

SUMMARY
The reactions catalyzed by the delta-5 and −6 desaturases (D5D/D6D), key enzymes responsible for highly unsaturated fatty acid (HUFA) synthesis, regenerate NAD+ from NADH. Here, we show that D5D/D6D provide a mechanism for glycolytic NAD+ recycling that permits ongoing glycolysis and cell viability when the cytosolic NAD+/NADH ratio is reduced, analogous to lactate fermentation. Although lesser in magnitude than lactate production, this desaturase mediated NAD+ recycling is acutely adaptive when aerobic respiration is impaired in vivo. Notably, inhibition of either HUFA synthesis or lactate fermentation increases the other, underscoring their interdependence. Consistent with this, a type 2 diabetes risk haplotype in SLC16A11 that reduces pyruvate transport (thus limiting lactate production) increases D5D/D6D activity in vitro and in humans, demonstrating a chronic effect of desaturase mediated NAD+ recycling. These findings highlight key biologic roles for D5D/D6D activity independent of their HUFA end-products and expand the current paradigm of glycolytic NAD+ regeneration.
Keywords: FADS1–3, SLC16A11, Delta-5-desaturase, Delta-6-desaturase, Polyunsaturated fatty acids, Highly unsaturated fatty acids, NAD+ recycling
In Brief
Kim et al. find that highly unsaturated fatty acid (HUFA) synthesis is a mechanism for glycolytic NAD+ recycling, analogous to lactate fermentation. This finding highlights a key biologic role for lipid desaturation independent of the HUFA end-products and provides insight on genetic studies linking HUFA desaturation with human disease.
Graphical Abstract
INTRODUCTION
The delta-5 desaturase (D5D) and delta-6 desaturase (D6D), encoded by FADS1 and FADS2 respectively, are required for the synthesis of highly unsaturated fatty acids (HUFAs) which are then esterified into cellular lipids (Nakamura and Nara, 2004). These enzymes introduce double bonds at fixed positions from the carboxyl end during the successive elongation and desaturation of the essential fatty acid linoleic acid (18:2n-6) to arachidonic acid (20:4n-6), and α-linolenic acid (18:3n-3) to eicosapentaenoic (20:5n-3) and docosahexaenoic (22:6n-3) acid. The FADS1 and FADS2 genes are oriented head-to-head on chromosome 11, and common variants in this region associated with alterations in HUFA content in circulating lipids (Gieger et al., 2008; Rhee et al., 2013) have also been associated with fasting glucose and type 2 diabetes (Dupuis et al., 2010; Fujita et al., 2012), anthropometric traits (Fumagalli et al., 2015), and cancer risk (Wei et al., 2014; Zhang et al., 2014). In addition, we have found that lipid HUFA content can change rapidly, increasing in plasma triacylglycerols (TAGs) within two hours of various glycolytic stimuli, including oral glucose ingestion, sulfonylurea administration, and exercise (Rhee et al., 2011). Why HUFA synthesis is so dynamic, and why genetic variation in this response has such a broad impact on human metabolic and proliferative phenotypes is unknown.
In glycolysis, glucose metabolism is coupled to the reduction of cytosolic nicotinamide adenine dinucleotide (NAD+) to NADH. Under aerobic conditions, the transfer of electrons into mitochondria and ultimately to the mitochondrial electron transport chain (ETC) can regenerate NAD+, whereas the cytosolic reduction of pyruvate to lactate can regenerate NAD+ when mitochondrial respiration is impaired. In either case, the flow of electrons from cytosolic NADH to an available electron acceptor restores cytosolic NAD+ and permits ongoing glycolysis. Notably, the reactions catalyzed by D5D and D6D also recycle NADH to NAD+. These enzymes contain an N-terminal cytochrome b5 domain that is required for electron transfer from NADH cytochrome b5 reductase and the substrate fatty acid to molecular O2, yielding a product fatty acid with an additional double bond, H2O, and NAD+ (Cho et al., 1999a; Cho et al., 1999b; Napier et al., 2003). In addition, D5D and D6D are endoplasmic reticulum (ER) membrane spanning enzymes, with catalytic domains that face the cytoplasmic pool of NAD+ and NADH (Fujiwara et al., 1984; Park et al., 2015; Park et al., 2012; Watanabe et al., 2016).
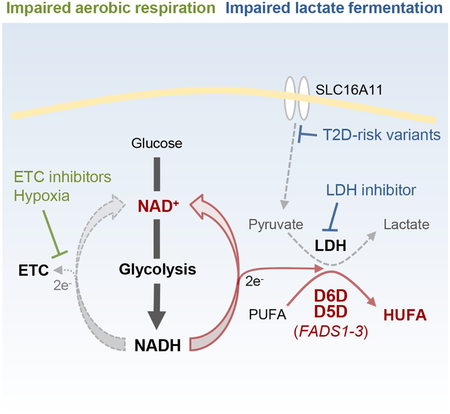
Here, we test the hypothesis that HUFA production provides a mechanism for glycolytic NAD+ recycling, analogous to lactate fermentation. These studies utilize liquid chromatography-mass spectrometry (LC-MS) based profiling methods that readily differentiate cellular lipids on the basis of double bond content (Jain et al., 2014; Rhee et al., 2011). In addition, they capitalize on the recent development of key tools for studying cytosolic NAD+ recycling, including a genetically encoded fluorescent sensor of the cytosolic NAD+/NADH ratio (Zhao et al., 2015), a recombinant NADH oxidase isolated from Lactobacillus brevis (Titov et al., 2016), and the use of alpha-ketobutyrate (AKB) to drive lactate dehydrogenase (LDH) activity (Sullivan et al., 2015). Together, these studies highlight D5D and D6D as an alternative to LDH for the flow of reducing equivalents generated during glycolysis, in vitro and in vivo. Finally, we demonstrate how this mechanism converges with the recent description of SLC16A11 as a pyruvate transporter, shedding light on the association between type 2 diabetes risk variants in SLC16A11 and altered lipid metabolism (Rusu et al., 2017; Williams et al., 2014).
RESULTS
Inhibition of Aerobic Respiration Increases Cellular HUFA Content
To test if inhibition of aerobic respiration and the subsequent increase in glycolysis impacts cellular lipid HUFA content, we treated mouse renal epithelial cells (IMCD3) with the mitochondrial ETC complex I inhibitor rotenone and profiled lipids using LC-MS. Along with the expected increase in media glucose consumption and lactate secretion (Figure 1A), twenty-four hour rotenone treatment causes a preferential increase in media HUFAs, e.g. arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid (Student’s t-test, P < 0.001 for all) (Figure 1B). Within cells, the increase in HUFA content is captured as a pattern of greater increases in highly unsaturated lipids (Figures 1C–1F). This pattern is evident among cholesterol esters, phosphatidylcholines, diacylglycerols and most dramatically TAGs (Figure 1F), where HUFA-containing TAGs such as TAG 54:9, TAG 56:10, and TAG 58:11 increase >50-fold following rotenone treatment (Bonferroni adjusted P < 0.05 for all; for each TAG, the first number denotes the total number of carbons and the second number denotes the total number of double bonds in the three acyl chains). Although different combinations of three fatty acyl chains can sum to these carbon and double bond totals, by definition these TAGs contain HUFAs in order to reach these high total double bond counts; we have also confirmed this experimentally using MS/MS fragmentation (Rhee et al., 2011). In contrast to the large increases in HUFA-containing TAGs, which represent a small fraction of total cellular TAGs, total triglycerides only increase 1.8-fold (Student’s t-test, P = 0.001) with rotenone treatment (Figure 1G and S1A). Similar findings were observed with different cell types and other methods of inhibiting aerobic respiration such as oligomycin treatment (mitochondrial ATP synthase inhibitor) and hypoxia (Figure S1B–S1D).
Figure 1. Inhibition of aerobic respiration increases lipid HUFA content in vitro.

(A-G) Effect of 24h rotenone (Rot, 200 nM) treatment of IMCD3 cells on (A) media glucose consumption and lactate secretion; (B) media free fatty acid levels, including linoleic acid (LA), alpha-linolenic acid (α-LA), gamma-linolenic acid (γ-LA), arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA); intracellular (C) cholesterol esters, (D) phosphatidylcholines, (E) diacylglycerols, and (F) triacylglycerols (TAGs) measured by LC-MS, differentiated along the x-axis on the basis of total acyl chain carbon and double bond content; and (G) total TAGs measured by colorimetric assay. Values are means ± SEM; *P < 0.05, **P < 0.01; ***P < 0.001; n = 3.
See also Figure S1.
Inhibition of Aerobic Respiration Increases D5D and D6D Activity
Whereas the decrease in β-oxidation that results from impaired aerobic respiration would be expected to cause some TAG accumulation, the magnitude and pattern of HUFA-containing TAG increases following rotenone treatment suggest a specific increase in HUFA synthesis as well. Indeed, this rotenone induced increase in HUFAs occurs rapidly, within 2 hours (Figure 2A), and is not associated with an increase in Fadsl or Fads2 mRNA or D5D protein levels (Figure 2B). In addition, rotenone does not change the distribution of D5D within the cell, with immunofluorescence demonstrating the enzyme’s diffuse expression throughout the ER (Figures 2C and S2A–S2D). We confirmed that the increase in HUFA-containing lipids following rotenone is due to an increase in D5D and D6D activity by measuring the conversion of isotope-labeled substrate (linoleic acid-13C18) to isotope-labeled products (gamma-linolenic acid-13C18 and arachidonic acid-13C18) (14.5-fold and 4.4-fold increase at 24h, respectively, Student’s t-test, P <0.001 for both) (Figures 2D and 2E). In turn, enzyme inhibition with either CP-24879 (dual D5D and D6D inhibitor) or SC-26196 (D6D inhibitor) prevents the rotenone induced increase in HUFA-containing TAGs (Figures 2F, S2E, S2F, and Table S1) (Obukowicz et al., 1998a; Obukowicz et al., 1998b). Thus, our data demonstrate that inhibition of aerobic respiration increases cellular lipid HUFA content by increasing D5D and D6D activity.
Figure 2. Inhibition of aerobic respiration increases D5D and D6D activity.

(A) Time course of rotenone effect on cellular TAGs.
(B) Top, western blot of D5D in IMCD3 cells following 2, 6, 12, and 24h of rotenone (200 nM) or vehicle. Representative gel from one of two independent experiments. Bottom, qPCR of Fads1 and Fads2 in IMCD3 cells following 6h rotenone (200 nM) or vehicle.
(C) Confocal micrograph of D5D (green), the ER marker reticulon 4 (RTN4, red), or merged image (yellow) in IMCD3 cells following 6h of 200 nM rotenone or vehicle.
(D-E) Isotope-labeled (D) free fatty acid products and (E) substrate of D5D and D6D activity in cultured media of IMCD3 cells following 6h and 24h of rotenone (200 nM) or vehicle.
(F) Intracellular TAGs in IMCD3 cells following 24h of rotenone (200 nM) with or without the desaturase inhibitors SC-26196 (10 μM) or CP-24879 (30 μM). Values denote means ± SEM; ns, P > 0.05.; **P < 0.01; ***P < 0.001; n = 3.
D5D and D6D Activity Increase in Response to Decreased Cytosolic NAD+/NADH Ratio
We next investigated the mechanism by which impaired aerobic respiration regulates D5D and D6D activity. The reactions catalyzed by D5D and D6D regenerate NAD+ from NADH (Figure 3A). Inhibition of aerobic cellular respiration increases cytosolic NADH levels, decreasing the cytosolic NAD+/NADH ratio; by contrast, rotenone does not alter the NADP+/NAPDH ratio (Figure 3B) (Zhao et al., 2011). To determine whether this decrease in cytosolic NAD+/NADH is responsible for the increase in D5D and D6D activity, we tested the effect of increasing the NAD+/NADH ratio using LbNOX, a recombinant NADH oxidase isolated from Lactobacillus brevis (Titov et al., 2016). This enzyme has a very low Km for O2, is compartment-specific (i.e. cytosolic), and is highly selective for NADH over NADPH. Using SoNar (Zhao et al., 2015), a genetically encoded fluorescent sensor for cytosolic NAD+/NADH, we confirmed that LbNOX expression causes a sustained increase in the cytosolic NAD+/NADH ratio (Figure 3C). Further, whereas LbNOX expression has no impact on FADS1 or FADS2 mRNA or D5D protein levels (Figure 3D), its impact on cytosolic NAD+/NADH is sufficient to reduce TAG HUFA content (Figure 3E). In cells treated with rotenone, LbNOX expression attenuates both the decrease in cytosolic NAD+/NADH (Figure 3C) and increase in HUFA-containing lipids (Figure 3F).
Figure 3. Increasing the cytosolic NAD+/NADH ratio with an NADH oxidase reduces D5D and D6D activity.

(A) Schema for D5D and D6D mediated desaturation and NAD+ recycling.
(B) Effect of rotenone on total cellular NAD+/NADH and NADP+/NAPDH ratios. NAD+/NADH ratio (left) and NADP+/NADPH ratio (right) measured using an enzymatic assay of cell lysates from IMCD3 cells treated with rotenone (200 nM) or vehicle for 6 hours.
(C) Effect of rotenone (100 nM) treatment of HeLa cells, with or without LbNOX expression, on cytosolic NAD+/NADH measured using the ratio of fluorescence intensities excited at 485 nm and 430 nm in SoNar expressing cells.
(D) qPCR (left) of FADS1 and FADS2 in HeLa cells with or without LbNOX expression (+/− Dox); western blot (right) of D5D in HeLa cells with or without LbNOX expression (+/− Dox), 6h following rotenone (200 nM) or vehicle.
(E and F) Effect of LbNOX expression on HeLa cell TAGs measured by LC-MS (E) relative to vehicle, or (F) with or without rotenone (100 nM at 6h). Values are means ± SEM; ns, P > 0.05; ***P < 0.001; n = 3 for all experiments except n = 10 for (C).
An alternative method to increase the cytosolic NAD+/NADH ratio is supplementation with alpha-ketobutyrate (AKB) or pyruvate (King and Attardi, 1989; Sullivan et al., 2015). Both molecules are substrates for lactate dehydrogenase (LDH), accepting electrons from NADH to regenerate NAD+ and produce either alpha-hydroxybutyrate (AHB, from AKB) or lactate (from pyruvate) (Figure 4A). An important feature of AKB is that it acts only as an electron acceptor and does not directly contribute carbon molecules to metabolism, e.g. fatty acid synthesis (Sullivan et al., 2015). In cells treated with AKB, AHB levels increase 110-fold (Student’s t-test, P< 0.001), and this conversion is further augmented to >340-fold (Student’s t-test, P< 0.001) in the presence of rotenone (Figure 4B). In turn, increasing LDH activity with either AKB or pyruvate attenuates both the decrease in cytosolic NAD+/NADH ratio (Figures 4C, S3A, and S3B) and the increase in HUFA-containing TAGs following rotenone treatment (Figures 4D and S3C–E); under experimental conditions where the cytosolic NAD+/NADH is normalized for the entire duration of the experiment, the effect of rotenone on HUFA TAG synthesis is rescued completely (Figures S3F and S3G). By contrast, AKB supplementation has minimal impact on tricarboxylic acid cycle intermediate levels, which decrease ~90% following treatment with rotenone (Figure 4E). These findings are consistent with AKB modulating HUFA synthesis through its impact on cytosolic NAD+ recycling rather than a direct effect on mitochondrial metabolism. In isotope studies, AKB reduces the conversion of linoleic acid-13C18 to gamma-linolenic acid-13C18 (0.66-fold at 24h, Student’s t-test, P < 0.001) and arachidonic acid-13C18 (0.40-fold at 24h, Student’s t-test, P = 0.002) following rotenone, without reducing the overall consumption of linoleic acid-13C18 (Figures 4F and 4G).
Figure 4. Modulating the cytosolic NAD+/NADH ratio via LDH activity modulates D5D and D6D activity.

Schema for LDH mediated NAD+ recycling; alpha-ketobutyrate (AKB), alpha-hydroxybutyrate (AHB), NHI-2 (LDH inhibitor).
(B-G) Effect of rotenone (200 nM) treatment of IMCD3 cells, with or without AKB (ImM), on (B) intracellular AHB at 24h; (C) cytosolic NAD+/NADH measured with SoNar; (D) intracellular TAGs at 24h; (E) intracellular citrate, isocitrate, and succinate at 24h; and (F and G) isotope-labeled free fatty acids in media at 6h and 24h.
(H-I) Effect of NHI-2 (30 μM) and rotenone (100 nM) treatment of IMCD3 cells on (H) cytosolic NAD+/NADH measured with SoNar, where box plot shows upper and lower quartiles and whiskers show maximum and minimum values; and (I) intracellular TAGs at 24h. Values denote means ± SEM; ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; n = 3 for all experiments except n = 10 for (C, H).
See also Figure S3.
In contrast to LDH activation with AKB or pyruvate, treatment with the LDH inhibitor NHI-2 decreases the cytosolic NAD+/NADH ratio (Figure 4H) and significantly increases HUFA-containing TAGs (Figure 4I) (Calvaresi et al., 2013). The combination of NHI-2 and rotenone results in even greater increases in HUFA-containing TAGs than either alone, e.g. with Bonferroni adjusted P < 0.05 for TAG 54:9, TAG 56:10, and TAG 58:11 (Figure 4I). Collectively, these studies of ETC inhibition, NADH oxidase expression, and LDH activation and inhibition demonstrate the dynamic relationship between the cytosolic NAD+/NADH ratio and cellular HUFA synthesis.
Modulation of D5D and D6D Alters the Cytosolic NAD+/NADH Ratio, Lactate Production, and Cell Proliferation
Because our data implicate D5D and D6D in glycolytic NAD+ recycling, we next tested the effect of modulating D5D and D6D on cell metabolism. Transient overexpression of D5D and D6D increases the cytosolic NAD+/NADH ratio (Student’s t-test, P< 0.001 for both at 48 hours after transfection), whereas transient knock-down of D5D and D6D reduces the cytosolic NAD+/NADH ratio (Student’s t-test, P< 0.001 for both at 48 hours after transfection) (Figures 5A and S4A–S4D). Pharmacologic inhibition with either SC-26196 or CP-24879 also reduces the cytosolic NAD+/NADH ratio (Student’s t-test, P< 0.001 at 24 hours for both compounds), with a more significant reduction with the dual D5D/D6D inhibitor CP-24879 (Figure 5B). Blocking NADH oxidation with D5D/D6D inhibition would be expected to divert reducing equivalents elsewhere. Following treatment with CP-24789, we observe an increase in cellular ROS (Figure S4E) as well as a dose-dependent increase in media lactate and decrease in media pyruvate (Figure 5C). The latter finding demonstrates an increase in LDH mediated pyruvate consumption that partially compensates for the loss of desaturase mediated NAD+ regeneration. Consistent with this, augmenting LDH mediated NAD+ regeneration with AKB prevents the decrease in media pyruvate/lactate following CP-24879 treatment (Student’s t-test, P< 0.001) (Figure 5D).
Figure 5. D5D and D6D inhibition reduces cytosolic NAD+/NADH, increases LDH activity, and impairs cell proliferation.

(A-B) Cytosolic NAD+/NADH measured with SoNar in (A) HeLa cells transfected with FADS1 or FADS2 cDNA or shRNA targeting FADS1 or FADS2; and (B) IMCD3 cells treated with rotenone (200 nM) or the desaturase inhibitors SC-26196 (10 μM) and CP-24879 (30 μM).
(C-D) Media lactate, pyruvate, and/or pyruvate/lactate ratio from IMCD3 cells treated with (C) rotenone (200 nM) or increasing concentrations of CP-24879 (CP); and (D) CP-24789 (30 μM) with or without AKB (1 mM).
(E-H) Cell proliferation at 3 days for (E) IMCD3 cells treated with increasing concentrations of CP-24789; (F) IMCD3 cells treated with vehicle, CP-24789 (30 μM), CP-24789 (30 μM) and pyruvate (1 mM), CP-24789 (30 μM) and AKB (1 mM), rotenone (200 nM), and rotenone (200 nM) and AKB (1 mM); (G) HeLa cells transfected with shRNA targeting FADS1 and FADS2 or control; and (H) IMCD3 cells treated with CP-24789 (30 μM) with AKB (1 mM) and/or a mixture containing 10 μM each of arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA).
(I) Schema for D5D and D6D activity and NAD+ recycling in relation to glycolysis and lactate fermentation. Values are means ± SEM; ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; n = 20–30 for (A), n = 10 for (B), n = 3 for (C, D), and n = 4 for (E-H).
See also Figure S4.
In addition to decreasing the cytosolic NAD+/NADH ratio, CP-24879 causes a dose-dependent decrease in cell proliferation (Student’s t-test, P = 0.002 at 3μM, P< 0.001 at 10 μM, P< 0.001 at 30μM) (Figure 5E). The deleterious impact of D5D/D6D inhibition on cell growth is mitigated by relatively richer media conditions (Figure S4F and Table S2), an effect we hypothesize is due in part to greater pyruvate content (there is no difference in HUFA end- products between the different media). Indeed, co-treatment with either AKB or pyruvate significantly attenuates the antiproliferative effect of desaturase inhibition (Figure 5F). Similarly, AKB co-treatment rescues the decrease in cell proliferation observed with transient knockdown of D5D and D6D (Figure 5G). Importantly, AKB has a significantly greater salutary effect on cell proliferation than a mixture of arachidonic acid, eicosapentaenoic acid and docosahexaenoic acid in CP-24879 treated cells, highlighting the relatively greater importance of NAD+ regeneration than HUFA end-products for mediating the effects of D5D and D6D activity on cell growth (Figure 5H). Taken together, our findings show that the interaction between cytosolic NAD+/NADH and D5D and D6D activity is bidirectional, whereby decreasing cytosolic NAD+/NADH increases HUFA synthesis and decreasing HUFA synthesis decreases the cytosolic NAD+/NADH ratio, increases lactate production, and reduces cell growth (Figure 5I).
D5D and D6D Activity is Adaptive During Acute Inhibition of Aerobic Respiration In Vivo
Given the impact of desaturase mediated NAD+ regeneration in vitro, we next examined its role in vivo, using rotenone to inhibit aerobic respiration in C57BL/6J mice and then profiling tissue homogenates 10 hours later (Figure S5A). We found that rotenone increases TAGs in kidney, liver, and skeletal muscle (ANOVA, P < 0.0001 for each), but not heart, with greater increases in HUFA-containing TAGs (Figure 6A) than total triglycerides (Figure 6B). Notably, no changes were observed in tissue lactic acid levels (Figure 6C). The tissue specificity of these findings may in part be attributable to relative levels of desaturase expression as D5D is most strongly expressed in kidney and liver (Figure 6D). Rotenone does not impact D5D protein levels (Figure 6E), whereas Fadsl and Fads2 mRNA levels decrease following rotenone treatment (Figure 6F), consistent with the known feedback inhibition of HUFA end-products on gene expression.
Figure 6. Acute increase in HUFA synthesis is adaptive when aerobic respiration is impaired in vivo.

(A-C) Effect of 10 h rotenone (2.8 mg/kg, intraperitoneal) treatment in 11-week old C57BL/6J mice on (A) heart, kidney, liver, and skeletal muscle (S. Mus) tissue TAGs measured by LC-MS, (B)total tissue TAGs measured by colorimetric assay, (C) tissue lactate measured by LC-MS.
(D and E) Western blot of D5D for (D) different tissue samples from one vehicle (V, Veh) and one rotenone (R, Rot)-treated mouse and (E) kidney homogenates from all vehicle and rotenone-treated mice. Representative gels from one of two independent experiments.
(F) Kidney tissue Fadsl and Fads2 mRNA from all vehicle and rotenone-treated mice. Values are means ± SEM; ns, P> 0.05; *P <0.05; **P < 0.01; n = 3 mice per group.
(G-J) Effect of co-treatment of CP-24879 (5 mg/kg, intraperitoneal) and rotenone (1.7 mg/kg, intraperitoneal) in 11-week old C57BL/6J mice on (G) NAD+/NADH ratio in kidney tissue; (H) renal tubular damage assessed by H&E staining (asterisk: protein casts, arrow head: apoptotic epithelial cells, arrow: tubular necrosis); (I) plasma creatinine and blood urea nitrogen (BUN); and (J) plasma aminotransferases (ALT and AST). Values are means ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001; n = 3 mice per group.
See also Figure S5 and S6.
A similar increase in tissue lipid HUFA content is observed in kidney and liver homogenates from mice exposed to 8% hypoxia versus normoxia (Figures S5B–S5F) and we have also observed this pattern of HUFA rich TAG accumulation in mouse kidney tissue following ischemia reperfusion injury (Tran et al., 2016). Although cellular lipid accumulation in these contexts has traditionally been viewed as deleterious, our studies suggest that acute increases in HUFA synthesis may represent an adaptive mechanism to support cellular metabolism and viability. Indeed, co-treatment of animals with CP-24879 and rotenone results in a lower tissue NAD+/NADH ratio and significantly greater kidney injury than either CP-24879 or rotenone alone (Figures S5A and 6G–6I); a trend for similar findings was observed with liver injury as well (Figure 6J). These findings show how desaturase mediated NAD+ recycling supports tissue viability when aerobic respiration is acutely impaired. Finally, to extend our findings to a non-injury in vivo model, we show that metformin significantly increases HUFA-containing TAGs in kidney and liver at a dose known to reduce tissue NAD+ levels (Figure S6A) (Gui et al., 2016); these results were further corroborated in vitro, where metformin reduces the cytosolic NAD+/NADH ratio and increases cellular HUFA TAGs (Figures S6B and S6C).
D5D and D6D Activity are Chronically Increased in Individuals with the Type 2 Diabetes SLC16A11 Risk Haplotype
Inhibition of aerobic respiration results in large increases in glucose consumption and a marked reduction in the cytosolic NAD+/NADH ratio. To assess the interaction between cytosolic NAD+/NADH and D5D and D6D activity in a more physiologic context, we examined cells exposed to high (25 mM) versus normal glucose (5.5 mM) media for 24 hours. Exposure to high glucose results in a 3% decrease in the cytosolic NAD+/NADH ratio (Student’s t-test, P = 0.017) and increase in HUFA containing TAGs (ANOVA, P < 0.0001), although the changes are of lesser magnitude compared to rotenone treatment (Figures S7A and S7B). Further, LbNOX expression attenuates both the decrease in cytosolic NAD+/NADH (Figure S7A) and increase in HUFA-containing lipids (Figure S7C) in cells exposed to high glucose.
Recently, a type 2 diabetes-associated haplotype in SLC16A11 was identified that explains ~20% of the increased type 2 diabetes prevalence in Mexico (Williams et al., 2014). These studies showed that SLC16A11 functions as a pyruvate transporter, and that by lowering both SLC16A11 expression and localization to the cell membrane, type 2 diabetes risk variants reduce SLC16A11 transport activity (Rusu et al., 2017). Genetic perturbation of SLC16A11 induces rapid changes in cellular lipid levels, but the mechanism connecting plasma membrane solute transport to lipid metabolism is unclear. Here, we tested whether reduced pyruvate transport attributable to the type 2 diabetes risk variants reduces cytosolic NAD+/NADH ratio, as occurs with NHI-2 mediated LDH inhibition. We expressed SLC16A11 in HEK293 cells co-transfected with SoNar and compared the non-risk SLC16A11 reference protein to the type 2 diabetes-associated protein that contains four missense variants (V113I, D127G, G340S, and P443T) and one silent variant (L187L) (Figures 7A and 7B). We find that the type 2 diabetes variant-associated reduction in pyruvate transport is associated with a 4% fall in the cytosolic NAD+/NADH ratio (Student’s t-test, P < 0.001) (Figure 7C), as well as an increase in TAG HUFA synthesis that can be reversed with pyruvate supplementation (Figure 7D). To extend these findings to humans, we used LC-MS to profile plasma from 677 individuals in the Mexico City Diabetes Study who had been genotyped at the SLC16A11 locus; at the time of plasma sampling, no study subjects had developed diabetes mellitus. We find a clear pattern of increased TAG HUFA content among the 364 carriers of the SLC16A11 risk haplotype compared to 313 controls (Figure 7E), although the magnitude of differences is attenuated relative to the in vitro comparison. By contrast, there was no difference in body mass index, fasting glucose, total cholesterol, high density cholesterol, or total triglycerides (Figure 7F). In conjunction with our cellular studies of LDH activation, LDH inhibition, and SLC16A11 expression, these human data underscore the biochemical interplay between lactate and HUFA production during glycolysis and provide a mechanism for how a reduction in pyruvate transport across the cell membrane can directly and chronically modulate lipid metabolism (Figure 7G).
Figure 7. The SLC16A11 diabetes risk haplotype reduces cytosolic NAD+/NADH ratio and increases HUFA synthesis in humans.

(A) Western blot of SLC16A11T2D-V5 and SLC16A11REF-V5 in HEK293 cells, representative gel from one of two independent experiments.
(B) qPCR of SLC16A11 in HEK293 cells expressing empty vector, SLC16A11T2D-V5, or SLC16A11ref-V5.
(C) Cytosolic NAD+/NADH measured with SoNar in HEK293 cells expressing either SLC16A11T2D-V5 or SLC16A11REF-V5.
(D) Intracellular TAGs in HEK293 cells expressing SLC16A11T2D-V5 or SLC16A11REF-V5 (red), or SLC16A11T2D-V5 with or without 1mM pyruvate supplementation (blue). Values are means ± SEM; ***P < 0.001; n = 3 for (B, D), and n = 40 for (C).
(E) Plasma TAGs among carriers of the SLC16A11 risk haplotype (n=364) relative to controls (n=313). Values are means; *P<0.05; **P < 0.01.
(F) Mexico City Diabetes Study clinical characteristics at time of lipid profiling
(G) Schema for interaction between SLC16A11, cytosolic NAD+/NADH, and HUFA synthesis.
See also Figure S7.
DISCUSSION
In addition to modulating membrane fluidity, HUFAs can be released from membrane lipids and then converted to eicosanoids and other bioactive molecules that play diverse roles in health and disease (Funk, 2001; Wassall and Stillwell, 2009). As such, the significance of D5D and D6D activity have largely been viewed in terms of the biologic actions of their enzymatic end- products. Dietary intake and transcriptional control of FADS1 and FADS2 expression are established determinants of cellular HUFA content (Nakamura and Nara, 2004). Here, we show that changes in cytosolic NAD+ and NADH redox states also influence D5D and D6D activity, establishing a bidirectional link between glycolysis and polyunsaturated fatty acid desaturation. These findings alter the existing paradigm of NAD+ regeneration in glycolysis, and highlight a key biologic role for D5D and D6D action independent of their end-products.
Interest in FADS1 and FADS2 has been motivated by recent genome-wide association studies implicating desaturase activity in glycemic, growth, and proliferative phenotypes. More specifically, common variants that are associated with reduced desaturase expression are associated with decreased HUFA content in circulating lipids (Gieger et al., 2008; Rhee et al., 2013). These same alleles result in lower fasting glucose (Dupuis et al., 2010), lower risk of type 2 diabetes (Fujita et al., 2012), decreased height and weight among Inuits (Fumagalli et al.,2015), and decreased risk of colon cancer and laryngeal cancer in East Asians (Wei et al., 2014; Zhang et al., 2014). Some of these associations may be attributable to reduced synthesis of HUFA end-products; for example, reduced eicosanoid availability for cell signaling could have anti-proliferative effects (Wang and Dubois, 2010). Alternatively, our results raise the possibility that reduced D5D and D6D activity reduces cell proliferation by reducing the cytosolic NAD+/NADH ratio, as we show that AKB, but not a cocktail of HUFAs, reverses growth inhibition by dual D5D and D6D inhibition. Whether desaturase mediated NAD+ recycling plays a role in Warburg (aerobic) glucose metabolism in cancer cells requires further investigation (Koppenol et al., 2011)—interestingly, recent studies have shown that some cancer cells are net consumers of glucose and lactate, raising the possibility that alternative mechanisms to support glycolytic NAD+ recycling may be required (Faubert et al., 2017). Ultimately, both enzymatic activity and end-products are likely to be pertinent to how HUFA synthesis impacts cellular metabolism, and we acknowledge that our studies do not disentangle their relative contribution to the FADS-related phenotypes established by human genetics.
Interest in FADS1 and FADS2 has been further enhanced by lipidomic studies of human physiology. Using an earlier iteration of the platform utilized herein, we profiled plasma from individuals before and after oral glucose tolerance testing, oral sulfonylurea administration, and exercise treadmill testing (Rhee et al., 2011), using each individual as his or her own biologic control. In each case, we observed an increase in the HUFA content of circulating TAGs within two hours, before any changes in FADS1 or FADS2 expression might occur. These observations raised the question of why diverse glycolytic stimuli would acutely increase D5D and D6D activity, with particular interest in the liver where lipoproteins destined for release into plasma are assembled (Quehenberger and Dennis, 2011). Our findings here provide an explanation for the dynamic changes observed in humans, connecting glycolysis to HUFA synthesis through cytosolic NAD+ and NADH. We observe increases in D5D and D6D activity, with modest increases when glycolysis is stimulated with high glucose, and marked increases when glycolysis is stimulated with inhibition of mitochondrial respiration, including in the liver and kidney in vivo. D5D and D6D activity also increases with LDH inhibition, demonstrating that it is the cytosolic NAD+/NADH ratio, rather than glucose utilization per se, that triggers increased desaturase activity. The combined inhibition of mitochondrial respiration and LDH yields the most dramatic increases in cellular HUFA levels, outlining the capacity of this system for accommodating the flow of reducing equivalents diverted from their canonical pathways.
Among the tissues we examined, the expression of D5D and the subsequent increase in HUFA levels in mice treated with rotenone or exposed to hypoxia was greatest in liver and kidney, with no accompanying change in tissue lactate. Inhibition of desaturase activity further lowered the tissue NAD+/NADH ratio and enhanced the renal toxicity of rotenone (with a trend for increased hepatic toxicity). Thus, the accumulation of intracellular HUFA may be acutely adaptive by facilitating ongoing glycolysis when aerobic respiration is impaired. The relatively higher expression of D5D and D6D in kidney and liver is notable, as these two organs are responsible for the majority of whole-body lactate clearance under normal conditions, and thus have a high threshold for net lactate production (Kraut and Madias, 2014) (Bellomo, 2002).
The impact of desaturase mediated NAD+ regeneration is likely to be context dependent. As noted, prior GWAS have outlined the potential salutary consequences of decreased desaturase expression and HUFA synthesis, including lower fasting glucose and type 2 diabetes risk; conversely, a long term increase in desaturase activity contributes to increased metabolic disease. Here, we find that the SLC16A11 risk haplotype—associated with a significant increase in type 2 diabetes among Mexicans, and shown to reduce the protein’s pyruvate transport capacity at the plasma membrane by ~50%—results in a lower cytosolic NAD+/NADH ratio, with a subsequent increase in HUFA synthesis (Halestrap, 2013). This phenotype can be reversed with pyruvate supplementation, although it is unclear if the added pyruvate enters cells through SLC16A11 or through a different monocarboxylate transporter. Remarkably, the increase in TAG HUFA content observed in vitro is recapitulated in plasma obtained from Mexican individuals who have the SLC16A11 risk haplotype compared to controls. These results provide mechanistic insight into how coding variants in SLC16A11 alter lipid metabolism, i.e. that reduced LDH activity attributable to reduced pyruvate availability lowers the cytosolic NAD+/NADH ratio, triggering a sustained increase in HUFA synthesis. Why increased D5D and D6D action promotes type 2 diabetes requires more investigation, but may relate to an increase in intracellular TAG accumulation, for example in the liver or the beta cell. Whether the increase in HUFA content itself, for example through an effect on eicosanoid production or cell signaling, confers some harm in this context also warrants consideration.
Our results raise several questions. Stearoyl-CoA desaturase (SCD) performs a similar chemical reaction as D5D and D6D, but acts on saturated fatty acids, yielding monounsaturated fatty acids such as palmitoleic and oleic acid. However, our lipidomics data do not demonstrate an increase in SCD activity in response to inhibition of mitochondrial respiration or LDH. More work is required to explore the differential response of SCD versus D5D and D6D—however, it is notable that SCD lacks an N-terminal cytochrome b5-like domain present in D5D and D6D, and that NADPH, rather than NADH, is believed to be the major electron donor for the SCD-mediated enzymatic reaction (Guillou et al., 2004). D5D and D6D are responsible for the synthesis of both ω−6 and ω−3 HUFA. Increased enzymatic activity triggered by a reduction in the cytosolic NAD+/NADH ratio would not be expected to have a differential impact on their relative production, and indeed, we found increases of both, best appreciated with the increase in individual free fatty acids or cholesterol esters that contain a single acyl chain. However, more formal study of the relative production of ω−6 and ω−3 HUFA is warranted, as these molecules have disparate and sometimes antagonistic effects, for example in inflammation and hemostasis (Zarate et al., 2017).
Also of interest is the stoichiometry of glycolytic NAD+ regeneration via mitochondrial respiration, lactate fermentation, and HUFA synthesis. Clearly, this will depend on the experimental settings, including glucose, pyruvate, and polyunsaturated fatty acid availability, as well as glycolytic capacity and desaturase expression in the chosen cell line. For a given set of conditions, measurement of glucose consumption and end-products alone is not sufficient. As demonstrated by our lipidomics data, HUFA end-products are incorporated diffusely across cellular lipids, including some that are not readily measured, as well as secreted into media. Our results show that NADH produced during glycolysis can drive HUFA synthesis, but the entire pool of cytosolic NADH can contribute reducing equivalents, including cytosolic NADH generated from non-glycolytic dehydrogenase reactions. Finally, HUFA synthesis requires the alternating action of both desaturases and elongases (Jakobsson et al., 2006). Fatty acid elongation also requires NADH as a substrate, regenerating NAD+; thus it may be more appropriate to view the entirety of HUFA synthesis, including desaturation and elongation, as a mechanism of cytosolic NAD+ recycling. However, because average daily intake of the HUFA precursors linoleic and alpha-linolenic acids is ~0.07 mol/day for adults (U.S. Department of Agriculture, Agricultural Research Service, 2018), this mechanism can only account for 0.03~0.08 mol/day of NAD+ regeneration, assuming 16–36% of ingested lipid is converted to HUFA end-products (Burdge et al., 2002; Burdge and Wootton, 2002). By contrast, whole body lactate turnover is likely >20–80 fold greater (Bergman et al., 1999; Katz et al., 1993). Nevertheless, our data support a physiologically relevant role for desaturase mediated NAD+ recycling, particularly in select tissues such as kidney and liver, and in the context of chronic changes in cytosolic redox status as may occur with genetic variants in FADS1–3 or SLC16A11.
In sum, our findings advance knowledge of metabolism along several axes. First, they show that polyunsaturated fatty acid desaturation increases in response to a reduction in the cytosolic NAD+/NADH ratio. Second, they characterize this response as a mechanism for glycolytic NAD+ recycling that operates in parallel with lactate fermentation and is acutely adaptive in vivo when aerobic respiration is impaired. In this role, the importance of polyunsaturated fatty acid desaturation expands beyond the downstream biologic roles of HUFA end-products. Third, our results provide mechanistic insight on how variants in the monocarboxylate transporter SLC16A11 may impact cellular HUFA production, and thus contribute to the pathogenesis of type 2 diabetes. And finally, our results provide insight on human genetic studies linking HUFA desaturation with glycemic, growth, and cancer phenotypes as well as human physiologic studies demonstrating rapid changes in plasma HUFA levels.
Limitations of Study
Although we use established knowledge about PUFA consumption and lactate turnover to outline the greater magnitude of lactate fermentation, we do not systematically compare and quantitate the two processes in vivo. More work is required to fully characterize in which tissues and under what conditions HUFA synthesis makes a substantial contribution to glycolytic NAD+ recycling and to understand the functional consequences of increasing cellular HUFA content via this mechanism.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eugene P. Rhee (eprhee@partners.org). Certain materials are shared with academic and non-profit research organizations for research only under an MTA.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell Lines and Cell Culture
Mouse inner medullary collecting duct (IMCD3) and human (female) embryonic kidney (HEK) 293 cells were obtained from ATCC. IMCD3 cells were maintained in DMEM F-12 medium (30–2006, ATCC) and HEK293 cells were maintained in MEM medium (30–2003, ATCC), both containing 10% fetal bovine serum (FBS, HyClone) and 100 units/ml penicillin/streptomycin (Life Technologies). Doxycycline-inducible human (female) cervical carcinoma HeLa-LBNOX cells (provided by D. Titov and V. Mootha) were cultured in 10% FBS and 100 units/ml penicillin/streptomycin-containing DMEM medium (D5921, Sigma-Aldrich) supplemented with 1 mM pyruvate, 4 mM L-glutamine, 200 μg/mL geneticin and 1 μg/mL puromycin (all from Thermo Fisher Scientific). A week before experiments, geneticin and puromycin were removed from the culture media. All cells were grown at 37°C in a humidified atmosphere with 5% CO2. Cells with fewer than 20 passages were used.
Animal Studies
All studies were approved by the Institutional Animal Care and Use Committee of the Massachusetts General Hospital and conducted under their guidelines. Ten-week-old C57BL/6J male mice were purchased from the Jackson Laboratory. Mice were housed in a controlled environment with a 12/12-h light-dark cycle (7am to 7pm) and fed a normal chow diet, Prolab® Isopro® RMH 3000 (LabDiet). Mice were used for experiments one week after arrival. For rotenone treatment, mice underwent i.p. injection with 2.8 mg/kg rotenone (1.4 mg/ml stock) in DMSO or DMSO vehicle (n=3, each group). In another set of experiments, mice were co-treated with a single dose of rotenone (1.7 mg/kg, i.p.) and multiple doses of CP-24879 (5 mg/kg, i.p.) for 2 days (n=3, each group). Following euthanasia 10 hours after rotenone treatment, tissues were collected and immediately snap frozen in liquid nitrogen. Chow was removed 4 hours prior to sacrifice. For metformin treatment, mice were treated with metformin (500 mg/kg, i.p.) in saline or saline vehicle (n=3, each group) and chow was removed. Following euthanasia 4 hours after metformin treatment, tissues were collected and immediately snap frozen in liquid nitrogen. For hypoxia, mice were divided into two groups (n=4, each group) and were exposed to hypoxia (8% O2) or normoxia (21% O2) at ambient sea-level pressure as previously described (Jain et al., 2016). In brief, mice were placed in 60 liter plexiglass chambers. Each chamber was ventilated from an inlet using a continuous mixture of nitrogen and air. The mixture was adjusted to obtain the desired oxygen concentration using an oxygen analyzer (MiniOx I) placed at the outlet of the chamber. The oxygen analyzer was calibrated using a reference tank with a concentration of 8.55% O2 (Airgas). The flow of the mixture was 5 l/min and was adjusted to maintain a CO2 concentration below 1000ppm inside the chamber. The CO2 concentration in the chamber as well as the temperature and the humidity were monitored continuously using a dedicated infrared CO2 analyzer, thermometer, and humidity meter (Extech CO200 Monitor, Extech Instruments). To ensure fasting status at the time of sample collection, the chamber was briefly opened at 4 hours prior to sacrifice and chow was removed. Following euthanasia, tissues were collected and immediately snap frozen in liquid nitrogen.
Human Studies
Study participants in the Mexico City Diabetes Study, as well as genotyping using the Illumina OMNI2.5 array and exome-sequencing, have been previously described (Estrada et al., 2014; Williams et al., 2014). Informed consent was obtained from all subjects, and the study was approved by the Comité de Etica e Investigatión committee at The Centro de Estudios en Diabetes. In sum, plasma from a total of 677 individuals with genotyping, including 364 carriers of the SLC16A11 risk haplotype (mean age 52.6 ± 7.6; 40.4 % male) and 313 controls (mean age 52.6 ± 7.6; 37.4 % male), underwent lipid profiling as described below. No individuals at the time of lipid profiling had type 2 diabetes. An analysis of the influence of sex on the association between SLC16A11 status and TAG pattern was not performed because sex is not known to influence the association between SLC16A11 status and diabetes risk
METHOD DETAILS
In Vitro Experimental Conditions
Inhibition of mitochondrial respiration
IMCD3 and HEK293 cells were seeded at 200,000 cells per well in 6-well plates and incubated overnight. The following day, HEK293 cells were washed once with 2 mL of pyruvate-free MEM (M5650, Sigma-Aldrich) and then the media was changed to 2 mL of pyruvate-free MEM containing 10% dialyzed FBS (Thermo Fisher Scientific), penicillin/streptomycin and 2 mM L-glutamine premixed with either 200 mM rotenone or 0.1% DMSO. Similarly, IMCD3 cells were washed once with 2 mL of pyruvate-free DMEM (D5671, Sigma-Aldrich) and then the media was changed to 2 mL of pyruvate-free DMEM containing 10% dialyzed FBS, penicillin/streptomycin and 2 mM L-glutamine premixed with 200 nM rotenone, 5 nM oligomycin, 2 mM metformin, or 0.1% DMSO. For D5D/D6D inhibition, media was changed to2 mL of pyruvate-free DMEM containing 10% dialyzed FBS, penicillin/streptomycin and 2 mM L-glutamine premixed with 200 nM rotenone ± 10 μM SC-26196 or 200 nM rotenone ± 30 μM CP-24879. For modulation of cytosolic NAD+/NADH, media was changed to 2 mL of pyruvate- free DMEM containing 10% dialyzed FBS and penicillin/streptomycin premixed with 200 nM rotenone ± 1 mM alpha-ketobutyrate (AKB) or 200 nM rotenone ± 1 mM pyruvate. For LDH inhibition, media was changed to 2 mL of pyruvate-free DMEM containing 10% dialyzed FBS, penicillin/streptomycin and 2 mM L-glutamine premixed with 30 mM NHI-2 ± 100 nM rotenone. For all experiments, cells and media were collected for analysis at 24 hours unless otherwise specified.
Hypoxia
IMCD3 cells were seeded at 200,000 cells per well in 6-well plates and incubated overnight. The following day, cells were washed once with 2 mL of pyruvate-free DMEM and media was changed to 2 mL of pyruvate-free DMEM containing 10% dialyzed FBS, penicillin/streptomycin and 2 mM L-glutamine. For the induction of hypoxia, one plate of cells was placed in a hypoxia chamber (STEMCELL Technologies) containing a 1% O2 gas mixture and incubated at 37°C. At 1 hour, the chamber was re-gassed to eliminate O2 contained in the media or trapped in the plastic ware; a petri dish with 20 mL of water was placed inside the chamber to prevent excessive evaporation of culture medium. Control cells were placed outside of the hypoxia chamber, but within the same incubator. Cells were collected for analysis at 24 hours.
LbNOX expression
HeLa-LbNOX cells were seeded at 100,000 cells per well in 6-well plates and incubated overnight. The following day, cells were washed once with 2 mL of pyruvate-free DMEM and media was changed to 2 mL of pyruvate-free DMEM containing 10% dialyzed FBS, penicillin/streptomycin and 4 mM L-glutamine with or without 300 ng/mL doxycycline. Twenty-four hours after doxycycline addition, media was exchanged to 2 mL of pyruvate-free DMEM containing 10% dialyzed FBS, penicillin/streptomycin and 4 mM L-glutamine premixed with 100 nM rotenone ± 300 ng/mL doxycycline or 0.1% DMSO ± 300 ng/mL doxycycline. Cells were collected for analysis at 6 hours.
Dose-effect of CP-24879
IMCD3 cells were seeded at 200,000 cells per well in 6-well plates and incubated overnight. The following day, cells were washed once with 2 mL of pyruvate-free DMEM and media was changed to 2 mL of pyruvate-free DMEM containing 10% dialyzed FBS, penicillin/streptomycin and 2 mM L-glutamine premixed with 3 μM CP-24879, 10 μM CP-24879, 30 μM CP-24879, 200 nM rotenone, or 0.1% DMSO. Media was collected for analysis at 24 hours.
Transient transfection
HeLa and HEK293 cells were seeded at 100,000 and 300,000 cells per well in 6-well plates, respectively. The following day, media was replaced with fresh antibiotic-free MEM and cells were transfected with FuGENE HD transfection reagent (Promega). More specifically, cells were incubated with 1 μg of plasmid DNA encoding FADS1, FADS2, shFADS1, shFADS2, SLC16A11REF, or SLC16A11T2D and 3 μL of FuGENE HD transfection reagent for 24 hours (human FADS cDNA and shRNA expression vectors were purchased from OriGene Technologies). After transfection, cells were washed once with 2 mL of pyruvate-free MEM and then the media was changed to 2 mL of pyruvate-free MEM containing 10% dialyzed FBS and 2 mM L-glutamine. Cells were collected for analysis at 24 hours (2 days after the start of transfection).
Conversion of linoleic acid-13C18 to gamma-linolenic acid-13C18 and arachidonic acid-13C18
IMCD3 cells were seeded at 200,000 cells per well in 6-well plates and incubated overnight. The following day, cells were washed twice with 2 mL of pyruvate-free DMEM and media was changed to 2 mL of pyruvate-free DMEM containing 10 mM linoleic acid-13C18, 1 mg/mL fatty acid-free bovine serum albumin (Sigma-Aldrich), penicillin/streptomycin and 2 mM L-glutamine premixed with 0.1% DMSO ± 1 mM AKB, 200 nM rotenone ± 1 mM AKB, or 30 μM CP-24879. Media was collected for analysis at 6 and 24 hours. For D5D/D6D activity measurements after transient knockdown of FADS1 and FADS2 in HeLa cells, 0.5 μg of each pRS-shFADSl and pRS-shFADS2 vectors were transfected using Fugene HD for 24 hours. After transfection, cells were washed twice with 2 mL of pyruvate-free DMEM and media was changed to 2 mL of pyruvate-free DMEM containing 10 μM linoleic acid-13C18, 1 mg/mL fatty acid-free bovine serum albumin (Sigma-Aldrich) and 2 mM L-glutamine. Media was collected for analysis at 24 hours.
Total protein measurement
For each experimental condition, a duplicate experiment was performed for total protein quantitation. For these 6-well plates, cells were washed with 2 mL of cold PBS once, then scraped with 0.3 mL RIPA buffer containing Halt protease inhibitor cocktail (all from Thermo Fisher Scientific), and protein levels were measured using the DC Protein Assay Kit (Bio-Rad). Results of cell culture analyses, specifically LC-MS based measurement of lipids, fatty acids, lactate, pyruvate and other polar metabolites; total triglycerides; and media glucose were all normalized to protein levels.
LC-MS Analyses
Sample preparation and extraction
As described above, cells were grown in 6 well plates for LC-MS analyses. After vacuum aspiration of media, cells were washed with cold PBS (without Mg2+ and Ca2+) and extracted with either 800 μL isopropanol cooled to 4°C (for lipid profiling) or 800 μL of 80% methanol cooled to −80°C (for polar metabolite profiling). Cell debris and extraction solution were transferred to a 1.5 mL centrifuge tube and extracted for lh (at 4°C for lipid profiling, on dry ice for polar metabolite profiling), vortexed, centrifuged (10,000 rcf, 4°C, 10 min), and then supernatants were transferred to a fresh 1.5 mL centrifuge tube and stored at −80°C until analysis. For polar metabolite profiling, pellets were further extracted using 100 μL of 80% methanol and secondary extracts were pooled with primary extracts. For fatty acid-13C18 measurements of cultured media, 30 μL of media were extracted with 120 μL of 80% methanol cooled to 4°C; for pyruvate and lactate measurements, 30 μL of media were extracted with 120μL of 80% methanol containing 2 μg/mL l-lactate-d3. Extracted media were then vortexed, centrifuged (10,000 rcf, 4°C, 10 min), and supernatants were transferred to a fresh 1.5 mL centrifuge tube and stored at - 80°C until analysis. For mouse samples, snap frozen tissue samples were cut to ~60 mg and then homogenized in 4 volumes of ice-cold water using a Tissue Lyser II (Qiagen). For lipid profiling of mouse samples, 10 μL of tissue homogenate were extracted in 190μL of isopropanol containing 0.2 ng/μL l,2-dilauroyl-sn-glycero-3-phosphocholine, vortexed, and centrifuged (10,000 rcf, RT, 10 min), and supernatants were stored at −80°C until analysis. For lactate measurements in mouse samples, 30 μL of tissue homogenate were extracted in 170μL of 80% methanol containing 2 μg/mL l-lactate-d3, vortexed, and centrifuged (9,000 × g, 4°C, 10 min), and supernatants were stored at −80°C until analysis. For lipid profiling of human plasma samples, 10 μL of plasma were extracted in 190 μL of isopropanol containing 0.2 ng/μL 1,2- dilauroyl-sn-glycero-3-phosphocholine, vortexed, and centrifuged (10,000 rcf, RT, 10 min), and supernatants were stored at −80°C until analysis.
Lipid profiling
10μL of extracted cell lysate or 2 μL of extracted mouse tissue or human plasma were separated by reversed phase chromatography using an Acquity UPLC BEH C8 column (Waters); mobile phase A: 10 mM ammonium acetate in 95% water/5% methanol/0.1% formic acid; mobile phase B: 0.1% formic acid in methanol. The column was eluted isocratically with 20% mobile phase B for 1 minute followed by a linear gradient to 80% mobile phase B over 2 minutes, a linear gradient to 100% mobile phase B over 7 minutes, then 3 minutes at 100% mobile phase B. MS data were acquired with a Q Exactive Plus orbitrap mass spectrometer (Thermo Fisher Scientific) in the positive ion mode using electrospray ionization and full scan MS over m/z 200–1100 (Williams et al., 2014). Raw data were integrated and visually inspected using TraceFinder 3.3 software (Thermo Fisher Scientific). For each lipid analyte, the first number denotes the total number of carbons in the lipid acyl chain(s), and the second number (after the colon) denotes the total number of double bonds in the lipid acyl chain(s).
Fatty acid-13C18 measurement
2μL of extracted cultured media were separated using reverse phase chromatography using a 150 × 2.1 mm Acquity UPLC BEH Cl8 column (Waters); mobile phase A: 0.01% formic acid in water; mobile phase B: 0.01% acetic acid in acetonitrile. The column was eluted isocratically with 20% mobile phase B for 3 minutes followed by a linear gradient to 100% mobile phase B over 12 minutes, then 3 minutes at 100% mobile phase B. MS data were acquired with a Q Exactive hybrid quadrupole orbitrap mass spectrometer (Thermo Fisher Scientific) in the negative ion mode using electrospray ionization and full scan MS over m/z 70–850. Raw data were integrated and visually inspected using TraceFinder 3.3 software (Thermo Fisher Scientific), with monitoring for the loss of hydrogen at the following masses: 297.2933 (linoleic acid-13C18), 295.2777 (gamma-linolenic acid-13C18), and 321.2933 (arachidonic acid-13C18).
Polar metabolite profiling
Negatively charged polar analytes including alpha-hydroxybutyrate and citric acid cycle intermediates were measured in 10 μL of extracted cell lysates separated using a 150 × 2.0 mm Luna NH2 column (Phenomenex); mobile phase A: 20mM ammonium acetate, 20mM ammonium hydroxide in water; mobile phase B: l0mM ammonium hydroxide in 25% methanol/75% acetonitrile. The column was eluted isocratically with a linear gradient from 90% to 0% mobile phase B over 10 minutes, then 2 minutes at 0% mobile phase B. MS data were acquired with multiple reaction monitoring in the negative ion mode on a 5500 QTRAP MS (SCIEX). Raw data were integrated and visually inspected using MultiQuant software (version 2.1, SCIEX).
Lactate and pyruvate measurement
10 μL of extracted cultured media were separated using a 150 × 2.0 mm Luna NH2 column (Phenomenex); mobile phase A: 20mM ammonium acetate, 20mM ammonium hydroxide in water; mobile phase B: l0mM ammonium hydroxide in 10% water/22.5% methanol/67.5% acetonitrile. The column was eluted isocratically with a linear gradient from 100% to 0% mobile phase B over 10 minutes, then 2 minutes at 0% mobile phase B. MS data were acquired with multiple reaction monitoring in the negative ion mode on a TSQ Quantiva triple quadrupole MS (Thermo Fisher Scientific). Raw data were integrated and visually inspected using TraceFinder 2.1 software (Thermo Fisher Scientific). MRM transitions were as follows: pyruvate (87/43), lactate (89.1/43.1), lactate-d3 (92.1/45.1). Metabolite identifications were confirmed using synthetic mixtures of reference compounds.
Other Measurements
Total Triglyceride Measurement
In the comparison of 24h treatment of 200 nM rotenone versus 0.1% DMSO in IMCD3 cells, the same experiment as described for 6-well plates was repeated, except scaled for 1,000,000 cells at seeding in 10 cm culture dishes. Total triglycerides were measured using a TAG quantification kit (K622–100, Biovision). For mouse samples, 10 μL of tissue homogenate or plasma were extracted in 90 μL of 5.5% Nonidet P-40 solution (Thermo Fisher Scientific) and total TAG levels were measured as per the manufacturer’s instructions.
Measurement of Cytosolic NAD+/NADH Ratio Using SoNar
IMCD3 and HeLa-LbNOX cells were seeded at 600,000 cells in 10 cm culture dishes and incubated overnight. The following day, media was replaced with fresh antibiotic-free DMEM and cells were transfected with Fugene HD transfection reagent. More specifically, IMCD3 cells were incubated with 5 μg of pCDNA3.1-SoNar (provided by Y. Yang and J. Loscalzo) and 18 μL of Fugene HD transfection reagent for 24 hours, then transfected cells were resuspended with trypsin and replated at 20,000 cells per well on a 96-well clear bottom black microplate (Coming) in FluoroBrite DMEM (Life Technologies) containing 5% dialyzed FBS, 1 mM pyruvate and 2 mM L-glutamine. Twenty-four hours after transfected cells had been seeded on the 96-wells plate, cells were washed once with 100 μL of FluoroBrite DMEM and media was changed to 100 μL of FluoroBrite DMEM containing 2% dialyzed FBS and 2 mM L-glutamine. One hour later, the following compounds were added (final concentration): DMSO (0.1%), rotenone (1 nM to 200 nM), AKB (1 mM), rotenone (200 nM) + AKB (1 mM), pyruvate (1 mM), rotenone (200 nM) + pyruvate (1 mM), NHI-2 (30 μM), or metformin (2 mM). Dual-excitation ratios of the SoNar sensor were measured immediately afterwards and at the specified time points using an Envision multi-label reader (PerkinElmer) with 430 nm or 485 nm excitation and 535 nm emission for both excitation wavelengths. The procedure for LbNOX- expressing HeLa cells was the same, except that at the time of replating on the 96-well plate, media was also supplemented with or without 300 ng/mL doxycycline. For transient overexpression or knockdown of FADS1 and FADS2, 3 μg of each vector was co-transfected with 3 μg of pCDNA3.1-SoNar in HeLa cells, and subsequent steps were as described above. For SoNar measurements after transient overexpression of SLC16A11 variants in HEK293 cells, 3 μg of pLX304-SLC16A11REF or pLX304-SLC16A11T2D was co-transfected with 3 μg of pCDNA3.1-SoNar using Fugene HD as described above for 24 hours, then transfected cells were replated at 60,000 cells per well on a 96-well clear bottom black microplate in DMEM (D5921) containing 5% dialyzed FBS, 2 mM L-glutamine and MEM non-essential amino acids. Twenty- four hours after transfected cells had been seeded on the 96-wells plate, SoNar signals were measured.
Intracellular NAD+/NADH and NADP+/NADPH measurements
NAD (NAD+ and NADH) and NADP (NADP+ and NADPH) were measured using Biovision colorimetric kits (K337–100 and K347–100, respectively). To remove NADH or NADPH consuming enzymes, cell or mouse kidney tissue lysates were filtered through 10 kDa molecular weight cut off filters (Amicon Ultra, EMD Millipore) and the filtrate was subsequently used for further analysis.
Media Glucose Concentration
Media glucose levels were measured using a glucose colorimetric assay kit according to the manufacturer’s instructions (K606–100, Biovision).
Quantitative PCR
Total RNA was extracted using a RNeasy mini kit (Qiagen) and cDNA was synthesized using an AMV first-strand cDNA synthesis kit (Thermo Fisher Scientific). PCR reactions were performed using a CFX Connect Real-Time PCR machine (Bio-Rad) and SYBR green master mix (Thermo Fisher Scientific) according to the manufacturer’s instructions. The primer sequences are listed in Table S3.
Western Blots
A standard protocol was used for immunoblotting of cell lysates. In brief, samples were prepared using 4× Laemmli sample buffer (Bio-Rad), separated on a SDS polyacrylamide gel, transferred onto a 0.45 μm PVDF membrane (Thermo Fisher Scientific), and incubated with the primary and HRP-conjugated secondary antibodies. Immunoreactive proteins were visualized using a SuperSignal West Dura detection kit (Thermo Fisher Scientific). Following detection of D5D, FLAG-LbNOX, FLAG-D5D, FLAG-D6D, or V5-SLC16A11 the membrane was stripped using Restore™ western blot stripping buffer (Thermo Fisher Scientific) for 15 min, then reprobed with a α-tubulin or β-actin antibody for the detection of loading control. We were unable to identify a specific antibody for endogenous D6D.
Microscopy
IMCD3 and HeLa cells were grown on Lab-TEK II chamber slides (Nunc). A standard immunocytochemical method was used for immunostaining of D5D or subcellular marker proteins. In brief, cells were fixed in pre-cooled (−20°C) 100% methanol for 5 min at 4°C, then blocked with 0.2% BSA and 10% normal goat serum (NGS) in PBST (PBS + 0.05% Tween 20) for 1 hour at room temperature. For endogenous D5D visualization, cells were incubated with Alexa Fluor-488-labeled anti-D5D and Alexa Fluor-568-labeled anti-reticulon 4 or anti-calnexin or anti-β-actin in 5% NGS in PBST overnight at 4°C. The smooth ER marker reticulon 4 (Shibata et al., 2010; Shibata et al., 2006) antibody was labeled with Alexa Fluor-568 using the APEX antibody labeling kit (Thermo Fisher Scientific). For transient FADS1 and FADS2 overexpression, cells were stained with anti-FLAG and anti-β-actin. After incubating the primary antibodies, cells were further incubated with Alexa-Fluor-568-conjugated goat anti-mouse IgG H&L (Thermo Fisher Scientific) and Alexa Fluor-488-conjugated goat anti-rabbit IgG in 5% NGS in PBST for 1 hour at room temperature. Stained cells were mounted with ProLong diamond antifade mountant with DAPI (Thermo Fisher Scientific) and examined using a laser-scanning confocal microscope (LSM 510, Zeiss) or conventional fluorescence microscope (Eclipse Ni, Nikon).
Cell Proliferation
IMCD3 and HeLa cells were quantitated using the CyQUANT cell proliferation assay kit (Molecular Probes). Fluorescence measurements were made with an Envision multi-label reader with excitation at 485 nm and emission detection at 535 nm.
Mouse Histology and Blood Chemistry
Hematoxylin & eosin (H&E) staining was used to assess renal tubular damage. Plasma creatinine, blood urea nitrogen (BUN), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) were analyzed using a DRI-CHEM® 7000 Analyzer (Heska).
Cellular ROS Production
The levels of cellular ROS were determined in IMCD3 cells using chloromethyl-2’,7’- dichlorofluorescein diacetate (CM-H2DCFDA, Thermo Fisher Scientific), a cell permeable, non- fluorescent precursor of DCF. IMCD3 cells were incubated with 25 mM CM-H2DCFDA for 45 minutes. After loading the probe, cells were washed once with FluoroBrite DMEM and then the media was changed to FluoroBrite DMEM containing 2% dialyzed FBS and 2 mM L-glutamine premixed with 30 μM CP-24879, 200 μM tert-butyl hydroperoxide, or 0.1% DMSO. Fluorescence measurements were made with an Envision multi-label reader with excitation at 485 nm and emission detection at 535 nm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification
For comparative analysis of D5D expression in mouse kidney tissues, western blots images were quantified with ImageJ (NIH).
Statistical Analyses
Total triglycerides measured by colorimetric assay, as well as linoleic acid-13C18, gamma-linolenic acid-13C18, and arachidonic acid-13C18, glucose, other free fatty acids, lactate, pyruvate, other polar metabolite levels, qPCR results, NAD+/NADH ratio, cell proliferation, and ROS production were analyzed using Student’s t-tests. For comparison of individual TAGs measured by LC-MS, a Student’s t-test was performed. When noted, Bonferroni-adjustment was made for the total number of TAGs assayed, P-value/50. Tissue lysate lipid content measured by LC-MS was compared using two-way ANOVA. Statistical analyses were performed using Graph Pad Prism 6 and statistical parameters can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-D5D (FADS1) [EPR6898] | Abcam | Cat#ab126706; RRID: AB_11130088 |
| Anti-D5D [EPR6898], Alexa Fluor 488 conjugate | Abcam | Cat#ab214251 |
| Anti-D6D (FADS2), N-terminal | Abcam | Cat#ab170665 |
| Anti-D6D | Abcam | Cat#ab72189; RRID: AB_2041175 |
| Anti-Nogo A+B (reticulon 4) | Abcam | Cat#ab47085; RRID: AB_881718 |
| Anti-Calnexin [AF18] | Thermo Fisher Scientific | Cat#MA3–027; RRID: AB_2069043 |
| Anti-DYKDDDDK (FLAG) | Cell Signaling Technology | Cat#2368; RRID: AB_2217020 |
| Anti-V5 [D3H8Q] | Cell Signaling Technology | Cat#13202; RRID: AB_2687461 |
| Anti-β-actin [8H10D10] | Cell Signaling Technology | Cat#3700; RRID: AB_2242334 |
| Anti-α-tubulin [DM1A] | Cell Signaling Technology | Cat#3873; RRID: AB_1904178 |
| Goat anti-mouse IgG, Alexa Fluor 568 conjugate | Thermo Fisher Scientific | Cat#A11004; RRID: AB_141371 |
| Goat anti-rabbit IgG, Alexa Fluor 488 conjugate | Thermo Fisher Scientific | Cat#A11034; RRID: AB_2576217 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Human plasma samples | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rotenone | Sigma-Aldrich | Cat#R8875; CAS: 83-79-4 |
| Oligomycin A | Sigma-Aldrich | Cat#75351; CAS: 579-13-5 |
| Metformin | EMD Millipore | Cat#317240; CAS: 1115-70-4 |
| SC-26196 | Sigma-Aldrich | Cat#PZ0176; CAS: 218136-59-5 |
| CP-24879 | Sigma-Aldrich | Cat#C9115; CAS: 10141-51-2 |
| NHI-2 | Sigma-Aldrich | Cat#SML1463; CAS: 1269802-97-2 |
| α-Ketobutyrate | Sigma-Aldrich | Cat#K0875; CAS: 2013-26-5 |
| Sodium pyruvate | Thermo Fisher Scientific | Cat#11360070; CAS: 113-24-6 |
| Doxycycline | Sigma-Aldrich | Cat#D3447; CAS: 10592-13-9 |
| Arachidonic acid | EMD Millipore | Cat#181198; CAS: 506-32-1 |
| Docosahexaenoic acid | Sigma-Aldrich | Cat#D2534; CAS: 6217-54-5 |
| Eicosapentaenoic acid | EMD Millipore | Cat#324875; CAS: 10417-94-4 |
| Fatty acid-free bovine serum albumin | Sigma-Aldrich | Cat#A8806; CAS: 9048-46-8 |
| Sodium L-lactate-d3 solution | Sigma-Aldrich | Cat#616702; CAS: 867-56-1 (unlabeled) |
| Linoleic acid-13C18 | Cambridge Isotope Laboratories | Cat#CLM-6855; CAS: 60–33-3 (unlabeled) |
| Prostaglandin E2-d4 | Cayman Chemical | Cat#314010; CAS: 34210-10-1 |
| 1,2-dilauroyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids | Cat#850335; CAS: 18194-25-7 |
| Acetic acid, for LC/MS | Fisher Scientifics | Cat#A113-50; CAS: 64-19-7 |
| Formic acid, for LC/MS | Honeywell Fluka | Cat#56302; CAS: 64-18-6 |
| Ammonium acetate, for LC/MS | Sigma-Aldrich | Cat#73594; CAS: 631-61-8 |
| Ammonium hydroxide solution, for HPLC | Honeywell Fluka | Cat#17387; CAS: 1336-21-6 |
| Water, LC/MS grade | Fisher Scientifics | Cat# W6-4; CAS: 7732-18-5 |
| Methanol, LC/MS grade | Fisher Scientifics | Cat#A456-4; CAS: 67-56-1 |
| Isopropanol, LC/MS grade | Fisher Scientifics | Cat#A461-1; CAS: 67-63-0 |
| Acetonitrile, LC/MS grade | Fisher Scientifics | Cat#A955-4; CAS: 75-05-8 |
| tert-Butyl hydroperoxide | Sigma-Aldrich | Cat#458139; CAS: 75-91-2 |
| CM-H2DCFDA | Thermo Fisher Scientific | Cat#C6827; CAS: 4091–99-0 (H2DCFDA) |
| Critical Commercial Assays | ||
| DC protein assay kit | Bio-Rad | Cat#5000116 |
| Triglyceride quantification kit | BioVision | Cat#K622 |
| NAD/NADH quantitation kit | BioVision | Cat#K337 |
| NADP/NADPH quantitation kit | BioVision | Cat#K347 |
| Glucose assay kit | BioVision | Cat#K606 |
| RNesay mini kit | Qiagen | Cat#74104 |
| AMV first-strand cDNA synthesis kit | Thermo Fisher Scientific | Cat#12328 |
| SYBR green master mix | Thermo Fisher Scientific | Cat#A25741 |
| SuperSignal West Dura detection kit | Thermo Fisher Scientific | Cat#34076 |
| APEX Alexa Fluor 568 antibody labeling kit | Thermo Fisher Scientific | Cat#A10494 |
| CyQUANT cell proliferation assay kit | Thermo Fisher Scientific | Cat#C7026 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| mIMCD3 | ATCC | CRL-2123 |
| HEK293 | ATCC | CRL-1573 |
| Doxycycline-inducible HeLa-LbNOX cells | Titov et al., 2016 | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | The Jackson Laboratory | JAX: 000664 |
| Oligonucleotides | ||
| Primers for qPCR, see Table S3 | This paper | N/A |
| Recombinant DNA | ||
| pCMV6-Entry-empty | OriGene Technologies | Cat#PS100001 |
| pCMV6-Entry-human FADS1 | OriGene Technologies | Cat#RC229252 |
| pCMV6-Entry-human FADS2 | OriGene Technologies | Cat#RC223780 |
| pRS-scrambled non-effective shRNA control | OriGene Technologies | Cat#TR30012 |
| pRS-human shFADS1 | OriGene Technologies | Cat#TR313102; Tube ID: D |
| pRS-human shFADS2 | OriGene Technologies | Cat#TR313101; Tube ID: B |
| pLX304-empty | Rusu et al., 2017 | N/A |
| pLX304-SLC16A11REF-V5 | Rusu et al., 2017 | N/A |
| pLX304-SLC16A11T2D-V5 | Rusu et al., 2017 | N/A |
| Software and Algorithms | ||
| TraceFinder 3.3 (or 3.2) | Thermo Fisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30493 |
| MultiQuant 2.1 | SCIEX | https://sciex.com/products/software/multiquant-software |
| Prism 6 | GraphPad Software | https://www.graphpad.com/scientific-software/prism |
| ImageJ | NIH | https://imagej.nih.gov/ij |
| Other | ||
| Dialyzed FBS | Thermo Fisher Scientific | Cat#26400044 |
| Nonidet P-40 solution | Thermo Fisher Scientific | Cat#85124 |
| FuGENE HD transfection reagent | Promega | Cat#E2311 |
| MEM non-essential amino acids solution | Thermo Fisher Scientific | Cat#11140050 |
| Prolong diamond antifade mountant with DAPI | Thermo Fisher Scientific | Cat#P36966 |
| Amicon Ultra 10 kDa cut off filters | EMD Millipore | Cat#UFC501096 |
| 100 × 2.1 mm Acquity UPLC BEH C8 column | Waters | Cat#186002878 |
| 150 × 2.1 mm Acquity UPLC BEH C18 column | Waters | Cat#186002353 |
| 150 × 2.0 mm Luna NH2 column | Phenomenex | Cat#00F-4378-B0 |
Highlights.
Blocking ETC or lactate production reduces cytosolic NAD+/NADH and increases HUFAs
HUFA synthesis by D5D and D6D is a mechanism for glycolytic NAD+ recycling
D5D and D6D mediated NAD+ regeneration can be acutely adaptive in vivo
SLC16A11 and FADS1–3 may influence metabolic risk via impact on cytosolic NAD+/NADH
ACKNOWLEDGMENTS
We thank D. Titov and V. Mootha for providing LbNOX (Massachusetts General Hospital, Boston, MA), Y. Yang and J. Loscalzo for providing SoNAR (East China University, Shanghai, China; Brigham and Women’s Hospital, Boston, MA), and I. A. Rosales (Massachusetts General Hospital, Boston, MA) for assistance with mouse kidney histology. This work was supported by NIH U01 DK106981 and the Extramural Grant Program of Satellite Health Care, a not-for-profit renal care provider. The MCDS was supported by NIH R01 HL24799; Consejo Nacional de Ciencia y Tecnologi’a 2092, M9303, F677-M9407, 251M, 2005-C01–14502, and SALUD 2010–2151165; and Consejo Nacional de Ciencia y Tecnologi’a (CONACyT). The MCDS lipid profiling analyses were conducted as part of the Slim Initiative for Genomic Medicine, a project funded by the Carlos Slim Foundation in Mexico. All LC-MS data are stored at the Broad Institute and the Endocrine Unit, Massachusetts General Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bellomo R (2002). Bench-to-bedside review: lactate and the kidney. Crit Care 6, 322–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman BC, Wolfel EE, Butterfield GE, Lopaschuk GD, Casazza GA, Horning MA, and Brooks GA (1999). Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol 87, 1684–1696. [DOI] [PubMed] [Google Scholar]
- Burdge GC, Jones AE, and Wootton SA (2002). Eicosapentaenoic and docosapentaenoic acids are the principal products of alpha-linolenic acid metabolism in young men*. Br. J. Nutr 88, 355–363. [DOI] [PubMed] [Google Scholar]
- Burdge GC, and Wootton SA (2002). Conversion of alpha-linolenic acid to eicosapentaenoic, docosapentaenoic and docosahexaenoic acids in young women. Br. J. Nutr 88, 411–420. [DOI] [PubMed] [Google Scholar]
- Calvaresi EC, Granchi C, Tuccinardi T, Di Bussolo V, Huigens RW 3rd, Lee HY, Palchaudhuri R, Macchia M, Martinelli A, Minutolo F, et al. (2013). Dual targeting of the Warburg effect with a glucose-conjugated lactate dehydrogenase inhibitor. Chembiochem 14, 2263–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HP, Nakamura M, and Clarke SD (1999a). Cloning, expression, and fatty acid regulation of the human delta-5 desaturase. J. Biol. Chem 274, 37335–37339. [DOI] [PubMed] [Google Scholar]
- Cho HP, Nakamura MT, and Clarke SD (1999b). Cloning, expression, and nutritional regulation of the mammalian Delta-6 desaturase. J. Biol. Chem 274, 471–477. [DOI] [PubMed] [Google Scholar]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, et al. (2010). New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet 42, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada K, Aukrust I, Bjorkhaug L, Burtt NP, Mercader JM, Garcia-Ortiz H, Huerta-Chagoya A, Moreno-Macias H, Walford G, Flannick J, et al. (2014). Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 311, 2305–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Hara K, Shojima N, Horikoshi M, Iwata M, Hirota Y, Tobe K, Seino S, and Kadowaki T (2012). Variations with modest effects have an important role in the genetic background of type 2 diabetes and diabetes-related traits. J. Hum. Genet 57, 776–779. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Ishibashi T, and Imai Y (1984). Cytoplasmic location of linoleoyl-CoA desaturase in microsomal membranes of rat liver. Arch. Biochem. Biophys 233, 402–407. [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Moltke I, Grarup N, Racimo F, Bjerregaard P, Jorgensen ME, Korneliussen TS, Gerbault P, Skotte L, Linneberg A, et al. (2015). Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 349, 1343–1347. [DOI] [PubMed] [Google Scholar]
- Funk CD (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875. [DOI] [PubMed] [Google Scholar]
- Gieger C, Geistlinger L, Altmaier E, Hrabe de Angelis M, Kronenberg F, Meitinger T, Mewes HW, Wichmann HE, Weinberger KM, Adamski J, et al. (2008). Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet 4, e1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson SM, Freinkman E, Thomas CJ, and Vander Heiden MG (2016). Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab 24, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillou H, D’Andrea S, Rioux V, Barnouin R, Dalaine S, Pedrono F, Jan S, and Legrand P (2004). Distinct roles of endoplasmic reticulum cytochrome b5 and fused cytochrome b5-like domain for rat Delta6-desaturase activity. J. Lipid Res 45, 32–40. [DOI] [PubMed] [Google Scholar]
- Halestrap AP (2013). The SLC16 gene family - structure, role and regulation in health and disease. Mol. Aspects Med 34, 337–349. [DOI] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, et al. (2016). Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Ngoy S, Sheth SA, Swanson RA, Rhee EP, Liao R, Clish CB, Mootha VK, and Nilsson R (2014). A systematic survey of lipids across mouse tissues. Am. J. Physiol. Endocrinol. Metab 306, E854–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson A, Westerberg R, and Jacobsson A (2006). Fatty acid elongases in mammals: their regulation and roles in metabolism. Prog. Lipid Res 45, 237–249. [DOI] [PubMed] [Google Scholar]
- Katz SD, Bleiberg B, Wexler J, Bhargava K, Steinberg JJ, and LeJemtel TH (1993). Lactate turnover at rest and during submaximal exercise in patients with heart failure. J. Appl. Physiol 75, 1974–1979. [DOI] [PubMed] [Google Scholar]
- King MP, and Attardi G (1989). Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503. [DOI] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, and Dang CV (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337. [DOI] [PubMed] [Google Scholar]
- Kraut JA, and Madias NE (2014). Lactic acidosis. N. Engl. J. Med 371, 2309–2319. [DOI] [PubMed] [Google Scholar]
- Nakamura MT, and Nara TY (2004). Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu. Rev. Nutr 24, 345–376. [DOI] [PubMed] [Google Scholar]
- Napier JA, Michaelson LV, and Sayanova O (2003). The role of cytochrome b5 fusion desaturases in the synthesis of polyunsaturated fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 68, 135–143. [DOI] [PubMed] [Google Scholar]
- Obukowicz MG, Raz A, Pyla PD, Rico JG, Wendling JM, and Needleman P (1998a). Identification and characterization of a novel delta6/delta5 fatty acid desaturase inhibitor as a potential anti-inflammatory agent. Biochem. Pharmacol 55, 1045–1058. [DOI] [PubMed] [Google Scholar]
- Obukowicz MG, Welsch DJ, Salsgiver WJ, Martin-Berger CL, Chinn KS, Duffin KL, Raz A, and Needleman P (1998b). Novel, selective delta6 or delta5 fatty acid desaturase inhibitors as antiinflammatory agents in mice. J. Pharmacol. Exp. Ther 287, 157–166. [PubMed] [Google Scholar]
- Park HG, Park WJ, Kothapalli KS, and Brenna JT (2015). The fatty acid desaturase 2 (FADS2) gene product catalyzes Delta4 desaturation to yield n-3 docosahexaenoic acid and n-6 docosapentaenoic acid in human cells. FASEB J 29, 3911–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park WJ, Kothapalli KS, Reardon HT, Lawrence P, Qian SB, and Brenna JT (2012). A novel FADS1 isoform potentiates FADS2-mediated production of eicosanoid precursor fatty acids. J. Lipid Res 53, 1502–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quehenberger O, and Dennis EA (2011). The human plasma lipidome. N. Engl. J. Med 365, 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee EP, Cheng S, Larson MG, Walford GA, Lewis GD, McCabe E, Yang E, Farrell L, Fox CS, O’Donnell CJ, et al. (2011). Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J. Clin. Invest 121, 1402–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee EP, Ho JE, Chen MH, Shen D, Cheng S, Larson MG, Ghorbani A, Shi X, Helenius IT, O’Donnell CJ, et al. (2013). A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab 18, 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusu V, Hoch E, Mercader JM, Tenen DE, Gymrek M, Hartigan CR, DeRan M, von Grotthuss M, Fontanillas P, Spooner A, et al. (2017). Type 2 Diabetes Variants Disrupt Function of SLC16A11 through Two Distinct Mechanisms. Cell 170, 199–212 e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, and Rapoport TA (2010). Mechanisms determining the morphology of the peripheral ER. Cell 143, 774–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Voeltz GK, and Rapoport TA (2006). Rough sheets and smooth tubules. Cell 126, 435–439. [DOI] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, and Mootha VK (2016). Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, et al. (2016). PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Agriculture, Agricultural Research Service. (2018). Nutrient Intakes from Food and Beverages: Mean Amounts Consumed per Individual, by Gender and Age, What We Eat in America, NHANES 2015–2016.
- Wang D, and Dubois RN (2010). Eicosanoids and cancer. Nat. Rev. Cancer 10, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassall SR, and Stillwell W (2009). Polyunsaturated fatty acid-cholesterol interactions: domain formation in membranes. Biochim. Biophys. Acta 1788, 24–32. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Ohno M, Taguchi M, Kawamoto S, Ono K, and Aki T (2016). Identification of amino acid residues that determine the substrate specificity of mammalian membrane-bound front-end fatty acid desaturases. J. Lipid Res 57, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Yu D, Liu M, Wang M, Zhao M, Jia W, Ma H, Fang J, Xu W, Chen K, et al. (2014). Genome-wide association study identifies three susceptibility loci for laryngeal squamous cell carcinoma in the Chinese population. Nat. Genet 46, 1110–1114. [DOI] [PubMed] [Google Scholar]
- Williams AL, Jacobs SB, Moreno-Macias H, Huerta-Chagoya A, Churchhouse C, Marquez-Luna C, Garcia-Ortiz H, Gomez-Vazquez MJ, Burtt NP, Aguilar-Salinas CA, et al. (2014). Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate R, El Jaber-Vazdekis N, Tejera N, Perez JA, and Rodriguez C (2017). Significance of long chain polyunsaturated fatty acids in human health. Clin Transl Med 6, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Jia WH, Matsuda K, Kweon SS, Matsuo K, Xiang YB, Shin A, Jee SH, Kim DH, Cai Q, et al. (2014). Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat. Genet 46, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, Hu H, Chen X, Zhou HM, Huang X, et al. (2015). SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab 21, 777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Jin J, Hu Q, Zhou HM, Yi J, Yu Z, Xu L, Wang X, Yang Y, and Loscalzo J (2011). Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab 14, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.