observe differences in splenocyte apoptosis between Stat1ΔM/PMN and Stat1fl/fl mice upon CpG-ODN treatment. Interestingly, our data suggest that during MCMV infection, myeloid STAT1 drives extramedullary erythropoiesis and megakaryopoiesis. Replenishment of blood platelets during systemic inflammation depends on the activation of a stem cell-like megakaryocyte progenitor in the bone marrow (Haas et al., 2015). Splenic macrophages may activate these specialized progenitors or facilitate their proliferation and differentiation by shaping the local cytokine milieu (Müller-Newen et al., 2017). Macrophages are important constituents of the erythropoietic niche, termed erythroblastic islands (EIs), in bone marrow and spleen. Within EIs, macrophages regulate erythropoiesis by various adhesive signals and soluble factors (Chasis and Mohandas, 2008; Johns and Christopher, 2012), suggesting that STAT1 might promote extramedullary erythropoiesis through upregulating some of these factors and signals, although this clearly requires further investigation.
It has remained elusive whether STAT1 in myeloid cells requires cytokine-induced activation to promote EMH. We show that IFNγ signaling in myeloid cells is not required for MCMV-induced EMH, which is in line with the unimpaired splenomegaly and expansion of Ter119+ erythroid cells upon MCMV infection in spleens of mice with complete IFNGR1 deficiency (Jordan et al., 2013). Furthermore, we did not find evidence for involvement of type I IFNs and IL-27. Future studies will be required to determine whether this is due to redundancies in cytokine-induced responses, involvement of other STAT1-activating cytokines, or STAT1 functions that do not rely on cytokine-induced activation.
IFNγ is associated with the loss of bone marrow HSCs during severe anemia (de Bruin et al., 2014), which was recently attributed to IFNγ signaling in macrophages during aplastic anemia induced by sublethal irradiation and bulk splenocyte transfer (McCabe et al., 2018). We show that MCMV infection results in reduced bone marrow cellularity and erythroblast abundance in the absence of myeloid STAT1. This finding argues against a requirement for IFNγ signaling in macrophages for MCMV-induced bone marrow suppression and is consistent with earlier studies indicating that short-term exposure to IFNγ during microbial infection is not detrimental to the host (de Bruin et al., 2014).
Collectively, our study reveals STAT1-dependent activities of myeloid cells as essential components of the immune defense against MCMV. Moreover, we show that STAT1 in myeloid cells is an essential driver of EMH under stress conditions, in particular of the erythroid and megakaryocytic lineage, marking it as potential therapeutic target to treat bone marrow failure caused by CMV infection and possibly other diseases that involve EMH.
STAR ★ METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. rer. nat. Birgit Strobl (birgit.strobl@vetmeduni.ac.at).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Stat1ΔM/PMN (Stat1flox/flox/LysMCre) were described previously (Wallner et al., 2012). Ifnar1ΔM/PMN and Ifngr2ΔM/PMN mice were generated by crossing Ifnar1fl/fl (Kamphuis et al., 2006) or Ifngr2fl/fl mice (Lee et al., 2015) with LysMCre-mice (Clausen et al., 1999). Ifngr2fl/fl mice were kindly provided by Werner Müller (Technical University Braunschweig, Germany). IL-27Rα-deficient mice (Il27ra−/−/Wsx1−/−, B6N.129P2-Il27ratm1Mak/J) were from The Jackson laboratory. Littermate controls, wild-type mice and mice heterozygous for the Cre recombinase were used as controls for the experiments. Experiments were performed with age- and sex-matched (8 – 12 weeks old) mice. All mice were on C57BL/6 background and bred at the University of Veterinary Medicine Vienna under specific pathogen-free conditions according to Federation of European Laboratory Animal Science Associations (FELASA) guidelines. All animal experiments were approved by the institutional ethics and animal welfare committee and the national authority according to §§ 26ff. of Animal Experiments Act, Tierversuchsgesetz 2012 - TVG 2012 (BMWF 68.205/0243-II/3b/2011, BMWFW 68.205/0032-WF/II/3b/2014, BMWFW 68.205/0212-WF/V/3b/2016 and BMBWF-68.205/0134-V/3b/2018).
Cells
Mouse embryonic fibroblasts (MEFs) were isolated from C57BL/6N mice. Briefly, 13–14 days old embryos were dissected and, after removing heads and livers, minced with scissors and resuspend in PBS supplemented with penicillin (50 μg/ml) and streptomycin (50 U/ml). The suspension was passed through a 100 μm nylon cell strainer and centrifugated at 200 × g for 5 min. The pellet was washed with PBS two times and 5 × 106 cells were plated per 10 cm dish in DMEM (high glucose), 10% FCS (heat-inactivated), Pen/Strep (100 μg/ml and 100 U/ml), 2mM L-glutamine and 50μM β-mercaptoethanol. Cells were split when confluent or stored in liquid nitrogen in 10% DMSO in FCS.
Viruses
The murine cytomegalovirus (MCMV) used in this study was a salivary gland-derived (SG) Smith strain (American Type Culture Collection, ATCC® VR194). The Δm157-MCMV strain (Bubić et al., 2004) was tissue culture-derived (TC).
METHOD DETAILS
In vivo infection/CpG-ODN injection
Mice were infected intraperitoneally (i.p.) with SG-MCMV or Δm157-MCMV at the dose indicated. CpG-ODN was injected i.p. at a dose of 10 nmol. PBS was used as a control. In all cases the injected volume was 200 μl.
Plaque assay
Organs were mashed through a 100 μm nylon cell strainer followed by homogenization with a Wheaton tight-fitting pestle (Figure 1, Figure 3F) or organs were homogenized in PBS with an automated tissue homogenizer (1500 rpm, 90 s, MiniG, SPEX) (Figure 3G). Plaque assays were performed using primary MEFs (at passage numbers below 5 and grown to sub-confluence) using centrifugation enhancement (room temperature, 30 min, 110 × g) followed by 90 min incubation at 37°C, 5% CO2. Virus inoculum was removed and cells were overlaid with 1% of low-melting agarose in growth medium. MCMV plaques were counted under microscope 3 to 4 days later.
RT-qPCR for MCMV genome copies
Genomic DNA was isolated from homogenized tissues after proteinase K digestion using phenol/chloroform/isoamyl alcohol purification, followed by precipitation with isopropanol and washing with ethanol (70%). The DNA pellet was dissolved in TE-buffer and the DNA concentration was determined using Nano Drop. RT-qPCR was performed with primers for MCMV m54 (300 nM):
The RT-qPCR mixture was composed of 4mM MgCl2, 100 nM Evagreen® (Biotium), 1 U/rxn Hotfire Polymerase (Solis BioDyne), 200 μM dNTP mix and 1x Hotfire B buffer. PCR conditions were: 15 min at 95°C and 40 cycles 95°C for 20 s and 60°C for 1 min. For absolute quantification of the genome copies, a standard curve was obtained using serial dilutions of DNA from purified TC-MCMV.
Blood analysis and transaminase measurements
Differential blood analysis was performed with Scil Vet abc hematology analyzer. Blood was centrifugated at 3000 × g for 10 min and transaminases were measured in plasma using VetTest 8008.
Histology and immunohistochemistry
Organs were fixed in 10% formalin for 24 h and embedded in paraffin, cut into 4 μm sections, stained with hematoxylin and eosin (H&E) and scored by a trained pathologist, who was blinded for the groups. Scoring of liver pathology was performed as described (Tang-Feldman et al., 2011) and included interface activity, intralobular inflammation and necrosis, portal inflammation (0 = absent, 1 = mild, 2 = moderate, 3 = moderately severe, 4 = severe) and steatosis (0 = absent, 1 = present). The scoring of spleen pathology mainly focused on parameters of cell death, such as apoptosis, necrosis and cell proliferation in the marginal zone (0 = absent, 1 = mild, 2 = moderate, 3 = severe) as well as siderosis (0 = absent, 1 = mild/moderate, 2 = severe). For immunohistochemistry, in brief, de-paraffinized samples were rehydrated, endogenous peroxidase activity was blocked using 3% H2O2 and samples were subjected to antigen retrieval using 10 mM citrate buffer (pH 6, Vector labs). After several blocking steps, sections were incubated at 4°C overnight with either anti-mouse cleaved caspase 3 antibody (clone 5A1E, Cell Signaling Technologies) or anti-m123 antibody (Croma 101). After washing steps and incubation with a corresponding secondary antibody, visualization was achieved using the Vectastain ABC kit (Vector Labs), the slides were counterstained with hematoxylin and images were acquired using a Zeiss Axio imager microscope. Quantification of positive cells was performed by counting cells/field in 5 pictures per mouse (20-fold or 10-fold magnification), blinded for the groups.
RNA isolation and real-time qPCR
RNA was isolated using TRIzol reagent (Invitrogen) and reverse transcription was performed using iScript First Strand cDNA Synthesis Kit (Bio-Rad). Quantitative PCR for Ccl2 and Ccl5 was done with QIAGEN assays and the Evagreen® detection system. Ube2d2 was used as housekeeping gene with the following primers and FAM-labeled probe:
The RT-qPCR mixture was composed of 4mM MgCl2, 100 nM Evagreen® or FAM probe (for Ube2d2), 1 U/rxn Hotfire Polymerase (Solis BioDyne), 200 μM dNTP mix and 1x Hotfire B buffer. PCR conditions were: 15 min at 95°C and 40 cycles 95°C for 20 s and 60°C for 1 min.
In vivo NK cell depletion assay
NK cells were depleted using anti-NK1.1 antibody (clone PK136, InVivoMab). 300 μg of the antibody was injected i.p. 24 h before and 36 h after MCMV infection. Control groups were injected with 200 μL PBS. Spleens were collected 3 days after infection. Depletion efficiency was determined by FACS analysis of splenic NK cells (Figure S3B).
Colony forming unit assay
Five days after MCMV infection, splenocytes were isolated by mashing the spleens through a 100 μm cell strainer. Samples were centrifugated and erythrocyte lysis was performed. Cells were counted with Türk solution and 2 × 105 cells were plated in methylcellulose-based medium that included IMDM, 10% FCS (heat-inactivated), Pen/Strep (100 μg/ml and 100 U/ml), 2 mM L-glutamine and 50 μM β-mercaptoethanol with recombinant cytokines for mouse (Stem Cell Technology). Colonies were counted under microscope after 7 days incubation at 37°C, 5% CO2. BFU-E, blast-forming unit erythroid; CFU-G, colony-forming unit granulocyte; CFU-M, colony-forming unit macrophages; CFU-GEMM, colony-forming unit granulocyte, erythrocyte, monocyte, megakaryocyte.
Flow cytometry
Isolation of cells from spleen, bone marrow and liver
Splenocytes were isolated by mashing the spleens through a 100 μm cell strainer. Femurs were isolated from mice at the indicated time point, cleaned bones were crushed with mortar and pestle in PBS and mashed through a 100 μm cell strainer. Samples were centrifuged and erythrocyte lysis was performed in all staining panels, except for the erythroid compartment. Cells were counted (CASY cell counter, OMNI Life Science). Liver lymphocytes were isolated from euthanized mice after liver perfusion with 10 mL of PBS, livers were mashed through a 100 μm cell strainer and washed twice with PBS. Enrichment of lymphocytes was done with 37.5% Percoll solution and 2.5 × 106 cells (for extracellular staining) or 5 × 106 cells (for intracellular staining) per sample were used. Single stains were used to set up the compensation of fluorophores in FACS CantoTM, FACS DivaTM (BD Bioscience) and FlowJo® software (FLOWJO, LLC).
Staining of specific cell types
Cells were first stained with Fixable Viability Dye (APC Cy7, working solution 1:1000 in PBS) for 5 min at room temperature. Cells were washed with PBS and incubated with antibodies against surface markers at a dilution of 1:200 (except for CD11b, which was used at 1:400) for 20 min at 4°C in the dark. Staining panel 1 (Figure 4, Figure 5, Figure 6, Figure S4 and Figure S5): CD16/CD32 Fc block (unconjugated), CD3ε (eF450), CD19 (APC), CD11b (PerCP Cy5.5), Ly-6G (PE), NK1.1 (Pe Cy7) and F4/80 (FITC). Staining panel 2 (Figure 4, Figure 5 and Figure 6): CD16/CD32 Fc block (unconjugated), Ter119 (eF450) and CD44 (PE). Cells were washed with PBS and fixed with 2% PFA for 30 min at room temperature in the dark. Gating strategies are shown in Figures S2G, S4C and S4D.
Apoptosis (Annexin V/LD) staining
Staining was performed using Annexin staining kit according manufacturer’s instructions. Briefly, cells were stained with Fixable Viability Dye for 5 min at room temperature, washed with PBS and incubated with antibodies against surface markers 20 min at 4°C in the dark. Staining panel 1 (Figure 2 and Figure S2): CD16/CD32 Fc block (unconjugated), CD3 (eF450), CD19 (APC), CD11b (PerCP Cy5.5), NK1.1 (FITC) and Ly-6G (PE). Staining panel 2 (Figure 2 and Figure S2): CD16/CD32 Fc block (unconjugated), Ly-6C (eF450), CD3/CD19/NK1.1 (APC), CD11b (PerCP Cy5.5), Ly-6G (PE) and F4/80 (FITC). After staining, samples were washed with PBS, then with binding buffer and stained with Annexin V (PE Cy7) 15 min at room temperature, in dark. Samples were washed with binding buffer and fixed with intracellular fixation buffer (Intracellular Fixation & Permeabilization Buffer Set).
Intracellular staining for IFNγ
Cells were incubated for 20 min at 4°C in dark with following antibodies (Figure 3 and Figure S3): CD16/CD32 Fc block (unconjugated), CD3 (eF450), NK1.1 (FITC) and NKp46 (PE). Cells were washed with PBS and permeabilized with perm/wash buffer (Fixation/Permeabilization Solution Kit), according to the manufacturer’s instructions. Intracellular staining was done in perm/ wash buffer with anti-IFNγ (APC) antibody for 30 min at 4°C in the dark. Cells were washed and fixed with fixation solution.
Intracellular staining for Granzyme B
Splenocytes were incubated for 4 h in 1 mL of Brefeldin A/RPMI (1:1000 dilution) at 37°C, 5% CO2, stained with Fixable Viability Dye, washed with PBS and incubated 20 min at 4°C in the dark with following antibodies (Figure 3 and Figure S3): CD16/CD32 Fc block (unconjugated), CD3ε (eF450), NKp46 (PE) and NK1.1 (PeCy7). Cells were washed with PBS, fixed and permeabilized using Foxp3 Staining Kit according to the manufacturer’s instructions. Intracellular staining was performed in permeabilization buffer with anti-Granzyme B (APC) antibody for 30 min at 4°C in dark. Cells were washed with permeabilization buffer and then with PBS.
Intracellular staining for Ki67/DAPI
Cells were incubated for 20 min at 4°C in dark with following antibodies (Figure 3): CD16/CD32 Fc block (unconjugated), CD3e (APC), NKp46 (PE) and NK1.1 (PeCy7). Cells were washed with PBS, fixed for 20 min at room temperature using 200 μl Cytofix (BD, Fix and Perm). Fixed cells were permeabilized using BD perm washing buffer (1:10 diluted in MilliQ water) and subsequently incubated for 30 min at room temperature with 200 μl staining solution (1g/ml DAPI, 20 μl FITC conjugated anti-Ki67 antibody in PBS containing 0,1% Triton-X). Cells were washed with perm buffer and PBS. The gating strategy is shown in Figure S3C.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value of n (number of mice), N (number of experiments), the definition of central value and spread (mean ± SEM), and statistical significance are reported in the Figure Legends. Statistical analysis for PFU and genome copies of MCMV was done on log-transformed data. None of the data were removed from our statistical analysis as outliers. Data were considered to be statistically significant when p ≤ 0.05. In figures, asterisks represent statistical significances for comparisons between genotypes (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001) and hashtags represent statistical significances between MCMV-infected/CpG-ODN-treated mice and respective PBS controls (#p ≤ 0.05, ##p ≤ 0.01, ###p ≤ 0.001, ####p ≤ 0.0001). P values between 0.05 and 0.1 are indicated for the genotype comparisons. Statistical analysis was performed in GraphPad Prism 7 (Student’s t test and one-way ANOVA with Tukey’s multiple comparison test) or in R-studio IDE (two-way ANOVA, Figure 3G; https://www.rstudio.com/).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal antibody CD16/CD32 (Clone 93) | Thermo Fisher | Cat#14–0161-85; RRID:AB_467134 |
| Rat monoclonal antibody TER-119 (Clone TER-119) | Thermo Fisher | Cat#48–5921-82; RRID:AB_1518808 |
| Rat monoclonal antibody CD44 (Clone IM7) | Thermo Fisher | Cat# 12–0441-81; RRID:AB_465663 |
| Syrian hamster monoclonal antibody CD3e (Clone eBio500A2) | Thermo Fisher | Cat# 48–0033-82; RRID:AB_2016704 |
| Armenian hamster monoclonal antibody CD3e (Clone 145–2C11) | Thermo Fisher | Cat# 17–0031-82; RRID:AB_469315 |
| Rat monoclonal antibody CD19 (Clone eBio1D3) | Thermo Fisher | Cat# 17–0193-82; RRID:AB_1659676 |
| Mouse monoclonal antibody NK1.1 (Clone PK136) | Thermo Fisher | Cat# 25–5941-82; RRID:AB_469665 |
| Mouse monoclonal antibody NK1.1 (Clone PK136) | Thermo Fisher | Cat#11–5941-82; RRID:AB_465318 |
| Mouse monoclonal antibody NK1.1 (Clone PK136) | Thermo Fisher | Cat# 17–5941-82; RRID:AB_469479 |
| Rat monoclonal antibody CD335 (NKp46) (Clone 29A1.4) | Thermo Fisher | Cat# 12–3351-82; RRID:AB_1210743 |
| Rat monoclonal antibody CD11b (Clone M1/70) | Thermo Fisher | Cat# 45–0112-82; RRID:AB_953558 |
| Rat monoclonal antibody F4/80 (Clone BM8) | Thermo Fisher | Cat# 11–4801-82; RRID:AB_2637191 |
| Rat monoclonal antibody Ly-6G (Clone 1A8-Ly6g) | Thermo Fisher | Cat# 12–9668-82; RRID:AB_2572720 |
| Rat monoclonal antibody Ly-6C (Clone HK1.4) | Thermo Fisher | Cat# 48–5932-82; RRID:AB_10805519 |
| Rat monoclonal antibody Granzyme B (Clone NGZB) | Thermo Fisher | Cat# 50–8898-80; RRID:AB_11220478 |
| Rabbit Cleaved Caspase-3 (Asp175) (Clone 5A1E) | Cell Signaling Technology | Cat#9664S |
| Mouse m123/IE1 (Clone CROMA101) | Capri | Cat#HR-MCMV-08 |
| 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) | Sigma Aldrich | Cat#10236276001 |
| NK 1.1 (PK136) | InVivoMab | Cat# BE0036; RRID:AB_1107737 |
| Bacterial and Virus Strains | ||
| SG-MCMV (Smith strain) | American Type Culture Collection | ATCC® VR194 |
| Δm157-MCMV | Bubić et al., 2004 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Brefeldin A Solution (1000X) | Thermo Fisher | Cat# 00–4506-51 |
| Fixation/Permeabilization Solution Kit | BD Bioscience | Cat#554714 |
| CpG-ODN 1826 | Invivogen | Cat#tlrl-1826–5 |
| Foxp3 Staining Buffer Set Kit | Thermo Fisher | Cat#00–5523-00 |
| Red Blood Cell Lysing Buffer Hybri-Max | Sigma Aldrich | Cat#R7757–100ml |
| Methylcellulose-based medium with recombinant cytokines | Stem Cell Technologies | Cat#M3434 |
| (including EPO) for mouse cells | ||
| Fixable Viability Dye eFluor 780 | Thermo Fisher | Cat# 65–0865-18 |
| Intracellular Fixation & Permeabilization Buffer Set | Thermo Fisher | Cat#88–8824-00 |
| iScript cDNA Synthesis Kit | Bio Rad | Cat# 1708891 |
| HOT FIREPol DNA Polymerase | Solis BioDyne | Cat#01–08-01000 |
| Critical Commercial Assays | ||
| FITC Mouse Anti-Ki-67 Set | BD Bioscience | Cat#556026 |
| Annexin V Apoptosis Detection Set PE-Cyanine7 | Thermo Fisher | Cat#88–8130-72 |
| Experimental Models: Cell Lines | ||
| Mouse embryonic fibroblasts (MEFs) | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| WT (C57BL/6N) | The Jackson Laboratory | C57BL/6N |
| Stat1ΔM/PMN | Wallner et al., 2012 | N/A |
| Stat1fl/fl | Wallner et al., 2012 | N/A |
| Ifnar1fl/fl | Kamphuis et al., 2006 | N/A |
| Ifngr2fl/fl | Lee et al., 2015 | N/A |
| Stat1+/+ Cre (LysMCre cre/+) | Clausen et al., 1999 | N/A |
| Stat1−/− | Durbin et al., 1996 | N/A |
| Il27−/− (Wsx1−/−) | The Jackson Laboratory | B6N.129P2-Il27ratm1Mak/J |
| Oligonucleotides | ||
| m54-fwd-5′-CATCCGTTGCATCTCGTTG-3′, | Sigma Aldrich | N/A |
| m54 rev-5′-ACGTACATCGCTCTCTGCTG-3′ | Sigma Aldrich | N/A |
| Ube2d2-fwd 5′-AGG TCC TGT TGG AGA TGA TAT GTT-3′ | Sigma Aldrich | N/A |
| Ube2d2-rev 5′ -TTGGGAAATGAATTG TCA AGA AA-3′ | Sigma Aldrich | N/A |
| Ube2d2-probe 5′-CCA AAT GAC AGC CCC TAT CAG GGT GG-3′ | Sigma Aldrich | N/A |
| Ccl2 | QIAGEN | Cat# QT00167832 |
| Ccl5 | QIAGEN | Cat# QT01747165 |
| Software and Algorithms | ||
| R Studio | https://www.rstudio.com/ | N/A |
Highlights.
STAT1 signaling in myeloid cells restricts early MCMV replication
Myeloid STAT1 drives splenic EMH during MCMV infection and sterile inflammation
Myeloid STAT1 protects from MCMV-induced splenic macrophage, neutrophil, and NK cell apoptosis
MCMV induces EMH independently of IFNAR1 and IFNGR2 in myeloid cells and IL-27Rα
ACKNOWLEDGMENTS
This work was supported by the Austrian Science Fund (FWF; Infect-ERA project eDEVILLI FWF I-2187 to M.M.; DK-W1212 to M.M.; SFB-F6101, F6106, and F6107 to M.M., B.S., and V.S.; P26908-B20 and P29222-B28 to R.E.; and P-25642-B22 to B.S.), the NIH (1R01HL141513 and 1R01HL139641 to M. Bosmann), the Federal Ministry of Education and Research (01EO1503 to M. Bosmann), and Deutsche Forschungsgemeinschaft (BO3482/3–1 and BO3482/4–1 to M. Bosmann). We thank Claus Vogl for statistical support and Sandra Pilat-Carotta for sharing her expertise on erythrocyte development and differentiation. We are grateful to Luka Čičin-Šain and Lars Dölken for valuable discussions and to Tihana Tršan for critical reading of the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information contains five figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.017.
DECLARATION OF INTERESTS
The authors are responsible for the contents of this publication and declare no competing interests.
REFERENCES
- Alexandre YO, Cocita CD, Ghilas S, and Dalod M (2014). Deciphering the role of DC subsets in MCMV infection to better understand immune protection against viral infections. Front. Microbiol 5, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alliot C, Beets C, Besson M, and Derolland P (2001). Spontaneous splenic rupture associated with CMV infection: report of a case and review. Scand. J. Infect. Dis 33, 875–877. [DOI] [PubMed] [Google Scholar]
- Almeida-Porada GD, and Ascensăo JL (1996). Cytomegalovirus as a cause of pancytopenia. Leuk. Lymphoma 21, 217–223. [DOI] [PubMed] [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, Hill AB, and Lanier LL (2002). Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326. [DOI] [PubMed] [Google Scholar]
- Babić M, Krmpotić A, and Jonjić S (2011). All is fair in virus-host interactions: NK cells and cytomegalovirus. Trends Mol. Med 17, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT, King KY, Boles NC, Weksberg DC, and Goodell MA (2010). Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 465, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, and Casanova JL (2012). Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol 24, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst K, Frenz T, Spanier J, Tegtmeyer PK, Chhatbar C, Skerra J, Ghita L, Namineni S, Lienenklaus S, Köster M, et al. (2017). Type I interferon receptor signaling delays Kupffer cell replenishment during acute fulminant viral hepatitis. J. Hepatol Published online December 21, 2017. 10.1016/j.jhep.2017.11.029. [DOI] [PubMed] [Google Scholar]
- Brinkmann MM, Dağ F, Hengel H, Messerle M, Kalinke U, and Čičin-Šain L (2015). Cytomegalovirus immune evasion of myeloid lineage cells. Med. Microbiol. Immunol. (Berl.) 204, 367–382. [DOI] [PubMed] [Google Scholar]
- Brune W, Hengel H, and Koszinowski UH (2001). A mouse model for cytomegalovirus infection. Curr. Protoc. Immunol Chapter 19, Unit 19.17. [DOI] [PubMed] [Google Scholar]
- Bubić I, Wagner M, Krmpotić A, Saulig T, Kim S, Yokoyama WM, Jonjić S, and Koszinowski UH (2004). Gain of virulence caused by loss of a gene in murine cytomegalovirus. J. Virol 78, 7536–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasis JA, and Mohandas N (2008). Erythroblastic islands: niches for erythropoiesis. Blood 112, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SC, Liu HH, Chen CL, Chen PR, Liu MC, Lin SZ, and Chang KT (2015). Extramedullary hematopoiesis (EMH) in laboratory animals: offering an insight into stem cell research. Cell Transplant 24, 349–366. [DOI] [PubMed] [Google Scholar]
- Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, Pinho S, Leboeuf M, Noizat C, van Rooijen N, et al. (2013). CD169+ macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat. Med 19, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Förster I (1999). Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8, 265–277. [DOI] [PubMed] [Google Scholar]
- Crane MJ, Hokeness-Antonelli KL, and Salazar-Mather TP (2009). Regulation of inflammatory monocyte/macrophage recruitment from the bone marrow during murine cytomegalovirus infection: role for type I interferons in localized induction of CCR2 ligands. J. Immunol 183, 2810–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley-Bauer LP, Roback LJ, Wynn GM, and Mocarski ES (2014). Cytomegalovirus hijacks CX3CR1(hi) patrolling monocytes as immune-privileged vehicles for dissemination in mice. Cell Host Microbe 15, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels KA, Devora G, Lai WC, O’Donnell CL, Bennett M, and Welsh RM (2001). Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J. Exp. Med 194, 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin AM, Voermans C, and Nolte MA (2014). Impact of interferon-γ on hematopoiesis. Blood 124, 2479–2486. [DOI] [PubMed] [Google Scholar]
- Decker T, Stockinger S, Karaghiosoff M, Müller M, and Kovarik P (2002). IFNs and STATs in innate immunity to microorganisms. J. Clin. Invest 109, 1271–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte PJ, Echavarrìa M, Paparatto A, and Cacchione R (2003). [Spontaneous spleen rupture associated to active cytomegalovirus infection]. Medicina (B. Aires) 63, 46–48. [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, and Levy DE (1996). Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84, 443–450. [DOI] [PubMed] [Google Scholar]
- Ehninger A, Boch T, Uckelmann H, Essers MA, Müdder K, Sleckman BP, and Trumpp A (2014). Posttranscriptional regulation of c-Myc expression in adult murine HSCs during homeostasis and interferon-α-induced stress response. Blood 123, 3909–3913. [DOI] [PubMed] [Google Scholar]
- Einsele H, Mielke S, and Grigoleit GU (2014). Diagnosis and treatment of cytomegalovirus 2013. Curr. Opin. Hematol 21, 470–475. [DOI] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, and Trumpp A (2009). IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908. [DOI] [PubMed] [Google Scholar]
- Fodil-Cornu N, and Vidal SM (2008). Type I interferon response to cytomegalovirus infection: the kick-start. Cell Host Microbe 3, 59–61. [DOI] [PubMed] [Google Scholar]
- Frantzeskaki FG, Karampi ES, Kottaridi C, Alepaki M, Routsi C, Tzanela M, Vassiliadi DA, Douka E, Tsaousi S, Gennimata V, et al. (2015). Cytomegalovirus reactivation in a general, nonimmunosuppressed intensive care unit population: incidence, risk factors, associations with organ dysfunction, and inflammatory biomarkers. J. Crit. Care 30, 276–281. [DOI] [PubMed] [Google Scholar]
- Furusawa J, Mizoguchi I, Chiba Y, Hisada M, Kobayashi F, Yoshida H, Nakae S, Tsuchida A, Matsumoto T, Ema H, et al. (2016). Promotion of expansion and differentiation of hematopoietic stem cells by interleukin-27 into myeloid progenitors to control infection in emergency myelopoiesis. PLoS Pathog 12, e1005507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil MP, Bohn E, O’Guin AK, Ramana CV, Levine B, Stark GR, Virgin HW, and Schreiber RD (2001). Biologic consequences of Stat1-independent IFN signaling. Proc. Natl. Acad. Sci. U S A 98, 6680–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Plüddemann A, and Martinez Estrada F (2014). Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol. Rev 262, 36–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths P, Baraniak I, and Reeves M (2015). The pathogenesis of human cytomegalovirus. J. Pathol 235, 288–297. [DOI] [PubMed] [Google Scholar]
- Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, Wurzer S, Prendergast AM, Schnell A, Hexel K, et al. (2015). Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell 17, 422–434. [DOI] [PubMed] [Google Scholar]
- Hanson LK, Slater JS, Karabekian Z, Virgin HW 4th, Biron CA, Ruzek MC, van Rooijen N, Ciavarra RP, Stenberg RM, and Campbell AE (1999). Replication of murine cytomegalovirus in differentiated macrophages as a determinant of viral pathogenesis. J. Virol 73, 5970–5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry SC, Schmader K, Brown TT, Miller SE, Howell DN, Daley GG, and Hamilton JD (2000). Enhanced green fluorescent protein as a marker for localizing murine cytomegalovirus in acute and latent infection. J. Virol. Methods 89, 61–73. [DOI] [PubMed] [Google Scholar]
- Hirche C, Frenz T, Haas SF, Döring M, Borst K, Tegtmeyer PK, Brizic I, Jordan S, Keyser K, Chhatbar C, et al. (2017). Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Rep 19, 2345–2356. [DOI] [PubMed] [Google Scholar]
- Hokeness KL, Kuziel WA, Biron CA, and Salazar-Mather TP (2005). Monocyte chemoattractant protein-1 and CCR2 interactions are required for IFN-alpha/beta-induced inflammatory responses and antiviral defense in liver. J. Immunol 174, 1549–1556. [DOI] [PubMed] [Google Scholar]
- Hsu KM, Pratt JR, Akers WJ, Achilefu SI, and Yokoyama WM (2009). Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J. Gen. Virol 90, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns JL, and Christopher MM (2012). Extramedullary hematopoiesis: a new look at the underlying stem cell niche, theories of development, and occurrence in animals. Vet. Pathol 49, 508–523. [DOI] [PubMed] [Google Scholar]
- Johns JL, Macnamara KC, Walker NJ, Winslow GM, and Borjesson DL (2009). Infection with Anaplasma phagocytophilum induces multilineage alterations in hematopoietic progenitor cells and peripheral blood cells. Infect. Immun 77, 4070–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan S, Ruzsics Z, Mitrović M, Baranek T, Arapović J, Krmpotić A, Vivier E, Dalod M, Jonjić S, Dölken L, and Koszinowski UH (2013). Natural killer cells are required for extramedullary hematopoiesis following murine cytomegalovirus infection. Cell Host Microbe 13, 535–545. [DOI] [PubMed] [Google Scholar]
- Kamphuis E, Junt T, Waibler Z, Forster R, and Kalinke U (2006). Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 108, 3253–3261. [DOI] [PubMed] [Google Scholar]
- Kim CH (2010). Homeostatic and pathogenic extramedullary hematopoiesis. J. Blood Med 1, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffron AJ, Hummel M, Patterson BK, Yan S, Kaufman DB, Fryer JP, Stuart FP, and Abecassis MI (1998). Cellular localization of latent murine cytomegalovirus. J. Virol 72, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krmpotic A, Bubic I, Polic B, Lucin P, and Jonjic S (2003). Pathogenesis of murine cytomegalovirus infection. Microbes Infect 5, 1263–1277. [DOI] [PubMed] [Google Scholar]
- Kropp KA, Robertson KA, Sing G, Rodriguez-Martin S, Blanc M, Lacaze P, Hassim MF, Khondoker MR, Busche A, Dickinson P, et al. (2011). Reversible inhibition of murine cytomegalovirus replication by gamma interferon (IFN-γ) in primary macrophages involves a primed type I IFN-signaling subnetwork for full establishment of an immediate-early antiviral state. J. Virol 85, 10286–10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YH, Heslan JM, Poppema S, Elliott JF, and Mosmann TR (1996). Continuous administration of Il-13 to mice induces extramedullary hemopoiesis and monocytosis. J. Immunol 156, 3166–3173. [PubMed] [Google Scholar]
- Landolfo S, Gariglio M, Gribaudo G, and Lembo D (2003). The human cytomegalovirus. Pharmacol. Ther 98, 269–297. [DOI] [PubMed] [Google Scholar]
- Lee HM, Fleige A, Forman R, Cho S, Khan AA, Lin LL, Nguyen DT, O’Hara-Hall A, Yin Z, Hunter CA, et al. (2015). IFNγ signaling endows DCs with the capacity to control type I inflammation during parasitic infection through promoting T-bet+ regulatory T cells. PLoS Pathog 11, e1004635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, Lairson DR, Chan W, Du XL, Leung KS, Kennedy-Nasser AA, Martinez CA, Heslop HE, Brenner MK, and Krance RA (2011). Children with acute leukemia: a comparison of outcomes from allogeneic blood stem cell and bone marrow transplantation. Pediatr. Blood Cancer 56, 143–151. [DOI] [PubMed] [Google Scholar]
- Lisnić B, Lisnić VJ, and Jonjić S (2015). NK cell interplay with cytomegaloviruses. Curr. Opin. Virol 15, 9–18. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang J, Ginzburg Y, Li H, Xue F, De Franceschi L, Chasis JA, Mohandas N, and An X (2013). Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood 121, e43–e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewendorf A, and Benedict CA (2010). Modulation of host innate and adaptive immune defenses by cytomegalovirus: timing is everything. J. Intern. Med 267, 483–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh L, and Hudson JB (1981). Murine cytomegalovirus infection in the spleen and its relationship to immunosuppression. Infect. Immun 32, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucia HL, and Booss J (1981). Immune stimulation, inflammation, and changes in hematopoiesis. Host responses of the murine spleen to infection with cytomegalovirus. Am. J. Pathol 104, 90–97. [PMC free article] [PubMed] [Google Scholar]
- Marquardt A, Halle S, Seckert CK, Lemmermann NA, Veres TZ, Braun A, Maus UA, Förster R, Reddehase MJ, Messerle M, and Busche A (2011). Single cell detection of latent cytomegalovirus reactivation in host tissue. J. Gen. Virol 92, 1279–1291. [DOI] [PubMed] [Google Scholar]
- McCabe A, and MacNamara KC (2016). Macrophages: Key regulators of steady-state and demand-adapted hematopoiesis. Exp. Hematol 44, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe A, Smith JNP, Costello A, Maloney J, Katikaneni D, and MacNamara KC (2018). Hematopoietic stem cell loss and hematopoietic failure in severe aplastic anemia is driven by macrophages and aberrant podoplanin expression. Haematologica 103, 1451–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, et al. (1996). Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84, 431–442. [DOI] [PubMed] [Google Scholar]
- Mitrović M, Arapović J, Jordan S, Fodil-Cornu N, Ebert S, Vidal SM, Krmpotić A, Reddehase MJ, and Jonjić S (2012). The NK cell response to mouse cytomegalovirus infection affects the level and kinetics of the early CD8(+) T-cell response. J. Virol 86, 2165–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Newen G, Stope MB, Kraus T, and Ziegler P (2017). Development of platelets during steady state and inflammation. J. Leukoc. Biol 101, 1109– 1117. [DOI] [PubMed] [Google Scholar]
- Mutter W, Reddehase MJ, Busch FW, Bühring HJ, and Koszinowski UH (1988). Failure in generating hemopoietic stem cells is the primary cause of death from cytomegalovirus disease in the immunocompromised host. J. Exp. Med 167, 1645–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najjar I, and Fagard R (2010). STAT1 and pathogens, not a friendly relationship. Biochimie 92, 425–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro D (2016). Expanding role of cytomegalovirus as a human pathogen. J. Med. Virol 88, 1103–1112. [DOI] [PubMed] [Google Scholar]
- Noyes HA, Alimohammadian MH, Agaba M, Brass A, Fuchs H, Gailus-Durner V, Hulme H, Iraqi F, Kemp S, Rathkolb B, et al. (2009). Mechanisms controlling anaemia in Trypanosoma congolense infected mice. PLoS ONE 4, e5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawelec G, McElhaney JE, Aiello AE, and Derhovanessian E (2012). The impact of CMV infection on survival in older humans. Curr. Opin. Immunol 24, 507–511. [DOI] [PubMed] [Google Scholar]
- Presti RM, Popkin DL, Connick M, Paetzold S, and Virgin HW 4th. (2001). Novel cell type-specific antiviral mechanism of interferon gamma action in macrophages. J. Exp. Med 193, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe BM, Artwohl JE, Purcell JE, Lovaglio J, and Fortman JD (2011). Effects of weekly blood collection in C57BL/6 mice. J. Am. Assoc. Lab. Anim. Sci 50, 680–685. [PMC free article] [PubMed] [Google Scholar]
- Reddehase MJ, Weiland F, Münch K, Jonjic S, Lüske A, and Koszinowski UH (1985). Interstitial murine cytomegalovirus pneumonia after irradiation: characterization of cells that limit viral replication during established infection of the lungs. J. Virol 55, 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddehase MJ, Podlech J, and Grzimek NK (2002). Mouse models of cytomegalovirus latency: overview. J. Clin. Virol 25 (Suppl 2), S23–S36. [DOI] [PubMed] [Google Scholar]
- Sacher T, Mohr CA, Weyn A, Schlichting C, Koszinowski UH, and Ruzsics Z (2012). The role of cell types in cytomegalovirus infection in vivo. Eur. J. Cell Biol 91, 70–77. [DOI] [PubMed] [Google Scholar]
- Salazar-Mather TP, Lewis CA, and Biron CA (2002). Type I interferons regulate inflammatory cell trafficking and macrophage inflammatory protein 1alpha delivery to the liver. J. Clin. Invest 110, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Onai N, Yoshihara H, Arai F, Suda T, and Ohteki T (2009). Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat. Med 15, 696–700. [DOI] [PubMed] [Google Scholar]
- Sato A, Ooi J, Takahashi S, Tsukada N, Kato S, Kawakita T, Yagyu T, Nagamura F, Iseki T, Tojo A, and Asano S (2011). Unrelated cord blood transplantation after myeloablative conditioning in adults with advanced myelodysplastic syndromes. Bone Marrow Transplant 46, 257–261. [DOI] [PubMed] [Google Scholar]
- Schubert TE, Obermaier F, Ugocsai P, Männel DN, Echtenacher B, Hofstädter F, and Haerle P (2008). Murine models of anaemia of inflammation: extramedullary haematopoiesis represents a species specific difference to human anaemia of inflammation that can be eliminated by splenectomy. Int. J. Immunopathol. Pharmacol 21, 577–584. [DOI] [PubMed] [Google Scholar]
- Sing GK, and Ruscetti FW (1995). The role of human cytomegalovirus in haematological diseases. Baillieres Clin. Haematol 8, 149–163. [DOI] [PubMed] [Google Scholar]
- Sparwasser T, Hültner L, Koch ES, Luz A, Lipford GB, and Wagner H (1999). Immunostimulatory CpG-oligodeoxynucleotides cause extramedullary murine hemopoiesis. J. Immunol 162, 2368–2374. [PubMed] [Google Scholar]
- Stacey MA, Marsden M, Pham N TA, Clare S, Dolton G, Stack G, Jones E, Klenerman P, Gallimore AM, Taylor PR, et al. (2014). Neutrophils recruited by IL-22 in peripheral tissues function as TRAIL-dependent antiviral effectors against MCMV. Cell Host Microbe 15, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, and Cannon MJ (2006). Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin. Infect. Dis 43, 1143–1151. [DOI] [PubMed] [Google Scholar]
- Stoddart CA, Cardin RD, Boname JM, Manning WC, Abenes GB, and Mocarski ES (1994). Peripheral blood mononuclear phagocytes mediate dissemination of murine cytomegalovirus. J. Virol 68, 6243–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl B, Bubic I, Bruns U, Steinborn R, Lajko R, Kolbe T, Karaghiosoff M, Kalinke U, Jonjic S, and Müller M (2005). Novel functions of tyrosine kinase 2 in the antiviral defense against murine cytomegalovirus. J. Immunol 175, 4000–4008. [DOI] [PubMed] [Google Scholar]
- Tang-Feldman YJ, Lochhead GR, Lochhead SR, Yu C, and Pomeroy C (2011). Interleukin-10 repletion suppresses pro-inflammatory cytokines and decreases liver pathology without altering viral replication in murine cytomegalovirus (MCMV)-infected IL-10 knockout mice. Inflamm. Res 60, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay CH, and Welsh RM (1997). Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J. Virol 71, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeval JL, Cohen-Solal K, Tulliez M, Giraudier S, Guichard J, Burstein SA, Cramer EM, Vainchenker W, and Wendling F (1997). High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood 90, 4369–4383. [PubMed] [Google Scholar]
- Wallner B, Leitner NR, Vielnascher RM, Kernbauer E, Kolbe T, Kara-ghiosoff M, Rülicke T, Decker T, and Müller M (2012). Generation of mice with a conditional Stat1 null allele. Transgenic Res 21, 217–224. [DOI] [PubMed] [Google Scholar]
- Zimmerer JM, Lesinski GB, Radmacher MD, Ruppert A, and Carson WE 3rd. (2007). STAT1-dependent and STAT1-independent gene expression in murine immune cells following stimulation with interferon-alpha. Cancer Immunol. Immunother 56, 1845–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.