

Abstract
We have explored a method to convert a muraymycin biosynthetic intermediate 3 to an anticancer drug lead 2 for in vivo and thorough preclinical studies. Cu(OAc)2 forms a stable complex with the amide 4 and it prevents electrophilic reactions at the 2-((3-aminopropyl)amino)acetamide moiety. Under the present conditions, the desired 5”-primary amine was selectively protected with (Boc)2O to yield 6. The intermediate 6 was converted to 2 in two steps with 90% yield.
Graphical Abstract
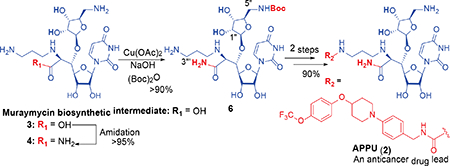
N-linked glycans play essential roles in many biological processes. Aberrant protein glycosylation is frequently observed in cancer cells. The integral membrane enzyme, N-acetylglucosaminephosphotransferase 1 (DPAGT1), plays a central role in N-linked glycoprotein biosynthesis, catalyzing the first step in the dolichol-linked oligosaccharide pathway. Certain cancer cells require an increase in N-linked glycosylation for their progression, thus, selective DPAGT1 inhibitors have therapeutic potential.1 The only inhibitors of DPAGT1 that have been reported to date are the antibiotic tunicamycin and its derivatives. Tunicamycins have been useful experimental tools to interrogate N-linked glycosylation and to examine the effects on protein folding.2 Tunicamycins inhibit growth across a wide range of mammalian cell lines in a non-selective fashion.1a In addition, physicochemical and biological properties of tunicamycins (e.g. water-solubility and narrow therapeutic window) are far from ideal as leads for the development of preclinical drugs. Therefore, new types of inhibitors are required. We have recently identified a strong DPAGT1 inhibitor, aminouridyl phenoxypiperidinbenzyl butanamide (APPB, 1), by screening of an aminoribosyl-uridine library generated based on muraymycin (Figure 1).1a,1b APPB selectively inhibits growth of solid tumors (e.g. KB, LoVo, SK-OV-3, MDA-MB-432S, HCT116, Panc-1, and AsPC-1) at low μM concentrations but does not inhibit growth of healthy cells at these same concentrations.1a APPB displayed a remarkably selective cytotoxicity (e.g. IC50 pancreatic cancer cells/ IC50 healthy cells: >1/100), metabolic stability (t1/2 >60 min.), and water solubility (~75 mg/mL for HCl salt). Thus, pharmacological studies of APPB and its related analogs using appropriate animal models are a focus of our continuing research efforts. APPB is a total synthetic product that is not feasible to access from reported natural products or muraymycin biosynthetic intermediates.3 Based on our structure-activity relationship studies of APPB, it was realized that a urea analogue, aminouridyl phenoxypiperidinbenzyl urea (APPU, 2 in Figure 1) shows equal biological activity to APPB without reducing the favorable physicochemical properties. Importantly, APPU has the potential to be synthesized from a biosynthetic intermediate of muraymycin (3) whose structure corresponds to de-N-methyl FR-900463.1d,4 Intermediate 3 can potentially be produced at large-scale by the disruption of the mur30 gene in the muraymycin-producing Streptomyces sp. NRRL 30471 (Figure 2).3 However, no synthetic method has been developed to convert the non-biologically active intermediate 3 to a pharmaceutically interesting product.5
Figure 1.

Structures of Anticancer DPAGT1 Inhibitors, APPB (1), APPU (2), and tunicamycin.
Figure 2.

Biosynthesis of Muraymycins in Streptomyces sp. NRRL30471.
In a semi-synthetic approach with 3, the most challenging transformation is the selective carbamation (temporary protection) of the primary amine (C5”-position) of the aminoribose moiety that allows functionalization at the other primary amine (C3”’-position). In this article, we report a semi-synthesis of APPU (2) from 3 by exploring transition metals that temporarily protect the 2-((3-aminopropyl)amino)acetamide system to achieve selective carbamation at the C5”-amine (Scheme 1).
Scheme 1.

Semi-synthetic Strategy of 1 from 2.
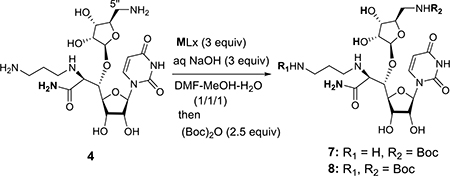
We have previously demonstrated amidation of protecting group free-amino acid derivatives with NH4Cl (excess), EDCI, glyceroacetonide-Oxyma, and NaHCO3 in water media.1d,6 Under the same condition, 37 was converted to the corresponding amide 4 in over 95% yield (Scheme 1). The amide 4 was examined in transition metal-mediated selective Boc-protection under aq NaOH in a mixture of DMF, MeOH, and H2O. Because of the strong coordination aptitude of group 4 elements to diamines, we screened Cu, Ni, Zn, V, Co, and Fe salts for selectively synthesizing the mono-Boc-protected triamine 7 in excellent yield. The transition-metal salts tested, selectivity of mono-/di-Boc (7/8), and isolation yield for 7 are summarized in Table 1. Reactions of 4 with a majority of the metal salts in Table 1 caused precipitation in a single solvent system (e.g. MeOH, H2O, or DMF); all reactions were carried out in a mixture of DMF-MeOH-H2O (1/1/1). Without addition of a transition metal salt, Boc-protection of 4 furnished a 1:1 mixture of 7 and 8 (entry 14).8 Boc-protection reactions in the presence of Zn salts, CoCl2, or Fe species provided 1:1 ratio of (7/8) with <50% isolation yield for 7 (entries 9–13). VCl3-mediated Boc-protection of 4 exhibited 7/8 selectivity of 4:1 and 7 was isolated in 40% yield after 12 h (entry 8). All Ni species tested gave rise to the desired mono-Boc product 7 in 85% yield with >19:1 (entries 5–7). Selective Boc-protection reactions mediated via the Cu species provided the only desired product 7 in 10–95% yield (entries 1–4). CuSO4- and CuBr2-mediated reactions did not progress toward completion even after 12 h (entries 1 and 3). On the other hand, selective Boc-protection performed with Cu(OAc)2 or CuCl2 provided 7 in 90–95% yield (entries 2 and 4). Kinetically, the Cu(OAc)2-mediated reaction is more favorable to the production of 7 than that with CuCl2; the reaction was completed in 1 h with Cu(OAc)2 (vs. 5 h with CuCl2). It may be attributed to the stronger coordination ability of CuCl2 to the C5”-amine, slowing down the electrophilic reaction with (Boc)2O. This hypothesis was proven correct by the successful reaction in the strictly controlled condition (1.0 equiv of CuCl2); 7 was synthesized in >90% in 1 h. We also examined selective Cbz-protection of 4 via the Cu(OAc)2-mediated condition developed in Table 1 (Scheme 2). In both cases (CbzCl and CbzOSu), 4 could be converted to the desired mono-Cbz protected product 9 in 95% without formation of di-Cbz by-product 10.
Table 1.
Selective Boc-protection of 4.a
 | ||||
|---|---|---|---|---|
| Entry | MLx | Time (h) | Selectivity (7 : 8)c | Yield for 7 (%)d |
| 1 | CuSO4 | 12 | 1 : 0 | 20 |
| 2 | Cu(OAc)2 | 12(1)b | 1 : 0 | 95 |
| 3 | CuBr2 | 12 | 1 : 0 | 10 |
| 4 | CuCl2 | 12(5)b,e | 1 : 0 | 90 |
| 5 | NiCl2•6H2O | 12(5)b | >19 : 1 | 85 |
| 6 | Ni(OAc)2•nH2O | 12(5)b | >19 : 1 | 85 |
| 7 | Ni(NO3)2•6H2O | 12(5)b | >19 : 1 | 85 |
| 8 | VCl3 | 12 | 4 : 1 | 40 |
| 9 | Zn(NO3)2•6H2O | 12(3)b | 1 : 1 | 45 |
| 10 | ZnCl2 | 12(3)b | 1 : 1 | 40 |
| 11 | CoCl2 | 12(3)b | 1 : 1 | 45 |
| 12 | FeSO4•H2O | 12(3)b | 1 : 1 | 40 |
| 13 | FeCl3•6H2O | 12(3)b | 1 : 1 | 40 |
| 14 | - | 12(1)b | 1 : 1 | 35 |
Reaction condition: 3 (1.0 equiv), MLx (3.0 equiv), and NaOH (1N, 4.0 equiv) in DMF-MeOH-H2O (1/1/1, 0.05M), after 30 min. (Boc)2O (2.5 equiv)
time in the parenthesis: time required for the reaction to be completed
Selectivity was determined via HPLC analysis for the reaction mixtures after 12h
Isolated yield
the same reaction with CuCl2 (1.0 equiv) provided 6 in >90 yield in 1 h.
Scheme 2.

Selective Cbz-protection of 4.
To obtain insight into mechanisms of the Cu-mediated selective Boc protection, we performed NMR studies for the Cu-4 complexes. Unfortunately, all Cu salts in Table 1 showed poor solubility in the complexation with 4 in conventional deuterated solvents (i.e. D2O, d6- DMSO, d6-acetone, and CD3OD). Thus, we synthesized two model compounds 11 and 12, and a mixture of 11 and 12 were applied to investigate complexation with the Cu-salts in solution. Complexation of 11 and 12 with CuCl2 resulted in less precipitate and higher-resolution NMR. 1H-NMR of a 1:1:1 mixture of 11, 12, and CuCl2 in D2O revealed that all carbon protons of the 2-((3-aminopropyl)amino)acetamide moiety of 11 are shifted to downfield; Δδ values were +0.73, +0.50, +0.26, and +0.05 for C2-H, C1’−2H, C2’−2H, and C3’−2H, respectively. In 13C-NMR of 11-CuCl complex, no chemical shifts were observed due to the complexation of 11 with CuCl2. Although participation of the carbonyl oxygen in 11-CuCl complex cannot be determined by these NMR experiments, coordination of the Cu-amide derivatives have previously been reported; based on X-ray and spectroscopic studies, the carbonyl groups of the primary amides are responsible for the formation of the stable Cu(II) complexes.9 In our NMR analyses, in the presence of CuCl2, the C5–2H in the aminoribose derivative 12 were not shifted (Δδ = 0), thus these data in solution-state may support that the C5”-amino group in 4 remains reactive in electrophilic reactions. Considering these facts, we speculate that the coordination geometries around the Cu(II) ion and 2-((3-aminopropyl)amino)acetamide moiety (highlighted in red) of 4 are as shown in 4-CuCl complex in Figure 3.
Figure 3.

Plausible 4-CuCl Complex Proposed Based on NMR Analyses.
Finally, we examined the urea-formation of the mono-Boc protected molecule 9 with the imidazole-carboxamide derivative 13 and deprotection to synthesize APPU (2). The coupling reaction between 9 and 13 was best performed in a mixed solvent system of DMF-CH2Cl2 (1/1) in the presence of Et3N to furnish the urea 14 in 90% yield. Deprotection of 14 with 30% TFA provided APPU•TFA (2-TFA). Purification of 2-TFA salt was performed by ion-exchange column (DOWEX 50Wx4, H+) to afford 2 in quantitative yield. The synthetic APPU (2) in Scheme 3 was determined by 1H- and 13C-NMR, MS, and HPLC to be identical to a sample previously synthesized via a total synthesis (see SI)1d.
Scheme 3.

Synthesis of APPU (1) from 9.
In conclusion, we have established the semi-synthesis of an anticancer agent, APPU (2), from the muraymycin biosynthetic intermediate 3. It was demonstrated that the 2-((3-aminopropyl)amino)acetamide moiety in the amide derivative 4 forms complexes with Cu species either in water or water-containing organic solvent systems. Such complexes can serve as protection of the primary amine at the C3”-position in 4, allowing us to perform selective carbamate- formation reactions of the primary amine at C5”-position. Synthetic pathways illustrated in Scheme 1 and 3 warrant a wide range of application of the biosynthetic intermediate 3 for development of novel agents to cure human diseases (i.e. infectious and oncology fields). We are currently studying disruption of the mur30 gene of Streptomyces sp. NRRL 30471 to improve production of 3 (Figure 2).10 Practicality of isolation and purification of 3 with the genetically modified Streptomyces sp. and a larger scale semi-synthesis of a DPAGT1 inhibitor with strong anticancer activity will be reported elsewhere.
Supplementary Material
Acknowledgment
We thank the National Institutes of Health (Grant GM114611). MK also thank University of Tennessee Health Science Center for generous financial supports (UTHSC College of Pharmacy, CORNET, and UTRF awards). NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant. Streptomyces sp. NRRL 30471 was acquired from USDA (NRRL Culture Collection).
Footnotes
Supporting Information Available Experimental procedures and copies of NMRs. This is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- (1).(a) Kurosu M Mol. Pharm. Org. Process Res 2018. 6, 141. [PMC free article] [PubMed] [Google Scholar]; (b) Mitachi K; Eslamimehr S; Kurosu SM; Kurosu M Abstracts of Papers, 256th ACS National Meeting & Exposition, Boston, MA, United States, 2018. [Google Scholar]; (c) Mitachi K; Aleiwi BA; Schneider CM; Siricilla S; Kurosu MJ Am. Chem. Soc 2016, 138, 12975. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mitachi K; Yun HG; Kurosu SM; Eslamimehr S; Lemieux MR; Klaić L; Clemons WM; Kurosu M ACS Omega 2018, 3, 1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Takatsuki A; Arima K; Tamura GJ Antibiot. 1971, 24, 215. [DOI] [PubMed] [Google Scholar]; (b) Gao Y; Feng HC; Walder K; Bolton K; Sunderland T; Bishara N; Quick M; Kantham L; Collier GR FEBS Lett. 2004, 563, 185. [DOI] [PubMed] [Google Scholar]; (c) Yuste-Checa P; Vega AI; Martin-Higueras C; Medrano C; Gamez A; Desviat LR; Ugarte M; Perez-Cerda C; Perez B PLoS One 2017, 12, e0179456/1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hou H; Sun H; Lu P; Ge C; Zhang L; Li H; Zhao F; Tian H; Zhang L; Chen T; Yao M; Li J Mol. Cancer Ther. 2013, 12, 2874. [DOI] [PubMed] [Google Scholar]; (e) Han X; Zhang X; Li H; Huang S; Zhang S; Wang F; Shi Y Oncotarget. 2015, 6, 38912. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Nami B; Donmez H; Kocak N; Exp Toxicol Pathol. 2016, 68, 419. [DOI] [PubMed] [Google Scholar]; (g) Akiyama T; Oishi K; Wullaert A PLoS One. 2016, 11, e0162448. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yoshida H; Matsui T; Yamamoto A; Okada T; Mori K Cell 2001, 107, 881. [DOI] [PubMed] [Google Scholar]; (i) Urano F; Wang X; Bertolotti A; Zhang Y; Chung P; Harding H; Ron D Science 2000, 287, 664. [DOI] [PubMed] [Google Scholar]; (j) Hitomi J; Katayama T; Taniguchi M; Honda A; Imaizumi K; Tohayama M Neurosci. Lett 2003, 357, 127. [DOI] [PubMed] [Google Scholar]; (k) Bravo R; Vicencio JM; Parra V; Troncoso R; Munoz JP; Bui M; Quiroga C; Rodriguez AE; Verdejo HE; Ferreira J; Iglewski M; Chiong M; Simmen T; Zorzano A; Hill JA; Rothermel BA; Szabadkai G; Lavandero SJ Cell Sci. 2011, 124, 2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) McDonald LA; Barbieri LR; Carter GT; Lenoy E; Lotvin J; Petersen PJ; Siegel MM; Singh G; Williamson RT J. Am. Chem. Soc 2002, 124, 10260. [DOI] [PubMed] [Google Scholar]; (b) Lin Cheng.; Chen W; Zhai L; Xu D; Huang T; Lin S; Zhou X; Deng Z Mol. BioSystems 2011, 7, 920. [DOI] [PubMed] [Google Scholar]; (c) Xu D; Liu G; Cheng L; Lu X; Chen W; Deng Z PloS one 2013, 8, e76068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ochi K; Ezaki M; Iwani M; Komori T; Kohsaka M Eurp. Patent Publication number: 0333177 A2, March 15, 1989. [Google Scholar]
- (5).Lin Y-I; Li Z; Francisco GD; McDonald LA; Davis RA; Singh G; Yang Y; Mansour TS Bioorg. Med. Chem. Lett 2002, 12, 2341.. [DOI] [PubMed] [Google Scholar]
- (6).(a) Wang Q; Wang Y; Kurosu M Org. Lett 2012, 14, 3372. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Y; Aleiwi BA; Wang Q; Kurosu M Org. Lett 2012, 14, 4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Isolation of 3; see Supporting Information.
- (8).The mon-Boc-protected molecule 7 and di-Boc-protected molecule 8 were synthesized via total chemical syntheses according to the procedure previously reported; see Supporting Information.
- (9).(a) Nonoyama K; Ojima H; Nonoyama M Inorganic. Chim. Acta 1976, 20, 127. [Google Scholar]; (b) Choi K; Young K; Yong S; Choo GH; Kim JG; Suh IH Acta Crystallog. Section C: Cryst. Struct. Comm 2001, C57, 1014. [DOI] [PubMed] [Google Scholar]
- (10).Dubeau M-P; Ghinet MG; Jacques P-É; Clermont N; Beaulieu C; Brzezinski R Appl. Env. Microbiol 2009, 75, 1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.