

An ultra-multiplexed microfluidic system uncovers signaling logic rules in neural stem cell differentiation and self-renewal.
Abstract
Dynamical control of cellular microenvironments is highly desirable to study complex processes such as stem cell differentiation and immune signaling. We present an ultra-multiplexed microfluidic system for high-throughput single-cell analysis in precisely defined dynamic signaling environments. Our system delivers combinatorial and time-varying signals to 1500 independently programmable culture chambers in week-long live-cell experiments by performing nearly 106 pipetting steps, where single cells, two-dimensional (2D) populations, or 3D neurospheres are chemically stimulated and tracked. Using our system and statistical analysis, we investigated the signaling landscape of neural stem cell differentiation and discovered “cellular logic rules” that revealed the critical role of signal timing and sequence in cell fate decisions. We find synergistic and antagonistic signal interactions and show that differentiation pathways are highly redundant. Our system allows dissection of hidden aspects of cellular dynamics and enables accelerated biological discovery.
INTRODUCTION
Cells operate in dynamic microenvironments where the type and concentration of signaling molecules are ever changing. The stem cell niche presents a range of signaling molecules and growth factors to maintain the stem cell pool. During development or injury, the chemical composition of the niche changes to allow differentiation into defined cell lineages. Signals received at different cell fate decision points determine differentiation trajectories (1). It is highly desirable to recapitulate these dynamic signaling environments in experiments to study stem cell behavior quantitatively, as well as in tissue regeneration applications.
Current live-cell analysis techniques are severely limited in creating and controlling complex dynamical microenvironments. Microfluidic cell culture has been proposed to improve time-consuming and labor-intensive tasks by automating operations (2–8) and to realize previously intractable experiments in dynamic cell culture (6). Individual devices for sorting, culturing, dynamically stimulating, imaging, tracking, and retrieving cells have been demonstrated; however, none of the current systems combine these capabilities. Further, the number of individual dynamic culture conditions created in previous microfluidic devices has been limited to less than 100 (7), limiting their utility in screening a large number of conditions in exploratory signaling and drug studies. In addition, maintaining long-term viable cultures of sensitive primary mammalian cells in microfluidic devices was so far elusive (8–11).
To address all these limitations and to build a universal system for dynamical cell control and analysis, we developed an ultra-multiplexed microfluidic system that combines multimode cell culture [single cell, two-dimensional (2D) monolayer, and 3D neurosphere], generation of dynamic chemical inputs, and 1500 individually addressable cell culture units on a single device (Fig. 1). Each of the 1500 culture chambers can be programmed to receive a different set of signaling molecules, growth factors, or drugs, whose composition and concentration can be automatically changed on-demand. Culture conditions including cell type, cell density, and support matrices can be predetermined for each independent chamber. Coupled with custom software for chip control and computational data processing, the system can perform programmed delivery of thousands of formulated fluidic inputs to designate on-chip culture units while monitoring and analyzing cellular responses via live-cell microscopy and end-point biochemical analysis methods (Fig. 1D). In a typical 1-week-long experiment, this system tracks ~30,000 individual cells cultured under 1500 dynamic individual conditions by performing ~106 pipetting steps with nanoliter precision, and creates millions of single-cell data points. These are capabilities well beyond manual, robotic, or other microfluidic systems in terms of labor, cost, and time.
Fig. 1. Ultra-multiplexed, automated cell culture system for dynamical live-cell analysis.

The microfluidic device contains 1500 independently programmable culture chambers. During a 1-week experiment, the device performs nearly 106 pipetting steps to create and maintain distinct culture conditions in each of the chambers. (A) Each chamber can execute a distinct dynamic culture program (combinations, timed sequences, sine waves, etc.) where the fluidic composition can be changed when desired, and dynamic processes (i.e., NF-κB localization or Hes5 expression) are tracked with single-cell resolution. An on-chip nanoliter multiplexer measures several fluids and mixes them at predetermined ratios to create complex chemical inputs. A peristaltic pump delivers inputs to any given chamber. For the combinatorial input scenario, several chemicals are mixed and delivered to the cells continuously. In sequential inputs, signaling molecules are changed with a programmed time interval (Δt = 1 day). a.u., arbitrary units. (B) The system can culture adherent or nonadherent cells in either suspension mode, monolayer populations, or 3D format using hydrogels. The novel two-layer geometry of the culture chambers allows diffusion-based media delivery to create a stable environment for cells, and provides the additional ability of single-cell tracking of even nonadherent cells during dynamical stimulation. (C) Left: Two-layer cell chamber design allows diffusion- or flow-based media delivery, 3D cell culture, immobilization of nonadherent cells by gravity, and automated cell retrieval. Middle: Fluid mechanical simulations indicate the flow rates for diffusion-based media delivery and cell retrieval via direct flow. Right: Each chamber is controlled by a network of dedicated channels and membrane valves that automate various cell culture procedures. (D) Cells can be immunostained in the chip. The system is integrated to a fluorescent microscope and can automatically track individual cells in time-lapse experiments. Single cells or populations of interest can be automatically retrieved from individual chambers for off-chip analysis or expansion. GFP, green fluorescent protein; RFP, red fluorescent protein. (E) Primary cells (e.g., mouse NSCs and human HSCs) and cell lines (e.g., Jurkat T cells and mouse fibroblasts) are viably cultured and maintained on chip for weeks. Growth rates equal or better than the well plate culture are achieved through frequent diffusion-based media delivery while maintaining an unperturbed microenvironment.
RESULTS
Cultivating a broad range of cells in dynamic microenvironments requires precise control of cell density, surface properties, support matrices, gas and fluidic exchange, media and growth factor delivery, and humidity. To realize this, we designed a simple two-layer culture chamber that creates a consistent microenvironment for long-term cellular studies (Fig. 1, B and C). The 3D culture chamber can deliver media and ligands to cells via diffusion, preventing cells from undesirable shear stress and displacement in live-cell tracking experiments (Fig. 1, C to E; fig. S1, A to F; and movies S1, S2, and S5). The use of diffusion-based media delivery is flow free and gentle and creates minimal mechanical disturbance to the cellular microenvironment (section S1). The culture chamber can be loaded with gels and other support matrices to enable 3D cell organization (Fig. 1, C and E, and fig. S1, G and H). Furthermore, on-demand cell retrieval from designated chambers is possible by automatic switching to flow-based media delivery (Fig. 1C; fig. S1, A to E; and movies S1 and S3). To formulate complex and dynamic chemical inputs on chip, we designed and integrated a new microfluidic chemical formulator. A wide range of time-varying chemical inputs with distinct characteristics (i.e., pulsed and sinusoidal inputs) can be generated from a few previously prepared fluid vials connected to the chip and can be delivered to live cells with subminute temporal resolution by diffusion or with subsecond resolution by regulated flow (Fig. 1A and movie S4).
The Achilles’ heel for microfluidic cell culture has been poor cell viability, which has been especially severe for sensitive primary mammalian cells (8, 9). To demonstrate general cell viability in our system, we first cultured mouse fibroblast cells and primary human hematopoietic stem cells (HSCs) in adherent and suspension culture modes, respectively, and stimulated them with either constant, pulsed, or sinusoidal formulations of the inflammatory cytokine tumor necrosis factor (TNF) to induce nuclear factor κB (NF-κB) signaling (Fig. 1A and figs. S2 and S3) (15). Dynamic stimulation and indexed tracking of nonadherent cells such as HSCs were not feasible before our study because of flow-based stimulation displacing cells and preventing single-cell tracking. We stimulated human HSCs with different concentrations of TNF as well as in combination with other cytokines [interferon-γ (IFNγ), interleukin-6 (IL-6), interferon gamma-induced protein 10 (IP10), and monocyte chemoattractant protein-1 (MCP-1)] and with dynamic (time-dependent) variations of selected cytokines. Viability for various cell types was further demonstrated by culturing suspension cells including the Jurkat T cell line and mouse HSCs (Fig. 1E and fig. S3C). In both cases, cells proliferated at similar, if not higher, rates than those in bulk experiments in traditional culture dishes.
We cultured primary mouse embryonic neural stem cells (NSCs) and stimulated them on our chip for weeks under three distinct modes: suspension as single cell or as neurosphere, adherent monolayer, and 3D in hydrogels (Figs. 1E and 2 and fig. S1, G and H). The number of adherent NSCs increases by 50% after 24 hours, comparable to traditional well plate culture. Meanwhile, the diameter of NSC spheres in suspension and 3D gel culture both doubled after 4 days of on-chip culture (Fig. 1E and movie S5). Notably, we observe that NSC spheres are often unresponsive to dynamic environmental changes as compared to monolayer cultured cells, suggesting that NSCs self-organize into a protective layered structure during suspension culture (figs. S1G and S4, A to E, and section S4). Thus, NSC differentiation and self-maintenance are assessed at the single-cell level during monolayer culture by tracking Hes5-GFP expression (indicative of self-renewing NSCs) and Dcx-RFP to label neuroblasts (indicating progress toward differentiation) (12, 13, 14, 16–19). NSC growth rate is another key feature we quantified to statistically assess NSC self-renewal.
To study early signaling events during mammalian forebrain development in vitro, we dynamically analyzed NSCs under combinatorial and time-varying signaling inputs in our system (Figs. 2 and 3). The six selected signaling molecules were identified through RNA sequencing of NSCs isolated from embryonic mouse brain tissue, whose receptors are highly expressed during mouse forebrain development. These factors were Jagged1, DLL1, EGF, PACAP, CXCL, and PDGF (20). The effect of these ligands on NSC differentiation and self-maintenance is not well understood. We hypothesized that the different combinations and temporal ordering of these ligands will lead to distinct cell fate outcomes (section S6). We therefore generated thousands of combinations and temporal sequences of these ligands on the chip and delivered them to NSCs cultured in different chambers while monitoring their differentiation and growth at the single-cell level by time-lapse microscopy (Fig. 2). Millions of single-cell data points were generated and quantified in these experiments. The stimulation input conditions are summarized in table S1. Control experiments in traditional 96-well plates under selected dynamic conditions were used to verify results obtained on the microfluidic device (figs. S4, F to H, and S5, A to E, and section S5).
Fig. 2. High-throughput dynamical analysis of NSC differentiation.
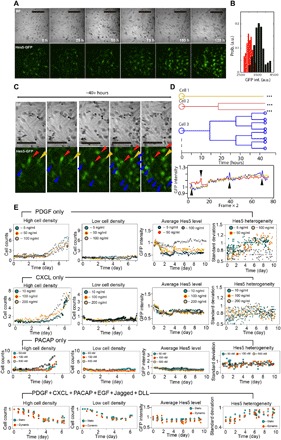
Millions of single-cell images are generated and automatically analyzed in live-cell signaling factor stimulation measurements, and few example datasets are shown here. (A) Time-lapse bright-field (BF) (top) and epifluorescence (bottom) images of NSCs cultured with PDGF (100 ng/ml). Scale bars, 100 μm. (B) Histogram of Hes5-GFP expression in NSCs before (red) and after (black) 1-week culture with PDGF (100 ng/ml). High levels of Hes5 in NSCs indicate maintenance of stem cell state, while reduced Hes5 indicates progress toward differentiation. (C) Enlarged bright-field (top) and corresponding epifluorescence (bottom) images of NSCs shown in (A), cultured on chip with PDGF (100 ng/ml). Selected cells were indicated by arrows and individually tracked over 40 hours during on-chip culture. (D) Lineage tracing (top) and Hes5-GFP expression level (bottom) for the three selected cells in (C). Distinct proliferation patterns were observed despite similar Hes5-GFP level. (E) We show examples of quantitative analysis of mouse NSC growth and Hes5 expression in different culture conditions. Each culture contains either a single ligand or a mixture of ligands that are highly expressed in developing mouse brain, including PDGF, CXCL, PACAP, EGF, Jagged, or DLL. Hes5 expression rate and variability significantly depend on signaling molecules present in culture chambers.
Fig. 3. High-throughput analysis of NSC dynamics reveals signaling logic rules in differentiation.
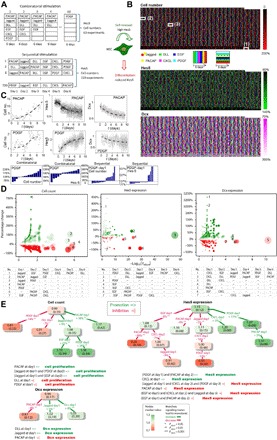
(A) NSCs were stimulated in two types of experiments: combinatorial stimulation and sequential stimulation. During combinatorial stimulation, the microfluidic device delivered all possible combinations of DLL, EGF, Jagged, PACAP, CXCL, and PDGF to distinct culture chambers and maintained these conditions for 6 days. During sequential stimulation, the environmental ligands were replaced daily during the 6-day experiments. Cell numbers and single-cell Hes5-GFP and Dcx-RFP expressions were recorded in each chamber over time. (B) Example datasets from one experiment. Signal-induced changes in NSC cell count (top), Hes5-GFP expression (middle), and Dcx expression (bottom) at day 6 are plotted as heat maps, together with the color-coded bars indicating the combinatorial and sequential signal inputs. The white squares show input conditions with the following ligand combinations and sequences: (1) DLL + EGF, (2) Jagged ≫ CXCL ≫ PDGF ≫ PACAP ≫ EGF ≫ DLL , and (3) CXCL ≫ PDGF ≫ PACAP ≫ EGF ≫ DLL ≫ Jagged. (C) Single-cell tracking reveals the dynamic variations in cell numbers and Hes5 and Dcx levels during 6 days of single ligand treatment, with PACAP or PDGF. The bottom row histograms show results of statistical analyses indicating the influence of PDGF on cell growth or Hes5 level in different experiments that also contain other ligands. Each bar represents a distinct culture experiment. (D) Statistical analysis of cell count and Hes5 and Dcx expression using all 720 sequential experiments via Wilcoxon rank-sum test. Y axes show the percentage change of cell numbers and Hes5 and Dcx expression compared to controls (i.e., the effect size) for each ligand input, and the x axes show the corresponding adjusted P value. The data are presented with colored bubbles, where the bubble’s diameter is proportional to the negative logarithm of adjusted P value, and Bubble’s color encodes percentage change (green for increase, red for decrease; stronger effect shown in more opaque color). Few selected inputs with high significance or large effect size are annotated with numbers 1 to 7 and are described in lower tables. (E) Decision trees are used to visualize the signaling paths toward NSC differentiation or self-maintenance, each of which shows a statistically significant monotonic increase (green paths) or monotonic decrease (red paths) in cell counts and Hes5 and Dcx expression. Each decision tree node includes a median value (color-coded as above) and median absolute deviation (in brackets) of measured values. On the connecting path between nodes, we show the decision attribute to be satisfied for splitting the tree, and the percentage change in cell count and Hes5 expression or Dcx expression (adjusted P value indicated by asterisks). Signaling logic rules resulting from the decision trees are listed below. Notations of “=>” and “=|” denote “promote” and “prohibit,” respectively.
High Hes5 expression in NSCs indicates maintenance of the stem cell state, while reduced Hes5 indicates progression toward differentiation (fig. S6, A to C and G to I, and section S7) (12, 21–26). Through on-chip immunostaining, we found that most NSCs that were stimulated with a single ligand did not fully differentiate to any distinct lineage within 6 days; however, combined stimulation with all six ligands made NSCs differentiate into neurons during this time period (fig. S5, F to J, and section S8). By evaluating NSC proliferation, Hes5 expression, and cell morphology, we found many culture conditions where the entire population progressed toward differentiation (Fig. 2, A to C, and fig. S6, D to F). Nevertheless, we also found substantial variability at the single-cell level in a given culture condition (Fig. 2, C to E; fig. S7, A and B; and section S10). Distinct proliferation patterns were observed despite similar Hes5-GFP levels (Fig. 2, D and E). Overall, NSCs proliferate at different rates depending on initial cell density, with higher densities leading to higher proliferation rates (Fig. 2E). Mean Hes5 expression levels and single-cell heterogeneity strongly depended on the signaling inputs received by each NSC culture.
To investigate the role of combinatorial or temporal ordering of signals in NSC maintenance or differentiation, we cultured and monitored NSCs under combinatorial and sequential applications of the six regulatory ligands (Fig. 3A; fig. S7, C to F; and table S1). For each independent NSC culture on the chip, we introduced either one ligand each day (sequential inputs) or a combination of the selected ligands over a 6-day period (combinatorial inputs). We measured the ratio of the ligand-treated cells to untreated controls to quantify changes in cell numbers, and Hes5-GFP and Dcx-RFP expression intensities. Each chip experiment consisted of 63 combinatorial and 720 sequential stimulation experiments, and experiments were repeated at least three times, resulting in nearly 3000 dynamic cultures (Fig. 3B). Each of the six ligands are used in various contexts repeatedly, and their effect on stem cell fate is measured directly in individual experiments and also by statistical analysis of all experiments that contain these ligands. Figure 3B shows raw data from a single experiment, which contains single ligand, combinatorial, and sequential stimulation conditions where cell numbers and Hes5 and Dcx expression are quantified in time-lapse measurements in single cells. In Fig. 3C, we show two example experiments: PACAP stimulation induces an increase in cell numbers and a decrease in Hes5 and Dcx levels over 10-day measurements. PDGF stimulation, however, increases both cell numbers and Hes5 levels but decreases Dcx levels in NSCs. In the lower panels of Fig. 3C, we plot the effect of platelet-derived growth factor (PDGF) on cell growth and Hes5 changes in different experiments that use PDGF along with other ligands. Each bar in the histogram shows the effect of PDGF in an individual experiment; in some experiments, PDGF increases the measured quantities, while in others, it leads to a decrease. These changes vary in a wide range, from negative to positive, indicating that the role of a ligand can be highly context dependent (27–30), which we further discuss in the following sections.
DISCUSSION
Our live-cell tracking measurements resulted in an extremely large, multidimensional dataset. To understand the effect of various stimulation conditions on NSC fate in such an experimental landscape, we subjected all experimental outputs to statistical analysis, i.e., Wilcoxon rank-sum test (see Materials and Methods). The effect size (i.e., percentage change in cell numbers and Hes5 and Dcx expression) and adjusted P values associated with each ligand input condition can be visualized using bubble plots (Fig. 3D). Several selected inputs that resulted in high significance or large effect size are annotated in lower tables in Fig. 3D and fig. S8. For example, the condition PACAP-day1 (annotated sequence no. 4) in the leftmost table overall increases cell numbers by 16% (adjusted P = 4.21 ×10−7). This result is achieved by our statistical test that analyzed all experiments that included the PACAP in day 1 of the stimulation protocol. Complete test results can be seen in table S2. Furthermore, we used multibranch decision trees to highlight the optimal signaling routes leading to a statistically significant monotonic increase/decrease in the measured cellular parameters (false discovery rate, 0.05). This led us to discover several nontrivial “cellular logic rules” describing the effect of various ligands on cell fate (Fig. 3E). For example, the condition Jagged-day1 increases cell numbers overall by 9% (adjusted P = 6.58 × 10−3), giving rise to a subgroup of experiments in which the median of normalized cell count is 1.08 (median absolute deviation, 0.35). If EGF is added on the following day, the median value increases to 1.33 (absolute deviation, 0.34; adjusted P = 5.37 ×10−4).
We further evaluated the results of our statistical analyses to understand the role of signaling molecules on NSC fate and uncovered several “signaling principles” that highlighted the importance of environmental context and signal timing in NSC differentiation and self-maintenance (Fig. 4 and fig. S7, F to H).
Fig. 4. Statistical analysis of dynamic stimulation experiments uncovers signaling principles in NSC differentiation and self-maintenance.

(A) Dynamic changes in cell number and Hes5 level plotted for two ligand combinations containing (DLL, EGF, PDGF) or (Jagged, EGF, PDGF). Dots are single-cell values, and dashed lines indicate population mean. The change in Hes5 expression can be directed from increase to decrease by only changing one ligand in the combination. (B) Comparison of two distinct sequential inputs highlights the importance of input sequence and timing. In both experiments, cells received the same six ligands, but in different orders. Changing the order of a single ligand (e.g., EGF from day 2 to day 6) directs NSCs to different cell fate. (C) Comparison of optimal and nonoptimal input ligand sequences that lead to monotonic changes in cell counts or Hes5 expression. Numbers in boxes indicate median value (color-coded) and median absolute deviation (in brackets) of cell count or Hes5 expression. ns, nonsignificant changes. Optimal paths are highlighted in green (increase) or red (decrease), while the alternatives paths are highlighted in black. (D) Increasing number of ligands in a stimulation experiment overall suppresses NSC proliferation, whereas reducing the ligand numbers enhances the stem cell pool. Including more ligands in experiments led to a reduction of the proliferation rate, while the Hes5 level remained relatively unchanged. Error bars indicate variability of individual experiments from the mean. (E) Synergy and antagonism between signaling molecules. The combination of two ligands may lead to enhanced (synergistic) or reduced (antagonistic) effect compared to experiments that use these molecules in isolation. At the top, synergy and antagonism for NSC ligands are defined. Rows at the bottom show actual molecules that are synergistic (green) or antagonistic (red) toward cell proliferation or Hes5 expression. Measured percent changes from controls are also indicated. (F) In sequential stimulation experiments, certain ligands assume context-dependent roles determined by timing of their introduction or the preconditions before use of that ligand. Boxes indicate the identity of ligands used in each day. X in brackets indicate that the exact identity of the ligand in that day does not change the outcome. (G) Multiple input conditions lead to similar change in cell numbers and Hes5 levels, suggesting redundancy in NSC signaling pathways. Example redundant pathways are color-coded. Numbers indicate percent change resulting from stimulation with ligands. (H) Cell fate toward differentiation or self-renewal may be decided by certain early signals, indicating early commitment toward self-renewal (PDGF-day1 ≫ DLL-day2 ≫ Jagged-day3) or differentiation (DLL-day1 ≫ PDGF-day2 ≫ CXCL-day3) (see table S2 for percentage change and P values associated with each condition).
First, dynamic single-cell tracking in combinatorial experiments shows that Hes5 expression can be directed from increase to decrease by only changing one ligand in the ligand combination (i.e., changing Jagged to DLL, in Fig. 4A). In sequential experiments where ligands were replaced on a daily basis, changing the temporal order of a single ligand can direct NSCs to different cell fates despite the fact that cells were overall exposed to the same set of ligands through the course of 6 days. For instance, delivering EGF on day 2 resulted in relatively higher Hes5 expression levels and unchanged cell numbers, indicating maintenance of the stem cell pool, whereas moving EGF to day 6 led to a significant reduction in Hes5 expression and cell number, indicative of differentiation (Fig. 4B).
Decision tree analysis found certain optimal routes for signal input sequences to achieve different outcomes. For example, one optimal route leading to NSC proliferation is Jagged-day1 ≫ EGF (or PDGF)-day2, increasing cell numbers by 9% (adjusted P = 6.58 × 10−3) in day 1 and by 24% in day 2 (adjusted P = 7.26 × 10−4). Any deviation from these routes (i.e., altering the applied ligands) is nonoptimal, resulting in either a decrease or no significant change of cell numbers. An example for Hes5 expression is the optimal route that reduces its expression by 17% by day 3 (adjusted P = 2.57 × 10−2) (Fig. 4C).
In addition, we found that increasing environmental complexity (i.e., increasing the number of ligands used in cultures) generally suppressed NSC proliferation; however, reducing the input complexity enhanced the stem cell pool. As Fig. 4D shows, including more ligands either in combination or sequentially led to an overall reduction of the proliferation rate compared to controls, while the Hes5 expression levels remained relatively unchanged.
We found that the combination of two ligands may induce enhanced (synergistic) or reduced (antagonistic) effects compared to experiments that use these ligands in isolation. Examples of these synergy and antagonism between ligands are shown in Fig. 4E. For instance, Jagged and DLL are synergistic toward reducing NSC numbers: The reduction of cell numbers under combined Jagged + DLL stimulation (−12%, adjusted P = 4.53 × 10−2) is larger at absolute scale than the sum of reductions under individual stimulation with either molecule (−6%; adjusted P = 4.40 × 10−2; nonsignificant, −2%). Other examples are also shown in Fig. 4E. On the other hand, Jagged and PACAP are antagonistic pairs in affecting cell proliferation, because combined stimulation with these molecules leads to a lesser reduction in cell numbers compared to the changes brought by their individual application (−6% for Jagged, adjusted P = 4.40 × 10−2; +14% for PACAP, adjusted P = 1.52 × 10−3).
Figure 4F shows the context-dependent role of several individual ligands, which is determined by the timing of its introduction or by the preconditions before the use of that ligand. For example, although PDGF on day 1 predominantly increases Hes5 expression by 11% (adjusted P = 2.94 × 10−56), using DLL before PDGF can revert this change to a 6% reduction (adjusted P = 4.44 × 10−3). If Jagged is applied before PDGF, the change of Hes5 expression is nonsignificant, although cell numbers increase. These context-dependent roles can be found in CXCL and Jagged (lower rows in Fig. 4F).
Another principle emerging from our analysis is the redundancy in NSC signaling pathways, reflecting the intrinsic flexibility of stem cells to respond to their dynamic environment and niche at the early times we measured. Figure 4G shows several examples of signaling routes leading to similar quantitative changes and cell fates. For example, the combinatorial inputs Jagged + DLL and Jagged + PACAP + CXCL + PDGF lead to very similar increases in cell numbers and Hes5 expression. Similarly, the sequential inputs DLL-day1 ≫ PDGF-day2 ≫ CXCL-day3 and EGF-day1 ≫ CXXL-day2 ≫ Jagged-Day3 ≫ PACAP-day4 lead to similar reduction in cell numbers and Hes5 expression. Similar redundant pathways are seen for both differentiation and self-renewal directions.
Last, our analyses show signs of early cellular commitment. Cells at certain early decision points can commit toward differentiation or self-renewal directions and tend to ignore subsequent signals they received. Figure 4H shows two examples: After cells received PDGF-day1 ≫ DLL-day2 ≫ Jagged-day3 in a row, signals that come after do not reverse the increase in cell numbers or Hes5 level, indicating self-renewal of stem cells. On the other hand, ligand sequence DLL-day1 ≫ PDGF-day2 ≫ CXCL-day3 initiates a strong decrease of cell number and Hes5 expression, regardless of many other signals that come after this sequence.
In this study, we presented an ultra-multiplexed microfluidic technology with unprecedented capabilities for high-throughput live-cell analysis under complex and dynamic signals. In record-breaking fashion, our microfluidic system mapped the signaling landscape of NSC differentiation in 3000 distinct microenvironments that mimic the dynamical stem cell niche. By statistical analysis and modeling of thousands of live-cell experiments, we identified cellular decision points and differentiation trajectories. Our microfluidic system greatly shortens the time span and improves the reproducibility of high-throughput screening processes with live cells. This technology allows the analysis of unprecedented combinatorial complexity, which may have relevance for the dynamic and regulated microenvironment of the tissue during homeostasis and regeneration.
MATERIALS AND METHODS
Design and fabrication of microfluidic chips
We designed and fabricated the microfluidic device according to the standard protocol, which is reported elsewhere (2). Briefly, we designed our two-layer device using AutoCAD (Autodesk Inc., San Rafael, CA, USA) and then printed the sketch on transparencies at 40-kdpi resolution (Fine Line Imaging, MN, USA). Molds for polydimethylsiloxane (PDMS) casting were produced using standard soft lithography. The channel network of the control layer, as well as the flow channels for the flow layer and culture chambers, was produced with either SU-8 3025 or SU-8 3075 (MicroChem, Westborough, MA, USA) on silicon wafers. For the flow layer, we additionally used AZ-50X (AZ Electronic Materials, Luxembourg) at valve positions. Photoresists were spun to a height of 25 μm for channels and 150 μm for culture chambers. To fabricate the chip, 72 g of PDMS (10:1 of monomer/catalyst ratio) was mixed, debubbled, and poured over the trimethylchlorosilane-treated patterned silicon wafer. The PDMS was then cured for 60 min at 80°C. Following plasma and alignment between flow and control layer, inlet holes were then punched after 2-hour thermal bonding. The chip was bonded to a PDMS-coated coverslip and cured for at least 12 hours at 80°C before use.
Chip setup, operation, and control
The glass slide carrying the microfluidic chip was cleaned and taped on a slide holder. Control channels were connected to miniature pneumatic solenoid valves (Festo, Switzerland) that were controlled with a custom MATLAB (MathWorks, USA) through graphical user interface (2). Optimal closing pressures of push-up PDMS membrane valves were determined individually for each chip, typically ranging from 25 to 30 psi. The cell culture chambers were treated with either fibronectin (0.25 mg/ml; Millipore, Austria) for 3T3 cell culture or polylysine (0.01%, Sigma-Aldrich) followed by laminin (1 to 2 mg/ml, Sigma-Aldrich) for adherent NSC culture. The remaining coating solution was flushed off from the chip using either phosphate-buffered saline or cell culture medium. Cell culture medium was prewarmed on chip for at least 1 hour before cell loading.
Cell culture and loading
For standard cell lines, we used Jurkat cells, RAW 264.7 macrophages p65−/− with p65-GFP and H2B-dsRed, as well as NIH 3T3 p65−/− cells with p65-dsRed and H2B-GFP for tracking and analysis of NF-κB activation. These cells were cultured according to the established protocols (31). To seed cells into the chip, adherent cells were harvested at 80% confluence with trypsin, resuspended, and loaded into chips through semiautomated loading program at cell densities from 104 to 106/ml depending on the desired cell density.
Murine HSCs were isolated with a FACSAria III flow cytometer (BD Biosciences) as Lin−/c-Kit+/Sca-1+/CD48−/CD150+/CD34− [lineage (Lin): CD3e/CD11b/CD19/CD41/B220/Gr-1/Ter119], which are approximately 50% pure HSCs. Macrophage colony-stimulating factor (M-CSF), a myeloid cytokine released during infection and inflammation, was used to induce HSC differentiation. Human CD34+ cells were isolated from mononuclear cells using the EasySep Human CD34 Positive Selection Kit (STEMCELL Technologies, Vancouver, BC, Canada). CD34+CD38−CD45RA−CD90+CD49f+ HSCs were sorted using a FACSAria III flow cytometer (BD Biosciences). Pro- and anti-inflammatory cytokines, including TNF, IFNγ, IL-6, IP10, and MCP-1, were introduced into the cellular environment as single ligand and in combination. Embryonic NSCs with Hes5-GFP and Dcx-RFP reporters were isolated at embryonic day 13.5 from a transgenic mouse carrying Hes5-GFP and Dcx-RFP using established protocol (14, 17, 18). The resulting primary cells were verified to carry both Hes5-GFP and Dcx-RFP after isolation and allowed to grow for few passages before use in the experiments (19). NSCs were cultured as neurospheres in culture media [Dulbecco’s Modified Eagle medium/Nutrient Mixture F-12 + GlutaMAX (Gibco, no. 31331-028); penicillin (10 U/ml); streptomycin (10 μg/ml); B-27 supplement (1:50); and fibroblast growth factor (0.02 μg/ml)]. As NSCs are sensitive to environmental variations, cell handling protocol before loading into the chip was examined systematically (including dissociation conditions and fluorescence-activated cell sorting). To obtain the optimal results, NSC spheres were collected and loaded into the chip 24 hours after fresh dissociation, where each sphere contains ~7 to 10 cells. To avoid potential artifacts due to prolonged in vitro culture, only NSCs within 10 passages were used in the study. In control experiments, transferring chip-cultured NSCs to a well plate showed the sphere-forming ability of Hes5-positive cells, validating Hes5 as a self-maintenance marker in our experiments (fig. S6, H and I) (12).
The environmental conditions were maintained using temperature control and incubator system (Live Cell Imaging Service GmbH, Basel, Switzerland) to strictly 37°C and >98% humidity and 5% CO2 during the experiment, and the PDMS chip was covered with a stage top incubator connected to a humidifier and a gas exchanger.
Live-cell fluorescence microscopy and data analysis
For image acquisition, a Nikon Ti-ECLIPSE microscope with an automated translational stage and a digital complementary metal-oxide semiconductor (CMOS) camera (ORCA-Flash4.0, Hamamatsu, Japan) was used. The stage and image acquisition was controlled via the NIS-Elements software (Nikon, Japan). Bright-field and fluorescence images were acquired and analyzed using a customized MATLAB program. The algorithm extracts single-cell traces including position, nuclear, and cytoplasm fluorescence level. For example, the 3T3 cell nuclear area in each image was identified via the fluorescent nuclear marker H2B-GFP, and then the mean value of the nuclear intensity of the p65-DsRed marker was measured and plotted as a function of time.
Wilcoxon rank-sum test and multiple-test correction
Experiments of sequential and combinatorial ligand additions consisted of 720 and 56 different conditions, respectively. Experiments for each individual treatment condition were repeated for three times. All datasets generated in this way were subjected to the statistical test described as follows. Denote a sequential condition as Sij = {ligand i is added on day j} and a combinatorial condition as Ci = {ligand i is present}, where ligand i = Jagged, DLL, EGF, PACAP, CXCL, PDGF and j = 1, 2, 3, 4, 5, 6. We were interested in investigating the ligands’ conditional effects by applying multiple treatments in specific order one after another, for sequential inputs (starting from day 1), and Cn| Ci, ⋯, Ck for combinatorial inputs. The controls are their negated counterparts or . We used the Wilcoxon rank-sum approach to examine whether the distributions of cell counts, Hes5 expression values, or Dcx expression values between these treatment/control comparisons are significantly different. No assumption about normality of the underlying distribution was used (32–34). The detailed steps for both sets of tests are as follows:
1. Pick a ligand condition for investigation and split measured group U accordingly into two subgroups: subgroup A that satisfies the condition and subgroup that does not.
2. Test whether the distributions of cell counts, Hes5 expression, or Dcx expression in these two subgroups are significantly different. The difference can be quantified by the median of the distribution of individual differences between randomly selected samples from subgroups A and . The effect size is defined as percentage change, i.e., , where ai ∈ A, uj ∈ U [all medians were computed using Hodge-Lehmann estimator (35)].
3. Repeat steps 1 and 2 for all investigated sample treatment sequences. Here, we controlled the false discovery rate and adjust P values of all tests using Benjamini-Hochberg procedure (36, 37).
Supplementary Material
Acknowledgments
Funding: This work was supported by the Swiss Initiative in Systems Biology (SystemsX). H.-L.T. thanks the EMBO Long-Term Fellowship (EMBO ALTF 858-2014) for the support. Author contributions: S.T., C.Z., and H.L.-T. designed and conducted the experimental study. A.R. and G.J. designed and performed statistical analysis. T.M. and V.T. provided mouse NSCs and performed NSC imaging in vivo. All authors contributed to the writing of the manuscript. Competing interests: S.T. is a founder, board member, and equity holder of BiomeSense Inc., a startup company developing automated microbiome measurement systems. C.Z. and S.T. are inventors on an international patent application related to the microfluidic technology described in this manuscript, filed by the University of Chicago (no. PCT/US2018/032727; filed on 15 May 2018). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav7959/DC1
Section S1. Temporal and spatial concentration distribution within the cell culture chambers
Section S2. Single 3T3 fibroblast cell culture and stimulation on chip
Section S3. Culture and stimulation of human and mouse HSCs on chip
Section S4. Culture and stimulation of NSC spheres on chip
Section S5. Combinatorial and sequential experiments performed in 96-well plates
Section S6. Extended discussion of high-throughput combinatorial and sequential input studies
Section S7. Hes5 expression as a valid marker for NSC stemness
Section S8. Immunostaining on chip and determining NSC phenotypes
Section S9. Statistical analysis of combinatorial and sequential results
Section S10. NSC single-cell tracking during combinatorial and sequential stimulation
Fig. S1. Experimental characterization of concentration variations during medium exchange.
Fig. S2. Assessment of the microfluidic system for dynamical cell culture and NF-κB signaling.
Fig. S3. Culture and stimulation of human HSCs on chip.
Fig. S4. Hes5 and Dcx expression regulating NSC cellular behavior.
Fig. S5. Combinatorial and sequential stimulation of six ligands regulating NSC self-renewal and differentiation.
Fig. S6. Correlation between Hes5 expression and NSC stemness.
Fig. S7. Combinatorial and sequential inputs regulating NSC proliferation, Hes5, and Dcx expression.
Fig. S8. Effect of various stimulation conditions on NSC cell fate subjected to statistical analysis.
Table S1. Microenvironment exposed to six single ligands and combinatorial and sequential ligand inputs (note: the order of the ligands in the table represents the order of ligands introduced into the microenvironments on daily bases).
Table S2. Statistical analysis results associated with sequential and combinatorial inputs of six ligands based on cell count measurements and Hes5 and Dcx expression level.
Movie S1. COMSOL simulation and time-lapse video of fluid exchange in a unit chamber on the chip.
Movie S2. Redistribution of GFP after medium exchange and all valves are closed.
Movie S3. Retrieval of adherent cells (3T3, left) and suspension-cultured cells (Jurkat, right) from the chip.
Movie S4. Stimulation of 3T3 cells by sinusoidal TNF-α inputs.
Movie S5. Cell tracking videos of NSC spheres (top) and monolayer (bottom).
REFERENCES AND NOTES
- 1.Hemberger M., Dean W., Reik W., Epigenetic dynamics of stem cells and cell lineage commitment: Digging Waddington’s canal. Nat. Rev. Mol. Cell Biol. 10, 526–537 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Unger M. A., Chou H.-P., Thorsen T., Scherer A., Quake S. R., Monolithic microfabricated valves and pumps by multilayer soft lithography. Science 288, 113–116 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Millet L. J., Gillette M. U., New perspectives on neuronal development via microfluidic environments. Trends Neurosci. 35, 752–761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lecault V., VanInsberghe M., Sekulovic S., Knapp D. J. H. F., Wohrer S., Bowden W., Viel F., McLaughlin T., Jarandehei A., Miller M., Falconnet D., White A. K., Kent D. G., Copley M. R., Taghipour F., Eaves C. J., Keith Humphries R., Piret J. M., Hansen C. L., High-throughput analysis of single hematopoietic stem cell proliferation in microfluidic cell culture arrays. Nat. Methods 8, 581–586 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Sackmann E. K., Fulton A. L., Beebe D. J., The present and future role of microfluidics in biomedical research. Nature 507, 181–189 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Jeong J. W., McCall J. G., Shin G., Zhang Y., Al-Hasani R., Kim M., Li S., Yong Sim J., Jang K.-I., Shi Y., Hong D. Y., Liu Y., Schmitz G. P., Xia L., He Z., Gamble P., Ray W. Z., Huang Y., Bruchas M. R., Rogers J. A., Wireless optofluidic systems for programmable in vivo pharmacology and optogenetics. Cell 162, 662–674 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gómez-Sjöberg R., Leyrat A. A., Pirone D. M., Chen C. S., Quake S. R., Versatile, fully automated, microfluidic cell culture system. Anal. Chem. 79, 8557–8563 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Mehling M., Tay S., Microfluidic cell culture. Curr. Opin. Biotechnol. 25, 95–102 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Millet L. J., Stewart M. E., Sweedler J. V., Nuzzo R. G., Gillette M. U., Microfluidic devices for culturing primary mammalian neurons at low densities. Lab Chip 7, 987–994 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Wu S., O’Lexy R., Xu M., Sang Y., Chen X., Yu Q., Gallagher K. L., Symplastic signaling instructs cell division, cell expansion, and cell polarity in the ground tissue of Arabidopsis thaliana roots. Proc. Natl. Acad. Sci. U.S.A. 113, 11621–11626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giobbe G. G., Michielin F., Luni C., Giulitti S., Martewicz S., Dupont S., Floreani A., Elvassore N., Functional differentiation of human pluripotent stem cells on a chip. Nat. Methods 12, 637–640 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Basak O., Taylor V., Identification of self-replicating multipotent progenitors in the embryonic nervous system by high Notch activity and Hes5 expression. Eur. J. Neurosci. 25, 1006–1022 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Basak O., Giachino C., Fiorini E., MacDonald H. R., Taylor V., Neurogenic subventricular zone stem/progenitor cells are notch1-dependent in their active but not quiescent state. J. Neurosci. 32, 5654–5666 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giachino C., Basak O., Taylor V., Isolation and manipulation of mammalian neural stem cells in vitro. Methods Mol. Biol. 482, 143–158 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Kellogg R. A., Tay S., Noise facilitates transcriptional control under dynamic inputs. Cell 160, 381–392 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Lugert S., Basak O., Knuckles P., Haussler U., Fabel K., Götz M., Haas C. A., Kempermann G., Taylor V., Giachino C., Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell 6, 445–456 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Sykova E., Forostyak S., Stem cells in regenerative medicine. Laser Ther. 22, 87–92 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behnan J., Grieg Z., Joel M., Ramsness I., Stangeland B., Neuroepigenetics Gene knockdown of CENPA reduces sphere forming ability and stemness of glioblastoma initiating cells. Neuroepigenetics 7, 6–18 (2016). [Google Scholar]
- 19.Couillard-Despres S., Winner B., Karl C., Lindemann G., Schmid P., Aigner R., Laemke J., Bogdahn U., Winkler J., Bischofberger J., Aigner L., Targeted transgene expression in neuronal precursors: Watching young neurons in the old brain. Eur. J. Neurosci. 24, 1535–1545 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Allen Institute for Brain Science, Allen Brain Atlas (Allen Institute for Brain Science, 2017). [Google Scholar]
- 21.Basak O., Beumer J., Wiebrands K., Seno H., van Oudenaarden A., Clevers H., Induced quiescence of Lgr5+ stem cells in intestinal organoids enables differentiation of hormone-producing enteroendocrine cells. Cell Stem Cell 20, 177–190.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Lugert S., Vogt M., Tchorz J. S., Müller M., Giachino C., Taylor V., Homeostatic neurogenesis in the adult hippocampus does not involve amplification of Ascl1high intermediate progenitors. Nat. Commun. 3, 670 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Ehm O., Göritz C., Covic M., Schäffner I., Schwarz T. J., Karaca E., Kempkes B., Kremmer E., Pfrieger F. W., Espinosa L., Bigas A., Giachino C., Taylor V., Frisén J., Chichung Lie D., RBPJκ-dependent signaling is essential for long-term maintenance of neural stem cells in the adult hippocampus. J. Neurosci. 30, 13794–13807 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giachino C., Basak O., Lugert S., Knuckles P., Obernier K., Fiorelli R., Frank S., Raineteau O., Alvarez-Buylla A., Taylor V., Molecular diversity subdivides the adult forebrain neural stem cell population. Stem Cells 32, 70–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori H., Ninomiya K., Kino-oka M., Shofuda T., Omedul Islam M., Yamasaki M., Okano H., Taya M., Kanemura Y., Effect of neurosphere size on the growth rate of human neural stem/progenitor cells. J. Neurosci. 84, 1682–1691 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Pastrana E., Silva-Vargas V., Doetsch F., Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 8, 486–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangan P. R., Harrington L. E., O’Quinn D. B., Helms W. S., Bullard D. C., Elson C. O., Hatton R. D., Wahl S. M., Schoeb T. R., Weaver C. T., Transforming growth factor-ß induces development of the TH17 lineage. Nature 441, 231–234 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Bettelli E., Carrier Y., Gao W., Korn T., Strom T. B., Oukka M., Weiner H. L., Kuchroo V. K., Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Flores G. V., Duan H., Yan H., Nagaraj R., Fu W., Zou Y., Noll M., Banerjee U., Combinatorial signaling in the specification of unique cell fates. Cell 103, 75–85 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Urbán N., Guillemot F., Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front. Cell. Neurosci. 8, 396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tay S., Hughey J. J., Lee T. K., Lipniacki T., Quake S. R., Covert M. W., Single-cell NF-κB dynamics reveal digital activation and analogue information processing. Nature 466, 267–271 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.M. Hollander, D. A. Wolfe, Nonparametric statistical methods, in Wiley Series in Probability and Mathematical Statistics (Wiley, 1973). [Google Scholar]
- 33.Mann H. B., Whitney D. R., On a test of whether one of 2 random variables is stochastically larger than the other. Ann. Math. Stat. 18, 50–60 (1947). [Google Scholar]
- 34.Wilcoxon F., Individual comparisons by ranking methods. Biometrics Bull. 1, 80–83 (1945). [Google Scholar]
- 35.E. L. Lehmann, H. J. M. D’Abrera, Nonparametrics: Statistical Methods Based on Ranks (Springer, ed. 1, 2006). [Google Scholar]
- 36.Benjamini Y., Hochberg Y., Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
- 37.Benjamini Y., Yekutieli D., The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188 (2001). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaav7959/DC1
Section S1. Temporal and spatial concentration distribution within the cell culture chambers
Section S2. Single 3T3 fibroblast cell culture and stimulation on chip
Section S3. Culture and stimulation of human and mouse HSCs on chip
Section S4. Culture and stimulation of NSC spheres on chip
Section S5. Combinatorial and sequential experiments performed in 96-well plates
Section S6. Extended discussion of high-throughput combinatorial and sequential input studies
Section S7. Hes5 expression as a valid marker for NSC stemness
Section S8. Immunostaining on chip and determining NSC phenotypes
Section S9. Statistical analysis of combinatorial and sequential results
Section S10. NSC single-cell tracking during combinatorial and sequential stimulation
Fig. S1. Experimental characterization of concentration variations during medium exchange.
Fig. S2. Assessment of the microfluidic system for dynamical cell culture and NF-κB signaling.
Fig. S3. Culture and stimulation of human HSCs on chip.
Fig. S4. Hes5 and Dcx expression regulating NSC cellular behavior.
Fig. S5. Combinatorial and sequential stimulation of six ligands regulating NSC self-renewal and differentiation.
Fig. S6. Correlation between Hes5 expression and NSC stemness.
Fig. S7. Combinatorial and sequential inputs regulating NSC proliferation, Hes5, and Dcx expression.
Fig. S8. Effect of various stimulation conditions on NSC cell fate subjected to statistical analysis.
Table S1. Microenvironment exposed to six single ligands and combinatorial and sequential ligand inputs (note: the order of the ligands in the table represents the order of ligands introduced into the microenvironments on daily bases).
Table S2. Statistical analysis results associated with sequential and combinatorial inputs of six ligands based on cell count measurements and Hes5 and Dcx expression level.
Movie S1. COMSOL simulation and time-lapse video of fluid exchange in a unit chamber on the chip.
Movie S2. Redistribution of GFP after medium exchange and all valves are closed.
Movie S3. Retrieval of adherent cells (3T3, left) and suspension-cultured cells (Jurkat, right) from the chip.
Movie S4. Stimulation of 3T3 cells by sinusoidal TNF-α inputs.
Movie S5. Cell tracking videos of NSC spheres (top) and monolayer (bottom).