

Abstract
Metformin is a widely prescribed drug used in the treatment of type II diabetes. While the drug has many mechanisms of action, most of these converge on AMP activated protein kinase (AMPK), which metformin activates. AMPK is a multifunctional kinase that is a negative regulator of mechanistic target of rapamycin (mTOR) and mitogen activated protein kinase (MAPK) signaling. Activation of AMPK decreases the excitability of dorsal root ganglion neurons and AMPK activators are effective in reducing chronic pain in inflammatory, post-surgical and neuropathic rodent models. We have previously shown that metformin leads to an enduring resolution of neuropathic pain in the spared nerve injury (SNI) model in male mice and rats. The precise mechanism underlying this long-lasting effect is not known. We conducted experiments to investigate the effects of metformin on SNI-induced microglial activation, a process implicated in the maintenance of neuropathic pain that has recently been shown to be sexually dimorphic. We find that metformin is effective at inhibiting development of neuropathic pain when treatment is given around the time of injury and that metformin is likewise effective at reversing neuropathic mechanical hypersensitivity when treatment is initiation weeks after injury. This effect is linked to decreased Iba-1 staining in the dorsal horn, a marker of microglial activation. Importantly, these positive behavioral and microglia effects of metformin were only observed in male mice. We conclude that the neuropathic pain modifying effects of metformin are sex-specific supporting a differential role for microglial activation in male and female mice.
Keywords: AMPK, neuropathic pain, microglia, pain, metformin
Graphical Abstract
Introduction
Chronic pain affects over 100 million Americans, more than the number of people suffering from heart disease, diabetes and cancer combined [1]. The problem of this epidemic of chronic pain is compounded by the fact that available analgesics are not efficacious for many patients over the long-term [2]. Among the many chronic pain types, neuropathic pain is common, affecting up to 10% of the population, and is particularly hard to treat. Front-line neuropathic pain medications have numbers needed to treat for 50% efficacy of between 5 and 10 [2]. New, more efficacious treatments for chronic pain in general, and neuropathic pain, are urgently needed [3]. Adenosine monophosphate-activated protein kinase (AMPK) is a fuel-sensing enzyme present in all mammals [4]. AMPK is a kinase that detects changes in the AMP/ATP ratio in all cells to regulate anabolic processes when cellular energy status is low [5]. Activation of AMPK decreases mTORC1 signaling as well as MAPK signaling leading to a general suppression of cap-dependent protein synthesis [4]. Peripheral nerve injury (PNI) leads to an induction of increased mTORC1 [6] and MAPK signaling in DRG neurons and activation of these signaling pathways has been linked to increased nociceptor excitability [7–10]. Many studies have now demonstrated that AMPK activation in nociceptive DRG neurons leads to a decrease in mTORC1 and MAPK signaling and a decrease in nociceptor excitability [11,12]. AMPK activators are also effective at reducing behavioral signs of pain in rodent models [11,12] and this is at least partially dependent on nociceptor-expressed AMPK because specific deletion of an α subunit of AMPK in these cells abrogates that anti-hyperalgesic effects of AMPK activators [13].
Metformin is a widely prescribed drug for type II diabetes. The drug activates AMPK in a liver kinase B1 (LKB1) dependent fashion and is therefore an indirect AMPK activator [14]. The precise mechanism through which metformin acts is still an area of controversy, however, the drug is widely viewed as safe, is effective for type II diabetes and could potentially be repurposed for the treatment of neuropathic pain. Metformin is effective in reversing SNI- and chronic constriction injury- (CCI) induced neuropathic pain in male mice and rats [8,15], is effective in reducing pain produced by surgical incision in male mice [16] and is also effective in preventing the development of inflammatory pain [13]. Metformin treatment can also prevent, but not reverse, the development of chemotherapy-induced peripheral neuropathy in female mice [17].
Microglia activation in the spinal dorsal horn has been linked to many types of chronic neuropathic pain in rodents [18,19]. Interestingly, a series of recent studies suggest that the effects of microglia on neuropathic pain in mice and rats are male sex-specific [20,21]. We hypothesized that the enduring effects of metformin that are observed in neuropathic pain models may be dependent on a reversal of microglial activation in the dorsal horn. This hypothesis would be consistent with recent findings in the CCI model in rats [15]. A corollary of this hypothesis is that metformin may be effective in male but not female mice. Our experiments demonstrate that metformin prevents, and reverses SNI-induced mechanical and cold hypersensitivity and that this effect is paralleled by decreased microglial activation. These effects were only observed in male mice. Our experiments have important implications for the potential repurposing of metformin as an anti-neuropathic pain drug as well as for understanding the mechanisms driving neuropathic pain.
Methods
Laboratory Animals
Animal procedures were approved by The University of Texas at Dallas Institutional animal care and use committee and were in accordance with National Institutes of Health Guidelines. All the experiments were performed male and female ICR mice obtained from Envigo at 4 weeks of age. Mice were housed in the University of Texas at Dallas Animal Care Facility for at least one week prior to the start of behavior testing and surgery. Animals had ad libitum access to food and water and were on a 12 hr non-inverted light/dark cycle. Experimenters were blinded to treatment groups in behavioral experiments. Mice were randomized to treatment groups using a random number generator and in such a manner that multiple treatment groups were always found within any individual cage of animals. Male and female mice were housed separately in groups of 4 per cage.
Behavioral Testing
Mechanical sensitivity was assessed using stimulation of the hindpaw of the mouse with calibrated von Frey filaments from Stoelting. We used 0.6, 1.0 and 1.4-gram filaments and measured the response frequency to 10 consecutive stimulations of the hindpaw with each filament with stimulations spaced by at least 5 sec following 45 minutes of habituation to the testing boxes. The response frequency for each filament force was recorded and graphed as a percentage. This method was adapted from previously studies [22,23]. Following this baseline testing, neuropathic pain was induced in half of the mice using the SNI surgery model. This surgery consisted of exposing and cutting the Peroneal and Tibial branches of the Sciatic nerve while leaving the Sural nerve intact [24]. The remaining mice received a sham surgery where the nerve was exposed but not cut. Two-weeks post-surgery, the withdrawal frequency test was repeated to ensure that mechanical hypersensitivity had indeed been produced. Following this test, groups of SNI mice and sham mice were treated with 200 mg/kg of Metformin (LKT Laboratories Inc.) dissolved in 0.9% saline through intraperitoneal (I.P.) injections (1/2” 30-gauge needle) once a day at 10 am for 7 consecutive days. For the prophylactic metformin treatment experiment, mice were given 200 mg/kg of metformin following baseline testing then SNI was performed after the 7th day of treatment. In another prophylactic experiment mice were treated with metformin for 7 days with injections starting on the day of SNI surgery. Vehicle treated mice for each experiment received daily injections of 0.9% saline solution for 7 consecutive days as well. Following injections, the withdrawal frequency test was performed at indicated time points post-SNI to determine persistent effects of metformin treatment, all behavioral studies were conducted at least 24 hours after the last injection of metformin. Cold allodynia was also measured using the acetone test [25]. To do this, the left hindpaw of the mouse was sprayed with 0.1 mL of acetone using a needle and syringe and the duration of reaction to the evaporative cooling stimulus was measured over the course of a minute. Behavioral tests were done in the same way for male and female mice.
Immunohistochemistry
After the end of behavioral testing in SNI or sham mice, the lumbar spinal cords and DRGs were removed, placed in 4% formalin overnight and transferred to 30% sucrose for cryoprotection for 24 hrs then mounted in Optimal Cutting Temperature (OCT) compound. Lumbar spinal cord and DRG sections were cut into 20 μm slices using a cryostat and mounted onto positively charged (Superfrost plus) slides for immunohistochemistry. An antigen retrieval step was performed using a 10 mM Citric Acid buffer solution pH 6.0 with .05% Tween 20 for 45 minutes at room temperature. Following 3 5-minute washes in 1X phosphate buffered saline (PBS), the slides were then put into a permeabilization solution containing 10% normal goat serum (NGS) and 0.2% Triton X 100 in PBS for 30 minutes. This was followed by another series of 5-minute washes in PBS and 1.5 hrs in a blocking solution containing 10% NGS and 0.01% Na azide in PBS. Following another PBS wash, the slides were incubated overnight in a primary antibody solution made from the blocking solution. The next day, the slides were washed again in PBS then incubated in a secondary antibody solution also made from the blocking solution for 1 hr. Following 3 more PBS washes and a wash in deionized H2O, Prolong Gold mounting media was used to mount coverslips. The primary antibodies used were Ionized calcium binding adaptor molecule 1 (IBA1) from Wako Chemicals (Cat 019–19741) for activated microglia for the spinal cord sections and at a concentration of 1:1000 with Alexa Fluor 488 goat anti-rabbit (GAR-488) secondary (1:1000) and isolectin B4 (IB4) conjugated to alexfluor-567 (1:300) from Life Technologies (Cat I21412) for non-peptidergic central projection labeling. We also used glial fibrillary acidic protein (GFAP) from Santa Cruz Biotechnology (Cat sc-33673) to stain astrocytes in spinal cord sections at a concentration of 1:1000. Imaging of the tissue sections was done using an Olympus Fluoview FV1200 laser scanning confocal microscope with scale bars indicating the level of magnification. Images were analyzed using the cell counter tool in ImageJ.
Neuron Culture
Dorsal root ganglia (DRG) were extracted aseptically from 5 to 12 4-week old male ICR mice per cell culture plate for each cellular imaging experiment and placed in Hank’s Buffered Salt Solution (HBSS, Invitrogen) on ice. The DRGs were dissociated enzymatically at 37 °C; first with collagenase A (1 mg/ml, Roche) for 25 min, then collagenase D (1 mg/ml, Roche) that included papain (30 μg/ml, Roche) for 20 min. Afterward, a trypsin inhibitor (1 mg/ml, Roche) that contained bovine serum albumin (BSA, Fisher, 1 mg/ml) was applied and the ganglia were mixed to allow for further dissociation with a polished Pasteur pipette. The tissue was then filtered through 70-μm nylon cell strainer (Falcon) and re-suspended in DMEM F-12 GlutaMax media (Invitrogen) that contained 10% fetal bovine serum (FBS, Hyclone) and 1% penicillin streptomycin (Pen-Strep). The media also contained NGF (10 ng/ml, Millipore) and 5-fluoro-2′-deoxyuridine + uridine (FRDU-U, 3.0 μg/ml + 7.0 μg/ml, Sigma) to reduce proliferation of glia and fibroblasts. Neurons were cultured for 7 days on 12-mm glass coverslips (#1 thickness, Chemglass) in a 24-well tissue culture plate (Falcon) coated with poly-d-lysine (Sigma) at 37 °C with 95% air and 5% CO2. On the day of the experiment, metformin was diluted into DMEM F-12 plus GlutaMax media and added directly onto the neurons at a concentration 20 mM for 1 hr.
Microglia isolation, Culture, and Treatment
Microglia from whole brain and spinal cord were isolated and cultured as described previously, with few modifications [26,27]. Mice were euthanized and whole brains and spinal cords were collected, placed in sterile dulbecco’s phosphate-buffered saline (DPBS), and then homogenized by passage through a 70-μm cell strainer in Dulbecco’s PBS (DPBS) supplemented with 0.2% glucose. Homogenates were centrifuged at 600 × g for 6 minutes at 10°C and resulting pellets were resuspended in sterile 70% isotonic Percoll (GE-Healthcare) at room temperature. A discontinuous sterile Percoll density gradient was layered on top of the 70% isotonic Percoll at 50%, 35%, and 0% (DPBS). The suspension was centrifuged at 2000 × g for 20 minutes and microglia were collected from the interphase between the 70% and 50% Percoll layers. Cells were washed with sterile DPBS and then resuspended in DMEM F-12 GlutaMax media (Invitrogen) that contained 5% fetal bovine serum (FBS, Hyclone) and 2 × penicillin streptomycin (Pen-Strep). Each isolation yielded ~5 × 105 viable cells from each mouse brain and spinal cord and roughly 100,000 – 150,000 cells per well were plated and cultured for two-three days on 12mm glass coverslips (#1 thickness, chemglass) in a 24-well tissue culture plate (Falcon) coated with poly-D-lysine (Sigma-Aldrich) at 37oC with 95% oxygen and 5% CO2. The day before the experiment, cells were serum-starved overnight (0% FBS media). On the day of the experiment, metformin (20 mM) was diluted into DMEM F-12 Glutamax and added directly onto the cells for 1 hour.
Immunocytochemistry (ICC) and digital image analysis
Following metformin treatment, the cells were washed with DPS and fixed with 10% formalin in phosphate buffered saline (PBS) for 30 minutes. Cells were blocked with 10% normal goat serum and labelled with Iba-1–635 (1:500; Wako Chemicals) and phospho-ACC (p-ACC, 1:1000; Cell Signaling Technologies) overnight at 4° C. Next, cells were washed and incubated with a flurochrome-conjugated secondary antibody (Alexa Fluor, anti-rabbit 488, invitrogen) and counterstained with a DNA stain, 4’,6-diamidino-2-phenylindole (DAPI) (Invitrogen) and mounted with Prolong Gold (Invitrogen). Neuron cultures were labelled with Anti-Peripherin antibody, Mouse monoclonal (1:500; Sigma Aldrich) and phospho-ACC (p-ACC, 1:1000; Cell Signaling Technologies) overnight at 4° C. Next, cells were washed and incubated with a flurochrome-conjugated secondary antibody (Alexa Fluor, anti-rabbit 488, invitrogen) and counterstained with a DNA stain, 4’,6-diamidino-2-phenylindole (DAPI) (Invitrogen) and mounted with Prolong Gold (Invitrogen). Images were taken on an Olympus Fluoview FV1200 laser scanning confocal microscope and analyzed using the co-localization tool within Olympus’ FV software. The intensity of each channel was adjusted so that only areas that contained a strong signal of 488 nm and 405 nm were visible. This adjusted imaged contained distinct puncta that could then be counted and analyzed using Graphpad prism 7xx. Results were reported as the average percent area in the positive threshold for all representative pictures.
Statistics
Data are shown as mean +/− standard error of the mean (SEM) and the number of animals or samples used in each analysis are given in figure legends. GraphPad Prism 7 was used to analyze data. Two-way ANOVAs were used to analyze von Frey data, one-way ANOVAs were used for acetone data and two tailed t-tests or one-way ANOVAs were used to analyze imaging data. Post-hoc tests used are described in figure legends. Significance level was set at α < 0.05.
Results
Metformin reverses SNI-induced mechanical and cold hypersensitivity in male mice.
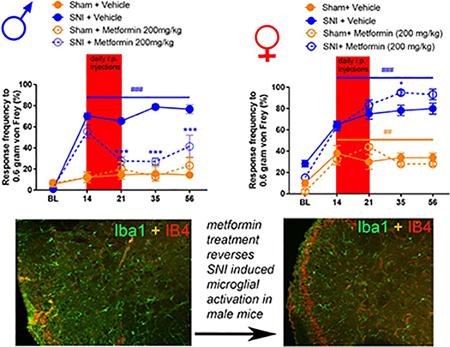
To assess the effects of metformin on established SNI-induced mechanical and cold hypersensitivity in male and female mice we obtained baseline responses for von Frey filament strengths of 0.6, 1.0 and 1.4 grams and then performed SNI or sham surgery. Mice were allocated to metformin (200 mg/kg) or saline treatment groups and then given injections via the I.P. injection route starting on day 14 after surgery. Starting at 21 days after surgery (after 7 days of metformin or vehicle treatment), and continuing at time points out to 56 days post-surgery, we monitored mechanical hypersensitivity weekly and then assessed cold hypersensitivity with the acetone test at day 56 post-surgery. Mice were then euthanized and tissues were taken for immunohistochemistry. In male mice, metformin reversed SNI-induced mechanical hypersensitivity to 0.6, 1.0 and 1.4 gram von Frey filaments and this effect persisted out to 56 days after SNI (32 days after the end of metformin treatment, Fig 1A-C). Additionally, we observed an increase in acetone response in SNI male mice that received saline treatment (Fig 1D) but this effect was not seen in SNI mice treated with metformin. These results suggest, as we have previously reported using different behavioral end-points [8,9], that metformin has an enduring effect on neuropathic mechanical and cold pain in male mice. We also compared the baseline response frequency to other timepoints for both the vehicle treated SNI and sham mice. For the sham mice, there was no difference between the baseline and the post-surgery timepoints. In the SNI vehicle treated mice there was a strong induction of neuropathic mechanical and cold hypersensitivity when compared to baseline, as expected. Therefore, in male mice, we do not observe any persistent changes in mechanical or cold sensitivity that are associated with the sham surgery.
Figure 1: Metformin treatment reverses SNI-induced mechanical and cold hypersensitivity in male mice:

A-C. SNI surgery caused mechanical hypersensitivity in male mice as measured using the withdrawal frequency test. After treatment with Metformin, the response frequency was decreased for all filament weights. D. Cold hypersensitivity was induced by SNI and the effect was reduced after metformin administration. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ###p<0.001 versus BL; N= 9 for all groups.
Female mice show robust responses to PNI but do not respond to metformin treatment.
We obtained baseline measures of mechanical sensitivity in female mice then performed SNI or sham surgery and confirmed the development of mechanical hypersensitivity 14 days after treatment. On day 14 mice were treated with metformin (200 mg/kg) or vehicle for 7 days and tested over the same time course that was used for male mice. Strikingly, 200 mg/kg metformin failed to reduce mechanical hypersensitivity in female mice at any time point (Fig 2A-C). Metformin also had no positive effect on cold hypersensitivity in female mice. Surprisingly, metformin exacerbated the acetone response in female mice with SNI (Fig 2D) but had no effect in sham mice demonstrating that this effect is not produced by metformin treatment alone. We also compared the baseline response frequencies for both the vehicle treated SNI and female sham mice. Unlike in male mice, female sham mice showed a significant mechanical hypersensitivity that persisted over the time course of the experiment (Fig 2A-C). This effect was slightly reduced by metformin treatment at the 1.4 gram filament strength.
Figure 2: Metformin treatment does not influence SNI-induced mechanical or cold hypersensitivity in female mice:

A-C. In female mice metformin failed to resolve mechanical hypersensitivity. Surgery (sham or SNI) induced a statistically significant difference from baseline for both groups ###p<.0001. D. A statistically significant increase in cold hypersensitivity was observed in the female SNI and the SNI + metformin group when compared to the vehicle group. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; N= 6 per group.
Metformin treatment reverses microglial staining intensity in the spinal cord in male but not female mice.
Following behavioral testing we removed spinal cords from male and female SNI and sham mice treated with metformin or vehicle 30 days earlier. Immunohistochemistry was preformed to examine Iba-1 and IB4 staining. In male SNI mice treated with vehicle we observed a clear increase in Iba-1 positive cells compared to sham vehicle treated mice (Fig 3A-E). Metformin treatment strongly reduced the number of Iba-1 positive cells in SNI mice (Fig 3B-C) This finding is consistent with recent data from male CCI rats treated with metformin [15] although the time courses of treatment and persistence of the reversal are different between these experiments. We also observed a loss of IB4 staining in the dorsal horn in SNI male mice treated with vehicle, as originally described in rats [28]. This absent band of staining in the male SNI vehicle group averaged 96.32 μm in length. This loss of IB4 staining was not evident in SNI male mice treated with metformin suggesting that this histochemical sign of PNI is also reversed by metformin treatment. It is notable that this change in IB4 staining in the dorsal horn induced by SNI was consistent with the time course of similar findings in a nerve constriction model in male rats [29].
Figure 3: Metformin reverses microglial activation in SNI male mice:

A-D. Representative immunohistochemistry images of the dorsal horn of male lumbar spinal cord at 20X magnification. Quantification of images shown in E. Metformin decreased SNI-induced microglia activation. SNI caused a loss of IB4 staining in the dorsal horn (A) that was reversed by metformin treatment (B) *p < 0.05; **p < 0.01; N= 5 per group.
Results in female mice were strikingly different. While the number of Iba-1 positive cells was increased by SNI (Fig 4A-C) there was no effect of metformin treatment. We also did not observe any loss of IB4 staining in female SNI mice suggesting that this change does not occur in female mice. The absolute number of Iba-1 positive cells in the dorsal horn of female mice was higher than in male mice in the sham condition, consistent with the development of mechanical hypersensitivity to sham surgery in female mice. Therefore, while metformin is able to reverse neuropathic mechanical and cold hypersensitivity and microglial reactivity in male mice, the same drug was without effect on all of these measures in female mice.
Figure 4: Metformin does not reverse microglial activation in SNI female mice:

A-D. Representative immunohistochemistry images of the dorsal horn of female lumbar spinal cord at 20X magnification. Quantification of images shown in E. SNI robustly stimulated microglial activation in female SNI mice, but metformin had no effect. A loss of IB4 staining was not observed in female SNI mice (A-B). ***p < 0.001; N= 5 per group.
SNI caused an increase in GFAP-positive cells in male mice but not females that was not affected by metformin treatment.
In addition to examining Iba-1, we also examined GFAP staining in the dorsal horn of male and female SNI mice following the behavioral experiments. Immunohistochemistry was performed to examine GFAP and IB4 staining in males and females. In male SNI mice treated with vehicle we observed a clear increase in GFAP positive cells compared to sham vehicle treated mice (Fig 5A-E). Metformin did not have an effect in reducing the amount of GFAP positive cells in SNI males (Fig 5B-D). This indicates that metformin treatment did not influence astrocyte proliferation.
Figure 5: Metformin does not reverse astrocyte proliferation in SNI male mice:

A-D. Representative immunohistochemistry images of the dorsal horn of male lumbar spinal cord at 20X magnification. Quantification of images shown in E. SNI robustly stimulated astrocyte proliferation in male mice, but metformin had no effect. ***p < 0.01; N= 5 per group.
As with the Iba-1 staining, we saw different results in the female tissue. While we did not see an effect of metformin on the number of GFAP-positive cells in female mice either, SNI also failed to cause a significant increase in the number of GFAP-positive cells when compared to sham mice (Fig 6A-E). These results are consistent with recent data from GFAP staining done in virgin female SNL mice [30] although the time courses of treatment and neuropathic model used are different between these experiments. This suggests that not only did metformin fail to have an influence on astrocytes in the spinal cord of female mice, but SNI does not appear to cause a proliferation of astrocytes in the lumbar spinal cord.
Figure 6: SNI did not result in an increases in astrocytes in the spinal dorsal horn of female mice:

A-D. Representative immunohistochemistry images of the dorsal horn of female lumbar spinal cord at 20X magnification. Quantification of images shown in E. SNI did not cause an increase in the number of astrocytes seen in the dorsal horn of female mice. N= 5 per group.
Prophylactic metformin treatment partially prevents development of neuropathy and microglial activation in male mice.
Having established that metformin reverses neuropathic pain selectively in male mice, we tested whether metformin could prevent development of SNI-induced neuropathic pain in male mice and whether any behavioral changes were correlated with spinal microglia. Naïve male ICR mice were treated with metformin (200 mg/kg) or saline for 7 days using two paradigms. In the first treatment paradigm male mice were given metformin starting 7 days prior to surgery and treatment was stopped the day of surgery. The rationale for the approach was to create a steady state plasma level of metformin to achieve strong AMPK activation at the time of injury. In the other paradigm, metformin treatment was started at the time of surgery and then continued for the ensuing 7 days. The first prophylactic treatment group showed decreased mechanical hypersensitivity at several time points after SNI (Fig 7A-C) but the magnitude of the effect was not as large as in the reversal treatment paradigm. Another set of male mice were treated in the second prophylactic treatment paradigm metformin (200 mg/kg) starting the day of surgery. We observed a significant decrease in mechanical hypersensitivity in the metformin treated mice when compared to the vehicle group (Fig 8A-C). We also tested cold allodynia using the acetone test in this cohort of mice and observed a significant decrease in response duration (Fig 8D) indicating that prophylactic treatment with metformin prevents the full development of mechanical and cold hypersensitivity but this treatment is most effective when given over the duration of neuropathic pain development rather than simply at the time of surgical injury.
Figure 7: Preemptive treatment with metformin for 7 days prior to SNI inhibits neuropathic pain in male mice:

Male mice were given metformin for 7 days prior to SNI. Mechanical hypersensitivity was inhibited at indicated time points after SNI. *p < 0.05; **p < 0.01; #p<0.05 versus BL. N= 6.
Figure 8: Treatment with metformin for the first 7 days after SNI inhibits neuropathic pain in male mice:

Male mice were given metformin for 7 days starting at the day of SNI surgery. Mechanical hypersensitivity was inhibited at the indicated time points after SNI. D. Metformin also decreased in cold hypersensitivity after SNI. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; N= 6 per group.
At 42 days post-SNI, in the second prophylactic metformin treatment paradigm, mice were sacrificed and their spinal cords were stained with Iba1 and IB4 as in the reversal experiment. We used the 7-day treatment starting at the time of surgical injury paradigm for this experiment because the behavioral effect was more robust. Metformin treatment caused a decrease in the number of Iba-1 cells in the dorsal horn compared to vehicle treatment (Fig 9A-C). Moreover, the loss of IB4 staining in the dorsal horn was evident in the vehicle treated mice but not in the metformin treated mice, consistent with our observations in the reversal treatment paradigm.
Figure 9: Early treatment with metformin blocks SNI-induced microglial activation in male mice:

A-B. Representative immunohistochemistry images of the dorsal horn of male lumbar spinal cord at 20X magnification. Quantification of images shown in C. Metformin decreased SNI-induced microglia activation. SNI caused a loss of IB4 staining in the dorsal horn (A) that was reversed by metformin treatment for the first 7 days after surgery (B). *p < 0.05; N= 5 per group.
Prophylactic metformin treatment had no effect on the development of neuropathy and microglial activation in female mice.
To further examine the apparent sexual dimorphism of metformin, we tested whether metformin could prevent development of SNI-induced neuropathic pain in female mice and whether any behavioral changes were correlated with spinal microglia. Naïve female ICR mice were treated with metformin (200 mg/kg) or saline for 7 days using the second prophylactic treatment paradigms previously described in which treatment began starting the day of surgery. Following drug treatment, all mice were tested for mechanical hypersensitivity 14 days post-surgery and at ensuing time points. Cold allodynia was also assessed. We observed no decrease in mechanical hypersensitivity or cold allodynia following metformin treatment compared to vehicle treatment (Fig 10A-D). At 42 days post-SNI, in the second prophylactic metformin treatment paradigm, mice were sacrificed and their spinal cords were stained with Iba1 and IB4 as in the reversal experiment. Metformin treatment had no effect on the number of Iba-1 cells in the dorsal horn compared to vehicle treatment (Fig 11A-E). Moreover, the loss of IB4 staining in the dorsal horn observed in the vehicle treated male mice was still not observed in any of the female mice, consistent with our observations in the reversal treatment paradigm.
Figure 10: Treatment with metformin for the first 7 days after SNI failed to inhibit neuropathic pain in female mice:

A-C. Female mice were given metformin for 7 days starting at the day of SNI surgery. Mechanical hypersensitivity was not inhibited at the indicated time points after SNI. D. Metformin also did not decrease cold hypersensitivity after SNI. N= 4 per group.
Figure 11: Early treatment with metformin does not block SNI-induced microglial activation in female mice:

A-B. Representative immunohistochemistry images of the dorsal horn of female lumbar spinal cord. Quantification of images shown in C. Metformin did not decrease SNI-induced microglia activation. Again, SNI did not cause a loss in IB4 staining in female mice that was seen in males following SNI. N= 6 per group.
Treatment of isolated male and female DRG neurons or microglia with metformin induces AMPK signaling.
DRGs were cultured from naïve male and female mice, and treated with 20 mM metformin or vehicle for 1 hr. For male and female DRG neurons, we observed a significant increase in p-ACC intensity with metformin treatment (Fig 12A-F) demonstrating that metformin induces AMPK activation in male and female DRG neurons.
Figure 12: Metformin induces AMPK activity in male and female DRG neurons in vitro.

Male and female neuron cultures were treated with metformin (20 mM) for 1 hr. A-B (male), D-E (female). Representative immunohistochemistry images of the DRG neurons at 40X magnification. Quantification of images shown in C (male) and F (female). Metformin increased p-ACC intensity in male and female neuron cultures. Only neurons that were positive for peripherin staining were analyzed. Maximum florescence refers to the maximum florescence intensity per neuron analyzed. **p < 0.01; ***p < 0.001. N= 20 images analyzed per group.
Microglia were cultured from naïve male and female mice and treated with 20 mM metformin or vehicle for 1 hr. As with the DRG neuron culture, we observed a significant increase in p-ACC intensity following metformin treatment in microglia for both male and female mice (Fig 13A-F). Therefore, metformin is able to activate AMPK in male and female DRG neurons and in microglia.
Figure 13: Metformin induces AMPK activity in male and female microglia in vitro.

Male and female microglia cultures were treated with metformin (20 mM) for 1 hr. A-B (male), D-E (female). Representative immunohistochemistry images of the DRG neurons at 40X magnification. Quantification of images shown in C and F. Metformin increased p-ACC intensity in male and female microglia cultures. Only microglia that were positive for Iba1 staining were analyzed. Maximum florescence refers to the maximum florescence intensity per microglial cell analyzed. *p < 0.05. N= 20 images analyzed per group.
Discussion
Our key result is that metformin has a differential effect on neuropathic pain and microglial activation in the spinal cord in male versus female mice. In male mice, metformin is able to both reverse and prevent the full expression of neuropathic pain and this is negatively correlated with microglial activation in the spinal cord. In female mice we did not observe any effect of metformin on neuropathic pain and metformin also failed to reverse microglial activation. These findings are consistent with a growing literature suggesting that microglia play a more important role in promoting chronic neuropathic pain in male than in female rodents [20,21]. Whether or not similar sex differences will be found in humans is an open question, however, there is now strong evidence linking neuro-inflammation to neuropathic pain in patients [31–33].
Another important finding from our experiments is that metformin did not appear to influence SNI-induced proliferation of astrocytes in the dorsal horn of the spinal cord of male mice. This suggests that a reversal of astrocyte proliferation is not required for a disease modification in neuropathic pain. Interestingly, neither SNI nor metformin treatment had an effect on GFAP-staining in the spinal cord for females suggesting that potential sex difference in astrocyte function in the maintenance of hypersensitivity caused by nerve injury. This is consistent with a previous finding from the Eisenach laboratory where astrocyte proliferation was also not seen in female rodents after nerve injury [30]. Given that there are clearly strong sexual dimorphisms in astrocyte and microglial function in neuropathic pain in mice and rats [20,30,34], future studies will assess if manipulation of sex hormones can alter the effect of metformin on neuropathic pain. Based on the existing literature we would expect to reveal that treatment of female ovariectomized mice with testosterone can lead to metformin efficacy for SNI mechanical hypersensitivity in female mice.
A critical question arising from our work is why metformin lacked effects in neuropathic pain in female mice. Metformin is obviously effective for type II diabetes treatment in male and female humans. This indicates that metformin is likely able to engage AMPK in many tissues in males and females. Metformin’s ability to increase AMPK activity in male and female neurons and microglia ex vivo further demonstrates that metformin can activate AMPK in relevant cell-types for our study in both male and female cells. However, metformin is a very water-soluble drug and cannot readily cross cell membranes without specific transporters, in particular the organic cation transporter OCT2 [35]. OCT2 shows sexually dimorphic expression in many tissues, in many species, with higher expression in males [36–39]. We are unaware of any studies showing dimorphisms in OCT2 expression in DRG or spinal cord or at the blood brain barrier. However, OCT2 expression is regulated by androgen receptors [38] and testosterone determines microglial contributions to neuropathic pain in mice [20]. Metformin is able to prevent chemotherapy-induced neuropathic pain in female mice [17]. However, chemotherapy neuropathic pain does not activate microglia but rather stimulates dorsal horn astrocytes [40–42]. Based on all of these factors, we propose that the most parsimonious explanation for our findings is a difference in pharmacokinetics for metformin between male and female mice. Since androgen receptors drive OCT2 expression [38,39], metformin may be incapable of activating AMPK in female microglia in vivo (as opposed to ex vivo) due to inability of metformin to cross the blood brain barrier. Another possibility is that AMPK activation in microglia inhibits androgen-driven processes that promote neuropathic pain in males [20,34] but AMPK activation in female microglia is incapable of acting on pathways in these cells that may promote neuropathic pain in females. A limitation of this work is that we have not been able to pinpoint the precise mechanism through which metformin has such strong actions on neuropathic pain in male, but not female mice. However, we argue that the bulk of the extensive evidence that we provide here is consistent with that difference hinging on an effect on microglia.
An important question for translation of these findings to the clinic relates to the doses and concentrations of metformin. Doses given to rodents are typically 100 – 200 mg/kg whereas the highest human dose of metformin fpr type II diabetes is 2 g/day for an average dose of ~ 30 mg/kg. It is difficult to equate metformin concentrations used in vitro to in vivo concentrations because of pharmacokinetic considerations and potential differences in transporter expression between in vivo and in vitro situations, which would be expected to have an impact on metformin apparent potency. We think that it is likely that higher doses than 2 g/day would be needed to achieve maximal efficacy in a human study on metformin for neuropathic pain. A recent study demonstrated that higher doses of metformin can be safely given to humans for the improvement of motor function in myotonic dystrophy [43]. This suggests that higher doses of metformin may also be used in human clinical trials for neuropathic pain, where we would expect greater efficacy in males based on the findings presented here.
Loss of IB4 staining in the dorsal horn following peripheral nerve injury has been characterized previous in rats and mice [28,29,44]. To the best of our knowledge, our work with metformin is the first report of a pharmacological reversal of this effect in the SNI model in mice. Both preemptive and reversal treatment was capable of reversing this loss of IB4 staining. The effect was accompanied by a decrease in spinal microglial activation in male mice. Interestingly, this effect did not emerge at all in female mice, even though there was robust activation of microglia in the spinal dorsal horn in these mice. The mechanism underlying the loss of IB4 staining in the dorsal horn of rodents after peripheral nerve injury are not known, but they apparently do not occur in female mice. We also observed a sustained mechanical hypersensitivity in female mice with sham SNI surgery that was not present in male mice. These are part of a growing abundance of major sexual dimorphisms in neuropathic and chronic pain mechanisms between male and female mice [20,21,45–47].
In conclusion, we have demonstrated that metformin has a disease modifying effect on male mice which includes a decrease in microglial activation in the dorsal horn of the spinal cord when given to reverse or to prevent neuropathic pain. This effect is remarkably long lasting as it persists for weeks after cessation of metformin treatment. We have also recently shown that metformin has disease-modifying effects on neuropathic pain-induced cognitive dysfunction in male mice [48]. Metformin does not appear to have any observable effects on neuropathic pain or microglial activation in female mice. Our findings contribute to a growing literature suggesting that metformin can be repurposed for the treatment of chronic pain [9,13,15–17,49–52] but suggest that the drug may only be effective in certain populations, when specific pain promoting mechanisms are engaged.
Table 1:
Statistical tests and values
| Test (Factor) | F (df1, df2) interaction F (df1, df2) time F (df1, df2) Drug treatment | P-Value | Adjusted p-value (Post-hoc comparison): BL, 14 days, 21 days, 35 days and 56 days |
|---|---|---|---|
| Two-way ANOVA (Fig 1A) | F (12, 155) = 10.46 F (4, 155) = 38.76 F (3, 155) = 103.7 |
PI <0.0001 Pt <0.0001 Pd <0.0001 |
SNI + Met vs. SNI + Veh: 0.9982, 0.1167, <0.0001, <0.0001, <0.0001 SNI + Met vs. Sham + Met: 0.9009, <0.0001, 0.6721, 0.1907, 0.0463 SNI + Met vs. Sham + Veh: 0.9548, <0.0001, 0.2056, 0.3366, 0.0008 SNI + Veh vs. Sham + Met: 0.8243, <0.0001, <0.0001, <0.0001, <0.0001 SNI + Veh vs. Sham + Veh: 0.9009, <0.0001, <0.0001, <0.0001, <0.0001 Sham + Met vs. Sham + Veh: 0.9982, >0.9999, 0.8243, 0.9859, 0.5141 |
| Twoway ANOVA (Fig 1B) | F (12, 155) = 11.74 F (4, 155) = 44.64 F (3, 155) = 105.2 |
PI <0.0001 Pt <0.0001 Pd <0.0001 |
SNI + Met vs. SNI + Veh: >0.9999, 0.9631, <0.0001, 0.0002, 0.0023 SNI + Met vs. Sham + Met: 0.7535, <0.0001, 0.8882, 0.0974, 0.0374 SNI + Met vs. Sham + Veh: 0.9991, <0.0001, 0.3610, 0.3869, 0.0374 SNI + Veh vs. Sham + Met: 0.9631, <0.0001, <0.0001, <0.0001, <0.0001 SNI + Veh vs. Sham + Veh: >0.9999, <0.0001, <0.0001, <0.0001, <0.0001 Sham + Met vs. Sham + Veh: >0.9999, >0.9999, >0.9999, >0.9999, >0.9999 |
| Two-way ANOVA (Fig 1C) | F (12, 154) = 5.76 F (4, 154) = 37.68 F (3, 154) = 90.99 |
PI <0.0001 Pt <0.0001 Pd <0.0001 |
SNI + Met vs. SNI + Veh: 0.1271, 0.7751, <0.0001, 0.0004, 0.2327 SNI + Met vs. Sham + Met: 0.8594, <0.0001, 0.9999, 0.9651, 0.0037 SNI + Met vs. Sham + Veh: 0.9997, <0.0001, 0.2647, 0.6104, 0.1382 SNI + Veh vs. Sham + Met: 0.9997, <0.0001, <0.0001, <0.0001, <0.0001 SNI + Veh vs. Sham + Veh: 0.8594, <0.0001, <0.0001, <0.0001, <0.0001 Sham + Met vs. Sham + Veh: >0.9999, >0.9999, 0.8594, >0.9999, 0.9997 |
| One-way ANOVA (Fig 1D) | F (3, 30) = 33.78 | P<0.0001 | SNI+ Met vs. SNI+ Veh: <0.0001 SNI+ Met vs. Sham+ Met: 0.0418 SNI+ Met vs. Sham+ Veh: 0.0749 SNI+ Veh vs. Sham+ Met: <0.0001 SNI+ Veh vs. Sham+ Veh: <0.0001 Sham+ Met vs. Sham+ Veh: 0.9919 |
| Two-way ANOVA (Fig 2A) | F (12, 90) = 7.708 F (4, 90) = 83.29 F (3, 90) = 146.3 |
PI <0.0001 Pt <0.0001 Pd <0.0001 |
SNI+ Met vs. SNI + Veh: 0.4916, >0.9999, 0.9923, 0.1291, 0.4916 SNI+ Met vs. Sham+ Met: 0.6379, <0.0001, <0.0001, <0.0001, <0.0001 SNI+ Met vs. Sham+ Veh: >0.9999, 0.0018, <0.0001, <0.0001, <0.0001 SNI + Veh vs. Sham+ Met: 0.0010, <0.0001, <0.0001, <0.0001, <0.0001 SNI + Veh vs. Sham+ Veh: 0.0867, 0.0006, <0.0001, <0.0001, <0.0001 Sham+ Met vs. Sham+ Veh: 0.9990, >0.9999, 0.5775, >0.9999, >0.9999 |
| Two-way ANOVA (Fig 2B) | F (12, 90) = 1.657 F (4, 90) = 113.6 F (3, 90) = 157.2 |
PI 0.0902 Pt <0.0001 Pd <0.0001 |
SNI+ Met vs. SNI + Veh: >0.9999, >0.9999, 0.6447, >0.9999, 0.6447 SNI+ Met vs. Sham+ Met: <0.0001, <0.0001, <0.0001, <0.0001, <0.0001 SNI+ Met vs. Sham+ Veh: 0.0014, <0.0001, <0.0001, <0.0001, <0.0001 SNI + Veh vs. Sham+ Met: <0.0001, <0.0001, <0.0001, <0.0001, <0.0001 SNI + Veh vs. Sham+ Veh: 0.0005, <0.0001, <0.0001, <0.0001, <0.0001 Sham+ Met vs. Sham+ Veh: >0.9999, >0.9999, >0.9999, 0.9505, 0.9505 |
| Two-way ANOVA (Fig 2C) | F (12, 90) = 1.806 F (4, 90) = 99.05 F (3, 90) = 114.5 |
PI 0.0589 Pt <0.0001 Pd <0.0001 |
SNI+ Met vs. SNI + Veh: 0.9992, >0.9999, 0.9769, >0.9999, >0.9999 SNI+ Met vs. Sham+ Met: <0.0001, <0.0001, <0.0001, <0.0001, <0.0001 SNI+ Met vs. Sham+ Veh: 0.0149, 0.0002, <0.0001, 0.0011, 0.0011 SNI + Veh vs. Sham+ Met: <0.0001, <0.0001, 0.0014, <0.0001, <0.0001 SNI + Veh vs. Sham+ Veh: 0.0002, <0.0001, 0.0182, 0.0011, 0.0099 Sham+ Met vs. Sham+ Veh: 0.7140, >0.9999, >0.9999, 0.0023, 0.0023 |
| One-way ANOVA (Fig 2D) | F (3, 18) = 31.34 | P<0.0001 | SNI + Met vs. SNI+ Veh: 0.0002 SNI + Met vs. Sham+ Met: <0.0001 SNI + Met vs. Sham+ Veh: <0.0001 SNI+ Veh vs. Sham+ Met: 0.0433 SNI+ Veh vs. Sham+ Veh: 0.0267 Sham+ Met vs. Sham+ Veh: 0.9957 |
| One-way ANOVA (Fig 3E) | F (3, 9) = 15.69 | P=0.0006 | SNI+ Met vs. SNI+ Veh: 0.0160 SNI+ Met vs. Sham + Met: 0.2254 SNI+ Met vs. Sham+ Veh: 0.1247 SNI+ Veh vs. Sham + Veh: 0.0014 SNI+ Veh vs. Sham+ Met: 0.0014 Sham + Veh vs. Sham+ Met: 0.9204 |
| One-way ANOVA (Fig 4E) | F (3, 12) = 87.54 | P<0.0001 | SNI+ Met vs. SNI+ Veh: 0.9950 SNI+ Met vs. Sham+ Met: <0.0001 SNI+ Met vs. Sham+ Veh: <0.0001 SNI+ Veh vs. Sham+ Met: <0.0001 SNI+ Veh vs. Sham+ Veh: <0.0001 Sham+ Met vs. Sham+ Veh: 0.9805 |
| One-way ANOVA (Fig 5E) | F (3, 11) = 19.07 | P=0.0001 | SNI+ Met vs. SNI+ Veh: 0.4476 SNI+ Met vs. Sham+ Met: 0.0006 SNI+ Met vs. Sham+ Veh: 0.8783 SNI+ Veh vs. Sham+ Met: 0.0043 SNI+ Veh vs. Sham+ Veh: 0.0030 Sham+ Met vs. Sham+ Veh: 0.0005 |
| One-way ANOVA (Fig 6E) | F (3, 10) = 3.231 | P=0.0693 | SNI+ Met vs. SNI+ Veh: 0.7526 SNI+ Met vs. Sham+ Met: 0.5263 SNI+ Met vs. Sham+ Veh: 0.4737 SNI+ Veh vs. Sham+ Met: 0.1040 SNI+ Veh vs. Sham+ Veh: 0.1027 Sham+ Met vs. Sham+ Veh: 0.9970 |
| Two-way ANOVA (Fig 7A) | F (6, 56) = 1.127 F (6, 56) = 8.379 F (1, 56) = 26.33 |
PI = 0.3587 Pt <0.0001 Pd<0.0001 |
SNI+ Met vs SNI+ Veh: 0.9914, 0.5675, 0.5675, 0.0531, 0.0285, 0.9914, 0.0148 |
| Two-way ANOVA (Fig 7B) | F (6, 56) = 0.5617 F (6, 56) = 7.403 F (1, 56) = 23.8 |
PI =0.7589 Pt <0.0001 Pd <0.0001 |
SNI+ Met vs SNI+ Veh: 0.9961, 0.0761, 0.2940, 0.0451, 0.7135, 0.4200, 0.5655 |
| Two-way ANOVA (Fig 7C) | F (6, 56) = 0.8278 F (6, 56) = 8.595 F (1, 56) = 28.91 |
PI=0.5534 Pt<0.0001 Pd<0.0001 |
SNI+ Met vs SNI+ Veh: 0.9233, 0.0215, 0.5389, 0.0118, 0.1757, 0.3928, 0.9233 |
| Two-way ANOVA (Fig 8A) | F (4, 30) = 4.082 F (4, 30) = 78.85 F (1, 30) = 50.88 |
P=0.0093 P<0.0001 P<0.0001 |
SNI+ Met vs SNI+ Veh: >0.9999, 0.0033, 0.0033, 0.0012, 0.0004 |
| Two-way ANOVA (Fig 8B) | F (4, 30) = 7.94 F (4, 30) = 172.9 F (1, 30) = 59.79 |
P=0.0002 P<0.0001 P<0.0001 |
SNI+ Met vs SNI+ Veh: 0.6879, <0.0001, 0.0016, <0.0001, 0.0004 |
| Two-way ANOVA (Fig 8C) | F (4, 30) = 6.346 F (4, 30) = 157.5 F (1, 30) = 56.54 |
P=0.0008 P<0.0001 P<0.0001 |
SNI+ Met vs SNI+ Veh: >0.9999, 0.0002, 0.0106, <0.0001, 0.0008 |
| Unpair ed T-test (Fig 8D) | F, DFn, Dfd = 1.18, 3, 3 | P= 0.0430 | SNI+ Met vs SNI+ Veh: 0.0430 |
| Unpair ed T-test (Fig 9C) | F, DFn, Dfd= 4.41, 3, 3 | P= 0.0192 | SNI+ Met vs SNI+ Veh: 0.0192 |
| Two-way ANOVA (Fig 10A) | F (6, 36) = 0.3987 F (6, 36) = 144.8 F (1, 36) = 2.513e-013 |
P=0.8749 P<0.0001 P>0.9999 |
SNI + Met vs. SNI + Veh: >0.9999, >0.9999, >0.9999, >0.9999, >0.9999, >0.9999, >0.9999. |
| Two-way ANOVA (Fig 10B) | F (6, 36) = 0.641 F (6, 36) = 249 F (1, 36) = 0.3919 |
P=0.6967 P<0.0001 P=0.5352 |
SNI + Met vs. SNI + Veh: >0.9999, >0.9999, >0.9999, >0.9999, >0.9999, >0.9999, >0.9999. |
| Two-way ANOVA (Fig 10C) | F (6, 36) = 0.3442 F (6, 36) = 264.7 F (1, 36) = 0.9423 |
P=0.9086 P<0.0001 P=0.3382 |
SNI + Met vs. SNI + Veh: >0.9999, >0.9999, >0.9999, >0.9999, >0.9999, >0.9999, >0.9999. |
| Unpair ed T-test (Fig 10D) | F, DFn, Dfd= 16.53, 3, 2 | P= 0.1151 | SNI+ Met vs. SNI+ Veh: 0.6617 |
| Unpair ed T-test (Fig 11C) | F, DFn, Dfd= 1.729, 5, 5 | P> 0.9999 | SNI+ Met vs. SNI+ Veh: 0.5627 |
| Unpair ed T-test (Fig 12C) | F, DFn, Dfd= 1.292, 19, 19 | P <0.0001 | Met vs. Veh: <0.0001 |
| Unpair ed T-test (Fig 12F) | F, DFn, Dfd=1.309, 19, 19 | P= 0.0022 | Met vs. Veh: 0.0022 |
| Unpair ed T-test (Fig 13C) | F, DFn, Dfd=1.241, 23, 23 | P= 0.0250 | Met vs. Veh: 0.0250 |
| Unpair ed T-test (Fig 13F) | F, DFn, Dfd=1.127, 23, 23 | P= 0.0281 | Met vs. Veh: 0.0281 |
Acknowledgements:
This work was supported by NIH grants R01NS065926 (TJP), R01NS102161 (TJP), R01 GM102575 (TJP and GD), K22 NS096030 (MDB), and The University of Texas STARS program (TJP and GD).
Footnotes
Competing interests:
The authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- 1.Education IoMCoAPRCa. Relieving pain in america: A blueprint for transforming prevention, care, education, and research; Washington (DC), 2011. [PubMed] [Google Scholar]
- 2.Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol 2015;14:162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price TJ, Basbaum AI, Bresnahan J, Chambers JF, de Koninck Y, Edwards RR, Ji RR, Katz J, Kavelaars A, Levine JD, Porter L, Schechter N, Sluka KA, Terman GW, Wager TD, Yaksh TL, Dworkin RH. Transition to chronic pain: Opportunities for new therapeutics. Nature Reviews Neuroscience 2018:in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardie. Amp-activated/snf1 protein kinases: Conserved guardians of cellular energy. Nature Reviews Molecular Cell Biology 2007;8:774–785. [DOI] [PubMed] [Google Scholar]
- 5.Kahn BBAT, Carling D, Hardie DG. Amp-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metabolism 2005;1:15–25. [DOI] [PubMed] [Google Scholar]
- 6.Terenzio M, Koley S, Samra N, Rishal I, Zhao Q, Sahoo PK, Urisman A, Marvaldi L, Oses-Prieto JA, Forester C, Gomes C, Kalinski AL, Di Pizio A, Doron-Mandel E, Perry RB, Koppel I, Twiss JL, Burlingame AL, Fainzilber M. Locally translated mtor controls axonal local translation in nerve injury. Science 2018;359:1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melemedjian OK, Asiedu MN, Tillu DV, Peebles KA, Yan J, Ertz N, Dussor GO, Price TJ. Il-6- and ngf-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eif4f complex. J Neurosci 2010;30:15113–15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melemedjian OKAM, Tillu DV, Sanoja R, Yan J, Lark A, Khoutorsky A, Johnson J, Peebles KA, Lepow T, Sonenberg N, Dussor G, Price TJ. Targeting adenosine monophosphate-activated protein kinase (ampk) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Molecular Pain 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melemedjian OK, Khoutorsky A, Sorge RE, Yan J, Asiedu MN, Valdez A, Ghosh S, Dussor G, Mogil JS, Sonenberg N, Price TJ. Mtorc1 inhibition induces pain via irs-1-dependent feedback activation of erk. Pain 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moy JK, Khoutorsky A, Asiedu MN, Black BJ, Kuhn JL, Barragan-Iglesias P, Megat S, Burton MD, Burgos-Vega CC, Melemedjian OK, Boitano S, Vagner J, Gkogkas CG, Pancrazio JJ, Mogil JS, Dussor G, Sonenberg N, Price TJ. The mnk-eif4e signaling axis contributes to injury-induced nociceptive plasticity and the development of chronic pain. J Neurosci 2017;37:7481–7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price TJ, Das V, Dussor G. Adenosine monophosphate-activated protein kinase (ampk) activators for the prevention, treatment and potential reversal of pathological pain. Curr Drug Targets 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asiedu MN, Dussor G, Price TJ. Targeting ampk for the alleviation of pathological pain. EXS 2016;107:257–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russe OQ, Moser CV, Kynast KL, King TS, Stephan H, Geisslinger G, Niederberger E. Activation of the amp-activated protein kinase reduces inflammatory nociception. J Pain 2013;14:1330–1340. [DOI] [PubMed] [Google Scholar]
- 14.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase lkb1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005;310:1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge A, Wang S, Miao B, Yan M. Effects of metformin on the expression of ampk and stat3 in the spinal dorsal horn of rats with neuropathic pain. Mol Med Rep 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burton MD, Tillu DV, Mazhar K, Mejia GL, Asiedu MN, Inyang K, Hughes T, Lian B, Dussor G, Price TJ. Pharmacological activation of ampk inhibits incision-evoked mechanical hypersensitivity and the development of hyperalgesic priming in mice. Neuroscience 2017;359:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao-Ying QL, Kavelaars A, Krukowski K, Huo XJ, Zhou W, Price TJ, Cleeland C, Heijnen CJ. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PLoS One 2014;9:e100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watkins LR, Milligan ED, Maier SF. Glial activation: A driving force for pathological pain. Trends in neurosciences 2001;24:450–455. [DOI] [PubMed] [Google Scholar]
- 19.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nature reviews Neuroscience 2009;10:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, Mogil JS. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 2015;18:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taves S, Berta T, Liu DL, Gan S, Chen G, Kim YH, Van de Ven T, Laufer S, Ji RR. Spinal inhibition of p38 map kinase reduces inflammatory and neuropathic pain in male but not female mice: Sex-dependent microglial signaling in the spinal cord. Brain Behav Immun 2016;55:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laird JM, Martinez-Caro L, Garcia-Nicas E, Cervero F. A new model of visceral pain and referred hyperalgesia in the mouse. Pain 2001;92:335–342. [DOI] [PubMed] [Google Scholar]
- 23.Pitcher MH, Price TJ, Entrena JM, Cervero F. Spinal nkcc1 blockade inhibits trpv1-dependent referred allodynia. Mol Pain 2007;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Decosterd CJW. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000:149–158. [DOI] [PubMed] [Google Scholar]
- 25.Allchorne AJ, Broom DC, Woolf CJ. Detection of cold pain, cold allodynia and cold hyperalgesia in freely behaving rats. Molecular Pain 2005;1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burton MD, Rytych JL, Amin R, Johnson RW. Dietary luteolin reduces proinflammatory microglia in the brain of senescent mice. Rejuvenation research 2016;19:286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norden DM, Fenn AM, Dugan A, Godbout JP. Tgfbeta produced by il-10 redirected astrocytes attenuates microglial activation. Glia 2014;62:881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey AL, Ribeiro-da-Silva A. Transient loss of terminals from non-peptidergic nociceptive fibers in the substantia gelatinosa of spinal cord following chronic constriction injury of the sciatic nerve. Neuroscience 2006;138:675–690. [DOI] [PubMed] [Google Scholar]
- 29.Lorenzo LE, Magnussen C, Bailey AL, St Louis M, De Koninck Y, Ribeiro-da-Silva A. Spatial and temporal pattern of changes in the number of gad65-immunoreactive inhibitory terminals in the rat superficial dorsal horn following peripheral nerve injury. Mol Pain 2014;10:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutierrez S, Hayashida K, Eisenach JC. The puerperium alters spinal cord plasticity following peripheral nerve injury. Neuroscience 2013;228:301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, Hill E, Hsu S, Izquierdo-Garcia D, Ji RR, Riley M, Wasan AD, Zurcher NR, Albrecht DS, Vangel MG, Rosen BR, Napadow V, Hooker JM. Evidence for brain glial activation in chronic pain patients. Brain 2015;138:604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albrecht DS, Granziera C, Hooker JM, Loggia ML. In vivo imaging of human neuroinflammation. ACS Chem Neurosci 2016;7:470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Albrecht DS, Ahmed SU, Kettner NW, Borra RJH, Cohen-Adad J, Deng H, Houle TT, Opalacz A, Roth SA, Melo MFV, Chen L, Mao J, Hooker JM, Loggia ML, Zhang Y. Neuroinflammation of the spinal cord and nerve roots in chronic radicular pain patients. Pain 2018;159:968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mapplebeck JCS, Dalgarno R, Tu Y, Moriarty O, Beggs S, Kwok CHT, Halievski K, Assi S, Mogil JS, Trang T, Salter MW. Microglial p2×4r-evoked pain hypersensitivity is sexually dimorphic in rats. Pain 2018 [DOI] [PubMed] [Google Scholar]
- 35.Kimura NMS, Tanihara Y, Ueo H, Okuda M, Katsura T, Inui K. Metformin is a superior substrate for renal organic cation transporter oct2 rather than hepatic oct1. Drug Metabolism and Pharmacokinetics 2005;20:378–386. [DOI] [PubMed] [Google Scholar]
- 36.Urakami Y, Nakamura N, Takahashi K, Okuda M, Saito H, Hashimoto Y, Inui K. Gender differences in expression of organic cation transporter oct2 in rat kidney. FEBS Lett 1999;461:339–342. [DOI] [PubMed] [Google Scholar]
- 37.Alnouti Y, Petrick JS, Klaassen CD. Tissue distribution and ontogeny of organic cation transporters in mice. Drug Metab Dispos 2006;34:477–482. [DOI] [PubMed] [Google Scholar]
- 38.Asaka J, Terada T, Okuda M, Katsura T, Inui K. Androgen receptor is responsible for rat organic cation transporter 2 gene regulation but not for roct1 and roct3. Pharm Res 2006;23:697–704. [DOI] [PubMed] [Google Scholar]
- 39.Groves CE, Suhre WB, Cherrington NJ, Wright SH. Sex differences in the mrna, protein, and functional expression of organic anion transporter (oat) 1, oat3, and organic cation transporter (oct) 2 in rabbit renal proximal tubules. J Pharmacol Exp Ther 2006;316:743–752. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Yoon SY, Zhang H, Dougherty PM. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of paclitaxel-induced painful neuropathy. J Pain 2012;13:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon SY, Robinson CR, Zhang H, Dougherty PM. Spinal astrocyte gap junctions contribute to oxaliplatin-induced mechanical hypersensitivity. J Pain 2013;14:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makker PG, Duffy SS, Lees JG, Perera CJ, Tonkin RS, Butovsky O, Park SB, Goldstein D, Moalem-Taylor G. Characterisation of immune and neuroinflammatory changes associated with chemotherapy-induced peripheral neuropathy. PLoS One 2017;12:e0170814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bassez G, Audureau E, Hogrel JY, Arrouasse R, Baghdoyan S, Bhugaloo H, Gourlay-Chu ML, Le Corvoisier P, Peschanski M. Improved mobility with metformin in patients with myotonic dystrophy type 1: A randomized controlled trial. Brain 2018;141:2855–2865. [DOI] [PubMed] [Google Scholar]
- 44.Casals-Díaz L, Vivó M, Navarro X. Nociceptive responses and spinal plastic changes of afferent c-fibers in three neuropathic pain models induced by sciatic nerve injury in the rat. Experimental Neurology 2009;217:84–95. [DOI] [PubMed] [Google Scholar]
- 45.Mogil JS, Bailey AL. Sex and gender differences in pain and analgesia. Prog Brain Res 2010;186:141–157. [DOI] [PubMed] [Google Scholar]
- 46.Lopes DM, Malek N, Edye M, Jager SB, McMurray S, McMahon SB, Denk F. Sex differences in peripheral not central immune responses to pain-inducing injury. Sci Rep 2017;7:16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Megat S, Shiers S, Moy JK, Barragan-Iglesias P, Pradhan G, Seal RP, Dussor G, Price TJ. A critical role for dopamine d5 receptors in pain chronicity in male mice. J Neurosci 2018;38:379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiers S, Pradhan G, Mwirigi J, Mejia G, Ahmad A, Kroener S, Price T. Neuropathic pain creates an enduring prefrontal cortex dysfunction corrected by the type ii diabetic drug metformin but not by gabapentin. J Neurosci 2018;38:7337–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor A, Westveld AH, Szkudlinska M, Guruguri P, Annabi E, Patwardhan A, Price TJ, Yassine HN. The use of metformin is associated with decreased lumbar radiculopathy pain. Journal of pain research 2013;6:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma J, Yu H, Liu J, Chen Y, Wang Q, Xiang L. Metformin attenuates hyperalgesia and allodynia in rats with painful diabetic neuropathy induced by streptozotocin. Eur J Pharmacol 2015 [DOI] [PubMed] [Google Scholar]
- 51.Ling YZ, Li ZY, Ou-Yang HD, Ma C, Wu SL, Wei JY, Ding HH, Zhang XL, Liu M, Liu CC, Huang ZZ, Xin WJ. The inhibition of spinal synaptic plasticity mediated by activation of amp-activated protein kinase signaling alleviates the acute pain induced by oxaliplatin. Exp Neurol 2017;288:85–93. [DOI] [PubMed] [Google Scholar]
- 52.Wang S, Kobayashi K, Kogure Y, Yamanaka H, Yamamoto S, Yagi H, Noguchi K, Dai Y. Negative regulation of trpa1 by amp-activated protein kinase in primary sensory neurons as a potential mechanism of painful diabetic neuropathy. Diabetes 2017 [DOI] [PubMed] [Google Scholar]