

Abstract
Epoxide hydrolases (EHs) are enzymes involved in the metabolism of endogenous and exogenous epoxides, and the development of EH inhibitors has important applications in the medicine. In humans, EH inhibitors are being tested in the treatment of cardiovascular diseases and show potent anti-inflammatory effects. EH inhibitors are also considerate promising molecules against infectious diseases. EHs are functionally very well studied, but only a few members have its three-dimensional structures characterized. Recently, a new EH from the filamentous fungi Trichoderma reseei (TrEH) was reported, and a series of urea or amide-based inhibitors were identified. In this study, we describe the crystallographic structures of TrEH in complex with five different urea or amide-based inhibitors with resolutions ranging from 2.6 to 1.7 Å. The analysis of these structures reveals the molecular basis of the inhibition of these compounds. We could also observe that these inhibitors occupy the whole extension of the active site groove and only a few conformational changes are involved. Understanding the structural basis EH interactions with different inhibitors might substantially contribute for the study of fungal metabolism and in the development of novel and more efficient antifungal drugs against pathogenic Trichoderma species.
Keywords: Trichoderma reesei, epoxide hydrolase, inhibitors, crystal structure
1. Introduction
Epoxide hydrolases (EHs) are essential enzymes found in all living organisms [1, 2] and catalyze the hydrolysis of epoxides, which open the epoxide groups converting them to the corresponding vicinal trans-dihydrodiols [1, 3]. Epoxide hydrolases are involved in various processes in different organisms. Naturally, these enzymes generally have three main functions: detoxification, catabolism, and regulation of signaling molecules [2]. Particularly, fungal EHs have been described to participate in the process of detoxification of xenobiotics, preventing their reaction with proteins or DNA, which may reduce the toxic effects of these molecules [4, 5].
Urea or amide-based EH inhibitors are molecules that have a urea or amide groups mimicking the epoxide moiety and bind to the catalytic site of most EHs, preventing the catalysis [6]. Several of these compounds have already been shown to be effective as a therapeutic strategy in the treatment of cardiovascular diseases since they reduce significantly the blood pressure in rats [7]. In addition, these molecules also have several other important bioactivities since they are effective against neuropathic diabetic pain in rodent models [8]; inhibit vascular smooth muscle cell proliferation [9]; show potent anti-inflammatory effects [10]; and have positive effects against equine laminitis [11].
EH inhibitors are also considered interesting molecules for controlling pathogens since several EHs have been described to be essential for the microorganisms, including Mycobacterium tuberculosis (the causative agent of tuberculosis), which has an essential EH involved in the biosynthesis of mycolic acids, an important lipid class from the mycobacterial cell wall, which is a key virulence factor [12]. Biswal et al. [13] reported that an EH from M. tuberculosis are involved in a detoxification pathway and could be a potential drug target for the development of antitubercular, however, further studies about in vivo activity of these EH inhibitors are necessary. The study from Spillman et al. [14] showed that two EHs from Plasmodium falciparum play an important role in the infection process of human cells, suggesting that inhibitors for these enzymes could be used against malaria. However, studies about the role of EH inhibitors in the process of malaria infection have so far not reported. An EH from Pseudomonas aeruginosa, an opportunistic and nosocomial bacteria, helps the microorganism to establish itself into the human lungs cavity [15]. Therefore further studies about the inhibition of this EH are necessary. However, there is a complete lack of studies about the activity of these EH inhibitors against pathogenic fungi.
Recently, our group reported the 3D-structure of a soluble EH from Trichoderma reesei QM9414 (TrEH) [16] and we also have identified potent and effective inhibitors against this enzyme (figure 1, compounds 2-5). In addition, these compounds inhibited the fungal growth in more than 60% [17]. In this study, we have used T. reesei as a microorganism model for the Trichoderma genus, although it is also widely used as an industrial host organism for protein production [18]. Generally, Trichoderma species are saprophytic filamentous fungi with worldwide distribution in the soil and organic material, but worryingly, some Trichoderma species are also described as an emerging fatal pathogen in immunocompromised patients, as T. longibrachiatum [19], T. harzianum [20] and T. pseudokoningii [21]. These opportunistic species of Trichoderma are clinically relevant pathogens and contribute to increase the morbidity and mortality, mainly in immunocompromised patients infected with HIV [22].
Figure 1. Structure of molecules identified as TrEH inhibitors.
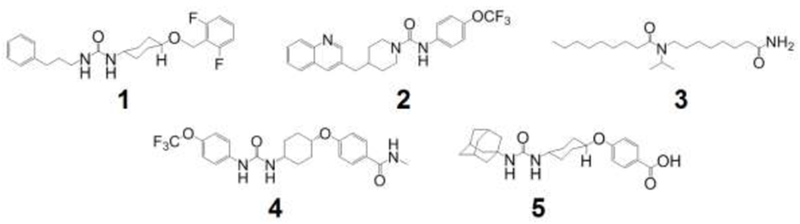
Molecules 2-5 were identified and the IC50 was determined by De Oliveira et al [17]. All inhibitors have an urea or amide group that might mimic an epoxide group in the active site of the enzyme.
Herein, we report the 3D-structures of TrEH in complex with five different urea or amide-based inhibitors at resolution between 1.7 to 2.6 Å, which provide insights into the structural base of the specificity and inhibitory mechanisms that may be used in the development of more EH specific inhibitors for the treatment of infections caused by pathogenic Trichoderma species.
2. Material and methods
2.1. Cloning, overexpression and purification
The cloning, overexpression and purification of TrEH has been published previously [23]. Briefly, the ORF corresponding to TrEH has been cloned and inserted into a pPROEX-HTa plasmid (Life Technologies, USA), which was used to transform competent E. coli BL21 cells, and the overexpression was carried out by the induction using IPTG. For the purification, the soluble fraction of the bacterial lysate was loaded onto a His-Trap Chelating column connected to an ÄKTA FPLC System (GE Healthcare, USA). The molecular mass and purity of the protein were determined by SDS–PAGE under denaturing conditions. The concentration of the purified protein was estimated following the method described by Whitaker and Einar-granum [24].
2.2. Inhibitors identification and IC50 determination
An high-throughput screening assay to identify TrEH inhibitors as well as the method used to determine the respectively IC50 was described by de Oliveira et al [17]. Briefly, we screened almost three thousands molecules synthesized as described by Shen and Hammock [6] with scaffolds based on urea, amide or carbamate-based inhibitors against TrEH. The inhibitory constants were measured using the TrEH ([Enzyme]final = 112.5 ng/mL), a fluorescent substrate (cyano(6-methoxy-naphthalen-2-yl)methyl oxiran-2-ylmethyl carbonate; PHOME) at [Substrate]final = 22.5 μM and urea or amide-based inhibitors (1 nM ≤ [Inhibitor]final ≤ 50 μM). These data were measured using a Gemini EM fluorescent plate reader (Molecular Devices, USA), with the excitation wavelength of 330 nm and an emission wavelength of 465 nm. The structures of the inhibitors are given in table 1, and boldface numbers throughout the text refer to these compounds.
Table 1.
IC50 for TrEH inhibitors.
| Inhibitor structure | N° | IC50 (nM)* | Reference |
|---|---|---|---|
| 1 | 29,0 ± 8,7 | Data from this study | |
| 2 | 99,0 ± 8,5 | [17] | |
| 3 | 138,8 ± 14,4 | [17] | |
| 4 | 259,3 ± 71,3 | [17] | |
| 5 | 357,9 ± 57,6 | [17] |
Results are averages of triplicate experiments.
2.3. Crystallization
The crystallization of TrEH was performed following the protocol established previously [16] with few modifications. Briefly, concentrated TrEH (13 mg/mL) was subjected to the vapor diffusion crystallization method through hanging drop crystallization technique using a condition constituted by 50 mM 3-morpholinopropane-1 -sulfonic acid (MOPS), pH 6.5, 40 mM potassium bromide and 44.6% PEG 4000. Previously in the co-crystallization experiments, TrEH was incubated for 30 minutes on ice in the presence of 10 mM of the compounds 1-5 (Figure 1).
2.4. Data processing and structure resolution
Crystals of TrEH in complex with different inhibitors were diffracted at PETRA-III/DESY (Hamburg, Germany) or at Laboratório Nacional de Luz Síncrotron (LNLS) (Campinas, Brazil). All the X-ray diffraction data were processed and analyzed using XDS [25], which were scaled by the program Aimless [26] from the CCP4i suite [27]. The structures of the TrEH in complex with different inhibitors were solved by molecular replacement using the program Phaser [28] using as a search model the structure of TrEH in apo form (PDB entry: 5URO). The refinement of the structures was carried out using the program phenix.refine [29] from PHENIX suite [30] and the visual inspection and manual building were performed by the program COOT [31]. The stereochemistry quality of the structures was verified using the program MolProbity [32]. To identify the protein-ligand interactions, we have used the program LIGPLOT [33]. PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC was used to prepare high-quality figures.
3. Results and discussion
3.1. Inhibitors identification and IC50 determination
Recently our research group described the screening of about three thousands compounds to identify the molecules with high inhibitory activity against TrEH [17], and we have identified four effective urea or amide-based TrEH inhibitors (Figure 1. molecules 2-5). In this work, we have identified and determined the IC50 for another further urea-based TrEH inhibitor synthesized as described in a review made by Shen and Hammock [6]. This compound has a similar inhibitory activity to the previously identified molecules (Table 1). 1, 2, 4 and 5 are urea-based inhibitors have several aromatic groups in their structure and, additionally, 5 has a further adamantane group at the extremity nearer the end of the urea moiety. The compound 3 is the unique amide-based inhibitor and it has a long aliphatic chain with a terminal amide function, probably mimicking epoxy fatty acids, which have already been described as a substrate of TrEH [17].
3.2. TrEH structure in complex with inhibitors
Crystals for TrEH in complex with inhibitors are generally thin plates belonging to the space group P21212. These crystals diffracted ranging between 1.7 to 2.6 Å resolution with a single monomer in the asymmetric unit. The 3D structures of TrEH in complex with the urea-based inhibitors 1, 2, 4 and 5 and amide-based 3 were determined by molecular replacement using as a search model the structure of T. reesei soluble epoxide hydrolase in apo form (TrEH; PDB entry 5URO) [16]. The X-ray data, crystallographic statistics and stereochemistry structure analysis are in the Supplementary Material Table 1.
TrEH has two domains, an α/β hydrolase domain constituted by a central β-sheet surrounded by α-helices and a cap domain which has seven α-helices and long loop regions. The superposition of all structures of TrEH in complexes with inhibitors indicated that the binding of inhibitors does not cause large conformational changes with an R.M.S.D. of Cα for all structures of about 0.5Å, corroborating the hypothesis that this enzyme is not very flexible [16] (Figure 2).
Figure 2. Superposition of TrEH structures in complex with 5 different inhibitors.
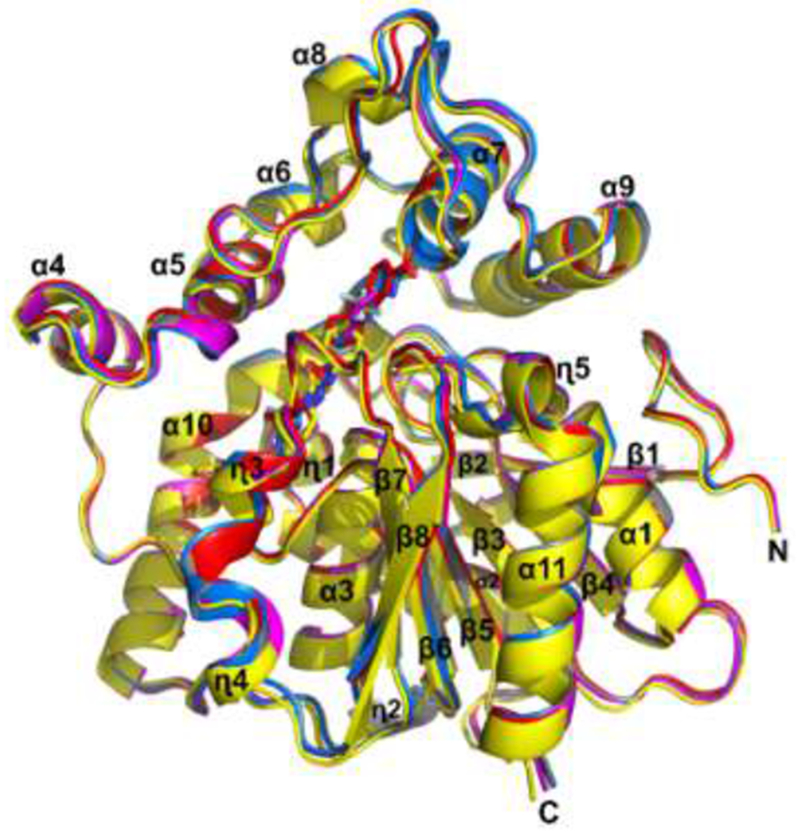
TrEH in complex with inhibitor 1 (pink), TrEH in complex with inhibitor 2 (yellow), TrEH in complex with inhibitor 3 (blue), TrEH in complex with inhibitor 4 (gray), TrEH in complex with inhibitor 5 (red).
All inhibitors interact through hydrogen bonds with D116, Y167 and Y252 (yellow amino acids from figure 3), which are key residues involved in the catalytic mechanism of TrEH [16]. This observation is in agreement with the study by Shen and Hammock [6], which indicates that the urea and amide groups could be used in EH inhibition studies since they bind to the catalytic site of EHs. In addition, all inhibitors also have interactions with residues of the catalytic site groove, including W46, W117, Q168, M193 and H313 (Figure 3). Additionally, most of the inhibitors (three or four) interact with H144, A288, F205, F165 and W314 (Figure 3). Inhibitors 2 and 5 also perform interactions with L289. Specifically, the Inhibitors 2, 3, 4 and 5 interact with A120, T141, M293 and G195, respectively. The hydrophobic interaction involving tryptophan, phenylalanine and histidine residues, which form π-interactions with different rings of the inhibitors 1-5. Figure 3 shows the π-stacking interactions of: H144 with 1,3-difluorobenzene of the compound 1, quinoline of the compound 2 and benzene of the compound 4; F165 and W117 with benzene of the compound 1 and 4, pyridine of compound 2 and adamantane of compound 5; and W314 with benzene of the compound 1 and 5.
Figure 3. Interaction of inhibitors 1-5 to the active site of TrEH.
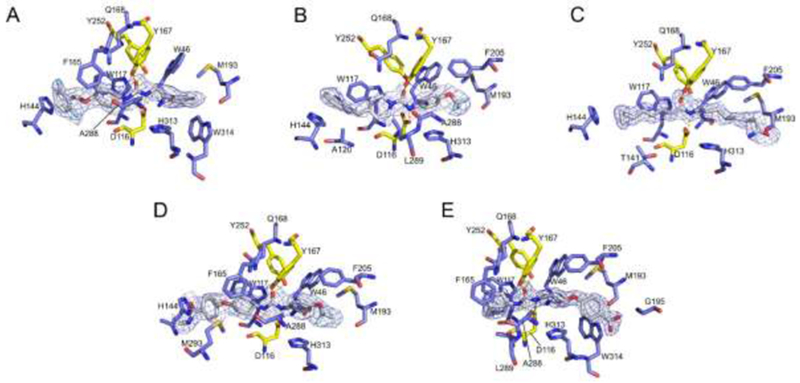
TrEH in complex with inhibitors: compound 1 (A), compound 2 (B), compound 3 (C), compound 4 (D), compound 5 (E). Residues that are hydrogen interacting with inhibitors are represented in yellow and those that are performing hydrophobic interaction are represented in blue.
Differently of the other compounds, 3 is an aliphatic molecule and consequently is unable to perform π-interaction, however, this compound still performs hydrophobic contacts as observed in the other compounds. In addition, this compound has a terminal amide group that might be mimicking epoxy fatty acids substrates as we have demonstrated that this enzyme is active against several fatty acids, such as those formed by arachidonic acid, linolenic acid, eicosapentaenoic acid and docosahexaenoic acid [17].
Analysis of the interactions of TrEH in complex with inhibitor 5 indicates that the amino acids W117, F165 and Q168 undergo significant conformational changes, including rotations in their side chains (17°, 30° and 18° for W117, F165, and Q168, respectively) (Figure 4) to adjust the interaction with the large adamantane moiety of this inhibitor in comparison to the apo structure. In contrast, other inhibitors, as 3, did not cause extensively conformational changes on TrEH and only the side chain of F165 rotates, allowing hydrophobic interactions with the inhibitor. However, as most of these inhibitors did not have bulky moieties as observed in 5, the rotation of FI 65 is only about 18° (Figure 4).
Figure 4. Conformational changes of amino acids caused by the interactions with inhibitors 3 and 5.
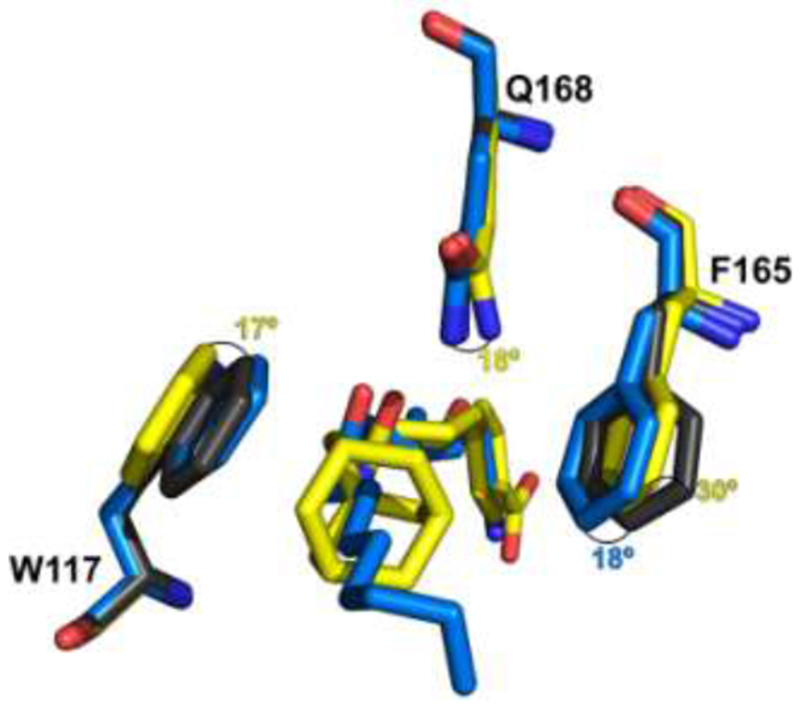
Conformational changes for tryptophan117, phenylalanine165 and Glutamine168 for the apo structure (Gray; PDB:5URO) in comparison to the inhibitors 3 (blue) and 5 (yellow). The numbers in blue and yellow show the rotation changes in degrees of the apo structure in comparison with structures in complex with inhibitor 3 and 5, respectively.
Interesting, all inhibitors identified to have high affinity for TrEH are sufficiently long in size to occupy the whole active site groove, extending to both entries of the cavity (Figure 5). Therefore, the size of the molecule could be a key characteristic for inhibitory activity, that could be reached at high affinity through the optimization of hydrophobic interactions of different groups in both sides of the urea or amide groups.
Figure 5. Active site surface of TrEH in complex with inhibitors.
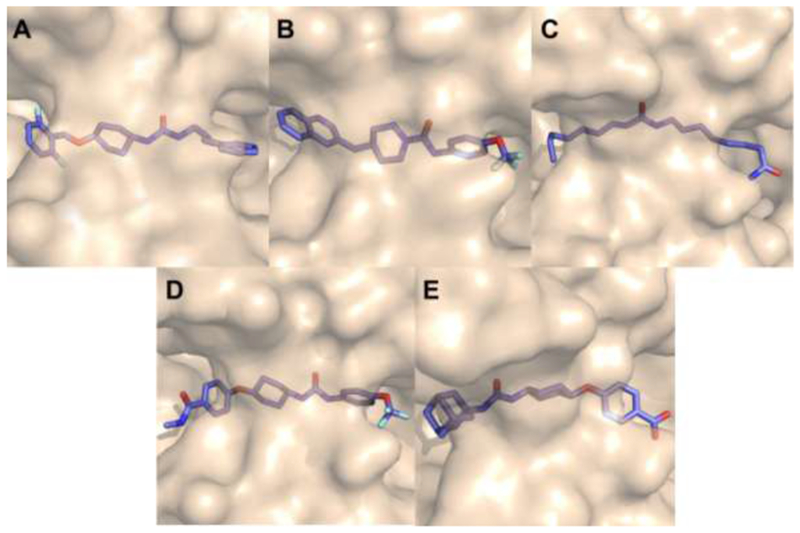
TrEH in complex with inhibitors: compound 1 (A), compound 2 (B), compound 3 (C), compound 4 (D), compound 5 (E).
In addition, urea or amide-based EH inhibitors have also been studied in an EH from M tuberculosis [13]. Interestingly, structures of EH from M. tuberculosis in complex with the best inhibitors for this enzyme have extensions in both sides of the urea or amide groups much smaller than the best compounds that inhibit TrEH. However, the IC50 of the best compound against the EH from M. tuberculosis is 19 nM, which is very similar to those observed for TrEH. Wilson et al. [16] report that the active site pocket volume of TrEH is much larger than M. tuberculosis EH (PDB: 2ZJF) [13] and consequently the size of the cavity might be an important factor that should be involved in the specificity of different molecules and that consequently have strong impact on the IC50 of the molecules.
The crystallographic structures of human EH in complex with different compounds of this series have also been determined [34]. The catalytic cavity volume of human EH is larger than TrEH [16], but a structure of human EH in complex with an inhibitor very similar to the inhibitor 4 described in this work was also reported. The difference between the two ligands is an amide group in place of the carboxyl group, which may indicate that this is an inhibitor that does not occupy the whole catalytic cavity. In Figure 6, we can observe the comparison of the structure around the active site of the EH from T. reesei, Human [34] and M tuberculosis [13], and the presence of a loop (highlighted in red) that delimits the size of the active site in different EHs and the urea or amide-based inhibitors.
Figure 6. Structure around the active site.
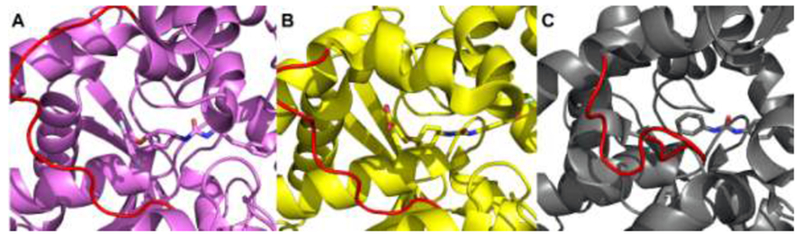
TrEH in complex with inhibitor 1 (A), Human EH in complex with inhibitor – PDB: 6AUM [34] (B), EH from the Mycobacterium tuberculosis in complex with inhibitor – PDB: 2ZJF [13](C). The loop in red delimits the size of the active site.
4. Conclusion
We have obtained five structures at a resolution ranging between 1.7 Å and 2.6 Å of a soluble epoxide hydrolase from the filamentous fungi T. reesei in complex with five urea or amide-based inhibitors. However, one of them is a new compound and it has been identified and characterized in this work and the other further inhibitors were previously determined by our research group [17]. In this same work, we demonstrated that these compounds (1 mM) could inhibit the fungus growth in more than 60% indicating that they could be used in the development of new drugs against pathogenic fungi. The structures of TrEH in complexes with different inhibitors show that the inhibitors do not cause drastic conformational changes and that these molecules occupy the whole extension of the active site groove.
All the TrEH inhibitors perform hydrogen bonds with the same amino acids (D116, Y167 and Y252). The hydrophobic interactions are variable from an inhibitor to another, but the most important ones are the π-stacking interactions between inhibitors and H144, F165, W117 and W314. The comparison of T. reesei, human and M. tuberculosis EH structures in complex with urea-based inhibitors shows that a Loop in the Cap domain delimits the size of the active site and consequently has a strong impact in the specificity of inhibitors against different enzymes.
The structural insights obtained in this work provide useful information into the inhibitory mechanism and interactions of urea or amide-based compounds with the amino acid residues of the active site of TrEH that may contribute in the drug development of efficient molecules against pathogenic Trichoderma species.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Environmental Health Sciences (NIEHS-R01 ES002710), the Superfund Program NIEHS (P42 ES 04699), and São Paulo Research Foundation (FAPESP-2014/24107-1; 2017/25705-8; 2015/09188-8 and 2018/00351-1). G.S.O., P.P.A. and J.A.R are supported by scholarships from FAPESP (2015/03329-9 and 2016/12859-4), FAPESP (2013/15906-5) and CNPq (142311/2016-2), respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that they have no conflict of interest.
5. References
- [1].Fretland AJ, Omiecinski CJ, Epoxide hydrolases: biochemistry and molecular biology, Chemico-biological interactions 129(1-2) (2000) 41–59. [DOI] [PubMed] [Google Scholar]
- [2].Morisseau C, Hammock BD, Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles, Annu Rev Pharmacol Toxicol 45 (2005) 311–33. [DOI] [PubMed] [Google Scholar]
- [3].Decker M, Arand M, Cronin A, Mammalian epoxide hydrolases in xenobiotic metabolism and signalling, Archives of toxicology 83(4) (2009) 297–318. [DOI] [PubMed] [Google Scholar]
- [4].Smit MS, Fungal epoxide hydrolases: new landmarks in sequence-activity space, Trends in biotechnology 22(3) (2004) 123–129. [DOI] [PubMed] [Google Scholar]
- [5].Wackett L, Gibson D, Metabolism of xenobiotic compounds by enzymes in cell extracts of the fungus Cunninghamella elegans, Biochemical Journal 205(1) (1982) 117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shen HC, Hammock BD, Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications, J Med Chem 55(5) (2012) 1789–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL, Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids, Circ Res 87(11) (2000) 992–8. [DOI] [PubMed] [Google Scholar]
- [8].Inceoglu B, Wagner KM, Yang J, Bettaieb A, Schebb NH, Hwang SH, Morisseau C, Haj FG, Hammock BD, Acute augmentation of epoxygenated fatty acid levels rapidly reduces pain-related behavior in a rat model of type I diabetes, Proc Natl Acad Sci U S A 109(28) (2012) 11390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH, Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation, Proc Natl Acad Sci U S A 99(4) (2002) 2222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD, Soluble epoxide hydrolase is a therapeutic target for acute inflammation, Proc Natl Acad Sci U S A 102(28) (2005) 9772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guedes AG, Morisseau C, Sole A, Soares JH, Ulu A, Dong H, Hammock BD, Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis, Vet Anaesth Analg 40(4) (2013) 440–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Madacki J, Laval F, Grzegorzewicz A, Lemassu A, Zahorszka M, Arand M, McNeil M, Daffe M, Jackson M, Laneelle MA, Kordulakova J, Impact of the epoxide hydrolase EphD on the metabolism of mycolic acids in mycobacteria, J Biol Chem 293(14) (2018) 5172–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Biswal BK, Morisseau C, Garen G, Cherney MM, Garen C, Niu C, Hammock BD, James MN, The molecular structure of epoxide hydrolase B from Mycobacterium tuberculosis and its complex with a urea-based inhibitor, J Mol Biol 381(4) (2008) 897–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Spillman NJ, Dalmia VK, Goldberg DE, Exported Epoxide Hydrolases Modulate Erythrocyte Vasoactive Lipids during Plasmodium falciparum Infection, MBio 7(5) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Flitter BA, Hvorecny KL, Ono E, Eddens T, Yang J, Kwak DH, Bahl CD, Hampton TH, Morisseau C, Hammock BD, Liu X, Lee JS, Kolls JK, Levy BD, Madden DR, Bomberger JM, Pseudomonas aeruginosa sabotages the generation of host proresolving lipid mediators, Proc Natl Acad Sci U S A 114(1) (2017) 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wilson C, De Oliveira GS, Adriani PP, Chambergo FS, Dias MVB, Structure of a soluble epoxide hydrolase identified in Trichoderma reesei, Biochim Biophys Acta 1865(8) (2017) 1039–1045. [DOI] [PubMed] [Google Scholar]
- [17].de Oliveira GS, Adriani PP, Wu H, Morisseau C, Hammock BD, Chambergo FS, Substrate and inhibitor selectivity, and biological activity of an epoxide hydrolase from Trichoderma reesei, Molecular biology reports (2018) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zafra G, Cortes-Espinosa DV, Biodegradation of polycyclic aromatic hydrocarbons by Trichoderma species: a mini review, Environ Sci Pollut Res Int 22(24) (2015) 19426–33. [DOI] [PubMed] [Google Scholar]
- [19].Myoken Y, Sugata T, Fujita Y, Asaoku H, Fujihara M, Mikami Y, Fatal necrotizing stomatitis due to Trichoderma longibrachiatum in a neutropenic patient with malignant lymphoma: a case report, Int J Oral Maxillofac Surg 31(6) (2002) 688–91. [DOI] [PubMed] [Google Scholar]
- [20].Guarro J, Antolin-Ayala MI, Gene J, Gutierrez-Calzada J, Nieves-Diez C, Ortoneda M, Fatal case of Trichoderma harzianum infection in a renal transplant recipient, J Clin Microbiol 37(11) (1999) 3751–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gautheret A, Dromer F, Bourhis JH, Andremont A, Trichoderma pseudokoningii as a cause of fatal infection in a bone marrow transplant recipient, Clin Infect Dis 20(4) (1995) 1063–4. [DOI] [PubMed] [Google Scholar]
- [22].Walsh T, Groll A, Hiemenz J, Fleming R, Roilides E, Anaissie E, Infections due to emerging and uncommon medically important fungal pathogens, Clinical Microbiology and Infection 10 (2004) 48–66. [DOI] [PubMed] [Google Scholar]
- [23].de Oliveira GS, Adriani PP, Borges FG, Lopes AR, Campana PT, Chambergo FS, Epoxide hydrolase of Trichoderma reesei: Biochemical properties and conformational characterization, Int J Biol Macromol 89 (2016) 569–74. [DOI] [PubMed] [Google Scholar]
- [24].Whitaker JR, Granum PE, An absolute method for protein determination based on difference in absorbance at 235 and 280 nm, Anal Biochem 109(1) (1980) 156–9. [DOI] [PubMed] [Google Scholar]
- [25].Kabsch W, Xds, Acta Crystallographica Section D: Biological Crystallography 66(2) (2010) 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Evans PR, Murshudov GN, How good are my data and what is the resolution?, Acta Crystallographica Section D: Biological Crystallography 69(7) (2013) 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, Overview of the CCP4 suite and current developments, Acta Crystallographica Section D 67(4) (2011) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software, Journal of applied crystallography 40(4) (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD, Towards automated crystallographic structure refinement with phenix. refine, Acta Crystallographica Section D: Biological Crystallography 68(4) (2012) 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, Hung L-W, Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard, Acta Crystallographica Section D: Biological Crystallography 65(6) (2009) 582–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta Crystallographica Section D: Biological Crystallography 60(12) (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [32].Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallographica Section D: Biological Crystallography 66(1) (2010) 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wallace AC, Laskowski RA, Thornton JM, LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions, Protein engineering, design and selection 8(2) (1995) 127–134. [DOI] [PubMed] [Google Scholar]
- [34].Kodani SD, Bhakta S, Hwang SH, Pakhomova S, Newcomer ME, Morisseau C, Hammock BD, Identification and optimization of soluble epoxide hydrolase inhibitors with dual potency towards fatty acid amide hydrolase, Bioorganic & medicinal chemistry letters 28(4) (2018) 762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.