

INTRODUCTION
Approximately 20% of patients with metastatic lung adenocarcinoma have somatic activating mutations in the epidermal growth factor receptor (EGFR) gene, EGFR.1 Patients with EGFR-mutant lung adenocarcinomas have a 70% response rate to first-line EGFR–tyrosine kinase inhibitor (TKI) therapy (ie, erlotinib, gefitinib, or afatinib).2 EGFR T790M is the dominant resistance mechanism to earlier-generation EGFR-TKIs.3 Osimertinib is approved by the US Food and Drug Administration for the treatment of EGFR-mutant lung cancers that have an acquired EGFR T790M after failure of a previous EGFR-TKI4 and now is approved in the first-line setting as well.5
Response to osimertinib eventually is followed by progression with known resistance mechanisms, including small-cell transformation3,6; acquired EGFR mutations, including G796/C797, L792 and L718/G7197; and non-EGFR–mediated resistance, including alterations/amplification in MET, HER2, BRAF, MEK, KRAS, and PIK3CA.8-11 There have been rare reports of acquired fusions, including RET, BRAF, and FGFR, as mechanisms of resistance to EGFR-TKI.12,13 This case series capitalizes on multimodality molecular analyses, which include next-generation sequencing (NGS) with Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT)1,14 and ArcherDx (Boulder, CO)15 platforms, immunohistochemistry (IHC), and fluorescent in-situ hybridization (FISH), to evaluate three cases of acquired resistance16 to osimertinib: one acquired RET fusion and two ALK rearrangements.
ACQUIRED ALK REARRANGEMENTS IN EGFR-MUTANT LUNG CANCER AFTER OSIMERTINIB
Case 1
A 65-year-old woman, who was a former 9–pack-year smoker, presented with a 3 cm × 2 cm lingular primary mass, hilar adenopathy, and a liver lesion (staging: T2pN2cM1b). The biopsy revealed adenocarcinoma with a 15-bp EGFR exon 19 deletion (exon19del). The patient received erlotinib and continued this treatment for 19 months before progression occurred. Plasma circulating tumor DNA (ctDNA) and the tumor biopsy were positive for EGFR T790M; notably, NGS showed no ALK rearrangement. The patient transitioned to treatment with osimertinib and necitumumab (ClinicalTrials.gov identifier NCT02496663)17 for 9 months until progression developed in the lungs. A biopsy noted an adenocarcinoma with IHC results positive for ALK and EGFR exon19del (Fig 1). NGS with ArcherDx and MSK-IMPACT confirmed an acquired EML4-ALK fusion (EML4 exons 1 through 6 fused with ALK exons 20 through 29; c.667+516:EML4_c.3173-415:ALKinv) in addition to the EGFR exon19del and T790M mutations. The patient started combination treatment with osimertinib 80 mg daily and crizotinib 200 mg twice daily, remained on treatment with stable disease (by RECIST version 1.1), and continued to receive clinical benefit from treatment, with no report of toxicity. Subsequent imaging demonstrated oligoprogression in the target lesion with stable disease in nontarget lesions. The patient underwent radiation to the oligoprogressive site and has remained on combination therapy with continued disease control. (Figs 2A and 3B; Appendix Table A1).
Fig 1.

Representative immunohistochemistry (IHC) of the pre- and post-osimertinib biopsies for patient case 1 at ×20 magnification. The preosimertinib biopsy was a fine-needle aspiration (FNA) and was partially fragmented on staining, whereas the acquired resistance (AR) sample was a core-needle biopsy. The pretreatment EGFR exon19del staining was performed with epidermal growth factor receptor (EGFR)–E746; the post-treatment IHC stained more diffusely for EGFR exon19del, likely related to the EGFR amplification (fold change: 3.0) noted on next-generation sequencing with MSK-IMPACT. The post-treatment biopsy showed new staining for ALK (clone D5F3) compared with the pretreatment sample, which was negative. The IHC data suggest the presence of the ALK rearrangement and EGFR mutation within the same cell (however, this cannot be confirmed by targeted next-generation sequencing because of technical limitations).
Fig 2.

Representative radiologic images that illustrate the response to combination therapy in patients with acquired ALK-rearranged disease as evaluated by RECIST version 1.1. (A) Osimertinib and crizotinib in patient case 1 after one cycle (28 days) of treatment; image shows overall stable disease (0% best response). (B) Osimertinib and alectinib in patient case 2 after one cycle (28 days) of treatment; image shows regression of the dominant right upper lobe mass (25% reduction).
Fig 3.
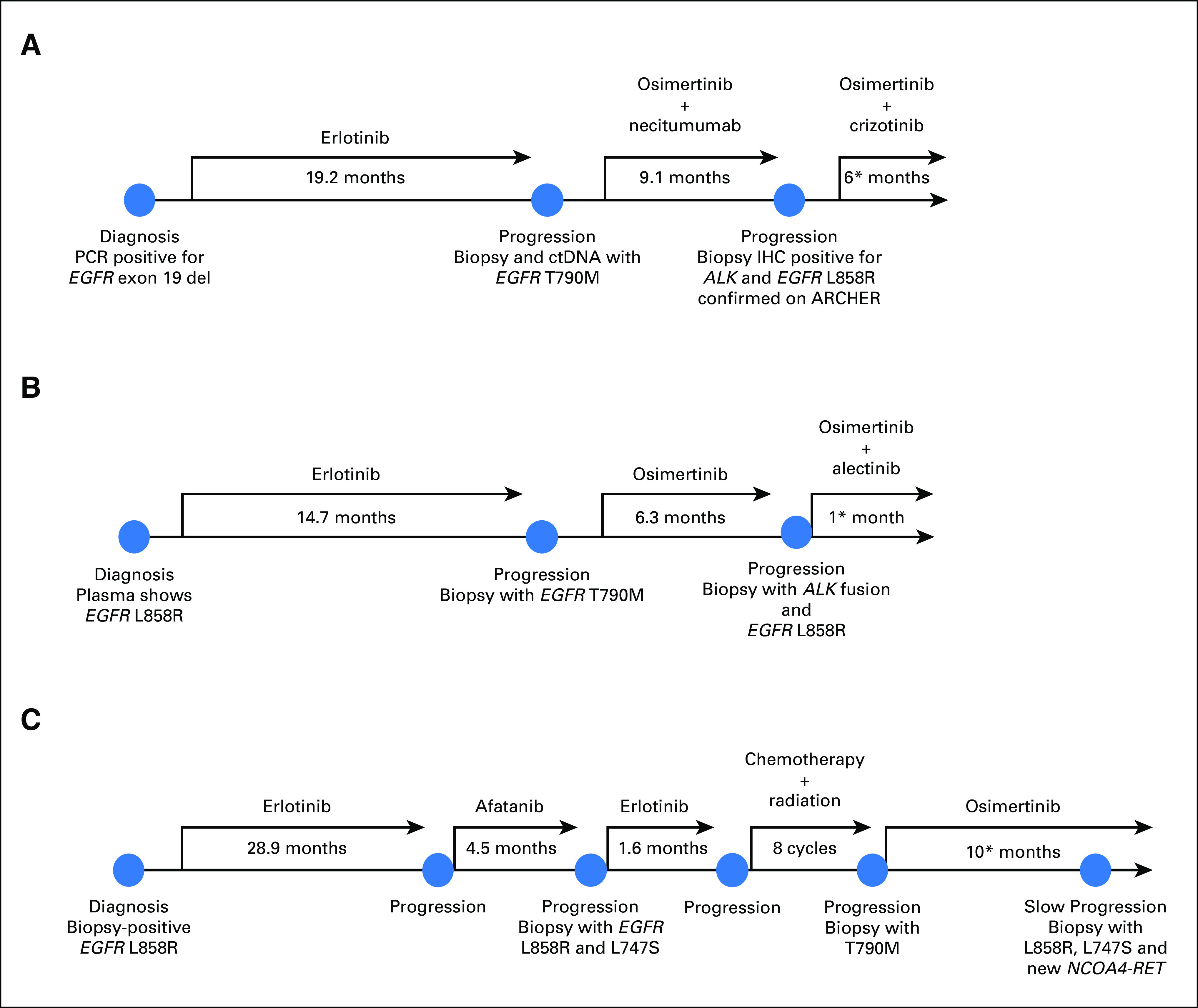
Description of clinical course and pertinent molecular and immunohistochemical findings for the three patient cases presented. (*) Treatment ongoing as of April 1, 2018.
Case 2
A 68-year-old woman, who was never a smoker, presented with a 9-cm right apical lung mass, right hilar lymphadenopathy, and a left temporal lobe mass (staging T4N1M1b). Biopsy of the lung mass demonstrated adenocarcinoma, and ctDNA showed EGFR L858R (insufficient tissue for IHC). She started treatment with erlotinib and continued it for 15 months until she experienced progression in a right supraclavicular lymph node. A biopsy revealed an EGFR T790M mutation on digital polymerase chain reaction, and NGS confirmed EGFR L858R and T790M mutations. (Of note, no ALK rearrangement was detected on NGS or FISH.) The patient started treatment with osimertinib and had an initial disease response, but oligoprogression occurred in the lung after 6 months. A lung biopsy found EGFR L858R mutation and ALK positivity on IHC. MSK-IMPACT confirmed the known EGFR L858R and T790M mutations as well as the new EML4-ALK rearrangement (EML4 exons 1 through 2 fused to ALK exons 20 through 29; c.208+4890:EML4_c.3173-373:ALKinv; Fig 3B; Appendix Table A2). The patient started treatment with alectinib 300 mg twice daily and osimertinib 80 mg daily; the first interval scan showed a decrease in the dominant right lung mass (25% reduction by RECIST version 1.1) as well as ongoing clinical benefit and no report of toxicity at the last follow-up. (Fig 2B).
ACQUIRED RET REARRANGEMENT IN EGFR-MUTANT LUNG CANCER AFTER OSIMERTINIB
Case 3
A 78-year-old man, who was never a smoker, presented with a 4-cm right lung mass and a liver lesion. A biopsy of the lung revealed adenocarcinoma; IHC was positive for EGFR L858R (staging T2aN2M1b), and this was confirmed by polymerase chain reaction. He started treatment with erlotinib and continued it for 29 months until progression developed in the lungs. A subsequent biopsy showed his original EGFR L858R and a newly acquired L747S mutation. He started combination treatment with carboplatin and pemetrexed along with palliative radiation to the right middle lobe. A repeat biopsy then showed an acquired EGFR T790M mutation in addition to EGFR L747S and L858R mutations, and he started osimertinib. The biopsy tissue was retrospectively analyzed with ArcherDx and was negative for gene fusion products. The patient developed oligoprogression after 16 months of osimertinib treatment and underwent local radiation. A right lung biopsy showed the known EGFR L747S and L858R mutations as well as a new NCOA4-RET fusion on MSK-IMPACT; the fusion was confirmed by ArcherDx. The patient has remained on osimertinib treatment as a result of the slow, asymptomatic progression (Fig 3A; Appendix Table A3).
EXPRESSION OF RET REARRANGEMENTS IN AN EGFR-MUTANT CELL LINE RESULTS IN OSIMERTINIB RESISTANCE
To determine if RET rearrangements confer resistance to osimertinib, we expressed two RET fusions in PC9 cells (del19 EGFR; ATCC, Manassas, VA) and generated stable cell lines using previously described techniques.18 CCDC6-RET was chosen because of prior work, which showed that the fusion partner did not influence RET activity in cell lines.19,20 Each condition was assayed in triplicate in at least two independent experiments. The PC9 cells were used to assess the effect of RET fusions on sensitivity of growth and apoptosis (caspase 3/7 activity) in the presence of osimertinib and cabozantinib. Expression of RET and phosphorylated RET were confirmed via Western blot (Fig 4A). Osimertinib treatment reduced EGFR and ERK1/2 phosphorylation in PC9-pCX4.1–empty cells (Fig 4B). However, ERK1/2 phosphorylation was not sensitive to osimertinib treatment in PC9-pCX4.1–CCDC6-RET cells but was diminished with cabozantinib treatment (Fig 4B). The presence of a RET fusion did not alter the ability of osimertinib to inhibit EGFR phosphorylation (Fig 4B). These results suggest that RET rearrangements can cause bypass activation of growth-promoting pathways in cells that express oncogenic EGFR.
Fig 4.
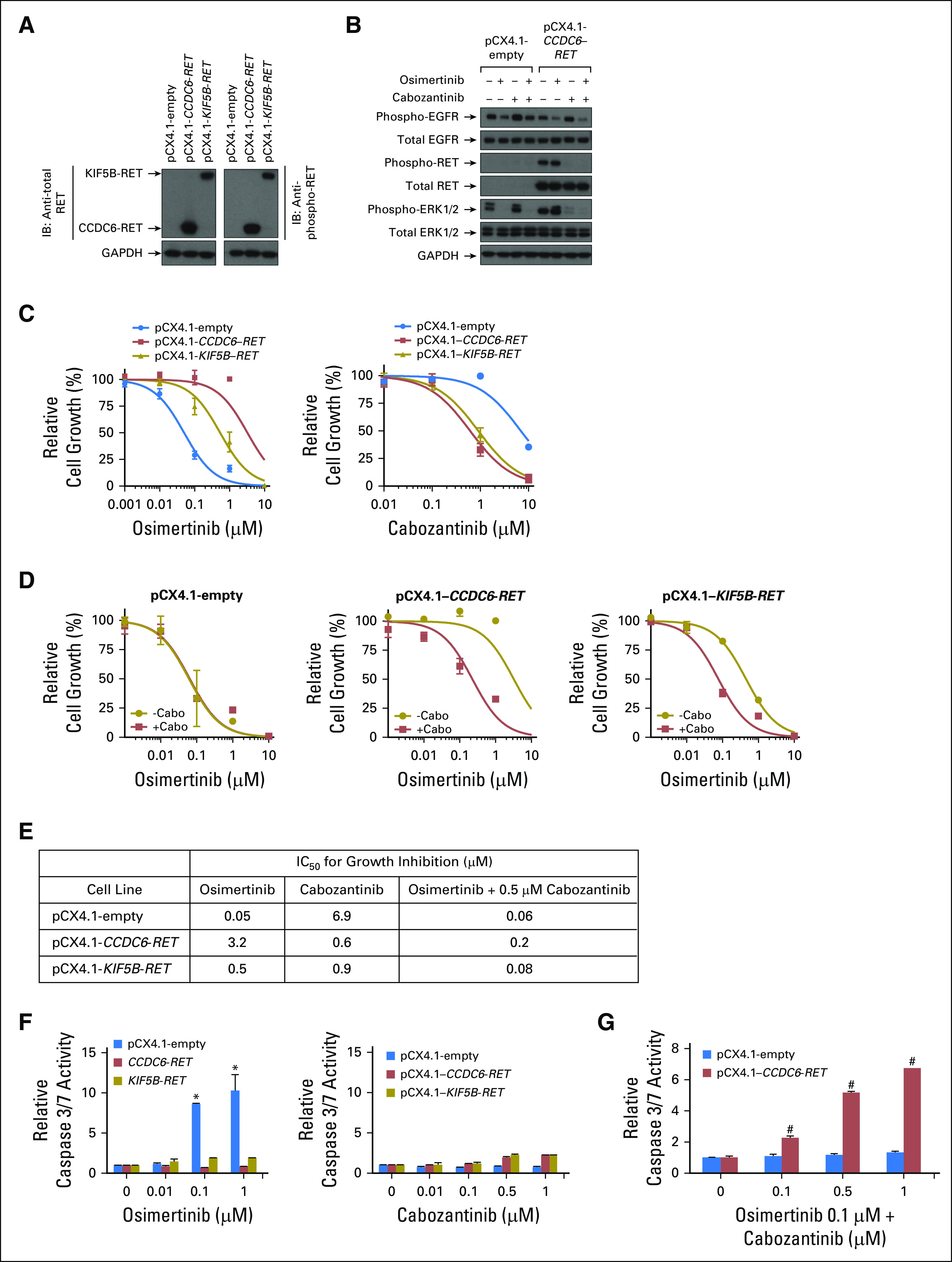
Expression of RET fusions that induce resistance to osimertinib with a concomitant increase in sensitivity to cabozantinib. PC9 cells were infected with lentivirus that harbored an empty plasmid (pCX4.1-empty) or pCX4.1 with either CCDC6-RET or KIF5B-RET complementary DNA (cDNA), and cells that expressed the plasmids were selected to generate stable cell lines. (A) Cell extracts were immunoblotted (indicated by IB) for (left panel) total RET (left panel) or (right panel) phosphorylated RET. (B) Cell lines were treated with either osimertinib 0.05 µM or cabozantinib or 0.25 µM for 1 h; cell extracts were prepared and then immunoblotted for the indicated proteins. (C) Cells were treated with increasing concentrations (as indicated on the figure) of (left panel) osimertinib or (right panel) cabozantinib for 96 h, and then growth was determined. (D) Cells were treated with the indicated concentrations of osimertinib in the presence of cabozantinib 0.5 µM for 96 h, and then growth was determined. (E) 50% inhibitory concentration (IC50) values were determined by nonlinear regression of growth data. (F) Cells were treated with the indicated concentrations of (left panel) osimertinib or (right panel) cabozantinib for 48 hours, and then caspase 3/7 enzymatic activity determined. (G) PC9 cells that expressed either pCX4.1-empty vector or pCX4.1–CCDC6-RET were treated with the indicated concentrations of cabozantinib in the presence of osimertinib 0.1 µM for 48 h, and then caspase 3/7 enzymatic activity was determined. Results are expressed as the fold change in caspase 3/7 activity compared with the corresponding cell line treated with osimertinib 0.1 µM alone. All experiments were conducted at least two times, and data represent the mean ± standard deviation. (*) P < .05 compared with untreated control. (†) P < .05 compared with the corresponding cell line treated with osimertinib 0.1 µM only. All data were analyzed by two-way Anova with the Tukey multiple comparison test. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; −cabo, without cabozanitinib; +cabo, with cabozanitinib.
Growth of PC9 cells that stably expressed CCDC6-RET or KIF5B-RET rearrangements was at least 10-fold less sensitive to osimertinib than PC9-pCX4.1–empty cells were (50% inhibitory concentrations described in Fig 4E). PC9 cells that expressed RET fusions acquired sensitivity to cabozantinib (Figs 4C [right panel] and 4E) when compared with PC9-pCX4.1–empty cells. Sensitivity to osimertinib was restored when cells were treated with cabozantinib and osimertinib (Fig 4D). Expression of RET fusions in PC9 cells prevented osimertinib-induced activation of caspase 3/7, similar to the effect on growth rate (Fig 4F [left panel]). Treatment of control-group PC9 cells or PC9-RET cells with cabozantinib did not lead to activation of caspase 3/7 (Fig 4F [left panel]). However, a combination of cabozantinib and osimertinib led to caspase 3/7 activation in PC9-pCX4.1–CCDC6-RET cells (Fig 4G). In contrast, cabozantinib treatment did not have an additive effect on osimertinib-induced caspase 3/7 activity in PC9-pCX4.1–empty cells (Fig 4G). These results suggest that RET rearrangements can induce resistance to osimertinib in cells that have EGFR mutations and that response to osimertinib is restored when it is used in combination with cabozantinib.
In conclusion, osimertinib is now a first-line treatment for metastatic EGFR-mutant lung cancers.5 Although potential resistance mechanisms to osimertinib have been identified, these have primarily been observed after later-line osimertinib in the setting of EGFR T790M mutations and have focused on ctDNA.9-11 Many alterations identified as potential resistance mechanisms are seen concurrently with EGFR in pretreatment samples, including amplifications of MET and HER2 as well as PIK3CA mutations, which makes the comparison with pretreatment tissue critical to identify truly acquired alterations.13 As NGS becomes standard of care, it is feasible and fruitful to molecularly profile tumors broadly before treatment and at progression to identify acquired alterations that mediate resistance.
It remains unclear whether mechanisms of resistance to first-line osimertinib will differ from mechanisms of resistance to later-line osimertinib or earlier-generation EGFR-TKIs. Osimertinib is a potent mutant-EGFR inhibitor that inhibits EGFR T790M, so there is a potential to see novel on-target21,22 and off-target23-25 resistance mechanisms. This is corroborated by the low frequency (15% to 25%) of EGFR C797S and other acquired EGFR alterations after osimertinib use compared with 60% frequency of acquired EGFR T790M after use of earlier-generation EGFR-TKIs.9,13,22 We may see new acquired alterations not previously seen or seen rarely with earlier-generation EGFR-TKIs. Our functional studies support the notion that acquired RET fusions can overcome the inhibitory effect of osimertinib by sustained activation of proliferation through MAPK signaling. Moreover, although cabozantinib can inhibit growth of EGFR- and RET-mutant/rearranged cell lines, a combination of osimertinib and cabozantinib induced apoptosis.
At our institution, among 174 patients with EGFR-mutant lung cancer in whom NGS was performed on tumor tissue after progression developed during treatment with erlotinib or afatinib, we found no ALK or RET fusions. To date, we have obtained biopsies from a small sample of patients (n = 14) after progression developed during osimertinib treatment; we found two ALK fusions and one RET fusion, which suggests a difference in the prevalence of these fusions in these two acquired resistance settings (Fisher’s exact test P < .001). It is unknown whether this potential enrichment of acquired fusions is related to the more potent EGFR inhibition of osimertinib or to the later-line setting after multiple lines of EGFR inhibition. To our knowledge, this is the first report to document the clinical benefit of osimertinib combined with another agent to target an acquired mutation in a different oncogene. After the dominant resistance mechanisms to osimertinib are identified, prospective testing of combination therapies will be needed in the first-line setting to prevent or delay resistance and in the second-line setting to reverse or overcome acquired resistance.
Appendix
Table A1.
Detailed Molecular and Immunohistochemical Characterization of Patient Case 1 Presented

Table A2.
Detailed Molecular and Immunohistochemical Characterization of Patient Case 2

Table A3.
Detailed Molecular and Immunohistochemical Characterization of Patient Case 3

Footnotes
Supported in part by the National Cancer Institute of the National Institutes of Health Grants No. T32 CA009207 and P30 CA008748.
AUTHOR CONTRIBUTIONS
Conception and design: Michael Offin, Romel Somwar, Gregory J. Riely, Mark G. Kris, William Travis, Alexander Drilon, Maria E. Arcila, Marc Ladanyi, Helena A. Yu
Collection and assembly of data: Michael Offin, Romel Somwar, Natasha Rekhtman, Ryma Benayed, Jason C. Chang, Andrew Plodkowski, Allan J.W. Lui, Juliana Eng, Marc Rosenblum, Bob T. Li, Gregory J. Riely, Mark G. Kris, William Travis, Alexander Drilon, Maria E. Arcila, Helena A. Yu
Provision of study material or patients: Natasha Rekhtman, Juliana Eng, Bob T. Li, Charles M. Rudin, Mark G. Kris, William Travis, Helena A. Yu
Data analysis and interpretation: Michael Offin, Romel Somwar, Natasha Rekhtman, Allan J.W. Lui, Marc Rosenblum, Bob T. Li, Charles M. Rudin, Mark G. Kris, William Travis, Alexander Drilon, Maria E. Arcila, Marc Ladanyi, Helena A. Yu
Administrative support: Mark G. Kris
Financial support: Mark G. Kris
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Michael Offin
No relationship to disclose
Romel Somwar
Research Funding: Helsinn Healthcare
Travel, Accommodations, Expenses: Helsinn Healthcare
Natasha Rekhtman
No relationship to disclose
Ryma Benayed
No relationship to disclose
Jason C. Chang
No relationship to disclose
Andrew Plodkowski
No relationship to disclose
Allan J.W. Lui
No relationship to disclose
Juliana Eng
No relationship to disclose
Marc Rosenblum
No relationship to disclose
Bob T. Li
Consulting or Advisory Role: Roche, BIosceptre International, ThermoFisher Scientific, Mersana, Guardant Health
Research Funding: Roche (Inst), Genentech (Inst), Illumina (Inst), BioMed Valley Discoveries (Inst), AstraZeneca (Inst), GRAIL (Inst)
Gregory J. Riely
Research Funding: Novartis (Inst), Roche (Inst), Genentech (Inst), Millenium (Inst), GlaxoSmithKline (Inst), Pfizer (Inst), Infinity Pharmaceuticals (Inst), ARIAD (Inst)
Patents, Royalties, Other Intellectual Property: Patent application submitted covering pulsatile use of erlotinib to treat or prevent brain metastases (Inst)
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Charles M. Rudin
Consulting or Advisory Role: Bristol-Myers Squibb, Abbvie, Seattle Genetics, Harpoon Therapeutics, Genentech, Roche, AstraZeneca
Mark G. Kris
Consulting or Advisory Role: AstraZeneca, Regeneron
William Travis
Speakers' Bureau: Genentech
Alexander Drilon
Consulting or Advisory Role: Ignyta, Loxo, TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Genentech, Roche, Takeda, Helsinn Therapeutics, BeiGene
Maria E. Arcila
Consulting or Advisory Role: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca, Invivoscribe, Raindance Technologies
Marc Ladanyi
Honoraria: Merck (I)
Consulting or Advisory Role: National Comprehensive Cancer Network, Boehringer Ingelheim, AstraZeneca (Tagrisso RFP Advisory Committee)
Research Funding: Loxo (Inst)
Helena A. Yu
Consulting or Advisory Role: AstraZeneca
Research Funding: Astellas Pharma, Incyte, Lilly Oncology, Novartis, Daiichi, AstraZeneca
REFERENCES
- 1.Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596–609. doi: 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jänne PA, Wang XF, Socinski J, et al. Randomized phase II trial of erlotinib (E) alone or in combination with carboplatin/paclitaxel (CP) in never or light former smokers with advanced lung adnocarcinoma: CALGB 30604 trial. J Clin Oncol. 2012;30:2063–2069. doi: 10.1200/JCO.2011.40.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu H, Arcila ME, Rekhtman N, et al. Analysis of mechanisms of acquired resistance to EGFR TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med. 2017;376:629–640. doi: 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N Engl J Med. 2018;378:113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 6.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z, Yang N, Ou Q, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non–small-cell lung cancer patients. Clin Cancer Res. 2018;24:3097–3107. doi: 10.1158/1078-0432.CCR-17-2310. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Li L, Han R, et al. Clinical analysis by next-generation sequencing for NSCLC patients with MET amplification resistant to osimertinib. Lung Cancer. 2018;118:105–110. doi: 10.1016/j.lungcan.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 9.Yang JC, Ahn MJ, Kim DW, et al. Osimertinib in pretreated T790M-positive advanced non–small-cell lung cancer: AURA study phase II extension component. J Clin Oncol. 2017;35:1288–1296. doi: 10.1200/JCO.2016.70.3223. [DOI] [PubMed] [Google Scholar]
- 10.Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piotrowska Z, Thress KS, Mooradian M, et al. MET amplification (amp) as a resistance mechanism to osimertinib. J Clin Oncol. 2017;35(suppl) abstr 9020. [Google Scholar]
- 12.Iams W, Chae Y. Acquired resistance to osimertinib by CCDC6-RET fusion in a patient with EGFR T790M–mutant metastatic lung adenocarcinoma. J Thorac Oncol . 2017;12:S2249–S2250. [Google Scholar]
- 13.Yu HA, Suzawa K, Jordan E, et al. Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res. 2018;24:3108–3118. doi: 10.1158/1078-0432.CCR-17-2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 16.Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non–small-cell lung cancer. J Clin Oncol. 2010;28:357–360. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.National Cancer Institute https://clinicaltrials.gov/ct2/show/NCT02496663 Osimertinib and necitumumab in treating patients with EGFR-mutant stage IV or recurrent non–small-cell lung cancer who have progressed on a previous EGFR tyrosine kinase inhibitor ( NCT02496663)
- 18.Li GG, Somwar R, Joseph J, et al. Antitumor activity of RXDX-105 in multiple cancer types with RET rearrangements or mutations. Clin Cancer Res. 2017;23:2981–2990. doi: 10.1158/1078-0432.CCR-16-1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okamoto K, Kodama K, Takase K, et al. Antitumor activities of the targeted multi-tyrosine kinase inhibitor lenvatinib (E7080) against RET gene fusion–driven tumor models. Cancer Lett. 2013;340:97–103. doi: 10.1016/j.canlet.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Arai S, Kita K, Tanimoto A, et al. In vitro and in vivo anti-tumor activity of alectinib in tumor cells with NCOA4-RET. Oncotarget. 2017;8:73766–73773. doi: 10.18632/oncotarget.17900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klempner SJ, Mehta P, Schrock AB, et al. Cis-oriented solvent-front EGFR G796S mutation in tissue and ctDNA in a patient progressing on osimertinib: A case report and review of the literature. Lung Cancer (Auckl) 2017;8:241–247. doi: 10.2147/LCTT.S147129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small-cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ou SI, Horn L, Cruz M, et al. Emergence of FGFR3-TACC3 fusions as a potential bypass resistance mechanism to EGFR tyrosine kinase inhibitors in EGFR-mutated NSCLC patients. Lung Cancer. 2017;111:61–64. doi: 10.1016/j.lungcan.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iams W, Chae Y. Acquired resistance to osimertinib by CCDC6-RET fusion in a patient with EGFR T790M–mutant metastatic lung adenocarcinoma. J Thorac Oncol. 2017;12:S2249–S50. [Google Scholar]
- 25.Ho CC, Liao WY, Lin CA, et al. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. 2017;12:567–572. doi: 10.1016/j.jtho.2016.11.2231. [DOI] [PubMed] [Google Scholar]