

Abstract
Objective:
Regulatory T cell (Treg) adoptive cell therapy has great potential to treat autoimmune disease. Currently, very little is known about how these cells impact inflamed tissues. The purpose of this study was to elucidate how autologous Treg therapy influences tissue inflammation in human autoimmune disease.
Methods:
We describe a patient with systemic lupus erythematosus (SLE) with active skin disease that received adoptive Treg therapy. We comprehensively quantify Tregs and immune activation in peripheral blood and skin with time post-treatment.
Results:
Deuterium tracking of infused Tregs revealed the transient presence of cells in peripheral blood accompanied by increased percentages of highly activated Tregs in diseased skin. Flow cytometric analysis and whole transcriptome RNA sequencing (RNAseq) revealed that Treg accumulation in skin was associated with a marked attenuation of the IFNγ pathway and a reciprocal augmentation of the IL-17 pathway. This phenomenon was more pronounced in skin relative to peripheral blood. To validate these findings, we found that Treg adoptive transfer in a murine model of skin inflammation also resulted in a pronounced skewing away from Th1 immunity and towards IL-17 production.
Conclusions:
We report the first case of a patient with SLE treated with autologous adoptive Treg therapy. Taken together, our results suggest that this treatment results in increased activated Tregs in inflamed skin with a dynamic shift from Th1 to Th17 responses.
INTRODUCTION
Regulatory T cells (Tregs) play an essential role in the establishment and maintenance of immune tolerance. Recent studies suggest that it may be possible to harness the immunoregulatory potential of these cells to treat autoimmune disease, prevent allograft rejection and attenuate graft-vs-host disease (1,2). These studies have employed either low-dose interleukin-2 (IL-2) as a means of pharmacologically augmenting Tregs (3) or ex vivo expansion and subsequent infusion of autologous Tregs (4–6). While prior studies have demonstrated augmentation of Treg numbers and function in peripheral blood, little is known about how these cells affect peripheral tissues. Specifically, it is unknown whether adoptively transferred Tregs modify the immune microenvironment in inflamed organs in human autoimmune disease.
Systemic Lupus Erythematosus (SLE) is a systemic autoimmune disease affecting multiple tissues. It has been suggested that Tregs are both quantitatively and qualitatively defective in patients with SLE (7). Adoptive transfer of Tregs in murine models of SLE has been shown to be efficacious (8) and recently, Treg augmentation with low-dose IL-2 appeared to show clinical benefit in a small cohort of SLE patients (9). Over the past decade, we have developed a robust protocol to isolate and expand Tregs from a variety of patient populations. In an initial phase I study in Type 1 Diabetes, we demonstrated that Tregs could be isolated to great purity using CD4, CD25 and CD127 cell surface markers, expanded in vitro over 2 weeks to 300–2000 fold and administered safely to patients in increasing doses from 0.05 to 2.6 × 108 cells (4). Importantly, we showed that a subset of stable Tregs could be observed in the blood for at least one year post-transfer. In this study, we sought to examine the safety and immune effects of Treg therapy in patients with SLE with prominent cutaneous disease. The presence of cutaneous involvement in these patients provided an opportunity to examine the effects of Treg therapy at the site of tissue injury. Although the recruitment and enrollment of patients was curtailed due a variety of clinical parameters and practical considerations, in this manuscript we describe the effects seen in one treated patient, highlighting the potential for systemic Treg therapy to alter the inflammatory infiltrates in inflamed skin.
RESULTS
Clinical Data
In 2016, we initiated an FDA-approved Phase 1 dose-escalating trial as part of the Autoimmunity Centers of Excellence consortium to evaluate the safety and immune parameters of treatment with autologous, ex vivo expanded Tregs in patients with cutaneous manifestations of lupus (NCT02428309). The trial, originally slated to enroll 9 subjects, was not completed due to screen failures and enrollment challenges and was ultimately limited to a single recruited patient. At the time of entry into the trial, the patient, a 46-year-old African American woman, had multiple active discoid lupus skin lesions (Supplemental Figure 1). She was taking stable doses of the following medications: hydroxychloroquine 200 mg twice a day, mycophenolate mofetil 1 gram twice a day, prednisone 5 mg/day, thalidomide 50 mg every other day, and aspirin 325 mg/day. Her baseline laboratory evaluation was notable for an erythrocyte sedimentation rate (ESR) of 30 mm/hour, anti-dsDNA antibody titer of 231 IU/ml, normal complement C3 and low complement C4. The Cutaneous LE Disease Area and Severity Index (CLASI) activity score was 21, the Systemic Lupus Erythematosus Disease Activity Index (SELENA SLEDAI) was 8, the Physician’s Global Assessment (PhGA) was 1.125 (scale of 0–3 inches), and the Patient’s Global Assessment (PGA) was 1.0 (scale of 0–3 inches). One week following infusion of 1 × 108 Tregs, the CLASI was unchanged. At week 12, the CLASI activity score was 19, SELENA SLEDAI was 8, PhGA was 1.0, PGA was 0.375, and serology was virtually unchanged from baseline (ESR - 34 mm/hour, anti-DNA - 238 IU/ml, complement C3 normal, complement C4 low). For the remainder of the 48-week follow-up period, the patient’s discoid lupus, overall clinical status and serologies remained stable.
Tregs and Cytokines in Peripheral Blood
The patient was infused with ex vivo expanded autologous Tregs that were highly suppressive in vitro and greater than 99% demethylated at the TSDR region of the Foxp3 gene locus, indicating Treg functionality and stability, respectively (Supplemental Figure 2) (10). On day −14, phlebotomy was conducted on the study subject. Approximately 400 mls of peripheral blood were drawn, and CD4+CD25+CD127lo/- Tregs cells were purified and expanded as previously described (4). During the ex vivo expansion period, the proliferating Tregs were cultured in the presence of 2H2 labelled, “deuterated” glucose, which incorporated into the deoxyribose moiety in replicating DNA through the de novo purine nucleotide synthesis pathway (11). Isotopic enrichment of the purine deoxyribonucleosides in Tregs was assessed by gas chromatography/mass spectrometry. The subject was treated with Tregs wherein approximately 60% of the DNA was enriched for the 2H-label without affecting the viability and integrity of the cells (data not shown). On day 0, the subject was infused with 1 × 108 deuterium-labeled, ex vivo expanded, Tregs. Expansion yielded >7.0×108 highly pure CD4+ T cells with >94% of these cells expressing high levels of CD25 and Foxp3 (Supplementary Figure 3), a degree of expansion and stability within the range of other studies (4). This is especially important as previous studies have suggested a reduced number of peripheral Tregs in the circulation of lupus patients and hypothesized that the cells were less functional than in healthy individuals, in part due to intrinsic defects, and in part due to ongoing medications (7). Importantly, the Tregs transfused into the subject were functionally competent in an in vitro suppression assay, had stable Foxp3 expression based on a DNA demethylation analysis and did not produce inflammatory cytokines at levels above that seen in other expanded Treg preparations (data not shown).
Peripheral blood was drawn at baseline (directly before Treg transfusion), day 1, and weeks 1, 2, 4, 8,12, and 24 for mechanistic studies. PBMCs were isolated from whole blood and either freshly sorted for deuterium analysis or cryopreserved for batched flow cytometric analysis. Both Tregs (CD4+CD127loCD25hi) and effector T (Teff) cell (CD4+CD25loCD127hi) populations were FACS-purified at specific times and analyzed for isotope detection by mass spectrometry (Figure 1A). At day 1 post-infusion, deuterium-labeled Tregs comprised ~1.5% of the total circulating Treg population. Isotope-labeled Tregs steadily decreased in peripheral blood over time and were undetectable after week 4 (Figure 1A). Throughout the entire study period, deuterium labeling was only detected in Tregs and not in CD4+ Teff cells.
Figure 1. Adoptively transferred Tregs are transiently detected in peripheral blood and alter T cell cytokine production.
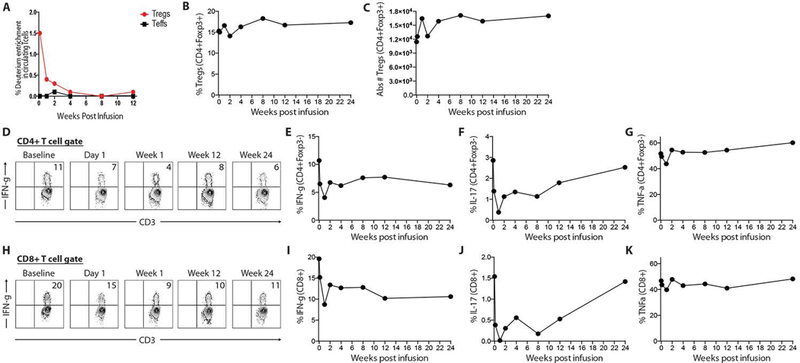
(A) Deuterium analysis of CD4+ Tregs and Teff cells in peripheral blood. (B & C) Total Treg percentages and absolute numbers in peripheral blood. (D) Representative flow cytometric plots of intracellular IFNg expression from CD4+ Teff cells in peripheral blood. (E, F, G) Quantification of intracellular IFNg, IL-17 and TNFa expression from CD4+ Teff cells. (H) Representative flow cytometric plots of intracellular IFNg expression from CD8+ T cells in peripheral blood. (E, F, G) Quantification of intracellular IFNg, IL-17 and TNFa expression from CD8+ T cells.
To determine whether Treg adoptive transfer influenced the Treg compartment and T cell cytokine production in peripheral blood, Tregs and intracellular cytokines from CD4+ Teff cells and CD8+ T cells were quantified by flow cytometry at specific times after infusion. Overall, Treg percentages and absolute cell numbers were unchanged throughout the study period (Figure 1B & 1C). There were no significant changes in Treg activation markers, such as CD25, CTLA-4, ICOS, Ki-67, CD98, and CD137 (data not shown). There was also no change in the CD4:CD8 ratio in peripheral blood (data not shown). To determine if the Treg infusion altered cytokine production in circulating T cells, PBMCs were stimulated with PMA/Iono and intracellular cytokine production quantified by flow cytometry. After Treg infusion, IFNγ production was reduced relative to baseline in both CD4+ Teff and CD8+ T cells throughout the entire study period (Figure 1D, 1E, 1H & 1I). Although very little IL-17A was observed in peripheral blood CD4+ Teff and CD8+ T cells, expression of this cytokine was slightly reduced following Treg infusion and returned to baseline levels by the end of the study period (Figure 1F & 1J). A similar trend was observed with TNFα (Figure 1G & 1K). Taken together, these results suggested that, although there were no appreciable changes in the cell function of most T cell subsets in peripheral blood, there was evidence of immediate reduction in IFNγ cytokine expression in both CD4+ and CD8+ T cells. Importantly, the reduction was observed during the time in which adoptively transferred Tregs were evident in the peripheral blood.
Tregs and Cytokines in Skin
Previous studies in animal models have suggested that a major impact of Treg function occurs at the site of inflammation. In an attempt to determine the effects of Treg therapy on immune subsets within the affected tissue, we quantified various T cell subsets and intracellular cytokine expression in diseased skin at baseline and 12-weeks post-Treg infusion. First, we examined the effect of the therapy on the Tregs in the tissue. The percentage of Tregs (Foxp3+) within the CD4+ T cell gate increased from 18% at baseline to 30% 12-weeks after Treg infusion (Figure 2A, 2B). In addition, we analyzed individual subsets of Tregs based on Foxp3hi versus Foxp3mid expression cells, as it has been shown in human tissues that these two populations differ markedly with respect to their suppressive capacity (12,13). Cells with the highest levels of Foxp3 are the most activated cells and thus the most suppressive. Foxp3hi Tregs increased from 3% at baseline to 16% 12-weeks after Treg infusion (Figure 2C); with no difference in Foxp3mid cells (Figure 2D). We could not determine whether the Foxp3hi cells contained the ex vivo expanded, adoptively transferred cells, as the mass spectrometry was not sensitive enough to pick up a signal in this small cell sample. It is thus unclear if the increase in Treg number and Foxp3 expression is a direct or indirect effect of the adoptive immunotherapy.
Figure 2. Accumulation of activated Tregs, attenuation of IFNg production and increased IL-17 production in skin after adoptive Treg cell therapy.
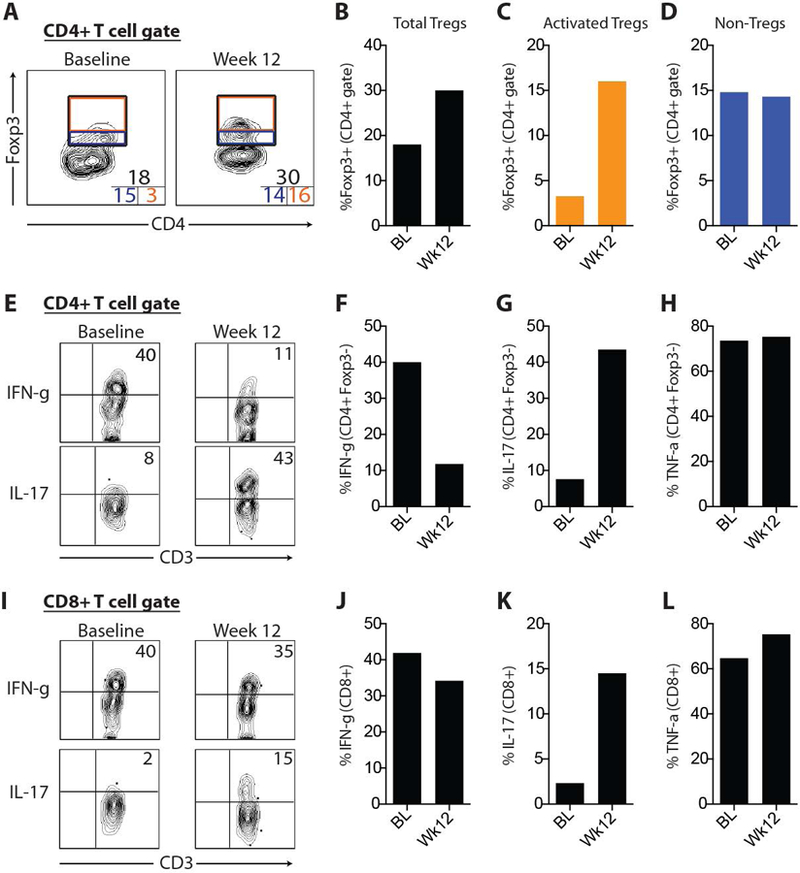
(A) Representative flow cytometric plots of Foxp3 expression from CD4+ T cells in skin. Numbers inset in corners represent total Tregs (top), Foxp3hi Tregs (i.e., “activated Tregs”; bottom right) and Foxp3mid Tregs (bottom left). (B, C, D) Quantification of data shown in part A. Representative flow cytometric plots of intracellular IFNg and IL-17 expression from CD4+ Teff cells (E) and CD8+ T cells (I) in skin with quantification of intracellular IFNg, IL-17 and TNFa expression from CD4+ Teff cells (F, G, H) and CD8+ T cells (J, K, L). BL, baseline.
The increased number of activated Tregs in skin was accompanied by a marked reduction in IFNγ production from CD4+ Teff cells (Figure 2E & 2F), with less effects on IFNγ-producing CD8+ T cells. Strikingly, there was a pronounced increase in IL-17A production in both CD4+ Teff cells and CD8+ T cells in skin 12-weeks after Treg infusion (Figure 2E, 2G, 2I & 2K). There were no appreciable differences in TNFα-producing T cells in skin 12-weeks after Treg infusion. Taken together, these results suggest that activated Tregs accumulate in diseased tissue after adoptive Treg infusion and are associated with an attenuation of IFNγ-producing Th1 cells and an increase in both Th17 cells and IL-17 producing CD8+ T cells.
Tissue whole transcriptome analysis
The above flow cytometric analyses were limited to examination of the T cell compartment. Thus, we determined the effects of adoptive Treg therapy changes at the tissue level using whole transcriptome RNAseq, with a focus on determining if the T cell cytokine profile was indicative of the whole tissue immune microenvironment. Freshly isolated diseased skin biopsies were taken at baseline and 12-weeks post-Treg infusion, RNA prepared and subjected to whole transcriptome analyses. We first looked at the relative expression of 9 well-documented and commonly accepted housekeeping genes (14). For each housekeeping gene, normalized counts between baseline and 12 weeks were compared. There was a slight trend towards increased expression of all genes in the 12-week samples; however, expression of these genes was quite similar between the 2 time points (Figure 3A). We next probed all differentially expressed genes (between baseline and 12-week samples) for genes documented to be associated with the IFNγ or IL-17 pathways (i.e., IFNγ and IL-17 response genes). Consistent with our flow cytometric data, we observed an overall attenuation of IFNγ response genes and a marked increase in IL-17 response genes (Figure 3B &3C). These results suggest that at the whole tissue level there is a reduction in IFNγ-mediated inflammation and an increase in IL-17-mediated gene expression in diseased skin 12-weeks post-Treg infusion.
Figure 3. Whole transcriptome analysis of skin tissue pre- and post-adoptive Treg cell therapy.
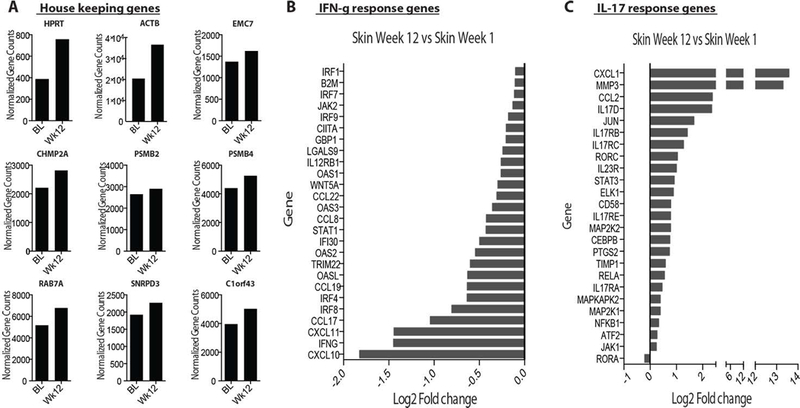
(A) Normalized gene counts (fragments per kilobase millions (FPKM) of 9 human housekeeping genes in baseline and week-12 diseased skin samples. Log2-fold changes of IFNg responses genes (B) and IL-17 response genes (C) between baseline and week-12 diseased skin samples.
Adoptively transferred Tregs suppress Th1 and augment Th17 responses in murine skin
Data from this single patient with cutaneous lupus suggested the interesting possibility that the adoptive transfer of ex vivo expanded, highly activated, Tregs results in attenuation of IFNγ-mediated inflammation and an augmentation in IL-17 responses. To explore these observations in the absence of additional patient enrollment, we examined whether adoptive transfer of Tregs resulted in a similar immune skewing in a well-characterized mouse model of skin inflammation. Topical application of Candida albicans (C. albicans) to abraded skin induces potent Th1 and Th17 immune response in skin and skin draining lymph nodes (SDLNs) in C57BL/6 mice (15–17). Importantly, this is not an infection model, as introduction of C. albicans in this context does not result in a productive infection, but instead is rapidly cleared by the host. Thus, this represents a robust mouse model of both Th1 and Th17 skin inflammation with which to test the effects of adoptively transferred Tregs. Immunodeficient RAG2 knock-out mice (RAG−/−) were adoptively transferred with autologous CD4+ Teffs alone or CD4+ Teffs with congenically marked autologous Tregs in a 1:1 ratio (Figure 4A). Fourteen days after adoptive transfer, dorsal skin was abraded and exposed to C. albicans. Seven days later, both the skin and SDLNs were harvested and intracellular cytokine production quantified by flow cytometry. Treg adoptive transfer resulted in a significant attenuation of IFNγ production from CD4+ Teffs in skin with a more limited effect on IFNγ production in SDLNs (Figure 4B, 4C, & 4D), similar to what was seen in the human PBMCs from the treated cutaneous lupus subject. Conversely, adoptively transferred Tregs resulted in a significant increase in IL-17A production from CD4+ Teffs in skin and SDLNs (Figure 4B, 4C, & 4D). These results suggest that adoptive transfer of autologous Tregs in a mouse model of skin inflammation results in a shift from Th1 to Th17 inflammation, that is most prominent in the inflamed tissue. Most importantly, the adoptive transfer of Tregs promoted the resolution of skin inflammation, as evidenced by reduced inflammatory infiltrate by routine histology (data not shown).
Figure 4. Treg adoptive transfer results in skewing away from Th1 and towards Th17 immune responses in a murine model of skin inflammation.
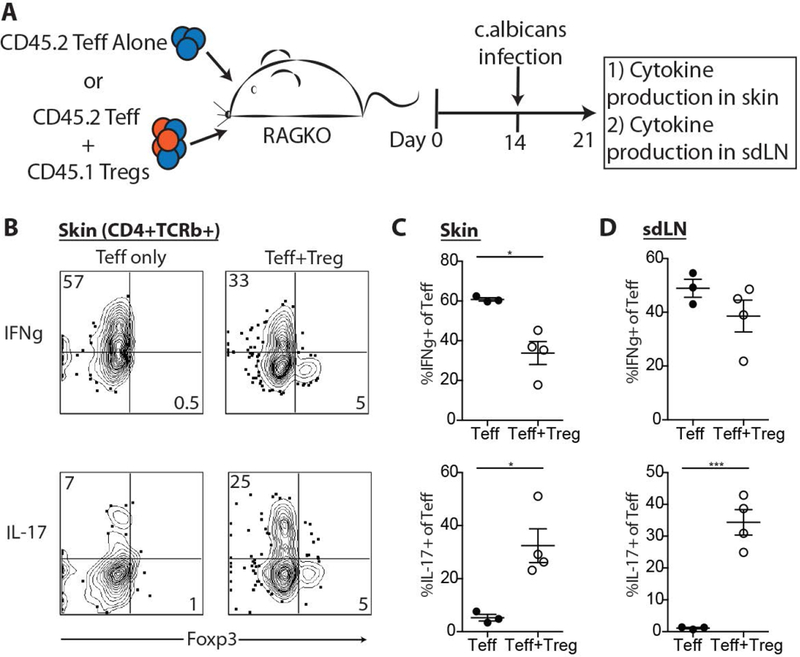
(A) CD4+ Teff cells were transferred alone or in combination with autologous Tregs to RAG2-deficient mice. The skin of recipient mice was abraded and exposed to c. albicans 14 days later. Seven days later, skin and SDLNs were harvest for cytokine expression by intracellular flow cytometry. Representative flow cytometric plots of IFNg (B) and IL-17 (E) production from skin infiltrating CD4+ Teff cells. Quantification of IFNg (C & D) and IL-17 (F & G) production from CD4+ Teff cells in skin and SDLNs. Results are representative of 3 replicate experiments. ***p<0.0005 and *p<0.05 by students t test.
Discussion
In this study, we set out to test the hypothesis that the adoptive transfer of autologous ex vivo expanded Tregs from patients with cutaneous lupus, would be achievable, safe and well-tolerated. Moreover, we compared an analysis of tissue samples at two time points from the study subject to determine the immune and biologic effects of Treg therapy. The results from this single subject suggest that there was indeed an alteration of T helper and cytotoxic T cell cytokines towards a Th17 phenotype after adoptive transfer. The resulting effects of this Treg transfer was corroborated by similar results in a non-infectious, inflammation model of candida in mice. Together, these results suggest that Treg therapy can alter the balance of Th1 and Th17 subsets in the local inflamed site, which may have an effect on clinical manifestations of the disease.
It must be noted up front that this was intended to be a dose-escalating study of the safety and mechanistic impact of the adoptive transfer of ex vivo expanded autologous Tregs in patients with cutaneous lupus. Unfortunately, for a number of logistical reasons, we were unable to enroll more than one subject in the study. Active skin manifestations in SLE are often accompanied by other more severe manifestations that were not compatible with the enrollment criteria for this trial. Thus, many potential subjects did not meet eligibility criteria for the trial. The original reason for focusing on patients with cutaneous lupus was the accessibility of skin biopsies to assess inflammation, as opposed to other tissues such as the kidney, which are considerably more difficult to obtain.
Thus, we were left with the recruitment of a single subject at the lowest proposed dose. At this dose, there was no demonstrable clinical impact. Nonetheless, there was indeed a set of intriguing mechanistic results that were observed. First, and importantly, we were able to isolate and expand Tregs from the patient. This is in and of itself an important observation, as several groups have noted that SLE patients have a suboptimal number of circulating Tregs and functional deficiencies in the cells. Moreover, these patients are often taking a variety of medications. This subject was taking hydroxychloroquine, mycophenolate mofetil and prednisone. Yet, the cells could be expanded in a fashion and with the stability seen in other disease settings (4).
It is important to note that, although there was significant engraftment of the adoptively transferred Tregs, as read out by the presence of deuterium labeling in Tregs isolated from the PBMCs on day 1, there was a rapid loss of labelled Tregs in the blood soon after transfer. This was most apparent when compared to previous studies using similar cell numbers for the adoptive immunotherapy ( (4) and unpublished results). There are a number of potential reasons for this outcome. Perhaps, the cells didn’t die but left the circulation, moving rapidly to the inflamed site. This remains possible but would be unique to this study when compared to the time course of cell number decay seen in Type 1 diabetes and organ transplant settings. An alternative explanation is that the cells have died rapidly after transfer. This might be due to the constellation of medications used by this subject, including the combination of anti-proliferative mycophenolate mofetil and steroid treatments. Alternatively, there is evidence that there is a defect in IL-2 production in this patient population. IL-2 is an essential growth factor for Tregs and the ex vivo expanded Tregs may be especially dependent on this cytokine for their survival. Recent studies have suggested that low dose IL-2 therapy in lupus patients results in an increase in endogenous Tregs and positive clinical effects (9). Thus, it might be logical to move forward with Treg therapy trials in lupus with a concomitant IL-2 therapy to maximize the effects of the individual therapies. In addition, vitamin D supplementation may augment the potential of adoptive Treg therapy, as this treatment has been shown to increase the Foxp3/IL-17A in SLE patients (18). Finally, it should be noted that it is unlikely that the rapid reduction in adoptively transferred Treg numbers in the blood was due to a dramatic change in cell surface phenotype, including loss of CD25 or increased CD127 expression. Mass spectrometry analyses showed that all of the deuterium label remained within the bona fide Treg subset, arguing against Treg instability for the decrease in labeled Tregs in the blood over such a short period of time.
One important observation in this patient was the increase in both the relative percentage of Tregs and the mean fluorescence intensity of the Foxp3 expression in the skin biopsy at 12 weeks versus baseline. It is possible that the cells identified in the tissue were derived from the adoptively transferred population, but the mass spectrometry analysis was not sensitive enough to detect deuterium in the sample. The alternative hypothesis is that the identified Tregs are infiltrating endogenous Tregs from circulation. There may be a constant infiltration of Tregs at sites of inflammation such as what occurred in this patient but normally these cells are compromised by cytokines in the milieu such as IL-6 and IL-12. The effects of the Tregs, either in the tissue or draining lymph nodes, in suppressing the Th1 environment (as seen in panel 3B, including STAT1, Th1 chemokines, etc.), may allow for a more stable resident Treg population in the inflamed site. Equally possible, the potential increase in the production of TGFβ at the site might promote both Tregs and Th17 cells, although this cytokine was not observed in the RNA gene expression analyses.
Finally, the most intriguing result of the current study in the single subject was the dramatic changes in T cell subset distribution in the inflamed skin of the patient. Not only were the number of IFN-γ-producing CD4 cells dramatically reduced but there was a concomitant increase in the number of IL-17a producing CD4 and CD8 cells in lesional skin. We do not know which of these changes was directly a consequence of the Treg therapy, as it is possible that the Th1/Th17 ratio was affected by the loss or increase in either population. However, several studies in mouse models have shown direct inhibition of IFN-γ producing cells by Tregs, especially Tregs that have a Th1-type phenotype (19), and it is possible that the loss of IFN-γ was involved in the relative expansion of the Th17 subset. In this regard, it was interesting to note that Th17 responses are not always pathogenic, as IL-17 is required for barrier homeostasis (20,21), and this patient’s clinical status did not get worse as a consequence of the Treg therapy. Moreover, similar results were observed in the mouse skin inflammation model of candida exposure, where Treg adoptive transfer led to a similar Th1 decrease and Th17 increase in the tissue, correlating with resolution of tissue inflammation. It will be critical moving forward to determine the precise mechanism(s) at play in this 3-cell subset interplay.
In summary, we attempted to conduct a full safety and immune biomarker study in patients with cutaneous lupus. However, the studies were incomplete due to recruitment challenges. Further studies need to be performed in other immune-mediated skin disorders to ascertain the reproducibility of these interesting and provocative results.
Supplementary Material
Acknowledgments
Financial support:
Supported by a grant from National Institutes of Allergy and Infectious Diseases (NIAID), Autoimmunity Centers of Excellence (UM1-AI110498).
Footnotes
Conflict of Interest:
Amy L Putnam: Patents pending on polyclonal Tregs.
Qizhi Tang: Juno sponsored research, Third Rock Venture consultation
Jeffrey A. Bluestone: Juno/Celgene, Bectin-Dickinson, Pfizer, Third Rock Ventures, and patents pending on polyclonal Tregs.
David Wofsy: Consulting relationships with Celgene and GlaxoSmithKline.
Michael Rosenblum: Consulting relationships with Celgene and TRex Bio.
References
- 1.Spence A, Klementowicz JE, Bluestone JA, Tang Q. Targeting Treg signaling for the treatment of autoimmune diseases. Curr Opin Immunol 2015;37:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang D, Tu E, Kasagi S, Zanvit P, Chen Q, Chen W. Manipulating regulatory T cells: a promising strategy to treat autoimmunity. Immunotherapy 2015;7:1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 2015;15:283–294. [DOI] [PubMed] [Google Scholar]
- 4.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 2015;7:315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 2011;117:3921–3928. [DOI] [PubMed] [Google Scholar]
- 6.Safinia N, Grageda N, Scottà C, Thirkell S, Fry LJ, Vaikunthanathan T, et al. Cell Therapy in Organ Transplantation: Our Experience on the Clinical Translation of Regulatory T Cells. Front Immunol 2018;9 Available at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.00354/full. Accessed April 16, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol 2015;45:344–355. [DOI] [PubMed] [Google Scholar]
- 8.Scalapino KJ, Daikh DI. Suppression of glomerulonephritis in NZB/NZW lupus prone mice by adoptive transfer of ex vivo expanded regulatory T cells. PloS One 2009;4:e6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med 2016;22:991–993. [DOI] [PubMed] [Google Scholar]
- 10.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009;30:899–911. [DOI] [PubMed] [Google Scholar]
- 11.Busch R, Neese RA, Awada M, Hayes GM, Hellerstein MK. Measurement of cell proliferation by heavy water labeling. Nat Protoc 2007;2:3045–3057. [DOI] [PubMed] [Google Scholar]
- 12.Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med 2016;22:679–684. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res 2017;27:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet TIG 2013;29:569–574. [DOI] [PubMed] [Google Scholar]
- 15.Igyártó BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 2011;35:260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kashem SW, Igyarto BZ, Gerami-Nejad M, Kumamoto Y, Mohammed JA, Jarrett E, et al. Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity 2015;42:356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 2015;43:515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marinho A, Carvalho C, Boleixa D, Bettencourt A, Leal B, Guimarães J, et al. Vitamin D supplementation effects on FoxP3 expression in T cells and FoxP3+/IL-17A ratio and clinical course in systemic lupus erythematosus patients: a study in a Portuguese cohort. Immunol Res 2017;65:197–206. [DOI] [PubMed] [Google Scholar]
- 19.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 2009;10:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floudas A, Saunders SP, Moran T, Schwartz C, Hams E, Fitzgerald DC, et al. IL-17 Receptor A Maintains and Protects the Skin Barrier To Prevent Allergic Skin Inflammation. J Immunol Baltim Md 1950 2017;199:707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song X, He X, Li X, Qian Y. The roles and functional mechanisms of interleukin-17 family cytokines in mucosal immunity. Cell Mol Immunol 2016;13:418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.