

Abstract
Atrial fibrillation (AF), the most prevalent arrhythmia, is often associated with enhanced inflammatory response. Emerging evidence point to a causal role of inflammatory signaling pathways in the evolution of atrial electrical, calcium handling and structural remodeling, which create the substrate of AF development. In this review, we discuss the clinical evidence supporting the association between inflammatory indices and AF development, the molecular and cellular mechanisms of AF, which appear to involve multiple canonical inflammatory pathways, and the potential of anti-inflammatory therapeutic approaches in AF prevention/treatment.
Keywords: atrial fibrillation, inflammation, inflammasome, TNFα, NFкB, IL-1β
INTRODUCTION
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia, serving as a primary risk factor of stroke, which is currently the 5th leading cause of death in the U.S. and 2nd in the world.[1,2] The AF prevalence continues to increase, affecting 6–12 million U.S. citizens by 2050 and nearly 18 million Europeans by 2060, which creates significant concerns as it propels toward epidemic status.[3] Despite the substantial progress that have been made in our current understanding of AF pathophysiology, the causes and perpetuators of AF including their underlying mechanisms are incompletely understood. Currently-available treatment options all have moderate effectiveness, likely because of our limited understanding of the mechanisms promoting initiation, progression and maintenance of AF.[4–6] There is a hope that a better understanding of the molecular basis AF will help to identify and implement safer and more efficient therapeutic approaches for AF management.
A variety of factors can increase the risk of AF including, but not limited to, genetic variants, smoking, alcohol drinking, aging, obesity, and inflammation, etc (Figure 1).[7,8] Emerging evidence demonstrates that inflammatory markers such as interleukin (IL)-6, IL-1 p, myeloperoxidase (MPO), and tumor necrosis factor-α (TNF-α) positively correlate with the progression of AF from paroxysmal (pAF) to persistent AF forms (perAF), and can predict the outcome of AF ablation.[9–13] There is an increasing evidence to suggest that inflammation is not a mere bystander of AF, but rather plays a causative role in its pathogenesis. This review discusses the clinical evidence supporting the association between inflammatory indices and AF development, the molecular and cellular mechanisms of AF, which appear to involve multiple canonical inflammatory pathways, and the potential of anti-inflammatory therapeutic approaches in AF prevention/treatment.
Figure 1. Fundamental mechanisms of atrial fibrillation (AF) and potential contribution of inflammation and inflammatory signaling to the evolution of AF-promoting ectopic activity and reentry.
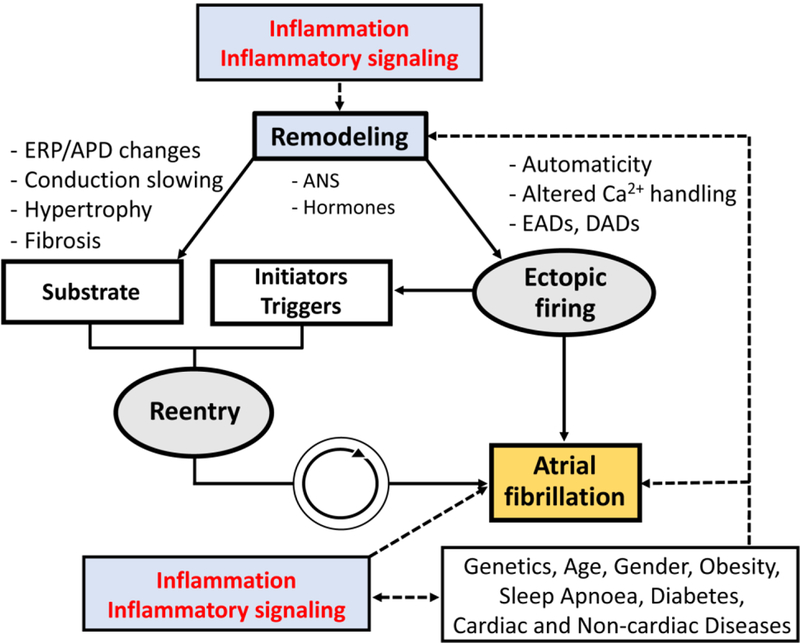
ANS, autonomic nervous system; APD, action potential duration; EADs, early afterdepolorizations; ERP, effective refractory period; DADs, delayed afterdepolorizations.
PATHOPHYSIOLOGY OF AF
The development of AF involves ectopic (triggered) activity and a reentrant substrate.[6,14] Figure 1 summarizes the fundamental AF-promoting mechanisms (Figure 1). Ectopic activity can maintain AF when occurring repetitively at high frequency and can act as a trigger, initiating reentry, the major AF-maintaining mechanism, in a vulnerable substrate characterized by electrical, autonomic, Ca2+-handling or structural abnormalities.[4,6] Ectopic activity results predominantly from abnormal automaticity or triggered activity due to early and delayed afterdepolarizations (EADs and DADs, respectively). Extensive prior work has identified a major role for Ca2+-handling abnormalities, mediated primarily by dysfunction of the major sarcoplasmic reticulum (SR) ryanodine type-2 Ca2+-release channel (RyR2) in the evolution of DADs and triggered activity.[6,15] The mechanisms of reentry depend on the interaction between effective refractory period (ERP), conduction velocity and local depolarization properties of the propagating wave-fronts (the source) and the surrounding tissue (the sink).[16] Fibrosis is an important cause of microreentrant circuits by promoting conduction block and slow, heterogeneous conduction.[17] In addition, ERP shortening due to electrical remodeling and enlargement of the left atrium (LA) also support reentry by increasing available space for reentrant circuits.[4] In more advanced forms of AF, electrical activity becomes increasingly complex with multiple concurrent reentry circuits and ectopic foci. Any persistent change in the function and/or structure of the atria constitutes atrial remodeling, which has many forms that promote the occurrence and/or maintenance of AF by acting on the fundamental arrhythmia mechanisms illustrated in Figure 1. Atrial remodeling increases the likelihood of ectopic firing or reentrant activity through a variety of potential molecular mechanisms, which were reviewed elsewhere.[6,15]There is emerging evidence for involvement of inflammatory signaling, particularly in atrial cardiomyocytes, in ectopic firing and reentry promoting atrial remodeling in AF, which will be discussed in detail below.[18–21]
INFLAMMATION & AF
Inflammation is an essential biological process that mammals utilize to defend injuries in order to maintain a homeostasis.[22,23] However uncontrolled inflammation might contribute to the pathophysiology of various diseases. Acute inflammation serves as the body’s controlled primary physiological response to noxious stimuli, as it immediately attempts to quell effects of the identified threat.[24] To facilitate this process, the body’s local vasculature is employed to administer bioactive signals and factors such as cytokines and chemokines, which encourage recruitment of innate immune cells, which coordinate to intercept, confine and neutralize the targeted threat. In the event that acute inflammation persists beyond removal of the original threat, the inflammatory response may transition into a more unstable state known as chronic systemic inflammation. Because the latter is a systemic condition it ultimately leads to dysregulation of homeostatic mechanisms causing harmful tissue remodeling, which supports the perpetuation of organ damage within the whole body.[22,24–27]
Growing evidence demonstrates a close association between inflammation and AF development, a trend that has been accurately verified since first reported by Bruins et al.[28,29] Inflammatory cytokines are well-recognized biomarkers that can predict the prevalence and prognosis of AF.[30] Biomarker profiling has become an important research area in AF, in hope to accurately predict the risk of AF in patients with and without history of AF, and predict the prognosis after AF ablation procedure.[31,32] Many studies demonstrated that a number of inflammatory cytokines can serve as biomarker to predict the incidence of AF and/or the outcome of AF ablation, including C-reactive protein (CRP), TNF-a, IL-6, IL-1 p, IL-8, and IL-10, which are summarized in Table 1. A detailed review on the role of cytokines as biomarkers of AF is provided by Harada et al. [30]
Table 1.
Summary of clinical studies demonstrating a correlation between inflammatory cytokines and AF development
| Biomarker | Results | Reference (PMID) |
|---|---|---|
| CRP | CRP increased in patients with permanent AF patients (vs. paroxysmal AF). | 11739301 |
| CRP levels can predict patient risk for AF development. | 14623805 | |
| Elevated CRP levels in post-op surgery patients correlate with greater risk of AF reoccurrence. | 29595637 | |
| TNF-α | Increase in the level of TNF-a correlates with the different stages of AF patients relative to those in sinus rhythm. | 23194937; 25746525 |
| IL-6 | IL-6 activation promotes CRP production and positively correlates with persistent/permanent AF development. | 25190079, 22096359, 26839066 |
| IL-6 serves as a risk factor and predictor of AF in patients with chronic kidney disease. | 26840403 | |
| IL-1β | Increased IL-1p serum levels associated with AF patients relative to sinus rhythm patients. | 22096359, 16053785, 20637189, 26283592, 20833691, 22684635 , 25425976 |
| IL-8 | IL-8 levels are elevated in clinically diagnosed AF patients. And the level of IL-8 varies according to AF duration. | 23194937 |
| IL-10 | IL-10 is elevated in persistent/permanent AF patients compared those with paroxysmal AF. | 20153266 |
CRP, C-reactive protein; IL-1β, interleukin-1β; IL-6, interleukin 6; TNF-α, tumor necrosis factor-a; PMID, PubMed identifier number.
INNATE INFLAMMATORY SIGNALING & AF
Innate immune cells play an important role in mediating inflammation, primarily via their ability to release pro-inflammatory cytokines. Infiltration of innate immune cells in the atrial myocardium has been observed in patients with AF. Leukocyte activation was increased in patients with perioperative AF,[33] and abundance of CD45-positive cells was higher in LA and right atria (RA) of AF patients.[34] Furthermore, among these immune cells, CD68-positive macrophages are more frequently observed in the atrial myocardium than the adaptive immune cells, such as CD3-positive T-lymphocytes.[35] TNF-α, nuclear factor κB (NFκB), and the ‘NACHT, LRR & PYD Domains-containing Protein 3’ (NLRP3) inflammasome are the best-characterized innate inflammatory signaling pathways. Although these inflammatory signaling pathways are primarily responsible for the maturation and release of cytokines, their potential cytokine-independent functions have also been linked to AF pathogenesis. Figure 2 summarizes the established and putative molecular mechanisms related to the innate inflammatory signaling pathways in AF pathogenesis (Figure 2), which are outlined below.
Figure 2. Putative molecular and cellular mechanisms of atrial fibrillation pathogenesis involving inflammatory pathways.
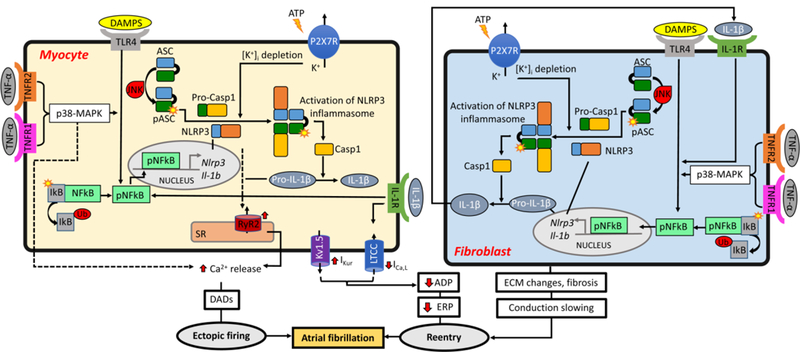
APD, action potential duration; ATP, adenosine triphosphate; EADs, early afterdepolorizations; ECM, extracellular matrix; ERP, effective refractory period; DADs, delayed afterdepolorizations; DAMPs, damage-associated molecular patterns; JNK, c-Jun N-terminal kinase; MAPK, activated protein kinase mitogen; P2X7R, P2X purinoceptor 7; SR, sarcoplasmic reticulum.
TNF-α signaling and AF
TNF -α, first identified in 1975, is considered as an endogenous mediator of inflammation, which is involved in a variety of cellular processes including activation of genes participating in inflammatory and immunoregulatory responses, proliferation, growth inhibition, and cell death.[36] The biological effects of TNF-α are mediated by two surface receptors, TNF receptor type-1 and type-2 (TNFR1 and TNFR2, respectively), both of which are expressed in cardiomyocytes (CMs), cardiac fibroblasts (CFs), and endothelial cells. Pathogen-associated molecular patterns (PAMPs) can stimulate both TNFR1 and TNFR2, and subsequently lead to the activation of transcription factors such as NFкB and activating protein 1 (AP-1),[36] which are two major downstream effectors of activated TNF-a. Increased TNF-α signaling may cause arrhythmia via multiple mechanisms. It has been shown that activation of TNF-α signaling can promote atrial electrical, structural, and contractile remodeling, all of which are hallmarks of the molecular pathophysiology of AF.[36] Cardiac-specific overexpression of TNF-α in mice can enhance the susceptibility to AF induction, which is associated with abnormal Ca2+ handling and decreased contractile function in atrial cardiomyocytes. [37] TNF-α signaling activation is also implicated in the endurance-exercise-induced AF model via the activation of NFкB and p38- MAPK pathway.[18] It has been demonstrated that the acute application of TNF-α to the pulmonary vein (PV) CMs can produce a variety of ionic changes including smaller L-type Ca2+ currents and related systolic Ca2+ transients, larger transient outward K+ currents (Ito), enhanced inward Na+-Ca2+-exchanger current (INCX), and a higher incidence of DADs, along with a transcriptional repression of sarcoplasmic reticulum ATPase (SERCA2a) expression.[19,23] In addition, elevated TNF-α levels in mice may promote atrial fibrosis, could slow conduction by altering the expression of connexin-40, and likely alters ECM by promoting the fibroblast-myofibroblast transition, thereby enhancing secretion of collagen and metalloproteinases.[18,37,38]
NFкB signaling and AF
NFкB is a transcription factor, which is inactive when it is bound to its endogenous inhibitor in cytoplasm (IB). In response to a variety of stress signals including the damage-associated molecular patterns (DAMPs), NFкB dissociates from IB and translocates to the nucleus, where it regulates transcription of its target genes including many inflammatory cytokines like TNF-α, IL-6, IL-8, and IL-1.[39] The most common mechanism activating NFкB is the toll-like receptor 4 (TLR4)-myeloid differentiation factor 88 (MyD88) pathway.[40,41] The expression of TLR4, MyD88, NFкB and phosphorylated (active) NFкB are all increased in LA of AF patients compared to sinus rhythm controls.[42]
Oxidative stress plays a significant role in AF pathophysiology,[13,43,44] and reactive oxidative species (ROS) are major activators of key signaling pathways and transcription factors including NFкB.[45] NFкB serves as a redox-sensitive transcription factor, which in response to oxidative stress, can suppress transcription of cardiac Na+ channels.[46] ROS detection of NFкB may also lead to modulation of other ion channels and transcription factors involved in AF development. Overall NFкB appears to serve as a nodal point in inflammatory signaling and might therefore represent a viable therapeutic target for AF treatment.[44]
NLRP3 inflammasome and AF
In the innate immunity, NLRP3 inflammasome is the most extensively studied member of NOD-like receptor (NLR) family.[47–49] NLRP3 is distinguished structurally from other NLR proteins by its N-terminal pyrin domain (PYD), which interacts with the PYD domain of the ASC [apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain (CARD)] adaptor molecule. ASC facilitates recruitment of the inactive zymogen precursor caspase-1 (pro-Casp1) to complete assembly of the functional NLRP3 inflammasome complex.[47–51] Once the NLRP3, aSc, and pro-Casp1 assemble, forming the inflammasome complex, it triggers the pro-Casp1 autocleavage to produce the active form of Casp-1. The active Casp-1 is an IL-1-converting enzyme that consists of a 10 kDa (p10) and a 20 kDa (p20) subunit. The active Casp-1 exhibits two distinct functions. It cleaves the inactive precursors of IL-1β and IL-18 (pro-IL-1β and pro-IL-18, respectively), thereby maturing the biologically active forms of IL-1β and IL-18. Macrophages and myofibroblasts can then secrete the mature cytokines, which serve as an inflammatory signaling-amplifier to perpetuate and spread the inflammatory response.[47–49,52] In addition, active Casp-1 can cleaves also gasdermin-D (GSDMD), releasing a 31 kDa N-terminal fragment (GSDMD-N), which forms the GSDMD-N pore through which IL-1β and IL-18 leave the cells to disseminate inflammation. If the number of pores in the plasma membrane increases above a certain threshold this can causes membrane rupture promoting a form of inflammatory cell death known as pyroptosis.[53– 55] Emerging evidence point to an important role of NLRP3 inflammasome in a number of cardiovascular diseases including ischemic cardiomyopathy, atherosclerosis, etc., which is likely attributed to its dual functions promoting both abnormal cytokine release and perhaps cell death. For instance, activation of NLRP3 inflammasome can promote the development of cardiomyopathy by 1) reducing the number of working CMs due to the Casp-1-mediated pyroptotic cell death, or 2) promoting structural remodeling by activation of cardiac fibroblasts or macrophages, ultimately leading to an enhanced release of cytokines and growth factors that can further remodel the ECM.[56–59]
There is clear evidence for a causal role of NLRP3 inflammasome activation in AF pathogenesis. We could recently demonstrate that the activity of NLRP3 inflammasome is increased in RA cardiomyocytes of patient with a history of pAF and perAF compared to sinus rhythm controls [77]. The activity of NLRP3 inflammasome was also enhanced in atrial samples of AF dogs and a mouse model of spontaneous AF induced by cardiac-specific overexpression of CREM-IbΔC-X.[21] Most important, the constitutive activation of cardiomyocyte NLRP3 inflammasome was sufficient to promote atrial ectopic activity and create a proarrhythmic substrate, enhancing the inducibility of AF. The cellular and molecular mechanisms underlying atrial ectopic activity and the reentrant substrate resulting from a specific activation of NLRP3 inflammasome in cardiomyocyte only included 1) aberrant RyR2-mediated SR Ca2+ release during diastole, 2) AP shortening likely due to the augmented ultra-rapid delayed-rectifier K+ current (IKur), 3) atrial hypertrophy, which might result from an increased level of myocyte- specific enhancer factor-2C (Mef2c), a well-known transcription activator associated with myocardial hypertrophy, and 4) atrial fibrosis. It remains to be determined whether these effects are exclusively mediated by IL-1β and IL-18 and whether pyroptosis also contributes to the evolution of the arrhythmogenic substrate. To the best of our knowledge our study is the first to demonstrate a causal link between NLRP3 inflammasome signaling and AF pathophysiology. The identification of the upstream mechanisms leading to NLRP3 inflammasome activation in AF, which need direct addressing in subsequent work, is expected to uncover novel therapeutic approaches for effective AF management.
POTENTIAL ANTI-INFLAMMATORY THERAPY FOR AF
Despite the advancements in understanding the mechanisms of AF and improved AF patient care, this clinical condition remains an ongoing socioeconomic burden. Since AF confers a fivefold increase in the risk for stroke, many therapy regimens incorporate a variety of blood thinner and anti-arrhythmia drugs to prevent thrombus formation.[60,61] Contemporary therapeutic options for AF treatment include: 1) cardioversion and defibrillation, 2) use of anti-arrhythmic drugs with limited efficacy and substantial toxicity [5], and 3) diverse atrial ablation procedures to isolate or destroy the arrhythmic sources.[60–62] However, AF patients may still face undesirable outcomes, such as pro-arrhythmic side-effects caused by the anti-arrhythmic drugs, or severe complication due to ablation, or recurrent AF after ablation.[62] Thus there is a clear unmet need for novel anti-AF therapeutics with improved efficacy and safety profiles.[62]
Frustaci et al. were the first to suggest a potential link between inflammation and AF and since then multiple pharmacological approaches targeting different inflammation signaling pathways are under evaluation in either animal studies or clinical trials.[63] Several antiinflammatory options that have been tested in animal studies might have anti-AF effects by either modulating the cellular determinants of ectopic activity and/or by targeting the determinants of the reentrant substrate (Figure 3). For example, in an exercise-induced AF model, TNF-α inhibition with etanercept, TNF-α gene ablation, or p38 inhibition, all prevented atrial structural remodeling and reduced AF-inducibility in response to exercise.[18] Inhibition of NLRP3 by MCC950 (a compound that can block the activation of NLRP3 inflammasome), by a shRNA (delivered by adeno-associated virus type-9), or by genetic ablation of Nlrp3 gene, reduced the susceptibility to pacing-induced AF in mice.[21] Of note, colchicine (a microtubule delopimerizing drug with anti-inflammatory effects) and low-dose methotrexate (an anticancer agent with pronounced anti-inflammatory properties), which are used for decades to treat chronic inflammatory diseases, also inhibit the NLRP3 inflammasome.[64,65] Although colchicine appears to effective against postoperative AF,[66] the potential beneficial effects of colcichine and methotrexate against other AF forms need prospective demonstration. In addition, a variety of NFкβ inhibitors are also available.[67,68] Although the effect of NFкP inhibition on AF development has not been determined, its protective role has been shown in animal models of cardiomyopathies [69,70]. Meanwhile, multiple clinical studies have been conducted to evaluate the efficacy of anti-inflammatory therapies in cardiomyopathies, which may pave the way for evaluating the therapeutic potential of anti-inflammatory strategies in AF patients. A recent clinical study (CANTOS) revealed that suppression of IL-1β, utilizing the cytokine neutralizing antibody canakinumab, can significantly reduce cardiac events in patients who are at risk.[71,72] Further investigation should address whether antibodies against IL-1β (canakinumab or gevokizumab) or IL-1α and IL-1β inhibitors such as anakinra and rilonacept can reduce the risk of AF in these patients. [65,73]
Figure 3. Proposed therapeutic strategies for AF by targeting inflammatory pathways.
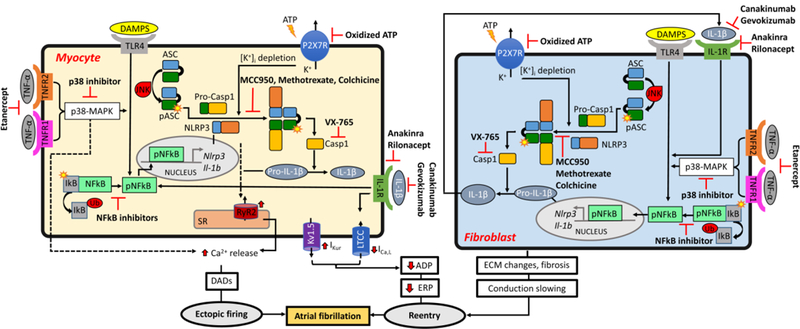
. APD, action potential duration; ATP, adenosine triphosphate; EADs, early afterdepolorizations; ECM, extracellular matrix; ERP, effective refractory period; DADs, delayed afterdepolorizations; DAMPs, damage-associated molecular patterns; JNK, c-Jun N-terminal kinase; MAPK, activated protein kinase mitogen; P2X7R, P2X purinoceptor 7; SR, sarcoplasmic reticulum.
As an alternative to targeting specific inflammatory regulators, some clinical studies suggest that inhibition of coagulation factors (e.g. activated factor-X; FXa), may also have anti-inflammatory effects. For example, non-valvular AF patients treated with FXa inhibitors demonstrated both reduced inflammation and coagulation relative to controls.[74] Clearly further prospective studies are needed to determine the potential anti-AF effects of oral anti-coagulants. It is possible that a three-pronged therapeutic approach utilizing anticoagulant, antiplatelet and anti-inflammatory drugs will be needed to combat AF and the related thrombogenesis in AF patients.[75] In addition, the abovementioned inflammatory signaling pathways mutually interact via transcriptional or posttranscriptional mechanisms, making it likely that inhibition of nodal points of their regulation will be required for effective treatment AF patients. Thus, future extensive work is needed to prove and validate the causal contribution of inflammation and inflammatory signaling to AF pathophysiology and whether anti-inflammatory agents could constitute a novel therapeutic approach to treat AF patients.
Highlights.
Atrial fibrillation (AF), the most prevalent arrhythmia, is often associated with enhanced inflammatory response.
Emerging evidence point to a causal role of inflammatory signaling pathways in the evolution of atrial electrical, calcium handling and structural remodeling, which create the substrate of AF development.
In this review, we discuss the clinical evidence supporting the association between inflammatory indices and AF development, the molecular and cellular mechanisms of AF, which appear to involve multiple canonical inflammatory pathways, and the potential of anti-inflammatory therapeutic approaches in AF prevention/treatment.
Acknowledgement
This work was supported by the NIH (R01-HL136389 to N.L. and D.D., and R01-HL131517 to D.D.), the American Heart Association (17PRE33660744 to L.S.J.), and the German Research Foundation DFG (Do 769/4–1 to D.D.).
Footnotes
Disclosure
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE
- [1].Feigin VL, Norrving B, Mensah GA. Global Burden of Stroke. Circ Res. 120 (2017) 439–448. [DOI] [PubMed] [Google Scholar]
- [2].Iwasaki YK, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology: implications for management. Circulation. 124 (2011) 2264–2274. [DOI] [PubMed] [Google Scholar]
- [3].Schnabel RB, Yin X, Gona P, Larson MG, Beiser AS, McManus DD, et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet (London, England). 386 (2015) 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res. 114, (2014) 1453–1468. [DOI] [PubMed] [Google Scholar]
- [5].Gheorghe-Andrei D, Dobrev D Antiarrhythmic drugs for atrial fibrillation: Imminent impulses are emerging. IJC Heart & Vasculature. 21 (2018) 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 114 (2014) 1483–1499. [DOI] [PubMed] [Google Scholar]
- [7].Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. The Journal of clinical investigation. 121 (2011) 2955–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D, et al. The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovascular research. 109 (2016) 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Qu YC, Du YM, Wu SL, Chen QX, Wu HL, Zhou SF. Activated nuclear factor-kappaB and increased tumor necrosis factor-alpha in atrial tissue of atrial fibrillation. Scand Cardiovasc J. 43 (2009) 292–297. [DOI] [PubMed] [Google Scholar]
- [10].Wu N, Xu B, Xiang Y, Wu L, Zhang Y, Ma X, et al. Association of inflammatory factors with occurrence and recurrence of atrial fibrillation: a meta-analysis. Int J Cardiol. 169 (2013) 62–72. [DOI] [PubMed] [Google Scholar]
- [11].Luan Y, Guo Y, Li S, Yu B, Zhu S, Li S, et al. Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. Europace. 12 (2010) 1713–1718. [DOI] [PubMed] [Google Scholar]
- [12].Rudolph V, Andrie RP, Rudolph TK, Friedrichs K, Klinke A, Hirsch-Hoffmann B, et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med. 16 (2010) 470–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, et al. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 7 (2010) 438–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Workman AJ, Kane KA, Rankin AC. Cellular bases for human atrial fibrillation. Heart Rhythm. 5 (2008) S1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Landstrom AP, Dobrev D, Wehrens XHT. Calcium Signaling and Cardiac Arrhythmias. Circ Res. 120 (2017) 1969–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Comtois P, Kneller J, Nattel S. Of circles and spirals: bridging the gap between the leading circle and spiral wave concepts of cardiac reentry. Europace. 7 Suppl 2 (2005) 10–20. [DOI] [PubMed] [Google Scholar]
- [17].Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 51 (2008) 802–809. [DOI] [PubMed] [Google Scholar]
- [18].Aschar-Sobbi R, Izaddoustdar F, Korogyi AS, Wang Q, Farman GP, Yang F, et al. Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFalpha. Nat Commun. 6 (2015) 6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee SH, Chen YC, Chen YJ, Chang SL, Tai CT, Wongcharoen W, et al. Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. 80 (2007) 1806–1815. [DOI] [PubMed] [Google Scholar]
- [20].Zhang Y, Wang YT, Shan ZL, Guo HY, Guan Y, Yuan HT. Role of inflammation in the initiation and maintenance of atrial fibrillation and the protective effect of atorvastatin in a goat model of aseptic pericarditis. Mol Med Rep. 11 (2015) 2615–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yao C, Veleva T, Scott L, Cao S, Li L, Chen G, et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. May 25 [Epub ahead of print] (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hunter P. The inflammation theory of disease. The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 13 (2012) 968–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kao YH, Chen YC, Cheng CC, Lee TI, Chen YJ, Chen SA. Tumor necrosis factoralpha decreases sarcoplasmic reticulum Ca2+-ATPase expressions via the promoter methylation in cardiomyocytes. Crit Care Med. 38 (2010) 217–222. [DOI] [PubMed] [Google Scholar]
- [24].Roth-Walter F, Jensen-Jarolim E, Stockinger H. Principles and Comparative Aspects of Adaptive Immunity2013. [Google Scholar]
- [25].Punchard NA, Whelan CJ, Adcock I. The Journal of Inflammation. J Inflamm (Lond). 1 (2004) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 160 (2015) 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Van Linthout S, Tschope C. Inflammation - Cause or Consequence of Heart Failure or Both? Curr Heart Fail Rep. 14 (2017) 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang H, Li J, Chen X, Wu N, Xie W, Tang H, et al. Association of Systemic Inflammation Score With Atrial Fibrillation: A Case-Control Study With Propensity Score Matching. Heart Lung Circ. 27 (2018) 489–496. [DOI] [PubMed] [Google Scholar]
- [29].Bruins P, te Velthuis H, Yazdanbakhsh AP, Jansen PG, van Hardevelt FW, de Beaumont EM, et al. Activation of the complement system during and after cardiopulmonary bypass surgery: postsurgery activation involves C-reactive protein and is associated with postoperative arrhythmia. Circulation. 96 (1997) 3542–3548. [DOI] [PubMed] [Google Scholar]
- [30].Harada M, Van Wagoner DR, Nattel S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ J. 79 (2015) 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jabati S, Fareed J, Liles J, Otto A, Hoppensteadt D, Bontekoe J, et al. Biomarkers of Inflammation, Thrombogenesis, and Collagen Turnover in Patients With Atrial Fibrillation. Clin Appl Thromb Hemost. (2018) 1076029618761006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hijazi Z, Aulin J, Andersson U, Alexander JH, Gersh B, Granger CB, et al. Biomarkers of inflammation and risk of cardiovascular events in anticoagulated patients with atrial fibrillation. Heart. 102 (2016) 508–517. [DOI] [PubMed] [Google Scholar]
- [33].Fontes ML, Mathew JP, Rinder HM, Zelterman D, Smith BR, Rinder CS, et al. Atrial fibrillation after cardiac surgery/cardiopulmonary bypass is associated with monocyte activation. Anesth Analg. 101 (2005) 17–23, table of contents. [DOI] [PubMed] [Google Scholar]
- [34].Chen MC, Chang JP, Liu WH, Yang CH, Chen YL, Tsai TH, et al. Increased inflammatory cell infiltration in the atrial myocardium of patients with atrial fibrillation. Am J Cardiol. 102 (2008) 861–865. [DOI] [PubMed] [Google Scholar]
- [35].Yamashita T, Sekiguchi A, Iwasaki YK, Date T, Sagara K, Tanabe H, et al. Recruitment of immune cells across atrial endocardium in human atrial fibrillation. Circ J. 74 (2010) 262–270. [DOI] [PubMed] [Google Scholar]
- [36].Ren M, Li X, Hao L, Zhong J. Role of tumor necrosis factor alpha in the pathogenesis of atrial fibrillation: A novel potential therapeutic target? Ann Med. 47 (2015) 316–324. [DOI] [PubMed] [Google Scholar]
- [37].Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM, et al. Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-{alpha}. Am J Physiol Heart Circ Physiol. 289 (2005) H1456–1467. [DOI] [PubMed] [Google Scholar]
- [38].Liew R, Khairunnisa K, Gu Y, Tee N, Yin NO, Naylynn TM, et al. Role of tumor necrosis factor-alpha in the pathogenesis of atrial fibrosis and development of an arrhythmogenic substrate. Circ J. 77 (2013) 1171–1179. [DOI] [PubMed] [Google Scholar]
- [39].Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annual review of immunology. 12 (1994) 141–179. [DOI] [PubMed] [Google Scholar]
- [40].Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 274 (1999) 10689–10692. [DOI] [PubMed] [Google Scholar]
- [41].Karki R, Igwe OJ. Toll-like receptor 4-mediated nuclear factor kappa B activation is essential for sensing exogenous oxidants to propagate and maintain oxidative/nitrosative cellular stress. PLoS One. 8 (2013) e73840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xu Q, Bo L, Hu J, Geng J, Chen Y, Li X, et al. High mobility group box 1 was associated with thrombosis in patients with atrial fibrillation. Medicine (Baltimore). 97 (2018) e0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Karam BS, Chavez-Moreno A, Koh W, Akar JG, Akar FG. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovascular Diabetology. 16 (2017) 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gao G, Dudley SC Jr. Redox regulation, NF-kappaB, and atrial fibrillation. Antioxid Redox Signal. 11 (2009) 2265–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zakkar M, Ascione R, James AF, Angelini GD, Suleiman MS. Inflammation, oxidative stress and postoperative atrial fibrillation in cardiac surgery. Pharmacol Ther. 154 (2015) 13–20. [DOI] [PubMed] [Google Scholar]
- [46].Liu M, Gu L, Sulkin MS, Liu H, Jeong EM, Greener I, et al. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol. 54 (2013) 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res. 8 (2015) 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. 1319 (2014) 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 265 (2015) 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. The Journal of clinical investigation. 119 (2009) 3502–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rivers-Auty J, Brough D. Potassium efflux fires the canon: Potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur J Immunol. 45 (2015) 2758–2761. [DOI] [PubMed] [Google Scholar]
- [52].Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, et al. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 4 (2015) 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase- 11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 526 (2015) 666–671. [DOI] [PubMed] [Google Scholar]
- [54].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 526 (2015) 660–665. [DOI] [PubMed] [Google Scholar]
- [55].Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 27 (2017) 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bracey NA, Beck PL, Muruve DA, Hirota SA, Guo J, Jabagi H, Wright JR Jr., MacDonald JA, Lees-Miller JP, Roach D, Semeniuk LM and Duff HJ. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp Physiol. 98 (2013) 462–473. [DOI] [PubMed] [Google Scholar]
- [57].Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF and Abbate A The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA. 108 (2011) 19725–19730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Valle Raleigh J, Mauro AG, Devarakonda T, Marchetti C, He J, Kim E, et al. Reperfusion therapy with recombinant human relaxin-2 (Serelaxin) attenuates myocardial infarct size and NLRP3 inflammasome following ischemia/reperfusion injury via eNOS-dependent mechanism. Cardiovascular research. 113 (2017) 609–619. [DOI] [PubMed] [Google Scholar]
- [59].Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol. 209 (2016) 215–220. [DOI] [PubMed] [Google Scholar]
- [60].Edgerton ZJ, Edgerton JR. A review of current surgical treatment of patients with atrial fibrillation. Proc (Bayl Univ Med Cent). 25 (2012) 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Copley DJ, Hill KM. Atrial Fibrillation: A Review of Treatments and Current Guidelines. AACN advanced critical care. 27 (2016) 120–128. [DOI] [PubMed] [Google Scholar]
- [62].Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nature reviews Drug discovery. 11 (2012) 275–291. [DOI] [PubMed] [Google Scholar]
- [63].Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 96 (1997) 1180–1184. [DOI] [PubMed] [Google Scholar]
- [64].Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res. 122 (2018) 1722–1740. [DOI] [PubMed] [Google Scholar]
- [65].Martinez GJ, Celermajer DS, Patel S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis. 269 (2018) 262–271. [DOI] [PubMed] [Google Scholar]
- [66].Lennerz C, Barman M, Tantawy M, Sopher M, Whittaker P. Colchicine for primary prevention of atrial fibrillation after open-heart surgery: Systematic review and meta-analysis. Int J Cardiol. 249 (2017) 127–137. [DOI] [PubMed] [Google Scholar]
- [67].Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 1799 (2010) 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Miller SC, Huang R, Sakamuru S, Shukla SJ, Attene-Ramos MS, Shinn P, et al. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem Pharmacol. 79 (2010) 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hammers DW, Sleeper MM, Forbes SC, Coker CC, Jirousek MR, Zimmer M, et al. Disease-modifying effects of orally bioavailable NF-kappaB inhibitors in dystrophin-deficient muscle. JCI Insight. 1 (2016) e90341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Maier HJ, Schips TG, Wietelmann A, Kruger M, Brunner C, Sauter M, et al. Cardiomyocyte-specific IkappaB kinase (IKK)/NF-kappaB activation induces reversible inflammatory cardiomyopathy and heart failure. Proceedings of the National Academy of Sciences of the United States of America. 109 (2012) 11794–11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sun Z, Zhou D, Xie X, Wang S, Wang Z, Zhao W, et al. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res Cardiol. 111 (2016) 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J. 162 (2011) 597–605. [DOI] [PubMed] [Google Scholar]
- [73].Buckley LF, Abbate A. Interleukin-1 blockade in cardiovascular diseases: a clinical update. Eur Heart J. 39 (2018) 2063–2069. [DOI] [PubMed] [Google Scholar]
- [74].Katoh H, Nozue T, Michishita I. Anti-inflammatory effect of factor-Xa inhibitors in Japanese patients with atrial fibrillation. Heart Vessels. 32 (2017) 1130–1136. [DOI] [PubMed] [Google Scholar]
- [75].Chi G, Jamil A, Radulovic M, Jamil U, Balouch MA, Marszalek J, et al. Dual antithrombotic plus adjunctive antiinflammatory therapy to improve cardiovascular outcome in atrial fibrillation patients with concurrent acute coronary syndrome: A triple-pathway strategy. Med Hypotheses. 114 (2018) 40–44. [DOI] [PubMed] [Google Scholar]