

Abstract
Sepsis is an extreme host response to infection that leads to loss of organ function and cardiovascular integrity. Mortality from sepsis is on the rise. Despite more than three decades of research and clinical trials, specific diagnostic and therapeutic strategies for sepsis are still absent. We report here the use of LFQ- and TMT-based quantitative proteomics to study the plasma proteome in five mouse models of sepsis. A knowledge-based interpretation of the data revealed a protein network with extensive connectivity through documented functional or physical interactions. The individual proteins in the network all have a documented role in sepsis and are known to be extracellular. The changes in protein abundance observed in the mouse models of sepsis have for the most part the same directionality (increased or decreased abundance) as reported in the literature for human sepsis. We have named this network the Plasma Proteome Signature of Sepsis (PPSS). The PPSS is a quantifiable molecular readout that could supplant the current symptom-based approach used to diagnose sepsis. This type of molecular interpretation of sepsis, its progression, and its response to therapeutic intervention is an important step in advancing our understanding of sepsis, and for discovering and evaluating new therapeutic strategies.
Keywords: Sepsis, mouse model, plasma proteomics, protein network, label-free quantification
1. Introduction
Sepsis is a severe host response to infection that can progress to septic shock, in which a severe pro-immune/inflammatory reaction provokes lymphocyte apoptosis, coagulopathy, multiple organ failure, and frequently death [1–6]. A recent rise in the worldwide mortality from septic shock, even in countries with optimal medical intervention, has had a substantial social and financial toll [4]. In developed countries, death from sepsis disproportionally affects newborns and the elderly, who share susceptibility to complications from an exacerbated immune response. These complications are thought to arise from immune exhaustion, making individuals prone to secondary nosocomial infections by bacteria, fungi and viruses [3, 4]. A massive 30-year effort to develop diagnostics and effective therapeutics for sepsis has largely failed, mainly due to a disconnect in pre-clinical findings and their effective translation in the clinic [5, 7–10]. The degree to which animal models of sepsis can be used to study processes relevant to the human disease has been questioned for a number of reasons [10,11]. Some of the questions regarding mouse models, like the original article indicating substantial differences in endothelial gene expression in human vs. murine sepsis, have since been refuted [12, 13]. Yet, there are other even more practical issues that can confound the interpretation of results from murine models of sepsis. Just as with sepsis in humans (primarily symptom-based), there isn’t a standard way at the molecular level of knowing the stage of sepsis other than the general health and appearance of the animals [11, 14, 15]. Better, well-characterized mouse models are therefore needed. Here we tackle some of these challenges using MS-proteomics to interrogate protein abundances in blood plasma from mouse models of septicemia commonly used in our laboratory, each infected with one of five different bacterial pathogens – S. enterica Typhimurium (ST), Streptococcus pneumoniae (SPN), Escherichia coli (EC), Staphylococcus aureus antibiotic-resistant (MRSA), and -sensitive (SA). The objective was to take the first step toward a protein network pharmacology approach [16] that could be used in sepsis mouse models in mechanistic and pharmacological studies.
To achieve this, we first used a label-free quantification (LFQ) shotgun proteomics approach to determine protein abundances in plasma from the SPN and ST sepsis mouse models and extracted a highly connected 84-protein network with known functions in sepsis. Protein abundances in the network obtained by LFQ were confirmed by TMT quantitative MS-proteomics in all five sepsis models (EC, MRSA, SA, SPN and ST). We call this network the Plasma Proteome Signature of Sepsis (PPSS). The PPSS is comprised of seven different functional sub-networks. This network-centric information can now be used as a foundational assay to study sepsis in pre-clinical mouse models at the molecular level.
2. Materials and Methods
The experimental strategy is summarized in Figure 1 and a detailed description of experimental protocols is provided in File S2.
Figure 1. Experimental workflow.
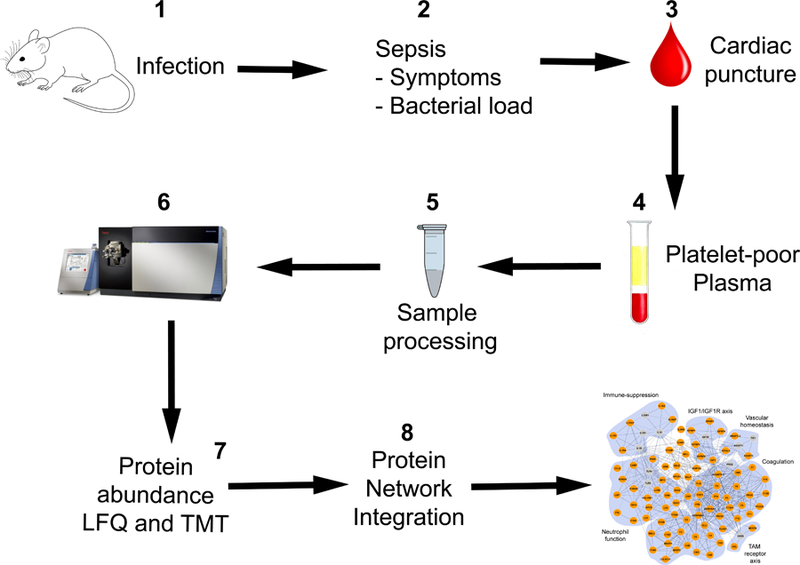
The experimental strategy entailed eight stages. 1. Mouse pathogen administration. 2. Sepsis monitoring and control of bacterial load in pre-septic and septic mice. 3. Blood collection by cardiac puncture for bacterial load determination and proteomics studies. 4. Platelet-poor plasma preparation from blood. 5. Sample processing for proteomics analysis. 6. Proteomics analysis by 2D-LC (high/low pH) and high-resolution MS/MS. 7. Protein abundance determination by LFQ in SPN and ST; and orthogonal validation of protein abundances by TMT in EC, MRSA, SA, SPN and ST. 8. Protein network integration from functional annotation databases and manual curation.
Mouse models of sepsis
Five pathogenic bacteria were used to infect mice and induce sepsis well characterized by our group [17]. Salmonella enterica Typhimurium strain 14028 (referred to as ST in the text) is Gram-negative and exhibits a repeating cycle of cellular internalization and shedding [18, 19]. Streptococcus pneumoniae D39 (SPN) is Gram-positive, lives extracellularly, typically colonizes the lung mucosa, and can become invasive [20, 21]. Escherichia coli strain ATCC 25922 (EC) colonizes the mucosal layer of the gastrointestinal tract and turns invasive during pathogenesis [22]. Staphylococcus aureus strain USA300 MRSA (MR) and Staphylococcus aureus strain Newman MSSA (SA) are both invasive bacteria that colonize nasopharyngeal epithelium [23, 24]. A detailed description of how C57BL/6J WT mice were infected, and how the pre-sepsis and sepsis phenotype was determined is based on prior work in our lab [25, 26] and found in File S2. Two biological replicates were used to collect MS-proteomics – three data collection technical replicates per biological sample. Control, pre-sepsis and sepsis samples from ST and SPN were analyzed by LFQ. Control and sepsis samples from EC, MR, SA, SPN and ST were analyzed by TMT.
Sample preparation for MS-proteomics
Platelet-poor plasma samples were prepared by blood centrifugation at low speed (4 °C); and handled for proteomics preparation according to recommended protocols stipulated by the Human Proteome Organization (HUPO) Plasma Proteome Project [27]. To minimize biological variability, samples analyzed in this study were a mixture of plasma from four mice, two from each gender: female and male. For each cohort (uninfected, pre-sepsis and sepsis), 30 μL of pooled plasma underwent four sample processing steps (File S2). 1) Depletion, 2) acetone precipitation (−20 °C), 3) cysteine reduction and alkylation and 4) digestion with a mixture of Trypsin/Lysine-C. After acetone precipitation, RapiGest/50 mM ammonium bicarbonate (pH 8.0A) was used in all steps. Peptide samples were stored at −80 °C until use for direct LFQ or TMT-labeling prior to MS/MS experiments. For TMT peptide labeling, we followed the recommendations given by the TMT manufacturer – https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0011639_TMT_Mass_Tagging_Reag_UG.pdf.
Shotgun MS-proteomics
We coupled a two-dimensional (2D)-liquid chromatography (LC)-tandem mass spectrometry (MS/MS) fractionation protocol to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). The first LC dimension was performed at pH 10 and the second one at pH 3, both in C18 reverse phase. Experimental details are found in File S2.
Protein identification and quantification
Protein identification and relative abundances were computed with MaxQuant 1.6.1.0 [28]. Relative protein abundances were calculated by the two normalization strategies available in MaxQuant: MaxLFQ [29] and intensity based absolute quantification (iBAQ) [30]. The values obtained by LFQ and iBAQ were highly consistent (Figure S1). We chose to use the LFQ values to discuss our results below. TMT data was analyzed in Maxquant with default parameters.
Computational reconstruction of the Plasma Proteome Signature of Sepsis (PPSS)
The protein network integration entailed an iterative use functional annotation repositories: Ingenuity Pathway Analysis (IPA)®, Metascape [31] and the REACTOME Pathway Knowledgebase [32]. To increase the quality of the data extracted, we refined the list of proteins by manual inspection of the literature. The quality of the list of proteins extracted was evaluated quantitatively using the “Search Tool for the Retrieval of Interacting Genes/Proteins” (STRING) [33].
3. Results and Discussion
Analysis of the Plasma Proteome in Sepsis
LFQ shotgun proteomics of plasma from control, pre-sepsis and sepsis samples (SPN and ST) produced 180 raw data files (30 each – 10 per technical replicate). Comparative violin plots showed that the overall distribution of normalized total ion intensities per protein in control and pre-sepsis samples was nearly indistinguishable, but different in the control vs. sepsis comparison (Figure S2). Based on these results, we focused on the sepsis vs. control experiments. After parsing, the protein search output contained 822 proteins with measurable total ion intensities in one or both sepsis vs. control comparisons (SPN and ST). These proteins had a 26% average sequence coverage and an average identification score of 98. Our dataset was composed of about 14,000 unique peptides (File S3). Individual total ion intensities per protein among the two control biological replicates were highly reproducible (Figure S3). The concordance among technical replicates was also very high (Figures S4 and S5). Two-sample T-test p-values were calculated, based on three technical replicates (injections) per sample in control and sepsis samples (Figure 2A-B). Proteins with statistically significant differences in abundance exhibited changes in the ratio of sepsis/control (SE/CTL) of > 0.6 (Log2)/1.5-fold, (natural number), with p-values (-Log10) ≥ 1.0.
Figure 2. Significant SE/CTL ratios in SPN and ST in LFQ data.
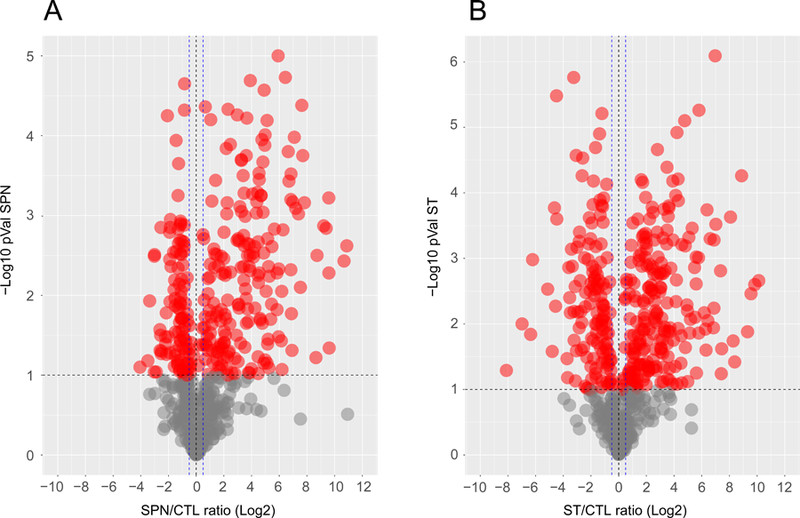
Volcano plots from (A) SPN and (B) ST data. Each dot represents a protein as the cross-correlation between the p-Value (-Log10) and the corresponding SE/CTL ratio Log2. Proteins with a p-Value ≥ 1.0 -Log10 are colored red. The black horizontal line indicates a cut-off of 1.0 for the p-Value. Statistically significant SE/CTL ratios are indicated by blue vertical dashed lines (+/− 0.5 SE/CTL (Log2) ratio).
Identification of a Plasma Proteome Signature of Sepsis (PPSS).
Proteins identified with significant SE/CTL ratios were used in the input for protein network integration as described above. We identified 84 plasma-secreted proteins known to be associated with human sepsis in the literature based on 159 citations (File S1). The sequence coverage per protein in the PPSS network (84 proteins) had an average coverage of 20% by LFQ. In the TMT experiments 66 of these proteins were identified and had an average coverage of ≥ 20%. Each protein was represented by at least two peptides (Table 1). These 84 proteins were highly connected in a STRING network (Figure 3A). We identified seven highly interconnected sub-networks, including neutrophil function, immune suppression, the Insulin Growth Factor Like/Insulin Grow Factor Receptor (IGF1-IGF1R) axis, vascular homeostasis, coagulation, the Tyro3, AXL and MER (TAM) receptor tyrosine kinase axis and complement. Changes in abundance determined by LFQ in SPN and ST were for the most part in accordance with those documented in the literature (Figure 3B, Table 1 and File S1). We suggest that this functionally interconnected network represents the Plasma Proteome Signature of Sepsis (PPSS). Two reviews on sepsis have suggested that there must be molecular hallmarks of this syndrome [1, 5]. Our findings are consistent with the idea that the PPSS is comprised of many such hallmarks, especially since the network encompasses key biological processes previously associated with human sepsis (Figure 3A).
Table 1.
PPSS protein ratios and sequence coverage
| PROTEIN | EC | MR | SA | SPN | ST | LFQ-SPN | LFQ-ST | LFQ coverarge | TMT coverage |
|---|---|---|---|---|---|---|---|---|---|
| Adamts13 | 0 | 0 | 0 | 0 | 0 | −1.27 | −0.47 | 12.6 | 1.6 |
| Adipoq | −0.82 | −1.01 | −0.72 | −0.37 | −0.69 | −0.16 | −1.56 | 26.7 | 24.7 |
| Angpt2 | 0.09 | 0.07 | 0.14 | −0.67 | 0.63 | 0 | 2.79 | 11.9 | 7.5 |
| Angptl3 | −1.5 | −0.2 | 0.36 | −0.1 | −0.03 | −3.01 | −0.49 | 35.8 | 9.9 |
| Apcs | 1.56 | 3.57 | 3.88 | 2.24 | 3.13 | 2.49 | 2.77 | 80.4 | 75.4 |
| Apoa1 | −0.63 | −0.92 | −1.01 | −0.27 | −0.89 | −0.95 | −1.31 | 87.5 | 82.6 |
| Axl | 0 | 0 | 0 | 0 | 0 | 1.65 | 2.19 | 16 | 0 |
| C1qa | −1.11 | 0.34 | 0.23 | −0.5 | 0.54 | −0.29 | 0.94 | 34.3 | 44.9 |
| C1qb | −1.17 | 0.29 | 0.21 | −0.45 | 0.68 | −0.43 | 1.07 | 36.8 | 43.1 |
| C1qc | −0.9 | 0.44 | 0.36 | −0.45 | 0.76 | −0.51 | 0.37 | 30.9 | 25.2 |
| C2 | 0.01 | 0.35 | 0.36 | 0.23 | 1.41 | 1.28 | 1.02 | 60 | 41.7 |
| C3 | 0.06 | 0.83 | 0.65 | 0.32 | 0.46 | 0 | 0.18 | 89.4 | 79.9 |
| C4b | −0.29 | 0.56 | 0.99 | 0.19 | 0.85 | 0.15 | 0.46 | 69.3 | 66.6 |
| C5 | −0.65 | −0.35 | −0.12 | −0.45 | −0.46 | −0.85 | −0.94 | 69.1 | 63.4 |
| C8a | −0.59 | −0.15 | −0.27 | −0.34 | −1.02 | −0.65 | −1.77 | 72.4 | 68.1 |
| C8b | −0.55 | −0.09 | −0.25 | −0.31 | −1.04 | −0.39 | −1.53 | 64.5 | 61.3 |
| C8g | −0.46 | 0.06 | −0.07 | −0.25 | −0.52 | −0.67 | −1.66 | 76.2 | 81.2 |
| C9 | −0.55 | −0.42 | −0.46 | −0.65 | −0.7 | −0.95 | −1.23 | 64.8 | 58.8 |
| Camp | 1.77 | 0.33 | 1.77 | 1.15 | 2.26 | 1.99 | 1.48 | 36 | 29.7 |
| Ccl8 | 0.15 | 1.19 | 1.22 | −0.4 | 1.96 | 0 | −2 | 58.8 | 58.8 |
| Cd14 | 2.79 | 1.89 | 1.26 | 0.47 | 3.62 | 6.27 | 7.36 | 43.4 | 53.8 |
| Clu | −0.25 | −0.27 | 0.17 | −0.28 | 0.63 | −0.22 | 0.25 | 50.9 | 47.1 |
| Colec11 | −0.57 | −0.59 | 0.06 | −0.67 | −1.58 | −2.25 | −3.61 | 27.6 | 13.6 |
| Crp | 1.31 | 0.92 | 0.51 | 0.8 | 0.87 | −0.01 | 0.39 | 54.2 | 74.7 |
| F10 | −0.5 | 0.1 | 0.09 | −0.23 | −0.65 | −0.56 | −1.14 | 36.4 | 43 |
| F11 | −0.3 | 1.16 | 1.3 | −0.22 | 1.83 | 1.46 | 1.59 | 34.6 | 33.2 |
| F12 | −0.28 | −0.48 | −0.36 | −0.37 | −0.19 | −0.54 | −0.23 | 44.2 | 33.5 |
| F13a1 | −0.74 | −1.04 | −1.03 | −0.63 | −1.79 | −1.6 | −3.01 | 60.4 | 56.7 |
| F13b | −0.78 | −0.89 | −0.83 | −0.69 | −2.05 | −1.92 | −3.41 | 53.1 | 47.8 |
| F2 | −0.45 | −0.06 | 0.1 | 0.07 | −0.43 | −0.79 | −1.27 | 56 | 51 |
| F5 | −0.14 | 0.43 | 0.7 | −0.05 | 0.29 | −0.26 | 0.35 | 40.3 | 38.6 |
| F7 | −0.52 | 0.91 | 1.03 | 0.4 | 0.09 | −0.82 | −0.37 | 35.9 | 37.9 |
| F8 | −0.2 | 1.13 | 1.33 | 0.36 | 0.62 | −0.78 | 0.74 | 8.1 | 4.4 |
| F9 | −0.38 | 0.64 | 0.79 | −0.12 | 0.47 | −0.47 | 0.35 | 41.8 | 28 |
| Fcn1 | −0.61 | −0.95 | −0.49 | −0.57 | −1.24 | −1.18 | −2.42 | 40.4 | 41.6 |
| Fth1 | 0.28 | 0.55 | 0.32 | −0.96 | 4.14 | 9.39 | 8.88 | 71.4 | 53.8 |
| Ftl1 | 0.29 | 0.09 | 0.34 | −0.88 | 3.89 | 4.82 | 4.29 | 58.5 | 65 |
| Grn | 1.72 | 2.26 | 2.23 | 1.31 | 2.79 | 5.12 | 5.79 | 59.1 | 47.2 |
| Gsn | −0.76 | −1.39 | −1.3 | −0.93 | −1.19 | −1.28 | −1.67 | 45.1 | 34.9 |
| Icam1 | 1.62 | 0.48 | 0.55 | 0.16 | 2.11 | 1.13 | 1.85 | 28.3 | 30.2 |
| Igf1 | −1.27 | −0.6 | −0.78 | −1.04 | −1.56 | −1.67 | −8.11 | 27.5 | 18.3 |
| Igfals | −1.39 | −1.71 | −1.09 | −0.82 | −2.1 | −1.87 | −3.35 | 55.9 | 44.3 |
| Igfbp1 | 1.73 | 2.03 | 1.47 | 0.81 | 4.31 | 0 | 0 | 54.8 | 39.7 |
| Igfbp2 | −0.44 | −0.52 | −1.47 | −0.98 | 0.6 | 0 | −3.65 | 59.3 | 36.4 |
| Igfbp3 | −0.93 | −0.6 | −0.62 | −0.62 | −1.69 | −3.02 | −4.47 | 39.7 | 39.7 |
| Igfbp4 | −0.42 | 0.7 | 0.58 | −0.68 | 0.09 | 0 | 0 | 33.1 | 25.2 |
| Igfbp5 | −1.07 | −1.22 | −0.64 | −0.83 | −0.25 | −2.42 | −6.22 | 45.4 | 19.2 |
| Il18bp | −0.4 | 0.08 | 0.03 | 0.32 | 1.75 | 2.58 | 3.57 | 33.2 | 19.2 |
| Il1r2 | 0.12 | 0.39 | 0.5 | −0.5 | 2.1 | 0.31 | 1.1 | 28 | 24.1 |
| Il1rap | −0.66 | −1.11 | −1.1 | −0.84 | −1.32 | −0.65 | −1.2 | 29.6 | 26.5 |
| Il1rn | 0.48 | 0.95 | 0.47 | −0.13 | 2.61 | 0 | 0 | 60.7 | 19.7 |
| Il2rg | 0.48 | −0.02 | −0.23 | −0.2 | 1.63 | 0.87 | 2.01 | 13 | 15.2 |
| Klkb1 | −0.83 | −1.56 | −1.61 | −0.83 | −1.82 | −1.1 | −2.21 | 63.5 | 63.3 |
| Kng1 | −0.19 | −0.08 | 0.44 | −0.07 | −0.07 | −0.73 | −0.35 | 59.5 | 52.8 |
| Lbp | 1.77 | 2.01 | 1.81 | 1.63 | 1.86 | 1.79 | 1.09 | 45.5 | 31.8 |
| Lcn2 | 3.36 | 4.02 | 3.55 | 2.05 | 4.29 | 6.7 | 6.88 | 46 | 43 |
| Ltf | 1.85 | 1.35 | 1.98 | 0.9 | 2.13 | 1.4 | 2.22 | 60 | 59.3 |
| Lyz2 | −0.12 | 0.83 | 1.5 | −0.46 | 1.93 | −0.81 | 1.01 | 45.3 | 27 |
| Masp1 | −0.68 | −0.26 | −0.13 | −0.18 | −0.39 | −1.22 | −0.6 | 43.8 | 41.9 |
| Masp2 | −0.59 | −0.17 | −0.07 | −0.45 | −0.85 | −1.12 | −0.61 | 62.3 | 51.5 |
| Mbl2 | −0.89 | −2.36 | −2 | −0.65 | −2.19 | −0.4 | −1.99 | 51.2 | 51.2 |
| Mertk | −0.41 | 0.17 | −0.54 | −0.32 | 0.65 | −0.94 | 0.87 | 11.7 | 7.2 |
| Mpo | 0.86 | 1.29 | 1.15 | 0.6 | 3.17 | 0 | 9.1 | 39 | 28.1 |
| Pf4 | −2.14 | −1.57 | −1.48 | −1.09 | −1.96 | 0.02 | −3.49 | 20 | 28.6 |
| Pglyrp1 | 1.18 | 2.15 | 1.28 | 1.57 | 2.47 | 0 | 1.48 | 45.6 | 34.6 |
| Pigr | 0.16 | −0.42 | 0.54 | −0.3 | 2.29 | 1.16 | 2.75 | 34.2 | 14 |
| Plg | −0.49 | −0.06 | −0.08 | −0.19 | −0.86 | −1.27 | −1.31 | 76.2 | 77.1 |
| Proc | −0.47 | 0.12 | 0.6 | 0.11 | −0.07 | −1.01 | −0.56 | 36.7 | 20.7 |
| Procr | 1.21 | 0.35 | 0.42 | −0.47 | 2.85 | 0.77 | 1.55 | 23.6 | 15.3 |
| Pros1 | −0.11 | 0.62 | 0.21 | −0.05 | 0.3 | 0.02 | −0.08 | 27.9 | 27.1 |
| Proz | −0.9 | 0.19 | 0.77 | 0 | 0.03 | −0.84 | −1.76 | 41.1 | 45.6 |
| S100a8 | 2.46 | 2.72 | 2.38 | 2.67 | 3.06 | 4.47 | 3.22 | 49.4 | 80.9 |
| S100a9 | 1.91 | 2.24 | 1.66 | 1.91 | 2.73 | 5.26 | 3.22 | 38.9 | 38.9 |
| Saa1 | 3.73 | 4.54 | 3.58 | 3.65 | 3.87 | 3.71 | 2.37 | 53.3 | 43.4 |
| Saa2 | 3.94 | 4.91 | 3.57 | 4.01 | 3.9 | 3.77 | 3.05 | 41 | 41.8 |
| Sele | 1.04 | 0.73 | 0.59 | 0.07 | 0.24 | 0 | 0 | 9.8 | 6 |
| Sell | −0.15 | −0.21 | −0.2 | −0.24 | 1 | −0.03 | 1.09 | 16.7 | 18.5 |
| Selp | 1.04 | 1.19 | 1.37 | 0.16 | 0.97 | −0.06 | −0.05 | 11.3 | 4 |
| Serpina1a | −0.53 | −0.52 | −0.83 | −0.51 | −0.97 | −1.1 | −1.21 | 18.2 | 14.3 |
| Serping1 | 0.32 | 0.55 | 0.58 | 0.06 | 0.83 | 0.58 | 0.36 | 58.9 | 48.4 |
| Tie1 | 0.16 | 0.66 | 0.63 | 0.26 | 1.11 | 0.96 | 2.19 | 13.6 | 1.6 |
| Vcam1 | 0.38 | −0.15 | −0.08 | −0.35 | 1.54 | 0.99 | 1.73 | 50.9 | 46.8 |
| Vtn | 0.11 | 0.08 | 0.24 | 0.14 | −0.6 | −0.39 | −1.49 | 56.5 | 51.9 |
| Vwf | 1.13 | 1.38 | 1.66 | 0.09 | 0.84 | 0.44 | 0.56 | 32.5 | 21.8 |
Figure 3. Evaluation of PPSS in STRING.

A) PPSS network in STRING showing only functional and physical interactions with high confidence cut-off values. Lines connecting proteins are functional or direct physical associations documented in the literature. The thickest lines are “very high confidence” (cutoff of 0.9–1.0). Thinner lines are “high confidence” (cutoff 0.7–1.0) and medium confidence (cutoff 0.4–1.0), respectively. The 84 proteins identified by MS-proteomics (colored orange). Proteins colored grey are important network hubs not identified in our data. B) PPSS Heatmap from LFQ data SE/CTL ratios (Log2) from SPN and ST samples.
The PPSS is Evident in Sepsis arising from Five Bacterial Pathogens
A 6-plex TMT strategy was used for two purposes. 1) As an orthogonal validation of LFQ-based protein abundance values determined in plasma from SPN and ST sepsis models. 2) To evaluate the SE/CTL ratio significance of the PPSS network in the plasma of sepsis arising from five different bacterial infections: two Gram-negative bacteria (EC and ST), and three Gram-positive bacteria (MR, SA and SPN). TMT experiments from two biological replicates per infection model showed for the most part, a reasonable correlation in SE/CTL ratios when compared to the LFQ values (Table 1, Figure S6 and File S4). When compared across sepsis models, a heatmap and correlation plots (Figures S6, S7 and File S4); and a principal components analysis (data not shown) did not show an evident pathogen-specific pattern.
Compared to the other sepsis models, the ST model had for the most part, concordant SE/CTL ratios but higher in magnitude. It should be mentioned that one potential limitation of this study is that pathogens were administered to mice by different routes of infection, and the course of sepsis proceeded at different rates (File S2). These experimental differences may account for the fact that the magnitude of change in abundance of a protein was often the largest in the ST model. This model is distinct from the other types of infection because progression to severe sepsis requires 8 days, as opposed to 48–96 hr. in the other models. We suspect that larger changes in abundance observed in the ST model are the result of a longer time period for the onset of severe sepsis, allowing for accumulation or clearance of factors from the blood.
PPSS subnetworks represent molecular readouts of well-defined mechanisms in sepsis
Despite this limitation, two subnetworks stand out in the LFQ and TMT data with highly concordant protein abundance changes in sepsis (Figures 4A): 1) an upregulation of the TLR4 response (CAMP [34], CD14 [35] and LBP [36]) (Figure 4B); 2) a loss of pro-survival signaling by downregulation of IGF1/IGF1R signaling axis [37, 38] (Figure 4C). TLR4-specific immune activation is an indicator of microbial infection [1–5]; and these protein abundance changes are well documented in the literature as indicators of sepsis initiation and severity. Likewise, the IGF1/IGFALS/IGF3/5 quaternary complex is a key functional component of the pro-survival IGF1-IGF1R signaling axis, and IGF1 is an indicator of sepsis severity in the clinic [39–42].
Figure 4. Immune-activation and immune-suppression sub-networks.
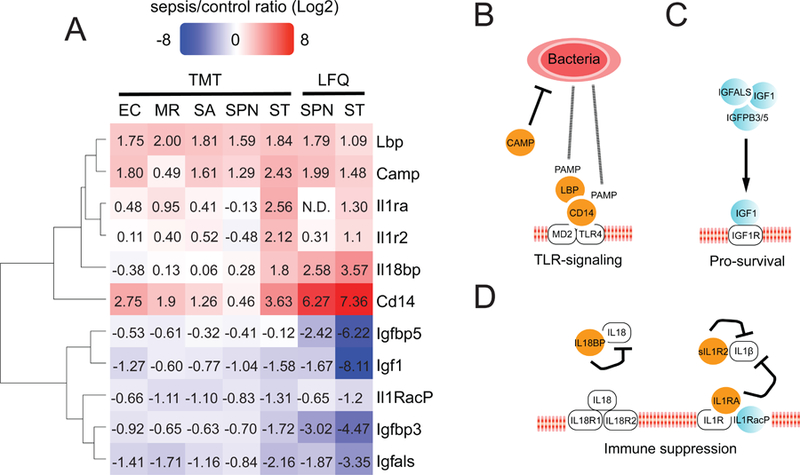
A) Heatmap showing LFQ and TMT SE/CTL (Log2) ratios for these subnetworks. B) Immune activation. The proteins CAMP, CD14 and LBP had consistent protein up-regulation values by LFQ and TMT across different infections. This is expected given that the TLR4-signaling axis is activated upon bacterial infection regardless of bacterial type. C) IGF1-IGF1R pro-survival signaling. A decrease in blood levels of IGF1 in clinical sepsis is an indicator of sepsis severity. D) Immune inhibition by soluble decoys. The up-regulation of IL-1Ra, IL-1R2 and IL-18BP in ST infection, in which immune activation was the highest.
We also found pathogen-specific differences at the sub-network level. The interleukin soluble decoys IL-1Ra, sIL1R2 and IL-18BP were found only upregulated in ST model (Figure 4A). A well-documented aspect of severe sepsis is that uncontrolled TLR4 immune activation correlates to a counterbalancing immune-suppression mediated by soluble interleukin decoys such as these (Figure 4D) [43]. Based on these results, the ST sepsis model may represent a valuable sepsis mouse model to study immune-suppression. Future targeted proteomics and biochemical studies will help corroborate these pathogen-specific results in connection with: 1) the well-documented TLR4-driven immune activation and interleukin-mediated immune-suppression [43]; 2) to recent reports in our group showing that the alkaline phosphatases IAP and TNAP decrease during sepsis in the ST and EC models (Gram-negative), but not in the MRSA and SPN (Gramm-negative) [17]. Due to their low abundance in plasma these two proteins were not identified in our datasets.
4. Concluding Remarks
Our results support five individual conclusions: i) the plasma proteome contains an 84-protein functionally connected network (the PPSS), all members of which have been implicated in human sepsis (File S1), ii) the PPSS has two-dimensions: protein identity of the proteins and protein abundance (Table 1), iii) the PPSS is evident in sepsis brought on by five separate pathogens, including two Gram-negative and three Gram-positive bacteria, iv) the PPSS can be further segregated into functional sub-networks including those involved in immune suppression, vascular homeostasis, coagulation, complement, neutrophil function, the IGF/IGF1R axis and the TAM receptor axis (Figure 3A), v) the MS-proteomics spectral evidence that led to the identification of the 84 proteins in the PPSS provides the “coordinates” (a spectral library), and will catalyze future hypothesis-driven targeted MS-proteomics.
These findings have important implications for understanding the molecular connections that drive the progression of sepsis and suggest a path toward assigning molecular criteria for assessing sepsis in pre-clinical animal models. To our knowledge, this is the first quantitative MS-proteomics study to compare the plasma proteome in five different mouse models of sepsis, each based on infection with different bacteria. An important aspect of the proteomic strategy was the use of both LFQ [29] and the orthogonal validation of protein abundance changes by 6-plex TMT [44]. Together the two approaches helped overcome their respective inherent limitations: gaps in identification because of the stochastic nature of LFQ, and compression of the magnitude change in TMT due to overlapping contaminants and MS/MS reporter signals [44]. Interpretation of the changes to the proteome were greatly assisted with pathway analysis tools like IPA, REACTOME and STRING, but even after discovering a functionally connected network with these tools, its size and statistical significance were enhanced with a substantial effort in manual curation, a strategy that has been emphasized by others [45–47].
Supplementary Material
A) SPN and B) ST. The scatter plots in the two panels show the cross-correlation of the SE/CTLT (Log2) ratios per protein (dot) – LFQ (y-axis) vs. iBAQ (x-axis). Red diagonal dashed line: linear regression trace. The cross-correlation dots across the y-axis are proteins with no value in LFQ but present in iBAQ.
LFQ data. This file contains three sheets named as follows. “Protein_Groups-Raw_values”: Protein Group parameters from the MaxQuant output. “Condensed_Protein_Groups_values”: condensed form of “Protein_Groups-Raw_values”. “iBAQ_LFQ-Ttest-values” SE/CTL (Log2) ratios and p-Values from the t-test calculations.
Same as in File S3, but for TMT data.
Violin plots comparing (Log10) LFQ total ion intensity per protein in SPN and ST tested by LFQ MS-proteomics – control, pre-sepsis and sepsis.
Protein total ion intensity (Log10) concordance to assess reproducibility among biological replicates. Red diagonal dashed line: linear regression trace.
LFQ data from SPN. Each dot correlates the normalized total ion intensity (Log10) per protein in-between technical replicates, in CTL and SE samples. Red diagonal dashed line: linear regression trace.
LFQ data from ST. Each dot correlates the normalized total ion intensity (Log10) per protein in-between technical replicates in CTL and SE samples. Red diagonal dashed line: linear regression trace.
Average from biological replicates. Red up-regulation. Blue down-regulation.
SE/CTL ratio cross-correlation across all infections.
References are in alphabetical order.
This file contains a detailed description of the protocol used to determine the pathogen doses and determination of pre-sepsis and sepsis symptoms.
Statement of significance.
The foremost challenge in the study of sepsis is the complexity of the syndrome, which involves numerous biological systems, cell types and several different organs. Because of this complexity, clinical definitions of sepsis are not molecular in nature [4, 8], rather they are based on patient symptoms. This confounds the rational development of new therapeutics and prognostic markers. Here MS-proteomics was used to characterize changes in the plasma proteome in five different models of murine sepsis. Our results are significant for four reasons: i) a comparison of changes to the plasma proteome from different murine models of sepsis has to our knowledge not yet been published, ii) the work identifies a functionally connected network of 84 proteins, the PPSS, that is common to all five murine models; each of these proteins has also been individually reported to be associated with human sepsis, iii) the high-quality proteomics spectra associated with the PSS will facilitate future hypothesis-driven targeted proteomics studies in mouse models of sepsis, iv) this new information can now be used as a foundational assay to assess the temporal progression of sepsis, and potentially determine how genetic or pharmacologic perturbations impact sepsis at the molecular level.
Acknowledgements
This research was funded by NIH grants HL131474 (J.W.S., J.D.M., J.D.E., and M.J.M.) and HL125352 (J.D.M.).
Abbreviations:
- 2D
Two-dimensional
- EC
Escherichia coli
- FDA
food and drug administration
- HUPO
human proteome organization
- iBAQ
intensity based absolute quantification
- IPA
ingenuity pathway analysis
- LC
liquid chromatography
- LFQ
label-free quantification
- MRSA
Staphylococcus aureus antibiotic-resistant
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- PPSS
plasma proteome signature of sepsis
- SA
Staphylococcus aureus antibiotic-sensitive
- SPN
Streptococcus pneumoniae
- ST
S. enterica Typhimurium
- STRING
Search Tool for the Retrieval of Interacting Genes/Proteins
- TMT
tandem mass tag
Footnotes
Conflict of interest disclosure
The authors declare no competing financial interest.
5. References
- [1].Rittirsch D, Flierl MA, Ward PA, Nat Rev Immunol 2008, 8, 776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Angus DC, van der Poll T, N Engl J Med 2013, 369, 840. [DOI] [PubMed] [Google Scholar]
- [3].Hotchkiss RS, Monneret G, Payen D, Nat Rev Immunol 2013, 13, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hotchkiss RS, Monneret G, Payen D, Lancet Infect Dis 2013, 13, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC, JAMA 2016, 315, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG, Nat Rev Immunol 2017, 17, 407. [DOI] [PubMed] [Google Scholar]
- [7].Reinhart K, Bauer M, Riedemann NC, Hartog CS, Clin Microbiol Rev 2012, 25, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, Sevransky JE, Sprung CL, Douglas IS, Jaeschke R, Osborn TM, Nunnally ME, Townsend SR, Reinhart K, Kleinpell RM, Angus DC, Deutschman CS, Machado FR, Rubenfeld GD, Webb SA, Beale RJ, Vincent JL, Moreno R, S. Surviving Sepsis Campaign Guidelines Committee including the Pediatric, Crit Care Med 2013, 41, 580. [DOI] [PubMed] [Google Scholar]
- [9].Marshall JC, Nat Rev Drug Discov 2003, 2, 391. [DOI] [PubMed] [Google Scholar]
- [10].Marshall JC, Trends Mol Med 2014, 20, 195. [DOI] [PubMed] [Google Scholar]
- [11].Dyson A, Singer M, Crit Care Med 2009, 37, S30. [DOI] [PubMed] [Google Scholar]
- [12].Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation, L. S. C. R. P. Host Response to Injury, Proc Natl Acad Sci U S A 2013, 110, 3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Takao K, Miyakawa T, Proc Natl Acad Sci U S A 2015, 112, 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Buras JA, Holzmann B, Sitkovsky M, Nat Rev Drug Discov 2005, 4, 854. [DOI] [PubMed] [Google Scholar]
- [15].Dejager L, Pinheiro I, Dejonckheere E, Libert C, Trends Microbiol 2011, 19, 198. [DOI] [PubMed] [Google Scholar]
- [16].Hopkins AL, Nat Chem Biol 2008, 4, 682. [DOI] [PubMed] [Google Scholar]
- [17].Yang WH, Heithoff DM, Aziz PV, Haslund-Gourley B, Westman JS, Narisawa S, Pinkerton AB, Millan JL, Nizet V, Mahan MJ, Marth JD, Cell Host Microbe 2018, 24, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Keestra-Gounder AM, Tsolis RM, Baumler AJ, Nat Rev Microbiol 2015, 13, 206. [DOI] [PubMed] [Google Scholar]
- [19].LaRock DL, Chaudhary A, Miller SI, Nat Rev Microbiol 2015, 13, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bogaert D, De Groot R, Hermans PW, Lancet Infect Dis 2004, 4, 144. [DOI] [PubMed] [Google Scholar]
- [21].Henriques-Normark B, Tuomanen EI, Cold Spring Harb Perspect Med 2013, 3. [DOI] [PMC free article] [PubMed]
- [22].Kaper JB, Nataro JP, Mobley HL, Nat Rev Microbiol 2004, 2, 123. [DOI] [PubMed] [Google Scholar]
- [23].Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr., Clin Microbiol Rev 2015, 28, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Krismer B, Weidenmaier C, Zipperer A, Peschel A, Nat Rev Microbiol 2017, 15, 675. [DOI] [PubMed] [Google Scholar]
- [25].Grewal PK, Aziz PV, Uchiyama S, Rubio GR, Lardone RD, Le D, Varki NM, Nizet V, Marth JD, Proc Natl Acad Sci U S A 2013, 110, 20218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yang WH, Heithoff DM, Aziz PV, Sperandio M, Nizet V, Mahan MJ, Marth JD, Science 2017, 358. [DOI] [PMC free article] [PubMed]
- [27].Rai AJ, Gelfand CA, Haywood BC, Warunek DJ, Yi J, Schuchard MD, Mehigh RJ, Cockrill SL, Scott GB, Tammen H, Schulz-Knappe P, Speicher DW, Vitzthum F, Haab BB, Siest G, Chan DW, Proteomics 2005, 5, 3262. [DOI] [PubMed] [Google Scholar]
- [28].Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M, J Proteome Res 2011, 10, 1794. [DOI] [PubMed] [Google Scholar]
- [29].Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M, Mol Cell Proteomics 2014, 13, 2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M, Nature 2011, 473, 337. [DOI] [PubMed] [Google Scholar]
- [31].Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, Yanguez E, Andenmatten D, Pache L, Manicassamy B, Albrecht RA, Gonzalez MG, Nguyen Q, Brass A, Elledge S, White M, Shapira S, Hacohen N, Karlas A, Meyer TF, Shales M, Gatorano A, Johnson JR, Jang G, Johnson T, Verschueren E, Sanders D, Krogan N, Shaw M, Konig R, Stertz S, Garcia-Sastre A, Chanda SK, Cell Host Microbe 2015, 18, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, Jupe S, Kalatskaya I, Mahajan S, May B, Ndegwa N, Schmidt E, Shamovsky V, Yung C, Birney E, Hermjakob H, D’Eustachio P, Stein L, Nucleic Acids Res 2011, 39, D691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C, Nucleic Acids Res 2017, 45, D362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Martin L, van Meegern A, Doemming S, Schuerholz T, Front Immunol 2015, 6, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Le Roy D, Di Padova F, Adachi Y, Glauser MP, Calandra T, Heumann D, J Immunol 2001, 167, 2759. [DOI] [PubMed] [Google Scholar]
- [36].Zweigner J, Gramm HJ, Singer OC, Wegscheider K, Schumann RR, Blood 2001, 98, 3800. [DOI] [PubMed] [Google Scholar]
- [37].Garlanda C, Riva F, Bonavita E, Gentile S, Mantovani A, Front Immunol 2013, 4, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rosen CJ, Pollak M, Trends Endocrinol Metab 1999, 10, 136. [DOI] [PubMed] [Google Scholar]
- [39].Bunn RC, Fowlkes JL, Trends Endocrinol Metab 2003, 14, 176. [DOI] [PubMed] [Google Scholar]
- [40].Lang CH, Krawiec BJ, Huber D, McCoy JM, Frost RA, Am J Physiol Regul Integr Comp Physiol 2006, 290, R963. [DOI] [PubMed] [Google Scholar]
- [41].Ashare A, Nymon AB, Doerschug KC, Morrison JM, Monick MM, Hunninghake GW, Am J Respir Crit Care Med 2008, 178, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hunninghake GW, Doerschug KC, Nymon AB, Schmidt GA, Meyerholz DK, Ashare A, Am J Respir Crit Care Med 2010, 182, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Papastathi C, Mavrommatis A, Mentzelopoulos S, Konstandelou E, Alevizaki M, Zakynthinos S, Growth Horm IGF Res 2013, 23, 98. [DOI] [PubMed] [Google Scholar]
- [44].Rauniyar N, Yates JR 3rd, J Proteome Res 2014, 13, 5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jensen LJ, Saric J, Bork P, Nat Rev Genet 2006, 7, 119. [DOI] [PubMed] [Google Scholar]
- [46].Winnenburg R, Wachter T, Plake C, Doms A, Schroeder M, Brief Bioinform 2008, 9, 466. [DOI] [PubMed] [Google Scholar]
- [47].Huang JK, Carlin DE, Yu MK, Zhang W, Kreisberg JF, Tamayo P, Ideker T, Cell Syst 2018, 6, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) SPN and B) ST. The scatter plots in the two panels show the cross-correlation of the SE/CTLT (Log2) ratios per protein (dot) – LFQ (y-axis) vs. iBAQ (x-axis). Red diagonal dashed line: linear regression trace. The cross-correlation dots across the y-axis are proteins with no value in LFQ but present in iBAQ.
LFQ data. This file contains three sheets named as follows. “Protein_Groups-Raw_values”: Protein Group parameters from the MaxQuant output. “Condensed_Protein_Groups_values”: condensed form of “Protein_Groups-Raw_values”. “iBAQ_LFQ-Ttest-values” SE/CTL (Log2) ratios and p-Values from the t-test calculations.
Same as in File S3, but for TMT data.
Violin plots comparing (Log10) LFQ total ion intensity per protein in SPN and ST tested by LFQ MS-proteomics – control, pre-sepsis and sepsis.
Protein total ion intensity (Log10) concordance to assess reproducibility among biological replicates. Red diagonal dashed line: linear regression trace.
LFQ data from SPN. Each dot correlates the normalized total ion intensity (Log10) per protein in-between technical replicates, in CTL and SE samples. Red diagonal dashed line: linear regression trace.
LFQ data from ST. Each dot correlates the normalized total ion intensity (Log10) per protein in-between technical replicates in CTL and SE samples. Red diagonal dashed line: linear regression trace.
Average from biological replicates. Red up-regulation. Blue down-regulation.
SE/CTL ratio cross-correlation across all infections.
References are in alphabetical order.
This file contains a detailed description of the protocol used to determine the pathogen doses and determination of pre-sepsis and sepsis symptoms.