

Cerebellar development is a highly regulated process involving numerous factors acting with high specificity, both temporally and by location. Part of this process involves extensive proliferation of cerebellar granule neuron precursors (CGNPs) induced by Sonic Hedgehog (SHH) signaling, but downstream effectors of mitogenic signaling are still being elucidated.
KEYWORDS: Sonic Hedgehog, hypoxia-inducible factor 1a, medulloblastoma, reactive oxygen species, stem cells
ABSTRACT
Cerebellar development is a highly regulated process involving numerous factors acting with high specificity, both temporally and by location. Part of this process involves extensive proliferation of cerebellar granule neuron precursors (CGNPs) induced by Sonic Hedgehog (SHH) signaling, but downstream effectors of mitogenic signaling are still being elucidated. Using primary CGNP cultures, a well-established model for SHH-driven proliferation, we show that SHH-treated CGNPs feature high levels of hypoxia-inducible factor 1α (HIF1α), which is known to promote glycolysis, stemness, and angiogenesis. In CGNPs cultured under normoxic conditions, HIF1α is posttranslationally stabilized in a manner dependent upon reactive oxygen species (ROS) and NADPH oxidase (NOX), both of which are also upregulated in these cells. Inhibition of NOX activity resulted in HIF1α destabilization and reduced levels of cyclin D2, a marker of CGNP proliferation. As CGNPs are the putative cells of origin for the SHH subtype of medulloblastoma and aberrant SHH signaling is implicated in other neoplasms, these studies may also have future relevance in the context of cancer. Taken together, our findings suggest that a better understanding of nonhypoxic HIF1α stabilization through NOX-induced ROS generation can provide insights into normal cell proliferation in cerebellar development and SHH-driven cell proliferation in cancers with aberrant SHH signaling.
INTRODUCTION
The cerebellum, or “little brain,” comprises only ∼10% of the volume of the human brain but contains over half of the neurons of the brain (1). While cerebellar roles in coordination of sensory input and motor control have been established, roles in cognitive function are also being discovered, further emphasizing the importance of this part of the brain (2). The development of the cerebellum involves periods of rapid growth and maturation controlled by a myriad of intricate signaling pathways (3). Much of this cellular proliferation occurs postnatally, and the human cerebellum is not completely developed until many months after birth (4). The highly proliferative nature of cerebellar development renders this site susceptible to cellular transformation and provides an excellent model system for research of central nervous system (CNS) tumors (2). One example is medulloblastoma (MB), which is the most common solid pediatric malignancy of the CNS. Notably, approximately 30% of all cases of medulloblastoma are characterized by perturbations in the expression of Sonic Hedgehog (SHH) pathway components (5). Elucidating the processes underlying early cerebellar development and aberrant SHH signaling is crucial in understanding both physiologic and pathophysiologic SHH-driven proliferation.
Cerebellar granule neuron precursors (CGNPs), neural progenitors arising in the rhombic lip, are central to cerebellar maturation while also being the putative cells of origin for SHH MB (2, 6, 7). At birth, the mouse cerebellum consists of three layers: the external granule layer (EGL), where CGNPs first reside, the molecular layer (MOL), where Purkinje neurons localize, and the internal granule layer (IGL), where CGNPs ultimately translocate. SHH ligand is secreted by the Purkinje neurons and is required for CGNPs to undergo rapid proliferation in the EGL before migrating through the MOL to the IGL, where they terminally differentiate and mature into glutamatergic interneurons (8, 9). Binding of SHH to its receptor, Patched (PTCH), activates target genes in CGNPs that drive proliferation and inhibit differentiation (7, 10, 11). Importantly, many of these target genes have also been implicated in SHH medulloblastoma, including N-MYC, GLI1, and YAP (12–14). Primary CGNPs derived from postnatal day 5 (P5) mice are a well-established model for dissecting SHH mitogenic signaling (12, 15). These cells can be cultured and treated with exogenous SHH to maintain them in a proliferative state for up to 72 h.
Hypoxia-inducible factor 1 (HIF-1) is a heterodimeric helix-loop-helix transcription factor which acts as a master regulator of gene expression in response to tumor oxidative stress across a broad array of developmental pathways and cancers (16–18). HIF-1-driven gene expression has been extensively studied in cancer and regulates oxidative stress-induced metabolic reprogramming (increased glycolysis), stemness, angiogenesis, cell survival, invasion, metastasis, and therapy resistance (19–25). The rapid degradation of HIF-1-alpha (HIF1α) is the rate-limiting step in HIF-1 activation, and its stabilization has been an attractive area of research and a target for the development of novel therapies (26, 27).
Most research on HIF1α is done in regard to oxygenation status. Conventionally, under normoxic conditions, prolyl hydroxylases (PHDs) hydroxylate HIF1α, which is then sequestered and marked for proteasomal degradation by the von Hippel-Lindau (VHL) complex, a tumor-suppressive E3 ubiquitin ligase (28, 29). HIF1α is stabilized under hypoxic conditions when its hydroxylation-driven degradation is inhibited. Interestingly, PHDs can be inhibited by reactive oxygen species (ROS), which interfere with the ability of PHDs to tag HIF1α for degradation, leading to HIF1α stabilization independent of hypoxia (30–36). Because PHD-mediated hydroxylation of HIF1α requires ferrous iron (Fe2+) and elevated ROS within the cell favors the conversion to ferric iron (Fe3+), ROS can hinder PHD function (37).
Potential regulators of ROS within proliferating CGNPs include the NADPH oxidase (NOX) family of transmembrane proteins given their well-established enzymatic production of cellular ROS and the extensive literature describing them as essential modulators of signal transduction across various cell types (38, 39). The NOX family of proteins uniquely produce ROS as their primary function (40). Currently, five NOX homologues and two so-called dual oxidase (DUOX) enzymes featuring peroxidase activity in addition to oxidase function have been identified in humans; they vary in the amount/type/timing of ROS production and their organ-specific expression (41). NOX4 is the only member of the NOX family to display constitutive activation (42–44). Of note, PHD inhibition/HIF1α accumulation resulting from NOX activity has been studied in some cancer models (45). Extensive work has been done analyzing the relationship between NOX4- and HIF1α-driven tumor progression (38, 39, 46, 47). Little is known of how ROS, NOX, and HIF interact during cerebellar development, wherein the cells are rapidly proliferating, express stem cell markers, and exhibit a glycolytic phenotype (48, 49).
We used primary CGNP cultures to investigate potential interactions between SHH signaling and HIF1α. Our results indicate that mitogenic SHH signaling upregulates HIF1α but not at the mRNA level. Rather, we found that HIF1α is stabilized under nonhypoxic conditions. Along with sustained cell cycle progression, SHH pathway activation also increased ROS production in CGNPs. Our findings support NOX4-produced ROS downstream of SHH as a key contributor to HIF1α stabilization. Taken together, these observations suggest a role for ROS signaling in SHH-driven proliferation, including HIF1α stabilization. Moreover, these results also suggest therapeutic applications in SHH-driven cancers for targeting ROS production, which would destabilize HIF1α and promote cell cycle exit.
RESULTS
HIF1α is upregulated at the protein level in SHH-treated CGNPs.
We first aimed to determine the level of HIF1α in SHH-stimulated CGNPs, since these cells feature certain phenotypes attributable to HIF activity, including glycolysis and expression of stem cell markers such as nestin. To this end, CGNPs from P5 mice were harvested and vehicle- or SHH-treated for 48 h. Whole-cell protein lysates were generated and Western blotting was performed according to standard methods (see Materials and Methods). CGNPs that were exposed to SHH displayed higher levels of HIF1α protein than vehicle-treated CGNPs and CGNPs that were treated with both SHH and the Smoothened inhibitor cyclopamine (Fig. 1A). Cyclin D1 is used as a proliferation marker and confirms SHH pathway activation in CGNPs. Quantitative reverse transcription-PCR (RT-PCR) experiments revealed there was no biologically significant change in HIF1α gene expression compared to that of GLI1 as a positive control. (Fig. 1B). We surmise that HIF1α is active since we observe upregulation of its established target, PKM2, consistent with the glycolytic phenotype seen in CGNPs (48) and PHD2 and PHD3, downstream targets of HIF1α that are also elevated in CGNPs (Fig. 1C and D).
FIG 1.

HIF1α protein is upregulated in SHH-treated CGNPs. (A) CGNPs were harvested from P5 WT mice and incubated for 48 h in the presence of exogenous vehicle (Veh), SHH, or SHH plus cyclopamine (Cyc.). HIF1α levels were detected by immunoblotting and quantified using densitometry (ImageJ). Cyclin D1 levels served as a positive control of SHH pathway activation. (B) Hif1α transcripts are not significantly elevated in SHH-treated CGNPs in three sets of cDNA generated from separate CGNP RNA lysates. Gli1 is used as a positive control. (C) Transcripts of Pkm2, an HIF1α target, are elevated in SHH-treated CGNPs. (D) PHD2 and PHD3, downstream targets of HIF1α, are elevated in CGNPs. Data are shown as means ± standard errors of the means (SEM). n = 3; *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
As we observed no change in mRNA levels of HIF1α, we sought to determine changes at the translational level of control. We therefore treated CGNPs with a combination of SHH and the mTOR inhibitor rapamycin and observed a decrease in HIF1α over time (Fig. 2). Reduced phosphorylation of ribosomal protein S6 (rpS6) serves as confirmation of mTOR inhibition. This finding is consistent with previous work regarding mTOR and HIF1α, indicating that SHH-induced HIF1α elevation occurs posttranscriptionally (50). Rapamycin did not alter HIF1α mRNA expression. Even in the presence of SHH, treatment of CGNPs with the protein synthesis inhibitor cyclohexmide resulted in complete loss of HIF1α by 40 min (Fig. 2C and D).
FIG 2.

HIF1α is posttranslationally upregulated in SHH-treated CGNPs. (A) Primary CGNPs were harvested from P5 WT mice and incubated for 48 h in the presence or absence of exogenous SHH. HIF1α levels were detected by immunoblot analysis after treatment with rapamycin (Rapa; 10 nM) for the indicated time intervals. Decreased phosphorylation of rpS6 served as a positive control of mTOR inhibition with rapamycin. Cleaved caspase 3 (Cl. Casp. 3) levels were used to assess cell death. (B) Densitometric quantification (ImageJ) of HIF1α levels depicted in panel A. (C) Rapamycin treatment does not significantly affect HIF1α mRNA expression. (D) Cycloheximide (Chx) treatment (100 mg/ml) results in complete HIF1α degradation by 40 min. Data are shown as means ± SEM. n = 3; *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
SHH increases ROS production in CGNPs.
Because of HIF1α’s known interactions with oxygen regulation, we decided to investigate its relationship to reactive oxygen species when the SHH pathway is activated. To test the hypothesis that ROS activity is modified in response to SHH, we measured the relative concentration of ROS in CGNPs treated with exogenous SHH compared to the levels of ROS in the vehicle-treated CGNPs. We proceeded to thoroughly optimize a ROS assay using 5(and 6)-chloromethyl-2',7'-dichlorodihydro-fluorescein diacetate (CM-H2DCFDA) (Life Technologies). We also employed propidium iodide to gate out dead and dying cells according to the strategy shown in Fig. S1 in the supplemental material. SHH-treated cells consistently demonstrated a larger population of cells with higher ROS levels than vehicle- or SHH-plus-cyclopamine-treated cells. (Fig. 3). Combined with previous data, ROS induction resulting from SHH signaling follows the same pattern of increased HIF1α protein levels due to SHH stimulation, suggesting a role for ROS in HIF1α stabilization that we then further validated by treatment with the ROS scavenger N-acetyl-l-cysteine (NAC).
FIG 3.

Reactive oxygen species are elevated in SHH-treated CGNPs. CGNPs were harvested from P5 WT mice, treated with Veh, SHH, or SHH plus cyclopamine (SHH+Cyc), and collected after 48 h. They were then stained with CM-H2DCFDA and propidium iodide as described in Materials and Methods and analyzed by flow cytometry. Histograms from a representative experiment are shown with a marker showing the percentage of high-ROS events under each condition. Data are shown as means ± SEM. *, P < 0.05 (ratio-paired two-tailed t test; n = 3).
Scavenging of ROS reduces HIF1α stabilization in SHH-treated CGNPs.
With evidence linking SHH to higher levels of intracellular ROS production in CGNPs, we decided to scavenge these radicals using the antioxidant NAC in vitro and measured effects on HIF1α stabilization. CGNPs were cultured in the presence or absence of exogenous SHH (see Materials and Methods) in serum-free medium for 36 h before NAC (10 mM) was added and then replenished every 6 h for the indicated total times (Fig. 4). All cells were collected, lysed, and analyzed via Western blotting. SHH treatment alone showed the expected high levels of HIF1α and cyclin D1 protein, but levels of both diminished with NAC treatment over time (Fig. 4). These data support the involvement of SHH-induced ROS signaling in HIF1α hypoxia-independent regulation in our systems. As expected, NAC treatment resulted in reduced ROS levels (Fig. S1B).
FIG 4.
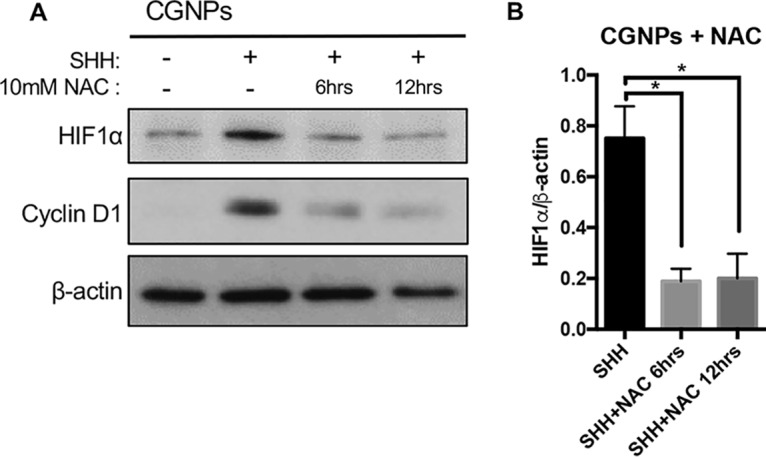
ROS scavenger NAC reduces HIF1α protein in SHH-treated CGNPs. CGNPs were harvested from P5 WT mice and incubated for 48 h in the presence or absence of exogenous SHH. Experimental wells were treated with 10 mM NAC every 6 h for up to 12 h. The cells were lysed and analyzed by Western blotting, and HIF1α levels were measured by densitometry (ImageJ). Data are shown as means ± SEM. n = 3; *, P < 0.05.
NADPH oxidase 4 is upregulated in SHH-treated CGNPs.
We next focused on identifying the source of ROS and decided to investigate the NADPH oxidase (NOX) family of proteins, as they are potent producers of nonmitochondrial ROS. After investigating several NADPH oxidases reported to be in the cerebellum, we found NOX4 upregulated in CGNPs downstream of SHH at the protein level but not at the mRNA level (Fig. 5A and B).
FIG 5.

NOX4 protein is elevated in SHH-treated CGNPs. (A) CGNPs were harvested from P5 WT mice and incubated for 48 h in the presence of exogenous Veh, SHH, or SHH plus cyclopamine (Cyc.). NOX4 protein levels were detected by Western blotting and quantified by densitometry (ImageJ). Cyclin D1 levels served as a positive control of SHH pathway activation. (B) Nox4 gene expression in CGNP lysates was performed by qPCR as described in Materials and Methods. Data are shown as means ± SEM. n = 3; *, P < 0.05.
NOX regulates HIF1α at the protein level and is necessary for proliferation.
We next wished to determine whether NOX inhibition, like ROS scavenging, would be sufficient to destabilize HIF1α. To this end, we used the NADPH oxidase inhibitor apocynin to see if SHH-related NOX4 activity could be linked to HIF1α’s dependency on ROS. We found HIF1α to be destabilized with several commonly used concentrations of apocynin (Fig. 6). Exposure to apocynin resulted in reduced ROS (Fig. S1C). We also screened a panel of short hairpin RNA (shRNA)-carrying lentiviruses predicted to target NOX4, and we observed that successful NOX4 knockdown was associated with reduction in HIF1α protein levels (Fig. 6B). Additionally, we found SHH-induced proliferation was attenuated in response to apocynin, as shown by the reduction in cyclin D2 at the protein level and GLI1 at the mRNA level (Fig. 6C) The observed reduction in GLI1 levels is in keeping with CGNPs losing their responsiveness to the SHH ligand as they are leaving the cell cycle (8).
FIG 6.

NOX4 regulates HIF1α protein and is necessary for proliferation. CGNPs were harvested from P5 WT mice and incubated for 48 h in the presence of exogenous Veh, SHH, or SHH plus apocyanine (Apo.). Several SHH-treated wells were exposed to increasing doses of the NADPH oxidase inhibitor apocynin for 12 h prior to lysis. (A) Western blot analysis of lysates obtained from the experiment. (B) CGNPs were harvested from P5 WT mice and incubated for 48 h in the presence of exogenous Veh, SHH, and scrambled shRNA carrying lentiviruses (Scr), Nox4-targeting shRNA lentiviruses produced using the clone numbers above the line or cyclopamine as indicated above the line. Successful knockdown of Nox4 was associated with reduced HIF1α protein levels. (C) qPCR analysis of RNA extracted from cells in the same experiment show a subsequent reduction in cell proliferation, as demonstrated by Gli1 expression. Fold inductions are shown with vehicle as the control. Data are shown as means ± SEM. n = 3; ****, P < 0.001.
DISCUSSION
The development of the cerebellum is a critical process mediated by SHH signaling. Mouse cerebellar development occurs largely postnatally and has become an important model for studying cerebellar development and SHH mitogenic signaling. Knowledge gained about this important signaling pathway and the downstream effects of SHH pathway activation can inform our understanding of cerebellar development and SHH-driven tumors. For example, medulloblastoma is the most common CNS malignancy among children, with approximately 30% of these tumors belonging to the Sonic Hedgehog molecular subclass. Investigating regulation of molecules driving overproliferation, such as inhibited differentiation, stemness, Warburg metabolism, and angiogenesis, may lead to identification of such therapeutic targets. In this context, HIF1α makes an attractive candidate due to the central role it plays in these hallmarks of cancer (19–25). Although HIF1α has been studied in the context of neural stem cells, it has not been explored in cerebellar progenitor cells, which have a defined cell fate yet express certain stem cell markers (51–53).
We employed an in vitro cerebellar granule neuron precursor (CGNP) primary culture system that allows us to closely study SHH mitogenic signaling and its downstream interactions/effectors. We first examined HIF1α status in these cells and found that SHH treatment resulted in HIF1α stabilization in the absence of mRNA increase. Since CGNPs reside adjacent to the meninges, which houses a highly oxygenated vascular network, we wished to investigate hypoxia-independent factors contributing to HIF1α stability. Accumulation of intracellular ROS has been shown to hinder the process by which HIF1α is degraded, independent of the oxygenation state (30–37, 54, 55). These reports are consistent with our finding that SHH pathway induction in CGNPs results in a significant increase in ROS levels along with marked HIF1α protein stabilization. Experiments aimed at directly sequestering ROS used the antioxidant NAC. Administering NAC to SHH-treated CGNPs resulted in diminished HIF1α levels.
The current literature supports the NOX family of proteins as being a significant regulator of intracellular ROS production in a multitude of cancers (38–41, 56, 57), and their activity has been linked to HIF1α stabilization (38, 39, 45–47). Indeed, we observed upregulation of NOX4 in CGNPs; there was no change in other NOX family members (data not shown). Taken together, our results indicate a role for NOX-generated ROS in hypoxia-independent HIF1α stabilization. Preliminary findings and other studies suggest that NOX4, one of the five human NOX homologues, is of particular interest within our paradigm (58–65).
Overall, our work supports a role for intracellular ROS in hypoxia-independent HIF1α stabilization in SHH-driven cerebellar progenitor cell proliferation (Fig. 7). These studies have important translational implications for developing reagents targeting HIF1α stabilization, such as novel antioxidant therapies, which could help elucidate novel developmental pathways and supplement the current standard of care in SHH-associated pathologies such as medulloblastoma, basal cell carcinoma, and a variety of adult solid tumors.
FIG 7.

NOX4 and ROS promote HIF1α stabilization. Shown is a schematic illustrating how NOX4 and ROS activity results in HIF1α protein accumulation in cells with elevated SHH activity.
MATERIALS AND METHODS
Animal studies.
Harvest of cerebellar granule neuron precursors from P4 or P5 neonatal wild-type (WT) mice and preparation of cerebella for cell culture or histological analysis were carried out in compliance with the Emory University institutional animal care and use committee guidelines, as was the harvest of tissue from NeuroD2-SmoA1 tumor-bearing mice. Mice used were not discriminated based on sex.
Cerebellar granule neuron precursor culture.
CGNP cultures were established as previously described (8). Cells were plated on poly-dl-ornithine (Sigma)-precoated plates. Where indicated, an SHH N-terminal fragment was used at a concentration of 3 μg/ml, and cyclopamine (R&D Systems) was used at 1 μg/ml. Cells were seeded in 10% fetal bovine serum (FBS) N2 medium for 2 h before being switched to serum-free N2 medium. Where indicated, NAC (Sigma), apocynin (Cayman Chemical), or rapamycin (Sigma) was administered in serum-free medium at the stated concentration for the given duration, up to 48 h of total culture. Notably, due to NAC’s short half-life, it was supplemented every 6 h during treatment. For infection with shRNA-carrying lentiviruses, CGNPs were plated in SHH for 24 h, and then the viral particles were added to the medium at a multiplicity of infection of 1. Particles were purchased from Sigma. The clones purchased were TRCN0000076583, TRCN0000076584, TRCN0000076585, TRCN000076586, and TRCN0000076587. A scrambled shRNA was also purchased.
Protein collection and immunoblotting.
Cells or tissues were collected, washed once with 1× phosphate-buffered saline (PBS), and then resuspended in complete lysis buffer. Whole-cell lysates were generated as previously described (8). To preserve HIF1α protein, special care was taken during collection with cells washed in ice-cold PBS and then collected by scraping in complete lysis buffer over ice. The Bradford assay and the Coomassie plus protein assay reagent (Thermo Scientific) were used to estimate protein concentrations. Twenty micrograms of protein from each sample was separated by SDS-PAGE on 8% or 10% gels and transferred to activated Immobilon polyvinylidene difluoride membranes (Millipore). Western blotting was carried out according to standard protocols. Primary antibodies used include anti-HIF1α (Novus Biologicals), anti-Nox4 (Abcam), anti-cleaved caspase-3 (Cell Signaling), anti-rpS6 (Cell Signaling), anti-cyclin D1 (Abcam), anti-cyclin D2 (Santa Cruz), anti-phospho-rpS6 (Cell Signaling), and anti-β-actin (Sigma). Horseradish peroxidase-conjugated secondary antibodies used were donkey anti-mouse (Jackson ImmunoResearch, Inc.) and goat anti-rabbit (Pierce) antibodies. Western blots were developed using ECL reagent (Thermo Scientific) and then exposing membranes to GE-Amersham chemiluminescence film for various periods of time to achieve optimal saturation. Quantification of relative protein levels was done by densitometry using ImageJ software (NIH).
RT-qPCR.
CGNP lysates were collected in TRIzol (Life Technologies), and RNA was extracted according to the manufacturer’s protocol. cDNA was then synthesized using the high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer’s protocol. Twenty nanograms of cDNA was added per well with the Bio-Rad SsoAdvanced universal SYBR green supermix and appropriate primer assay for the gene of interest. Primer assays purchased from Bio-Rad were for mRNA transcripts of GLI1, Hif1α, and Nox4 and the endogenous controls Actb and Gusb. With no available primer assay for Pkm2, custom primers for Pkm2 (forward, 5′-TCGCATGCAGCACCTGATT-3′; reverse, 5′-CCTCGAATAGCTGCAAGTGGTA-3′) (66) were ordered from Operon and added to the quantitative PCR (qPCR) assay at a concentration of 10 nM. The manufacturer’s protocol was followed, and qPCR was performed on a Bio-Rad CFX96 thermocycler with the fluorescent detection module. Analysis was done according to the ΔΔCT method (where is CT threshold cycle) of measuring relative gene expression. The runs were analyzed on a Bio-Rad CFX manager software, and three replicate runs were combined into gene studies adhering to MIQE standards.
Reactive oxygen species assay.
CGNPs were cultured as stated above in 6-well plates. At 48 h, CGNPs were washed in warm 1× PBS and harvested by trypsinization (250 μl of 0.05% trypsin). Ten percent FBS N2 medium was added to stop trypsinization, and cells were transferred to 1.5-ml tubes, where they were washed in warm 1× PBS, pelleted, and resuspended in warm 1× Hanks’ balanced salt solution (HBSS) and transferred to black 1.5-ml tubes, where 2 μl of 10 μM CM-H2DCFDA (Life Technologies) solution was added. Samples were placed in a cell incubator (37°C) with high relative humidity (95%) and controlled CO2 level (5%) in the dark for 45 min. CGNPs were resuspended by pipetting, propidium iodide was added to a concentration of 1 μg/ml, and 200-μl aliquots were added to a 96-well plate and analyzed by flow cytometry on a Beckman Coulter Cytoflex at the Emory University Pediatrics Flow Cytometry Core. CM-H2DCFDA was analyzed on the fluorescein isothiocyanate channel, and propidium iodide was analyzed on the peridinin chlorophyll protein channel. All conditions in each run were subject to the same gain settings. Once the CGNP population was gated in and doublets gated out, the remaining propidium iodide-positive population denoting dead cells was excluded. Five thousand events were then selected from the remaining live population under each condition, and a marker for the high-ROS population was created for each experimental condition covering the same range of signal strength in the FCS Express 6 software (De Novo Software).
Statistical analysis.
Statistical analysis was performed using Prism 6 (GraphPad Software, Inc.) or FCS Express 6 (De Novo Software) and Bio-Rad CFX manager for flow cytometry and qPCR analyses, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NINDS R01NS061070 (A.M.K.) and NCI Winship Cancer Institute P30 Center grant CA138292 (A.M.K.).
We thank Nicholas W. Eyrich’s Cancer Biology 4 + 1 master’s thesis committee (Mala Shanmugam and Keith Wilkinson) for valuable feedback on the project and Pablo Eyrich Freile for technical assistance.
N.W.E., C.R.P., M.H.R., and V.M. designed and carried out experiments, analyzed results, prepared figures, and wrote and edited the manuscript. A.M.K. oversaw the study, designed experiments, analyzed results, designed and edited figures, and wrote/edited the manuscript.
We have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00268-18.
REFERENCES
- 1.Wang VY, Zoghbi HY. 2001. Genetic regulation of cerebellar development. Nat Rev Neurosci 2:484–491. doi: 10.1038/35081558. [DOI] [PubMed] [Google Scholar]
- 2.Hatten ME, Roussel MF. 2011. Development and cancer of the cerebellum. Trends Neurosci 34:134–142. doi: 10.1016/j.tins.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leto K, Arancillo M, Becker EB, Buffo A, Chiang C, Ding B, Dobyns WB, Dusart I, Haldipur P, Hatten ME, Hoshino M, Joyner AL, Kano M, Kilpatrick DL, Koibuchi N, Marino S, Martinez S, Millen KJ, Millner TO, Miyata T, Parmigiani E, Schilling K, Sekerkova G, Sillitoe RV, Sotelo C, Uesaka N, Wefers A, Wingate RJ, Hawkes R. 2016. Consensus paper: cerebellar development. Cerebellum 15:789–828. doi: 10.1007/s12311-015-0724-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marzban H, Del Bigio MR, Alizadeh J, Ghavami S, Zachariah RM, Rastegar M. 2014. Cellular commitment in the developing cerebellum. Front Cell Neurosci 8:450. doi: 10.3389/fncel.2014.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM. 2012. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eberhart CG. 2008. Even cancers want commitment: lineage identity and medulloblastoma formation. Cancer Cell 14:105–107. doi: 10.1016/j.ccr.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behesti H, Marino S. 2009. Cerebellar granule cells: insights into proliferation, differentiation, and role in medulloblastoma pathogenesis. Int J Biochem Cell Biol 41:435–445. doi: 10.1016/j.biocel.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 8.Kenney AM, Rowitch DH. 2000. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 20:9055–9067. doi: 10.1128/MCB.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wechsler-Reya RJ, Scott MP. 1999. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22:103–114. doi: 10.1016/S0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 10.Dahmane N, Ruiz i Altaba A. 1999. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126:3089–3100. [DOI] [PubMed] [Google Scholar]
- 11.Wallace VA. 1999. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9:445–448. doi: 10.1016/S0960-9822(99)80195-X. [DOI] [PubMed] [Google Scholar]
- 12.Kenney AM, Widlund HR, Rowitch DH. 2004. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development 131:217–228. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- 13.Kimura H, Stephen D, Joyner A, Curran T. 2005. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene 24:4026–4036. doi: 10.1038/sj.onc.1208567. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, Taylor MD, Kenney AM. 2009. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev 23:2729–2741. doi: 10.1101/gad.1824509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guldal CG, Ahmad A, Korshunov A, Squatrito M, Awan A, Mainwaring LA, Bhatia B, Parathath SR, Nahle Z, Pfister S, Kenney AM. 2012. An essential role for p38 MAPK in cerebellar granule neuron precursor proliferation. Acta Neuropathol 123:573–586. doi: 10.1007/s00401-012-0946-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keith B, Johnson RS, Simon MC. 2011. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pouyssegur J, Dayan F, Mazure NM. 2006. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 18.Wang GL, Jiang BH, Rue EA, Semenza GL. 1995. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ajdukovic J. 2016. HIF-1–a big chapter in the cancer tale. Exp Oncol 38:9–12. doi: 10.31768/2312-8852.2016.38(1):9-12. [DOI] [PubMed] [Google Scholar]
- 20.Mimeault M, Batra SK. 2013. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J Cell Mol Med 17:30–54. doi: 10.1111/jcmm.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadri N, Zhang PJ. 2013. Hypoxia-inducible factors: mediators of cancer progression; prognostic and therapeutic targets in soft tissue sarcomas. Cancers (Basel) 5:320–333. doi: 10.3390/cancers5020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semenza GL. 2010. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semenza GL. 2011. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 1813:1263–1268. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semenza GL. 2016. Targeting hypoxia-inducible factor 1 to stimulate tissue vascularization. J Investig Med 64:361–363. doi: 10.1097/JIM.0000000000000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Semenza GL. 2010. Vascular responses to hypoxia and ischemia. Arterioscler Thromb Vasc Biol 30:648–652. doi: 10.1161/ATVBAHA.108.181644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burroughs SK, Kaluz S, Wang D, Wang K, Van Meir EG, Wang B. 2013. Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med Chem 5:553–572. doi: 10.4155/fmc.13.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Semenza GL. 2003. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 28.Mimura I, Nangaku M. 2010. The suffocating kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat Rev Nephrol 6:667–678. doi: 10.1038/nrneph.2010.124. [DOI] [PubMed] [Google Scholar]
- 29.Semenza GL. 2010. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klumpen E, Hoffschroer N, Zeis B, Gigengack U, Dohmen E, Paul RJ. 2017. Reactive oxygen species (ROS) and the heat stress response of Daphnia pulex: ROS-mediated activation of hypoxia-inducible factor 1 (HIF-1) and heat shock factor 1 (HSF-1) and the clustered expression of stress genes. Biol Cell 109:39–64. doi: 10.1111/boc.201600017. [DOI] [PubMed] [Google Scholar]
- 31.Lim S, Liu H, Madeira da Silva L, Arora R, Liu Z, Phillips JB, Schmitt DC, Vu T, McClellan S, Lin Y, Lin W, Piazza GA, Fodstad O, Tan M. 2016. Immunoregulatory protein B7-H3 reprograms glucose metabolism in cancer cells by ROS-mediated stabilization of HIF1alpha. Cancer Res 76:2231–2242. doi: 10.1158/0008-5472.CAN-15-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarkar R, Mukherjee S, Biswas J, Roy M. 2016. Phenethyl isothiocyanate, by virtue of its antioxidant activity, inhibits invasiveness and metastatic potential of breast cancer cells: HIF-1alpha as a putative target. Free Radic Res 50:84–100. doi: 10.3109/10715762.2015.1108520. [DOI] [PubMed] [Google Scholar]
- 33.Seo S, Seo K, Ki SH, Shin SM. 2016. Isorhamnetin inhibits reactive oxygen species-dependent hypoxia inducible factor (HIF)-1alpha accumulation. Biol Pharm Bull 39:1830–1838. doi: 10.1248/bpb.b16-00414. [DOI] [PubMed] [Google Scholar]
- 34.Shatrov VA, Sumbayev VV, Zhou J, Brune B. 2003. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood 101:4847–4849. doi: 10.1182/blood-2002-09-2711. [DOI] [PubMed] [Google Scholar]
- 35.Shida M, Kitajima Y, Nakamura J, Yanagihara K, Baba K, Wakiyama K, Noshiro H. 2016. Impaired mitophagy activates mtROS/HIF-1alpha interplay and increases cancer aggressiveness in gastric cancer cells under hypoxia. Int J Oncol 48:1379–1390. doi: 10.3892/ijo.2016.3359. [DOI] [PubMed] [Google Scholar]
- 36.Wang M, Kirk JS, Venkataraman S, Domann FE, Zhang HJ, Schafer FQ, Flanagan SW, Weydert CJ, Spitz DR, Buettner GR, Oberley LW. 2005. Manganese superoxide dismutase suppresses hypoxic induction of hypoxia-inducible factor-1alpha and vascular endothelial growth factor. Oncogene 24:8154–8166. doi: 10.1038/sj.onc.1208986. [DOI] [PubMed] [Google Scholar]
- 37.Taylor CT. 2008. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J 409:19–26. doi: 10.1042/BJ20071249. [DOI] [PubMed] [Google Scholar]
- 38.Jiang F, Zhang Y, Dusting GJ. 2011. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev 63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 39.Teixeira G, Szyndralewiez C, Molango S, Carnesecchi S, Heitz F, Wiesel P, Wood JM. 2016. Therapeutic potential of NADPH oxidase 1/4 inhibitors. Br J Pharmacol doi: 10.1111/bph.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bedard K, Krause KH. 2007. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 41.Brandes RP, Weissmann N, Schroder K. 2014. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med 76:208–226. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 42.Geiszt M, Kopp JB, Varnai P, Leto TL. 2000. Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci U S A 97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helmcke I, Heumuller S, Tikkanen R, Schroder K, Brandes RP. 2009. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid Redox Signal 11:1279–1287. doi: 10.1089/ars.2008.2383. [DOI] [PubMed] [Google Scholar]
- 44.von Lohneysen K, Noack D, Hayes P, Friedman JS, Knaus UG. 2012. Constitutive NADPH oxidase 4 activity resides in the composition of the B-loop and the penultimate C terminus. J Biol Chem 287:8737–8745. doi: 10.1074/jbc.M111.332494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li YN, Xi MM, Guo Y, Hai CX, Yang WL, Qin XJ. 2014. NADPH oxidase-mitochondria axis-derived ROS mediate arsenite-induced HIF-1alpha stabilization by inhibiting prolyl hydroxylases activity. Toxicol Lett 224:165–174. doi: 10.1016/j.toxlet.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 46.Meng D, Mei A, Liu J, Kang X, Shi X, Qian R, Chen S. 2012. NADPH oxidase 4 mediates insulin-stimulated HIF-1alpha and VEGF expression, and angiogenesis in vitro. PLoS One 7:e48393. doi: 10.1371/journal.pone.0048393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mondol AS, Tonks NK, Kamata T. 2014. Nox4 redox regulation of PTP1B contributes to the proliferation and migration of glioblastoma cells by modulating tyrosine phosphorylation of coronin-1C. Free Radic Biol Med 67:285–291. doi: 10.1016/j.freeradbiomed.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Bhatia B, Potts CR, Guldal C, Choi S, Korshunov A, Pfister S, Kenney AM, Nahle ZA. 2012. Hedgehog-mediated regulation of PPARgamma controls metabolic patterns in neural precursors and shh-driven medulloblastoma. Acta Neuropathol 123:587–600. doi: 10.1007/s00401-012-0968-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gogvadze V, Orrenius S, Zhivotovsky B. 2008. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 18:165–173. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 50.Abraham RT. 2004. mTOR as a positive regulator of tumor cell responses to hypoxia. Curr Top Microbiol Immunol 279:299–319. [DOI] [PubMed] [Google Scholar]
- 51.Candelario KM, Shuttleworth CW, Cunningham LA. 2013. Neural stem/progenitor cells display a low requirement for oxidative metabolism independent of hypoxia inducible factor-1alpha expression. J Neurochem 125:420–429. doi: 10.1111/jnc.12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.VanDlac AA, Cowan NG, Chen Y, Anderson RE, Conlin MJ, La Rochelle JC, Amling CL, Koppie TM. 2014. Timing, incidence and risk factors for venous thromboembolism in patients undergoing radical cystectomy for malignancy: a case for extended duration pharmacological prophylaxis. J Urol 191:943–947. doi: 10.1016/j.juro.2013.10.096. [DOI] [PubMed] [Google Scholar]
- 53.Roitbak T, Surviladze Z, Cunningham LA. 2011. Continuous expression of HIF-1alpha in neural stem/progenitor cells. Cell Mol Neurobiol 31:119–133. doi: 10.1007/s10571-010-9561-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rigiracciolo DC, Scarpelli A, Lappano R, Pisano A, Santolla MF, De Marco P, Cirillo F, Cappello AR, Dolce V, Belfiore A, Maggiolini M, De Francesco EM. 2015. Copper activates HIF-1alpha/GPER/VEGF signalling in cancer cells. Oncotarget 6:34158–34177. doi: 10.18632/oncotarget.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. 2007. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res 67:10823–10830. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 56.Heppner DE, van der Vliet A. 2016. Redox-dependent regulation of epidermal growth factor receptor signaling. Redox Biol 8:24–27. doi: 10.1016/j.redox.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roy K, Wu Y, Meitzler JL, Juhasz A, Liu H, Jiang G, Lu J, Antony S, Doroshow JH. 2015. NADPH oxidases and cancer. Clin Sci (Lond) 128:863–875. doi: 10.1042/CS20140542. [DOI] [PubMed] [Google Scholar]
- 58.Bonello S, Za¨Hringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, Kietzmann T, Go¨Rlach A. 2007. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol 27:755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- 59.Diebold I, Flugel D, Becht S, Belaiba RS, Bonello S, Hess J, Kietzmann T, Gorlach A. 2010. The hypoxia-inducible factor-2alpha is stabilized by oxidative stress involving NOX4. Antioxid Redox Signal 13:425–436. doi: 10.1089/ars.2009.3014. [DOI] [PubMed] [Google Scholar]
- 60.Helfinger V, Henke N, Harenkamp S, Walter M, Epah J, Penski C, Mittelbronn M, Schroder K. 2016. The NADPH oxidase Nox4 mediates tumour angiogenesis. Acta Physiol (Oxf) 216:435–446. doi: 10.1111/apha.12625. [DOI] [PubMed] [Google Scholar]
- 61.Hsieh CH, Shyu WC, Chiang CY, Kuo JW, Shen WC, Liu RS. 2011. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PLoS One 6:e23945. doi: 10.1371/journal.pone.0023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, Wang JJ, Yu Q, Chen K, Mahadev K, Zhang SX. 2010. Inhibition of reactive oxygen species by Lovastatin downregulates vascular endothelial growth factor expression and ameliorates blood-retinal barrier breakdown in db/db mice: role of NADPH oxidase 4. Diabetes 59:1528–1538. doi: 10.2337/db09-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Przybylska D, Janiszewska D, Goździk A, Bielak-Zmijewska A, Sunderland P, Sikora E, Mosieniak G. 2016. NOX4 downregulation leads to senescence of human vascular smooth muscle cells. Oncotarget 7:66429–66443. doi: 10.18632/oncotarget.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. 2007. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanaka M, Miura Y, Numanami H, Karnan S, Ota A, Konishi H, Hosokawa Y, Hanyuda M. 2015. Inhibition of NADPH oxidase 4 induces apoptosis in malignant mesothelioma: role of reactive oxygen species. Oncol Rep 34:1726–1732. doi: 10.3892/or.2015.4155. [DOI] [PubMed] [Google Scholar]
- 66.Panasyuk G, Espeillac C, Chauvin C, Pradelli LA, Horie Y, Suzuki A, Annicotte JS, Fajas L, Foretz M, Verdeguer F, Pontoglio M, Ferre P, Scoazec JY, Birnbaum MJ, Ricci JE, Pende M. 2012. PPARgamma contributes to PKM2 and HK2 expression in fatty liver. Nat Commun 3:672. doi: 10.1038/ncomms1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.