

Abstract
Numerous placebo-controlled studies have demonstrated the ability of ketamine, an NMDA receptor antagonist, to induce rapid (within hours), transient antidepressant effects when administered intravenously (IV) at subanesthetic doses (0.5 mg/kg over 40 min). However, the optimal antidepressant dose remains unknown. We aimed to compare to active placebo the rapid acting antidepressant properties of a broad range of subanesthetic doses of IV ketamine among outpatients with treatment-resistant depression (TRD). A range of IV ketamine doses were compared to active placebo in the treatment of adult TRD over a 3-day period following a single infusion over 40 min. This was an outpatient study conducted across six US academic sites. Outpatients were 18–70 years old with TRD, defined as failure to achieve a satisfactory response (e.g., less than 50% improvement of depression symptoms) to at least two adequate treatment courses during the current depressive episode. Following a washout period, 99 eligible subjects were randomly assigned to one of the five arms in a 1:1:1:1:1 fashion: a single intravenous dose of ketamine 0.1 mg/kg (n = 18), a single dose of ketamine 0.2 mg/kg (n = 20), a single dose of ketamine 0.5 mg/kg (n = 22), a single dose of ketamine 1.0 mg/kg (n = 20), and a single dose of midazolam 0.045 mg/kg (active placebo) (n = 19). The study assessments (HAM-D-6, MADRS, SDQ, PAS, CGI-S, and CGI-I) were performed at days 0, 1, 3 (endpoint), 5, 7, 14, and 30 to assess the safety and efficacy. The overall group × time interaction effect was significant for the primary outcome measure, the HAM-D-6. In post hoc pairwise comparisons controlling for multiple comparisons, standard dose (0.5 mg/kg) and high dose (1 mg/kg) of intravenous ketamine were superior to active placebo; a low dose (0.1 mg/kg) was significant only prior to adjustment (p = 0.02, p-adj = 0.14, d = −0.82 at day 1). Most of the interaction effect was due to differences at day 1, with no significant adjusted pairwise differences at day 3. This pattern generally held for secondary outcomes. The infusions of ketamine were relatively well tolerated compared to active placebo, except for greater dissociative symptoms and transient blood pressure elevations with the higher doses. Our results suggest that there is evidence for the efficacy of the 0.5 mg/kg and 1.0 mg/kg subanesthetic doses of IV ketamine and no clear or consistent evidence for clinically meaningful efficacy of lower doses of IV ketamine. Trial Registration: NCT01920555.
Subject terms: Drug discovery, Depression
Highlights
Question: What is the optimal, rapid antidepressant dose of intravenous (IV) ketamine, an NMDA receptor antagonist?
Findings: Our results suggest that there is evidence for the efficacy of the 0.5 mg/kg and 1.0 mg/kg subanesthetic doses of IV ketamine and no clear or consistent evidence for clinically meaningful efficacy of lower doses of IV ketamine. Most of the effect was due to differences at day 1.
Meaning: Our results suggest that there is a range of effective, subanesthetic doses of IV ketamine in TRD.
Introduction
Treatment-resistant depression (TRD) is a significant and common clinical challenge [1–3]. There are only four pharmacological treatments that are FDA (Food and Drug Administration) approved for adjunctive treatment in TRD patients: aripiprazole [4], quetiapine [5], olanzapine–fluoxetine combination [6], and brexpiprazole [7]. In addition, only three non-pharmacological therapies have been approved for TRD: transcranial magnetic stimulation [8], vagus nerve stimulation) [9], and electroconvulsive therapy [10]. There is a critical need for novel treatments for TRD patients.
Subanesthetic doses of ketamine— an N-methyl-d-aspartate (NMDA) receptor antagonist—have shown promise for the rapid treatment of TRD patients [11–14]. Over the last decade, a series of placebo-controlled studies have confirmed the ability of intravenous (IV) ketamine (0.5 mg/kg infusion) to provide significant symptom amelioration in TRD patients within a few hours, with symptoms typically returning within a period of days after discontinuation of the acute intervention [13, 14]. However, the exact mechanism of action of ketamine is not yet clear [15]. As pointed out in a recent review [13], nine meta-analyses of acute-phase randomized short-term trials of ketamine for depression have now reported statistically significant advantages of ketamine over placebo or active control conditions, across a variety of measures of depressive symptoms.
Though ketamine shows promise as a rapidly acting antidepressant, almost all the previous studies used standard IV doses of 0.5 mg/kg over 40 min; therefore, the optimal ketamine dose for the treatment of depression remains unknown. In this study, we aimed to assess the extent to which a single infusion of one of four different doses of ketamine was superior to active placebo in the treatment of TRD patients over 72 h (day 3), when added to stable, standard antidepressant therapies (clinical trial: NCT01920555). In addition, we explored whether there were significant differences in adverse event rates, dissociative symptoms, and blood pressure changes across this range of doses.
Methods
This was a double-blind, placebo-controlled study of the acute efficacy of IV ketamine or placebo added to ongoing antidepressant therapy (ADT) in the treatment of major depressive disorder (MDD) adults with TRD. Following a washout period for patients on prohibited psychotropic agents, 99 eligible subjects were randomly assigned to one of five 40 min infusion arms in a 1:1:1:1:1 fashion: a single dose of ketamine 0.1 mg/kg (n = 18), a single dose of ketamine 0.2 mg/kg (n = 20), a single dose of ketamine 0.5 mg/kg (n = 22), a single dose of ketamine 1.0 mg/kg (n = 20), and a single dose of midazolam 0.045 mg/kg (active placebo) (n = 19) (see Fig. 1) to minimize the unblinding risk due to adverse events (AEs), as in Murrough et al [16]. Prior to randomization, patients were grouped by body mass index (BMI) (group I: BMI ≤ 30; group II: BMI > 30), and were block randomized into each arm of the study, with the mg/kg ratio being maintained across all BMIs. The primary endpoint assessments were carried out over 3 days and all subjects were followed for 30 days to examine the benefit durability (see Fig. 1).
Fig. 1.

CONSORT diagram
The study assessments were performed at days 0, 1, 3, 5, 7, 14, and 30 to assess the safety and efficacy of all doses of ketamine compared to active placebo therapy in depressed patients demonstrating an inadequate response to at least 2 adequate ADTs during the current major depressive episode (TRD). This report focuses on the outcome during the acute phase of the study (days 0 through 3). This trial was conducted across six US academic sites (Massachusetts General Hospital, Baylor College of Medicine/Michael E. Debakey VA Medical Center, Icahn School of Medicine at Mount Sinai, Stanford University School of Medicine, University of Texas Southwestern, and Yale University) according to the US FDA guidelines and Declaration of Helsinki. Institutional Review Board (IRB)- and National Institute of Mental Health Data and Safety Monitoring Board (NIMH DSMB)-approved written informed consent was obtained from all patients.
All enrolled subjects were male and female outpatients between the ages of 18 and 70 years with a diagnosis of MDD in a current depressive episode of at least 8-week duration (as defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition-Text Revision (DSM-IV-TR™)). The diagnosis of MDD was supported by the Structured Clinical Interview for DSM-IV-Patient Edition (SCID-I/P). Furthermore, all subjects had TRD, defined as failure to achieve a subjective satisfactory response (e.g., less than 50% improvement of depression symptoms) to at least two adequate treatment courses during the current depressive episode (including the current ADT). All study participants with MDD were required to be on a stable (for at least 4 weeks) and adequate (according to the Massachusetts General Hospital (MGH) Antidepressant Treatment Response Questionnaire (ATRQ)) dose of ongoing ADT, with a total treatment duration of at least 8 weeks. Concurrent hypnotic therapy was allowed if the therapy had been stable for at least 4 weeks prior to screening and was expected to remain stable during the study. Patients were also allowed to continue treatment with benzodiazepines used for anxiety if therapy had been stable for at least 4 weeks prior to screening and expected to remain stable during the study. Patients on exclusionary concomitant psychotropic medications (e.g., opioids, tramadol, valproic acid, lamotrigine, carbamazepine, barbiturates, eszopiclone, stimulants, NMDA receptor antagonists such as memantine) were included only if they had been free of the exclusionary medication post-taper for five half-lives within the maximum screening period (28 days). Furthermore, subjects could be in concurrent psychotherapy, if stable. All subjects had a Montgomery–Asberg Depression Rating Scale [17] (MADRS) score of ≥20 at both the screen and baseline visits. All included patients were required to have a BMI between 18 and 35 kg/m2.
Major exclusion criteria were as follows: failure to achieve satisfactory response (e.g., less than 50% improvement of depression symptoms) to >7 treatment courses of a therapeutic dose of an ADT of at least 8-week duration in the current major depressive episode, MADRS total score of <20 at screening or baseline; a primary Axis I disorder other than MDD; current substance use disorder (abuse or dependence), with the exception of nicotine dependence, within 6 months prior to screening; and any history of ketamine or phencyclidine drug use. All subjects underwent urine drug testing at screening. Other major exclusion criteria included a history of bipolar disorder, schizophrenia, or schizoaffective disorders, or any history of psychotic symptoms in the current or previous depressive episodes. Furthermore, previous participants in research studies involving glutamatergic agents for depression were also excluded.
Following the in-person screen, the diagnosis and adequacy of treatment was confirmed by remote, independent raters from the MGH Clinical Trials Network and Institute (CTNI), via a teleconference administration of the Mood Disorders module of the SCID-I/P, MADRS, and the MGH ATRQ.
Assessments (outcome and safety measures)
All subjects were evaluated by the study clinicians with respect to the efficacy and safety measures described below. The 6-item Hamilton Depression Rating Scale (HAM-D-6) [18–21], with the time frame of the past 24 h, was administered as the primary outcome measure at each visit (days 0, 1, 3, 5, 7, 14, and 30) by the independent, remote MGH CTNI raters, as this version of the scale has been shown to be more sensitive to detect changes with treatment than the original 17-item version [21]. The intra-class correlation coefficient for the HAM-D-6 was >0.8 for all central raters, a reflection of the high inter-rater reliability. The follow-up sessions were conducted according to the method described by Fawcett et al. [22].
Secondary measures of depression were the MADRS, administered by the site clinicians, the self-rated Symptoms of Depression Questionnaire (SDQ) [23], the self-rated Positive Affect Scale (PAS), the self-rated Snaith-Hamilton Pleasure Scale (SHAPS) [24] and the global severity and improvement scales of the Clinical Global Impressions (CGI-S and CGI-I) [25]. These were administered at days 0, 1 (except for the MADRS), 3, 5, 7, 14, and 30, with a time frame of the past 24 h for the SDQ, PAS, SHAPS, CGI-S, and CGI-I, and of the past 3 days for the MADRS. Visual analogue scales (VAS) components assessing happy, sad, drowsy, irritated, alert, anxious, and restless [26, 27] were also administered with a time frame of the past 24 h, both immediately prior to starting and at 120 min post initiation of the infusion. We also administered at every visit the clinician-rated Columbia Suicide Severity Rating Scale (C-SSRS) [28], a measure of the spectrum of suicidal ideation and behavior. Dissociative symptoms during the infusion were measured using the Clinician-Administered Dissociative States Scale (CADSS) [29] at −5, 40, 80, and 120 min in relation to the start of the infusion. Blood pressure and heart rate were measured at time 0 (right before starting the infusion), and at 15–20-min intervals for 120 min following the infusion. Vital signs were measured at every study visit. Blood pressure was measured while the patient was supine and sitting. Observations were recorded pre-, post-, and during the infusion. To capture elevated systolic and diastolic blood pressure observations above a specified value (systolic >155, diastolic >99), each observation was dichotomously classified as in or out of range in our analyses.
At the screen visit and day 14, patients had a physical examination and at the screen visit, day 0, and day 3, they had blood drawn for chemistry and complete blood count (CBC) blood tests, and underwent electrocardiography (ECG) at day 0 and day 1. The presence of any potential side effect or adverse event were carefully documented at screen (for the past week) and at every subsequent visit (covering events since the last visit) using both the spontaneously reported adverse events and the Systematic Assessment for Treatment Emergent Events–Systematic Inquiry (SAFTEE-SI). The SAFTEE-SI [30] is a self-rated questionnaire assessing possible adverse events during the course of the trial. The time frame is the past 24 h. Reasons for premature discontinuation, including intolerable side effects, were recorded. Weight, oral temperature, and standing and supine pulse and blood pressure (Vital Signs) were recorded at each visit.
Statistical analyses
The primary aim was to demonstrate that any of the four doses of ketamine were superior to active placebo in reducing the HAM-D-6 score among TRD patients within 72 h, when added to stable antidepressant therapy. Secondary outcomes were changes in scores on the MADRS, CGI-S, CGI-I, SDQ, and PAS. To this end, we used a repeated-measures fixed-effects model for the observations taken at baseline, day 1 and day 3, with terms for visit (i.e., days 0, 1, and 3), and visit × treatment, the primary contrasts of interest being the difference between the four active treatments and placebo. To account for potential differences by site, we included SITE and its interaction terms with VISIT and TREATMENT in the original model, and removed non-significant terms one by one, leaving us with the basic VISIT, TREATMENT, and VISIT × TREATMENT that we report here. To conduct this analysis, we used PROC MIXED, with an unspecified variance covariance matrix. This analysis allowed the possibility of missed visits and is robust to data missing at random. To protect the family-wise error rate in the presence of multiple testing (i.e., each of four groups compared to placebo at days 1 and 3, resulting in 8 contrasts of interest), we used Holm’s sequentially rejective multiple test procedure [31].
We conducted two types of comparisons: 2-groups: superiority is demonstrated by a statistically significant greater decrease on the HAM-D-6 total score for patients receiving any ketamine dose versus active placebo therapy; 5-groups: comparing each of the four ketamine doses to the active placebo group, using Holm’s method to protect family-wise error rate in the presence of multiple testing. Specifically, Holm’s [31] method was applied to the model-produced differences of least squares for the a priori identified contrasts of interest. In the 2-group model, there were two such comparisons (i.e., the comparisons between the combined ketamine group vs. placebo at days 1 and 3); in the 5-group model, there were eight such comparisons (i.e., each of the four ketamine groups compared to placebo at days 1 and 3). The probability was deemed to be 80% that the study would detect a treatment difference between each ketamine dose and the active placebo if the effect size was greater than 1.
Secondary outcomes
We conducted comparisons of antidepressant efficacy of all doses of ketamine vs. active placebo on multiple measures of antidepressant efficacy, such as the MADRS, CGI-S, CGI-I, SDQ, and PAS. For the continuous secondary efficacy variables, the same approach as for the primary efficacy variable was used for the analyses.
Comparison of response rates on all of ketamine doses or active placebo at day 3 was carried out, with response being defined as a 50% or greater reduction in HAM-D-6 score from baseline. Differences in response rates were compared by performing an analysis using a generalized linear model of the repeated measures. A logit link function was used and the statistical inferences were based on generalized estimating equations (GEE).
We also tested if treatment effects on the primary outcome variable, HAM-D-6, were sustained through the follow-up period. To this end, we used the same model building approach as for the primary outcome analysis, but focused on assessments made on days 3–30. For parsimony, we modeled time linearly. In this analysis, the effect of interest was the main effect of TREATMENT.
We also compared changes in C-SSRS scores, and dissociative symptom levels, using CADSS scores, across all ketamine doses vs. active placebo. A correlation between depression outcome and dissociative symptoms was carried out based on any changes between 0 (−5 min) and 40, 80, and 120 min in the CADSS data. A correlation between depression outcome and C-SSRS scores was carried out.
Emergence of abnormal chemistry, CBC, or ECG conditions was tracked and tallied. Percentages of patients in the two treatment groups who experienced new or exacerbated spontaneously reported AEs were presented and analyzed overall and for each type of event.
Results
We initially planned to randomize 100 subjects, but we completed the study by randomizing 99 subjects, given the higher than expected retention. The retention was 96% at day 1, 95% at day 3 (endpoint), 95% at day 5, 92% at day 7, 88% at day 14, and 87% at day 32. Table 1 summarizes the sociodemographic and baseline clinical characteristics of the patients randomized to treatment. Characteristics were relatively similar across the five treatment groups, though it should be noted that, by chance, all the subjects assigned to the 0.2 mg/kg arm were whites, and there was a nearly 20% difference in the proportion of women between the five groups, with 40% women in the ketamine 1.0 mg/kg group, and 58% women in the midazolam group. On average, the 0.2 mg/kg group was more treatment resistant, as it had, on average, a history of one extra failed trial of antidepressants compared to the 0.5 mg/kg group.
Table 1.
Sample characteristics (n = 99)
| Ketamine 0.1 mg/kg n=18 | Ketamine 0.2 mg/kg n=20 | Ketamine 0.5 mg/kg n=22 | Ketamine 1.0 mg/kg n=20 | Midazolam 0.045 mg/kg n=19 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| M/% | SD | M/% | SD | M/% | SD | M/% | SD | M/% | SD | |
| Demographics and pharmacologically relevant variables | ||||||||||
| Age | 43.1 | 11.9 | 45.5 | 14.6 | 48.6 | 12.9 | 47.4 | 10.1 | 45.6 | 13.8 |
| BMI | 25.2 | 3.1 | 24.9 | 3.7 | 25.3 | 5.7 | 26.1 | 3.8 | 26.3 | 4.1 |
| Gender (% fem.) | 55.6 | 45.0 | 50.0 | 40.0 | 57.9 | |||||
| Hispanic (% yes) | 5.6 | 0.0 | 9.1 | 0.0 | 0.0 | |||||
| Race | ||||||||||
| White | 66.7 | 100.0 | 90.9 | 90.0 | 94.7 | |||||
| Asian | 16.7 | 0.0 | 4.6 | 5.0 | 0.0 | |||||
| Black | 16.7 | 0.0 | 0.0 | 5.0 | 0.0 | |||||
| Other | 0.0 | 0.0 | 4.6 | 0.0 | 5.3 | |||||
| Concomitant medications (% used) | ||||||||||
| Benzodiazepines | 50.0 | 50.0 | 45.5 | 45.0 | 31.6 | |||||
| Non-benzodiazepine hypnotics | 27.8 | 25.0 | 9.1 | 25.0 | 21.1 | |||||
| SSRIs | 44.4 | 60.0 | 54.6 | 50.0 | 52.6 | |||||
| SNRIs | 27.8 | 35.0 | 31.8 | 20.0 | 26.3 | |||||
| TCAs | 5.6 | 0.0 | 4.6 | 5.0 | 0.0 | |||||
| Other antidepressant(s) | 44.4 | 55.0 | 59.1 | 40.0 | 57.9 | |||||
| Clinical severity at baseline | ||||||||||
| No. of failed antidepressants for current episode | 3.3 | 1.3 | 3.7 | 1.6 | 2.7 | 1.2 | 2.9 | 1.2 | 2.9 | 1.4 |
| HAM-D-6 | 12.6 | 1.8 | 12.8 | 2.5 | 12.6 | 1.5 | 12.6 | 2.1 | 13.1 | 2.3 |
| MADRS | 33.8 | 5.9 | 34.5 | 8.5 | 31.6 | 3.9 | 32.7 | 5.9 | 33.6 | 7.1 |
| CGI-S | 5.0 | 0.8 | 5.2 | 0.7 | 4.9 | 0.6 | 5.2 | 0.8 | 5.0 | 0.7 |
| CGI-I | 3.9 | 0.3 | 4.1 | 0.2 | 4.1 | 0.7 | 4.0 | 0.5 | 4.2 | 0.6 |
| SDQ | 3.5 | 0.5 | 3.5 | 0.5 | 3.5 | 0.6 | 3.4 | 0.4 | 3.4 | 0.5 |
| PAS | 19.3 | 12.2 | 20.5 | 15.4 | 20.6 | 11.7 | 21.3 | 14.7 | 21.3 | 12.1 |
Benzodiazepines included Alprazolam, Clonazepam, Clorazepic acid, Diazepam, and Lorazepam; non-benzodiazepine hypnotics included Zaleplon, Zolpidem, and Trazodone
BMI body mass index, SSRIs selective serotonin reuptake inhibitors (incl. Fluoxetine, Citalopram, Escitalopram, Paroxetine, Sertraline, and Vilazodone), SNRIs serotonin–norepinephrine reuptake inhibitors (incl. Desvenlafaxine, Duloxetine, Venlafaxine, and Venlafaxine hydrochloride), TCAs tricyclic antidepressants (incl. Clomipramine and Nortriptyline), other antidepressants included Bupropion, Mirtazapine, Vortioxetine, HAM-D-6 Hamilton Rating Scale for Depression, 6-item version, MADRS Montgomery–Asberg Depression Rating Scale, CGI-S and CGI Clinical Global Impression of Severity and Improvement scales, SDQ Symptoms of Depression Questionnaire, PAS Positive Affect Scale
During the study, 4 subjects mistakenly received lower doses than they were randomized to receive due to calculation errors. One subject, assigned to the ketamine 0.1 mg/kg group, was underdosed by 33%, one subject, assigned to the midazolam 0.045 mg group, was underdosed by 32%, one subject assigned to the ketamine 0.2 mg/kg group, was underdosed by 11%, and one subject, assigned to ketamine 1.0 mg/kg group, was underdosed by 6.5%. As per recommendation by the NIMH DSMB, these 4 subjects were retained in the analyses, as originally randomized.
On the primary outcome measure, the HAM-D-6, there was a statistically significant day by group interaction effect (p = 0.0278) in the 2-group analysis between ketamine and active placebo (see Fig. 2a). Similarly, there was a statistically significant (p = 0.0391) day by group interaction effect in the 5-group analysis between ketamine (0.1, 0.2, 0.5, or 1.0 mg/kg) and active placebo (see Fig. 2b). Table 2 provides the pairwise comparisons of HAM-D-6 changes between ketamine doses and active placebo (midazolam 0.045 mg/kg), with Cohen’s effect sizes above 0.8 at day 1 for three doses (0.1, 0.5, or 1.0 mg/kg) and Cohen’s effect sizes above 0.4 at day 3 for three doses (0.1, 0.5, or 1.0 mg/kg). Only the 0.5 mg/kg and the 1.0 mg/kg dose remained statistically superior to placebo after adjusting for multiple comparisons, and only at day 1.
Fig. 2.
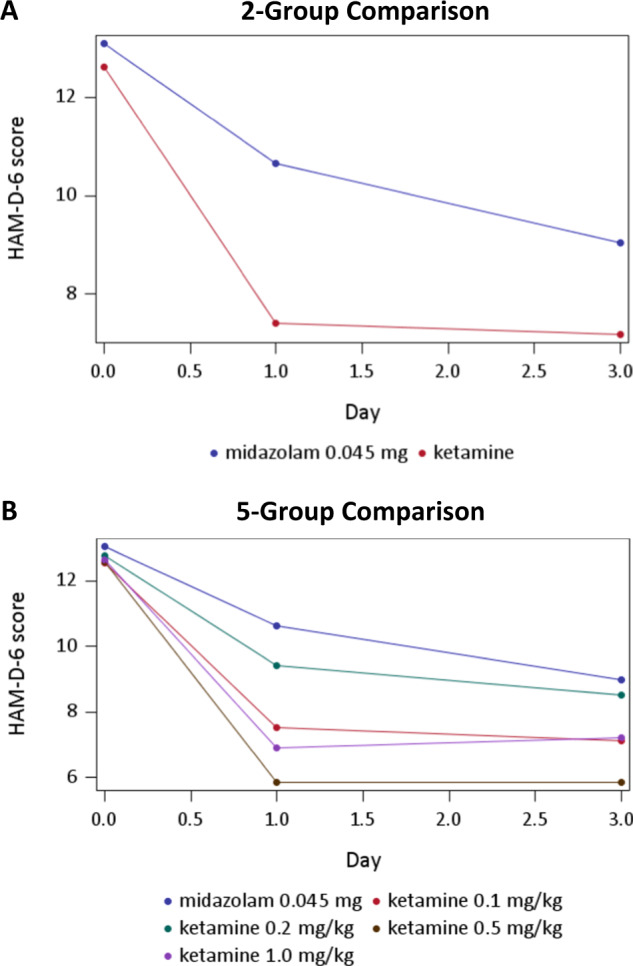
HAM-D-6 scores over the first 72 h of treatment; Fig. 2A reports the 2-group analysis; Fig. 2B reports the 5-group analysis;
Table 2.
Pairwise comparisons of hypothesized contrasts between ketamine groups vs. placebo for the HAM-D-6
| Model | Day | Dose group | Estimate | 95% CI | Raw p | Adj. p | Cohen’s d |
|---|---|---|---|---|---|---|---|
| 2-Group comparison | |||||||
| Day 1 | Combined | −3.25 | (−5.39, −1.11) | 0.00 | 0.01 | −0.86 | |
| Day 3 | Combined | −1.87 | (−4.14, 0.41) | 0.11 | 0.11 | −0.44 | |
| 5-Group comparison | |||||||
| Day 1 | 0.1 mg/kg | −3.18 | (−5.93, −0.43) | 0.02 | 0.14 | −0.82 | |
| Day 1 | 0.2 mg/kg | −1.13 | (−3.75, 1.49) | 0.39 | 0.79 | −0.40 | |
| Day 1 | 0.5 mg/kg | −4.79 | (−7.35, −2.24) | 0.00 | 0.00* | −1.21 | |
| Day 1 | 1.0 mg/kg | −3.76 | (−6.37, −1.15) | 0.01 | 0.04* | −0.95 | |
| Day 3 | 0.1 mg/kg | −2.04 | (−5.04, 0.95) | 0.18 | 0.72 | −0.49 | |
| Day 3 | 0.2 mg/kg | −0.36 | (−3.18, 2.46) | 0.80 | 0.80 | −0.12 | |
| Day 3 | 0.5 mg/kg | −3.21 | (−5.97, −0.44) | 0.02 | 0.14 | −0.71 | |
| Day 3 | 1.0 mg/kg | −1.84 | (−4.65, 0.96) | 0.20 | 0.72 | −0.44 | |
CI confidence interval; *Adj. p < 0.05
On the secondary outcomes, statistical significance of the group × time interaction effect was only achieved for the SDQ (p = 0.0105) and the PAS (p = 0.0341) in the 5-group comparison, and the PAS (p = 0.0332) and the CGI-S (p = 0.0204) in the 2-group comparison. For descriptive purposes, Table 3 presents information on the post hoc pairwise tests on these secondary outcomes regardless of the significance of the group × interaction effects, but focuses on comparisons that were statistically significant. Here it can be seen that, after adjustment for multiple comparisons, the 0.5 mg/kg dose was superior to placebo on all secondary outcomes, but only on day 1, with the exception of the MADRS, which was not assessed on day 1, and which did have a significant difference on day 3. The 1.0 mg/kg dose was also superior to placebo on day 1 on the CGI-S. Cohen’s d differences ranged from 0.94 to 1.27 for these effects.
Table 3.
Listing of statistically significant pairwise comparisons of each ketamine group to midazolam
| Measure | Day | Dose group | b | 95% CI | Raw p | Adj. p | Cohen’s d |
|---|---|---|---|---|---|---|---|
| MADRS | |||||||
| Day 3 | 0.5 mg/kg | −9.85 | (−16.56, −3.15) | 0.00 | 0.02* | −1.03 | |
| CGI-S | |||||||
| Day 1 | 0.5 mg/kg | −1.28 | (−2.02, −0.54) | 0.00 | 0.01* | −1.21 | |
| Day 1 | 1.0 mg/kg | −1.05 | (−1.81, −0.29) | 0.01 | 0.05* | −1.11 | |
| CGI-I | |||||||
| Day 1 | 0.5 mg/kg | −0.98 | (−1.64, −0.31) | 0.00 | 0.03* | −1.27 | |
| SDQ | |||||||
| Day 1 | 0.5 mg/kg | −0.61 | (−1.01, −0.21) | 0.003 | 0.0243* | −0.95 | |
| PAS | |||||||
| Day 1 | 0.5 mg/kg | 16.54 | (5.31, 27.77) | 0.0043 | 0.0347* | 0.94 | |
MADRS Montgomery–Asberg Depression Rating Scale, CGI-S and CGI-I Clinical Global Impression of Severity and Improvement scales, SDQ Symptoms of Depression Questionnaire, PAS Positive Affect Scale
*Adj. p < 0.05
Additionally, non-significant medium to large effects (i.e., d ≥ 0.50) were observed across all secondary outcomes (see Supplementary Table 1), and across three of the four doses (i.e., not for the 0.2 mg/kg dose), with effects ranging for the 0.1 mg/kg dose from d = 0.50 (day 1 on the CGI-I) to 0.85 (day 1 on the CGI-S), for the 0.5 mg/kg dose from 0.60 (day 3 on the CGI-I) to 0.75 (day 3 on the CGI-S), and for the 1.0 mg/kg dose from 0.51 (day 1 on the SDQ) to 0.62 (day 3 on the CGI-S). In line with outcomes on the primary outcome measure, effects tended to be larger for day 1 than day 3. Response rates (HAM-D-6 reduction from baseline score ≥50%) were 31% for 0.1 mg/kg, 21% for 0.2 mg/kg, 59% for 0.5 mg/kg, 53% for 1.0 mg/kg, and 11% for midazolam on day 1, and 47% for 0.1 mg/kg, 37% for 0.2 mg/kg, 57% for 0.5 mg/kg, 37% for 1.0 mg/kg, and 33% for midazolam on day 3 (Supplementary Figure 1). The group effect is significant for the 2-group comparison (p = 0.0237), due to differences at day 1 (adjusted p = 0.04224) but not day 3 (adjusted p = 0.4385), where the group effect is not significant for the 5-group comparison.
In examining trends over time during the follow-up period for the HAM-D-6, we found, after removal of the non-significant group × time interaction effect, a significant main effect for group in the 2-group (F(1, 92) = 4.20, p = 0.04), but not the 5-group (F(4, 89) = 1.76, p = 0.14) comparison, suggesting that participants treated with ketamine may have maintained lower scores on the HAM-D-6 throughout days 3–30, though notably this overall effect was no longer significant when comparing each ketamine dose to midazolam. Exploration of the per-dose comparisons suggest that the significant effect of the 2-group comparison may largely be due to effects in the 1.0 mg/kg group (see Supplementary Figure 2). In fact, the average HAM-D-6 scores across days 14 and 30 were 10.42 for 0.1 mg/kg, 10.14 for 0.2 mg/kg, 10.18 for 0.5 mg/kg, 8.26 for 1 mg/kg, and 11.08 for active placebo (midazolam). In both comparisons, there was a significant main effect of time (F(1, 92) = 18.93, p < 0.01; F(1, 89) = 18.87, p < 0.01), indicating that HAM-D-6 scores increased from days 3 to 30.
The scores on the CADSS during the infusion are reported in Supplementary Figure 3. There was a clear dose response 40 min after the infusion, with both 0.5 mg/kg and 1 mg/kg doses being significantly (p < 0.0001) greater than active placebo (midazolam 0.045 mg/kg); the lower ketamine doses were not significantly different from active placebo. There were no statistically significant correlations between CADSS scores 40 min after the infusion and HAM-D-6 scores at day 1 (r = −0.19; n = 94) and day 3 (r = −0.13; n = 92).
Regarding unblinding, both clinicians’ and participants’ guesses of treatment assignment were significantly related to actual treatment group (p < 0.01 for both), where both groups were able to correctly guess assignment to ketamine for the 0.5 mg/kg (100% and 77% guessed correctly by clinicians and participants, respectively) and the 1.0 mg/kg (95% correctly guessed by both) ketamine doses, but not for the 0.1 mg/kg (50%, 56%, respectively) and 0.2 mg/kg doses (55%, 45%, respectively). Assignment to placebo was guessed correctly 42% by clinicians and 37% by participants.
Supplementary Table 2 provides a list of spontaneously reported AEs divided among the five groups. When the four ketamine groups were combined and compared to the active placebo midazolam (Supplementary Table 3), there were no significant differences in rates of any of AEs. However, there were higher rates of specific AEs in the ketamine-treated patients compared to those treated with the active placebo midazolam: headache (11.3% vs. 0%), nausea (10% vs. 0%), vomiting (5% vs. 0%), and depression (3.8% vs. 0%). Of note, spontaneously reported suicidal ideation was reported by 2 of the ketamine-treated patients, but none of the active placebo-treated patients. On the other hand, the C-SSRS scores during treatment (see Supplementary Table 4) indicated non-significantly higher rates of wishing to be dead, non-specific active suicidal thoughts, and active suicidal ideation without intent to act on active placebo (midazolam) than ketamine.
Abnormally high hepatic chemistry values occurred in one patient (0.2 mg/kg ketamine) during his early termination visit (1.7 mg/dL total bilirubin, 47 IU/L aspartate transaminase, 103 IU/L alanine transaminase).
Blood pressure measurements above the cut-off values were rare during the medication infusion process, with systolic blood pressure of ≥155 being observed in 3.8% of the 1050 total observations, and diastolic blood pressure of ≥99 being observed in 1.2% of the observations. High systolic blood pressure readings occurred in a total of 21 participants, all of whom were in the ketamine groups. Prior to infusion, systolic blood pressure ≥155 was recorded in 2 patients, during infusion in 16 patients, and immediately following infusion in 8 patients. High diastolic blood pressure readings occurred in a total of 10 participants, all of whom were in the ketamine groups. Prior to infusion, diastolic blood pressure ≥99 was recorded in 1 patient, during infusion in 5 patients, and immediately following infusion in 5 patients. An examination of average blood pressure values over time (as averaged within phase) showed a phase × group interaction effect for both systolic (F(8, 94) = 11.14, p < 0.001) and diastolic blood pressure (F(8, 94) = 8.97, p < 0.001). These effects were driven by mean level differences during the infusion phase, where blood pressure values were higher in the ketamine 0.5 and 1.0 mg/kg groups compared to the active placebo midazolam, after adjusting for multiple comparisons. Examinations of the group means during the infusion process suggest a dose–response relationship, with increasingly higher blood pressure values, for systolic and diastolic, occurring in groups with increasingly higher ketamine dosages.
There was one serious adverse event that occurred during the trial. The participant attempted suicide by overdosing on day 11 and was subsequently evaluated by the study team and sent to the emergency room. The patient had received ketamine 0.2 mg/kg during the study.
Discussion
In this study, we aimed to assess the extent to which a broad range of subanesthetic IV ketamine doses were superior to active placebo (midazolam) therapy in the acute (72 h) treatment of TRD patients, when added to stable antidepressant therapy. The overall group × time interaction effect was significant for the primary outcome measure, the HAM-D-6. In post hoc pairwise comparisons controlling for multiple comparisons, standard dose (0.5 mg/kg) and high dose (1 mg/kg) of intravenous ketamine were superior to active placebo; a low dose (0.1 mg/kg) was significant only prior to adjustment (p = 0.02, p-adj = 0.14, d = −0.82 at day 1). Most of the interaction effect was due to differences at day 1. Our results suggest that there is evidence for the antidepressant efficacy of the 0.5 mg/kg and 1.0 mg/kg subanesthetic doses of IV ketamine and no clear or consistent evidence for clinically meaningful efficacy of lower doses of IV ketamine. The fact that the lowest dose of IV ketamine (0.1 mg/kg), which was no different from the active placebo in terms of blood pressure elevation and dissociative symptoms, was significantly more effective than active placebo prior to adjustment suggests that its effects were not enhanced because of functional unblinding. In fact, both clinicians and patients guessed correctly the assignment to ketamine 0.1 mg/kg only about half of the time (despite the fact that the probability of being assigned to ketamine was 80%), whereas the assignment to ketamine 0.5 mg/kg and 1 mg/kg was guessed correctly almost all the time, suggesting the possibility of unblinding for those higher doses.
There was a clear dose–response curve with respect to dissociative symptoms, as measured by the CADSS. At 40 min after the infusion start, both 0.5 mg/kg and 1 mg/kg doses had significantly greater CADSS scores than active placebo; this significant difference was not seen with the lower ketamine doses. Notably, there were no statistically significant correlations between changes in CADSS scores 40 min after the infusion and HAM-D-6 scores at Day 1 and Day 3, in contrast to the hypothesis by Luckenbaugh et al. [32]. Although rates of spontaneously reported AEs were not significantly different between ketamine-treated patients and active placebo patients, the elevated CADSS scores suggest the possibility of unblinding at the higher doses and may contribute to the numerically greater efficacy of 0.5 mg/kg of ketamine compared to 0.1 mg/kg. However, it does not explain the 0.5 mg/kg dose consistently having the highest numeric effect over the 1.0 mg/kg dose. Regarding blood pressure, the lowest ketamine dose (0.1 mg/kg) was less likely than the standard and high ketamine doses to lead to blood pressure elevations.
The dose of 0.2 mg/kg of ketamine, which was found to be effective in a recent trial [33], did not perform as well as the other doses, and this may be due simply to the relatively small sample sizes of each treatment group and perhaps to the fact that, on average, the 0.2 mg/kg group was more treatment resistant, as it had a history of one extra failed trial of antidepressants compared to the 0.5 mg/kg group.
Our longitudinal follow-up over 30 days shows that there is a prominent and rapid loss of efficacy for the single IV ketamine administration shortly after the day 3 timepoint, with little evidence of meaningful therapeutic benefit for most drug doses after day 5. Interestingly, there is a suggestion of greater sustained drug effect at the 1.0 mg/kg dose as far out as days 15–30, although the effect is rather modest. This unique finding is something that should be examined in future studies.
When the four ketamine groups were collapsed into one group and compared to active placebo (midazolam), there were no significant differences in rates of any of AEs. However, there were numerically higher rates of headache, nausea, vomiting, and depression among ketamine-treated patients. Of note, emergence of suicidal ideation was spontaneously reported by two of the ketamine-treated patients, but none of the active placebo-treated patients. On the other hand, the C-SSRS scores during treatment indicated non-significantly higher rates of wishing to be dead, non-specific active suicidal thoughts, and active suicidal ideation without intent to act on active placebo (midazolam) than on ketamine.
A methodological strength of our study was the use of an active placebo and of blinded, remote raters for the primary outcome efficacy ratings. However, it is possible that there may have been functional unblinding of the treatment arms to both the clinicians and subjects, especially with the 0.5 mg/kg and 1 mg/kg dose groups. This may have contributed to the results. Another limitation of our study was that each treatment group had a relatively small sample size (range of 18–22 patients per treatment arm) and the corresponding confidence intervals around our estimates of effect sizes for each dose are quite large [34], discouraging in-depth interpretation of observed small differences. In addition, the results may have been confounded by the variability in the degree of responsiveness to ketamine across the treatment groups, as suggested by Loo et al. [14]. Finally, our study cannot answer the question of whether raising doses in poor responders to the standard dose of 0.5 g/kg of ketamine is helpful and tolerated, or if lower doses are effective in patients who cannot tolerate the standard ketamine dose of 0.5 mg/kg.
Electronic supplementary material
Supplementary Table 1; Supplementary Table 2; Supplementary Table 3; Supplementary Table 4
Acknowledgements
This project was funded by the National Institute of Mental Health (NIMH) under Contract Rapidly-Acting Treatments for Treatment-Resistant Depression (RAPID) Number: HHSN271201100006I, to the Massachusetts General Hospital (Maurizio Fava and George Papakostas, co-principal investigators). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. We would like to thank Dr. Matthias Eikermann from MGH who served as a collaborator, and Drs. Mi Hillefors, Steven Zalcman, Adam Haim, and Galia Siegel from NIMH for their support which was absolutely critical to both the planning and the implementation of the study.
Compliance with ethical standards
Conflict of interest
LCC, MF, BBH, and HJ declare that they have no conflict of interest. CD: Dr. DeBattista has received grant support from Janssen, Neuronetics, St. Jude, and Biolite. He has served on the Advisory Board of Alkermes. CC: Dr. Cusin receives funding from NIMH (R01MH102279) and has received consulting fees from Janssen Pharmaceuticals, Takeda, Boehringer, Lundbeck. She has also participated in research funded by Janssen, Medtronic, Otsuka, Takeda. MF: reports 3-year disclosures as below: all lifetime disclosures can be viewed online at: http://mghcme.org/faculty/faculty-detail/maurizio_fava; research support: Alkermes, Inc., Johnson & Johnson, Axsome, Acadia Pharmaceuticals, Cerecor, Lundbeck Inc., Neuralstem, Otsuka, Taisho, Marinus Pharmaceuticals, BioHaven, Takeda, Vistagen, Relmada Therapeutics Inc., Stanley Medical Research Institute (SMRI), National Institute of Drug Abuse (NIDA); National Institute of Mental Health (NIMH), and PCORI. Dr. Fava has not done any personal consulting. Any consulting he has done has been on behalf of Massachusetts General Hospital. Stock/Other Financial Options: Equity Holdings: Compellis; PsyBrain, Inc. Royalty/patent, other income: patents for Sequential Parallel Comparison Design (SPCD), licensed by MGH to Pharmaceutical Product Development, LLC (PPD) (US_7840419, US_7647235, US_7983936, US_8145504, US_8145505); and patent application for a combination of Ketamine plus Scopolamine in Major Depressive Disorder (MDD), licensed by MGH to Biohaven. Patents for pharmacogenomics of Depression Treatment with Folate (US_9546401, US_9540691). Copyright for the MGH Cognitive & Physical Functioning Questionnaire (CPFQ), Sexual Functioning Inventory (SFI), Antidepressant Treatment Response Questionnaire (ATRQ), Discontinuation-Emergent Signs & Symptoms (DESS), Symptoms of Depression Questionnaire (SDQ), and SAFER; Lippincott, Williams & Wilkins; Wolkers Kluwer; World Scientific Publishing Co. Pte. Ltd. MPF: over the past 3 years, Dr. Freeman has received research support from: Takeda, JayMac, and Sage; she has served in advisory boards of: Janssen, Sage, JDS therapeutics, Sunovion, and Takeda; she has served in the Independent Data Safety and Monitoring Committee of Janssen (Johnson&Johnson); she has served as a medical editor for the GOED newsletter. Dr. Freeman is an employee of Massachusetts General Hospital, and works with the MGH National Pregnancy Registry [Current Registry Sponsors: Teva, Alkermes, Inc. (2016-Present); Otsuka America Pharmaceutical, Inc. (2008-Present); Forest/Actavis (2016-Present), Sunovion Pharmaceuticals, Inc. (2011-Present)]. As an employee of MGH, Dr. Freeman works with the MGH CTNI, which has had research funding from multiple pharmaceutical companies and NIMH. DFI: Dr. Ionescu is an employee of Janssen Pharmaceuticals and, within the past year, has received grant funding with salary support from the Brain and Behavior Research Foundation and the NIMH for ketamine research. DVI: In the past 3 years, Dr. Iosifescu has received consultation fees from Alkermes, Axsome, MyndAnalytics (CNS Response), Jazz, Lundbeck, Otsuka, Sunovion, and has received research support (through his academic institutions) from Alkermes, Astra Zeneca, Brainsway, LiteCure, Neosync, Roche, Shire. MJ: Dr. Jha has obtained contracted research support from Acadia Pharmaceuticals and Janssen Research & Development. SJM: over the past 3 years, Dr. Mathew has received consulting fees from Acadia, Alkermes, Allergan, Bracket, Cerecor, Fortress Biotech, Otsuka, and Valeant; he has received research support from: Janssen and NeuroRx; he has also received support from facilities and resources of the Michael E. Debakey VA Medical Center and the Johnson Chair for Research from Baylor College of Medicine. JM: in the past 3 years, Dr. Murrough has provided consultation services to Allergan, Fortress Biotech, Novartis, Janssen Research and Development, Genentech, and ProPhase, and has received research support from Avanir Pharmaceuticals. Dr. Murrough is named on a patent pending for neuropeptide Y as a treatment for mood and anxiety disorders as well as on patents pending for lithium to extend the antidepressant effect of ketamine and ketamine plus lithium as a treatment for suicidal ideation. The Icahn School of Medicine (employer of Dr. Murrough) is named on a patent and has entered into a licensing agreement and will receive payments related to the use of ketamine if it is approved for the treatment of depression. Dr. Murrough is not named on this patent and will not receive any payments. GIP: over the past 3 years, Dr. Papakostas has consulted to: Lundbeck, Sunovion, Brainsway, Pfizer, Boston Pharmaceuticals*, Novartis*, Acadia*, Axsome*, Genomind*, and Mylan* (*on behalf of Massachusetts General Hospital). He has received honoraria from: Lundbeck, Grunbiotics-Mylan, Takeda, Alkermes, Pfizer, Pharma Trade SAS, Asofarma, Sunovion, Brainsway, and Unilab Philippines. He has received research support from Neuralstem Inc and Tal Medical. GS: Dr. Sanacora has received consulting fees form Allergan, Alkermes, AstraZeneca, Biohaven Pharmaceuticals, Genentech, Janssen, Lundbeck, Merck, Navitor pharmaceuticals, Noven pharmaceuticals, Sage Pharmaceuticals, Takeda, Taisho Pharmaceuticals, Teva Pharmaceuticals and Vistagen Therapeutics over the last 36 months. He has also received additional research contracts from AstraZeneca, Bristol-Myers Squibb, Eli Lilly & Co., Johnson & Johnson, Hoffman La-Roche, Merck & Co., Naurex and Servier over the last 36 months. Free medication was provided to Dr. Sanacora for an NIH-sponsored study by Sanofi-Aventis. In addition, he holds shares in Biohaven Pharmaceuticals and is a co-inventor on a patent “Glutamate agents in the treatment of mental disorders” Patent number: 8778979. AFS: Dr. Schatzberg has served as a consultant for Alkermes, Avanir, Bracket, Lundbeck/Takeda, McKinsey, Myriad Genetics, Neuronetics, and Owl and as a speaker for Merck and Pfizer; he holds equity in Corcept (co-founder), Gilead, Incyte, Intersect ENT, Merck, Owl, Seattle Genetics, Titan, and Xhale; he has research funding from Janssen; and he is listed as an inventor on pharmacogenetic and mifepristone patents from Stanford University. MHT: Consulting/Advisory Board: Alkeremes Inc., Akili Interactive, Allergan Pharmaceuticals, Arcadia Pharmaceuticals, Avanir Pharmaceuticals, Brintellix Global, Bristol Myers Squibb, Caudex, Cerecor, Forest Pharmaceuticals, Global Medical Education Inc, Health Research Associates, Insys, Johnson & Johnson Pharmaceutical Research & Development, Lilly Research Laboratories, Lundbeck Research USA, Medscape, Merck & Co. Inc, Mitsubishi Pharma, MSI Methylation Sciences–Pamlab Inc., Navitor, Otsuka America Pharmaceutical Inc., One Carbon Therapeutics, Otsuka America Pharmaceutical Inc., Pfizer Inc, Takeda Global Research. Royalties: Janssen Research and Development LLC. Author Agreement: Janssen Asia Pacific, Oxford University Press. Honoraria: American Psychiatric Association. Grants: Agency for Healthcare Research and Quality (AHRQ), Cancer Prevention and Research Institute of Texas (CPRIT), National Institute of Mental Health (NIMH), National Institute of Drug Abuse (NIDA), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Center for Advancing Translational Sciences (NCATS), Johnson & Johnson, PCORI. STW: Dr. Wilkinson acknowledges support from the Agency for Healthcare Research and Quality (AHRQ; K12HS023000), the NIMH (T32MH062994), the Brain and Behavior Research Foundation (formerly NARSAD), the Robert E. Leet and Clara Guthrie Patterson Trust, the American Foundation for Suicide Prevention and the Yale Department of Psychiatry. Dr. Wilkinson has received consulting fees from Janssen Research & Development and from Guidepoint.
Electronic supplementary material
The online version of this article (10.1038/s41380-018-0256-5) contains supplementary material, which is available to authorized users.
References
- 1.Fava M. Diagnosis and definition of treatment-resistant depression. Biol Psychiatry. 2003;53:649–59. doi: 10.1016/S0006-3223(03)00231-2. [DOI] [PubMed] [Google Scholar]
- 2.Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am. 1996;19:179–200. doi: 10.1016/S0193-953X(05)70283-5. [DOI] [PubMed] [Google Scholar]
- 3.Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR* D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40. [DOI] [PubMed]
- 4.Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28:156–65. doi: 10.1097/JCP.0b013e31816774f9. [DOI] [PubMed] [Google Scholar]
- 5.Bauer M, El-Khalili N, Datto C, Szamosi J, Eriksson H. A pooled analysis of two randomised, placebo-controlled studies of extended release quetiapine fumarate adjunctive to antidepressant therapy in patients with major depressive disorder. J Affect Disord. 2010;127:19–30. doi: 10.1016/j.jad.2010.08.032. [DOI] [PubMed] [Google Scholar]
- 6.Tohen M, Case M, Trivedi MH, Thase ME, Burke SJ, Durell TM. Olanzapine/fluoxetine combination in patients with treatment-resistant depression: rapid onset of therapeutic response and its predictive value for subsequent overall response in a pooled analysis of 5 studies. J Clin Psychiatry. 2010;71:451–62. doi: 10.4088/JCP.08m04984gre. [DOI] [PubMed] [Google Scholar]
- 7.Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76:1224–31. doi: 10.4088/JCP.14m09688. [DOI] [PubMed] [Google Scholar]
- 8.O’Reardon JP, Solvason HB, Janicak PG, et al. Efficacy and safety of transcranial magnetic stimulation in the acute treatment of major depression: a multisite randomized controlled trial. Biol Psychiatry. 2007;62:1208–16. doi: 10.1016/j.biopsych.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 9.Bajbouj M, Merkl A, Schlaepfer TE, et al. Two-year outcome of vagus nerve stimulation in treatment-resistant depression. J Clin Psychopharmacol. 2010;30:273–81. doi: 10.1097/JCP.0b013e3181db8831. [DOI] [PubMed] [Google Scholar]
- 10.Trevino K, McClintock SM, Husain MM. A review of continuation electroconvulsive therapy: application, safety, and efficacy. J ECT. 2010;26:186–95. doi: 10.1097/YCT.0b013e3181efa1b2. [DOI] [PubMed] [Google Scholar]
- 11.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. [DOI] [PubMed]
- 12.Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64. [DOI] [PubMed]
- 13.Bobo WV, Voort JLV, Croarkin PE, Leung JG, Tye SJ, Frye MA. Ketamine for treatment-resistant unipolar and bipolar major depression: critical review and implications for clinical practice. Depress Anxiety. 2016;33:698–710. doi: 10.1002/da.22505. [DOI] [PubMed] [Google Scholar]
- 14.Loo CK, Gálvez V, O’Keefe E, Mitchell PB, Hadzi-Pavlovic D, Leyden J, et al. Placebo-controlled pilot trial testing dose titration and intravenous, intramuscular and subcutaneous routes for ketamine in depression. Acta Psychiatr Scand. 2016;134:48–56. doi: 10.1111/acps.12572. [DOI] [PubMed] [Google Scholar]
- 15.Wohleb E, Gerhard D, Thomas AS, Duman R. Molecular and cellular mechanisms of rapid-acting antidepressants ketamine and scopolamine. Curr Neuropharmacol. 2017;15:11–20. doi: 10.2174/1570159X14666160309114549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–42. [DOI] [PMC free article] [PubMed]
- 17.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- 18.Bech P, Allerup P, Gram L, Reisby N, Rosenberg R, Jacobsen O, et al. The Hamilton depression scale. Acta Psychiatr Scand. 1981;63:290–9. [DOI] [PubMed]
- 19.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Sullivan RL, Fava M, Agustin C, Baer L, Rosenbaum J. Sensitivity of the six‐item hamilton depression rating scale. Acta Psychiatr Scand. 1997;95:379–84. doi: 10.1111/j.1600-0447.1997.tb09649.x. [DOI] [PubMed] [Google Scholar]
- 21.Bech P, Boyer P, Germain J-M, Padmanabhan K, Haudiquet V, Pitrosky B, et al. HAM-D17 and HAM-D6 sensitivity to change in relation to desvenlafaxine dose and baseline depression severity in major depressive disorder. Pharmacopsychiatry. 2010;43:271–6. [DOI] [PubMed]
- 22.Fawcett J, Epstein P, Fiester SJ, Elkin I, Autry JH. Clinical management: imipramine/placebo administration manual. Psychopharmacol Bull. 1987;23:309–24. [PubMed] [Google Scholar]
- 23.Pedrelli P, Blais MA, Alpert JE, Shelton RC, Walker RS, Fava M. Reliability and validity of the Symptoms of Depression Questionnaire (SDQ) CNS Spectr. 2014;19:535–46. doi: 10.1017/S1092852914000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snaith R, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiatry. 1995;167:99–103. doi: 10.1192/bjp.167.1.99. [DOI] [PubMed] [Google Scholar]
- 25.Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse, and Mental Health Administration; 1976.
- 26.Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67:432–8. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63:1121–9. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Posner K, Oquendo MA, Gould M, Stanley B, Davies M. Columbia Classification Algorithm of Suicide Assessment (C-CASA): classification of suicidal events in the FDA’s pediatric suicidal risk analysis of antidepressants. Am J Psychiatry. 2007;164:1035–43. doi: 10.1176/ajp.2007.164.7.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bremner JD, Krystal JH, Putnam FW, et al. Measurement of dissociative states with the clinician-administered dissociative states scale (CADSS) J Trauma Stress. 1998;11:125–36. doi: 10.1023/A:1024465317902. [DOI] [PubMed] [Google Scholar]
- 30.Levine J, Schooler N. SAFTEE: a technique for the systematic assessment of side effects in clinical trials. Psychopharmacol Bull. 1986;22:343. [PubMed] [Google Scholar]
- 31.Holm S. A simple sequentially rejective multiple test procedure. Scand J Stat Theory Appl. 1979;6:65–70. [Google Scholar]
- 32.Luckenbaugh DA, Niciu MJ, Ionescu DF, Nolan NM, Richards EM, Brutsche NE, et al. Do the dissociative side effects of ketamine mediate its antidepressant effects? J Affect Disord. 2014;159:56–61. [DOI] [PMC free article] [PubMed]
- 33.Su TP, Chen MH, Li CT, et al. Dose-related effects of adjunctive ketamine in Taiwanese patients with treatment-resistant depression. Neuropsychopharmacology. 2017;42:2482–92. doi: 10.1038/npp.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leon AC, Davis LL, Kraemer HC. The role and interpretation of pilot studies in clinical research. J Psychiatr Res. 2011;45:626–9. doi: 10.1016/j.jpsychires.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1; Supplementary Table 2; Supplementary Table 3; Supplementary Table 4