

Abstract
Gastric cancer in young adults has been pointed out to comprise a subgroup associated with distinctive clinicopathological features, including an equal gender distribution, advanced disease, and diffuse‐type histology. Comprehensive molecular analyses of gastric cancers have led to molecular‐based classifications and to specific and effective treatment options. The molecular traits of gastric cancers in young adults await investigations, which should provide a clue to explore therapeutic strategies. Here, we studied 146 gastric cancer patients diagnosed at the age of 40 years or younger at the Cancer Institute Hospital (Tokyo, Japan). Tumor specimens were examined for Helicobacter pylori infection, Epstein‐Barr virus positivity, and for the expression of mismatch repair genes to indicate microsatellite instability. Overexpression, gene amplifications, and rearrangements of 18 candidate driver genes were examined by immunohistochemistry and FISH. Although only a small number of cases were positive for Epstein‐Barr virus and microsatellite instability (n = 2 each), we repeatedly found tumors with gene fusion between a tight‐junction protein claudin, CLDN18, and a regulator of small G proteins, ARHGAP, in as many as 22 cases (15.1%), and RNA sequencing identified 2 novel types of the fusion. Notably, patients with the CLDN18‐ARHGAP fusion revealed associations between aggressive disease and poor prognosis, even when grouped by their clinical stage. These observations indicate that a fusion gene between CLDN18 and ARHGAP is enriched in younger age‐onset gastric cancers, and its presence could contribute to their aggressive characteristics.
Keywords: diffuse type, genome, Helicobacter pylori, rearrangement, stomach
1. INTRODUCTION
Gastric cancer (GC) is the third most frequent cause of cancer‐related deaths worldwide.1 The incidence of GC increases after the age of 50 years and peaks in the 60s to 70s.2, 3, 4, 5, 6 Aging is one of the major factors for GC, like other cancers, yet a substantial subgroup of GCs occurs in younger individuals; this cohort is referred to as GC in young adults (GCYA).2, 7 A number of studies on GCYA report their universal clinicopathological features, an equal gender distribution, advanced disease with poor outcome, and diffuse‐type dominant histology.2, 3, 4, 5, 6, 7, 8, 9, 10 Whether there are common backgrounds explaining the early onset of GC awaits investigation. Whether GCYAs reveal higher malignant potential is also unclear, as a recent study reported that prognosis does not differ from the general population when patients were grouped by their clinical stage.13, 14 Treatment of GCYA is presently based on standardized up‐to‐date approaches established for GC in general.
The comprehensive molecular analyses of GC, including The Cancer Genome Atlas (TCGA), suggested 4 molecular subtypes: tumors positive for Epstein‐Barr virus (EBV), microsatellite instability (MSI), genomically stable (GS), and chromosomal instability (CIN). Among these subtypes, GS subtype tumors tend to reveal relatively early onset and diffuse‐type histology,19 suggesting that the molecular background of GCYA might relate to the GS subtype. In line with this possibility, a recent molecular analysis of GCYA reported that approximately two‐thirds of GCYA can be classified into the GS subtype.20 Gastric cancers with the GS subtype are barely sensitive to molecular‐targeted drugs, as they have few actionable alterations such as driver gene mutations or receptor tyrosine kinase (RTK) amplifications. Only 11% of GCYA tumors are positive for EBV or MSI‐high, the patient of which would benefit from immune checkpoint inhibitors.19 It is therefore crucial to investigate the molecular traits of GCYA to obtain a clue to explore therapeutic strategies.
In the present study, we studied a substantial number of GCYA cases encountered in the Cancer Institute Hospital (Tokyo, Japan) for their clinicopathological features and molecular characteristics. Our work identifies an enrichment of a fusion gene in the cohort that potentially plays a pathological role in GCYA patients.
2. MATERIALS AND METHODS
2.1. Patients and data collection
This study was retrospectively undertaken at the Cancer Institute Hospital of the Japanese Foundation for Cancer Research between January 2006 and September 2015. Patients who met all the following criteria were enrolled: (i) histologically proven stomach adenocarcinoma; (ii) diagnosed at the age of 40 years or younger; (iii) underwent gastrectomy, excluding additional surgery after endoscopic resection; (iv) received no prior chemotherapy or radiotherapy; and (v) provided informed consent to the use of their surgical specimen for the research. We excluded intramucosal diseases when tumor volume was insufficient for the analysis. Medical records were reviewed to obtain the clinical datasets such as age, sex, family history (cancer/GC), laboratory data, surgery, macroscopic type, TNM stage, recurrence, and outcomes. The clinical stage was determined based on the Japanese classification of gastric carcinoma (3rd English edition).21 Endoscopic findings were reviewed by board‐certified fellows of the Japan Gastroenterological Endoscopy Society, to diagnose the Helicobacter pylori (HP) infection testing the normal part of the stomach mucosa. The H&E stained slides were reviewed by certified pathologists to confirm the histological findings and evaluate the level of inflammation on the background stomach mucosa. Histological types were classified into intestinal and diffuse type based on the Lauren classification.22 The whole research design was approved by the institutional review board of the Cancer Institute of Japanese Foundation for Cancer Research (IRB No. 2013‐1128) and undertaken following the principles of the Declaration of Helsinki.
2.2. Tissue microarray assembly
Representative tumor areas were defined on H&E stained slides and corresponding tissue from surgical specimens were obtained and assembled on tissue microarray. These were used to examine gene splitting and fusion by FISH and to test positivity of MSI by immunohistochemistry (IHC). The MSI status was also examined using formalin‐fixed paraffin‐embedded (FFPE) specimens.
2.3. Evaluation of HP infection status
Helicobacter pylori infection status was examined through the following six criteria: (i) detection of bacterial body in normal gastric mucosa by HP Ab (Dako Polyclonal Rabbit Anti‐Helicobacter pylori; Dako) or H&E staining of FFPE section; (ii) elevated serum anti‐HP level (3 U/mL or higher); (iii) absence of endoscopically apparent regular arrangement of collecting venules in the lower gastric mucosa; (iv) history of HP eradication therapy; (v) positive for the urea breath test; and (vi) infiltration of inflammatory cells to gastric mucosa, atrophic gastritis, or intestinal metaplasia. Patients with at least 1 of these criteria were principally grouped as HP‐positive.
2.4. Evaluation for EBV and MSI
Positivity of EBV were examined by the EBV‐encoded small RNA‐in situ hybridization (EBER‐ISH) method, using BOND Ready‐to‐Use ISH EBER probe (PB0589; Leica, Newcastle, UK) and ISH with the automated BOND system (BOND‐MAX and BOND‐III systems; Leica) following the manufacturer's instructions. Expression of mismatch repair (MMR) genes including MLH1, MSH2, MSH6, and PMS2 was examined by IHC, in which the following Abs were used: MLH1 (ES05) (NCL‐L‐MLH1; Leica), MSH2 (G219‐1129) (556349; BD Biosciences, NJ, USA), MSH6 (EPR3945) (GTX62383; GeneTex, CA, USA) and PMS2 (A16‐4) (556415; BD). Tumor cells in the absence of at least 1 MMR protein were determined as MMR‐deficient. Normal epithelial cells and lymphocytes were used as an internal control for the evaluation.
2.5. Fluorescence in situ hybridization analyses and IHC
The FISH analyses were used to examine 18 genes relating the RTK signaling pathways (EGFR, ERBB2, MET, FGFR2, VEGFA, KRAS, and BRAF), cell cycle (CCND1, CCNE1, and cMYC), the RHOA pathway (RHOA and ARHGAP26/6), and genes known to have rearrangements (CLDN18, ALK, RET, ROS1, and NTRK‐1). Split‐FISH assays were carried out on FFPE specimens sliced to 4‐μm thickness, using bacterial artificial chromosome clone‐derived DNA probes. Cases positive for the split‐FISH assay were subjected to the fusion FISH assay to identify partner genes. Gene amplification was examined by FISH and IHC. A 5‐fold increase or more in FISH signals was tested for gene amplifications. Antibodies used in IHC included the Ventana I‐view PATHWAY anti‐human epidermal growth factor receptor‐2 (HER2) rabbit mAb (4B5) (Ventana, AZ, USA), anti‐epidermal growth factor receptor (EGFR) mouse mAb (2‐18C19) (pharmDx; Dako), and an anti‐MET (c‐MET) (EP1454Y) rabbit mAb (ab51067; Abcam, MA, USA). Scoring of HER2, EGFR, and MET were carried out based on Hofmann's criteria.23 Positivity was defined when complete or basolateral membrane reveal moderate (2+) or strong (3+) staining in more than 10% of cancer cells.
2.6. Reverse transcription‐PCR and DNA sequencing
Total RNA was extracted from FFPE specimens using the Recover All Total Nucleic Acid Isolation Kit for FFPE (Thermo Fisher Scientific, MA, USA). The extracted RNAs were processed for RT using specific primer (5′‐GAAGCCAATGCTCCAACT‐3′) and for PCR with DNA polymerase named PlatinumTaq High Fidelity (Thermo Fisher Scientific). Resulting PCR fragments were subcloned and sequenced using following primers: 5′‐TTGGGTCCAACACCAAAAAC‐3′ (forward) and 5′‐GAAGCCAATGCTGTCCAACT‐3′ (reverse).
2.7. RNA sequencing
Total RNA was extracted from frozen tissue by RNeasy Micro kit (Qiagen) and its quality was assessed with Bioanalyzer (Agilent). The RNA‐Seq libraries were prepared using TruSeq RNA Access Library Prep Kit‐ Set A (48 samples) and were sequenced on a DNA sequencer HiSeq 2500 (Illumina). The obtained data were screened for fusion genes using FusionCather software.24
2.8. Statistical analyses
Fisher's exact test was used for the comparison of category variables between 2 groups. Mann‐Whitney U test was used for the comparison of age, tumor diameter, and the number of metastatic lymph nodes (LNs) between 2 groups. Overall survival (OS) was estimated with the Kaplan‐Meier method. The data were analyzed as of December 7, 2018. Patients who were alive at this cut‐off date were treated to be censored. Overall survival was calculated from the date of surgery to death of any cause. The statistical differences of survival curves were assessed by log‐rank test, and cases where P < .05 were considered to be significant. All statistical analyses were carried out using GraphPad Prism version 7.03 for Windows (GraphPad Software, San Diego, CA, USA).
3. RESULTS
3.1. Patient characteristics
From January 2006 to September 2015, a total of 4809 patients with gastric adenocarcinoma underwent gastrectomy in our hospital. Among them, 178 patients (3.7%) were grouped as GCYA but 32 patients were excluded from the analysis due to insufficient access to their sample. Ultimately, 146 patients were enrolled to the present study (Table 1). The clinicopathological features of our cohort (women, 51.4%; diffuse type, 93.2%) was consistent with previous reports on GCYA.2, 3, 4, 5, 6, 7, 8, 9, 10
Table 1.
Characteristics of the gastric cancer in young adult (GCYA) cohort
| GCYA (n = 146) | |
|---|---|
| Median age, years (range) | 36 (16‐39) |
| Sex, n (%) | |
| Male | 71 (48.6) |
| Female | 75 (51.4) |
| Family history, n (%) | |
| Yes (cancer/gastric cancer) | 91/31 (62.3/21.2) |
| No | 46 (31.5) |
| Unknown | 9 (6.2) |
| Multiple cancer lesions, n (%) | |
| Yes | 7 (4.8) |
| No | 139 (95.2) |
| Location of primary tumor, n (%) | |
| Upper third | 41 (28.1) |
| Middle third | 65 (44.5) |
| Lower third | 40 (27.4) |
| Lauren classification, n (%) | |
| Intestinal | 10 (6.8) |
| Diffuse | 136 (93.2) |
| Gross appearance, n (%) | |
| Early | 72 (49.3) |
| Type 2 | 10 (6.8) |
| Type 3 | 40 (27.4) |
| Type 4 | 20 (13.7) |
| Others | 4 (2.7) |
| Depth of invasion, n (%) | |
| m/sm | 56 (38.4) |
| mp | 18 (12.3) |
| ss | 21 (14.4) |
| se/si | 51 (34.9) |
| LN metastasis, n (%) | |
| Absent | 76 (52.1) |
| Present | 70 (47.9) |
| Stage (JGCA 14th edition) | |
| Stage I/II | 91 (62.3) |
| Stage III/IV | 55 (37.7) |
JGCA, Japanese Gastric Cancer Association; LN, lymph node; m, mucosa; mp, muscularis propria; se, tumor penetration of serosa; si, tumor invasion of adjacent structures; sm, submucosa; ss, subserosa.
3.2. Helicobacter pylori infection
Among 146 GCYA patients, 45 patients (30.8%) were positive for HP infection, examined by elevated anti‐HP Ab levels (IgG) (>3 U/mL), positive for the urea breath test (>2.5‰), history of eradication therapy, and direct inspection of bacteria in the FFPE specimen stained by H&E and anti‐HP Ab. In addition, 86 patients (58.9%) revealed stomach mucosa with chronic inflammation, which is highly suggestive of HP infection. Among the remaining 15 patients, 7 patients were judged to be possibly HP‐negative but could not by evaluated sufficiently. Eight patients (5.8%) were judged to be HP‐negative, and 4 of them had tumors located at the esophagogastric junction or gastric cardia. These results indicate that inflammatory stomach mucosa causally related to HP infection is a strong background of GCYA (Figure 1).
Figure 1.
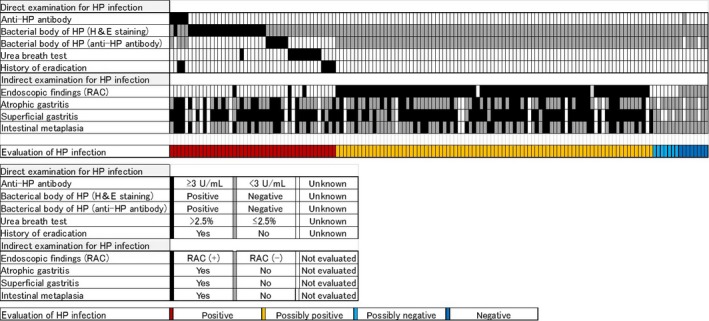
Evaluation for Helicobacter pylori (HP) infection. Infection was evaluated directly and indirectly (top panel), and color coded using the criteria indicated in the middle panel. Patients positive for any direct examination were grouped as “positive” (red), and positive for typical endoscopic findings, (ie, absence of regular arrangement of collecting venules [RAC]) were “possibly positive” (yellow). The remaining patients were considered as “possibly negative” (light blue) or “negative” (dark blue)
3.3. Molecular profiling of GCYA
Comprehensive molecular analyses of GCs19, 26 have led to molecular‐based classifications and offered specific and effective treatment options. Both EBV and MSI tumors are often sensitive to immunotherapy,27, 28 and thus have come to form independent entities among GCs.29 To investigate the molecular profile of GCYA, we first examined for EBV and MSI. There were 2 cases (1.4%) of EBV‐positive and 2 cases (1.4%) of MMR‐deficient tumors (Figure 2). The Cancer Genome Atlas analysis indicated that the amplification of genes related to the RTK signaling pathways were recurrently observed in the CIN subtype; these genes were ERBB2 (also known as HER2), KRAS, VEGFA, EGFR, FGFR2, and MET.19 We therefore classified tumors as CIN subtype when either gene amplification or overexpression of at least 1 gene associated with the RTK signaling pathway as the CIN subtype, and remaining cases as the GS subtype. In our GCYA cohort, RTK‐related genes were examined by amplification (FISH) (Figure 3A) and overexpression (IHC). Among 146 cases, only 20 (13.7%) were categorized to the CIN subtype, including HER2‐positive tumors in 8 cases. After excluding 7 invalid cases, 115 were grouped to the GS subtype, which corresponds to 78.8% of GCYA.
Figure 2.
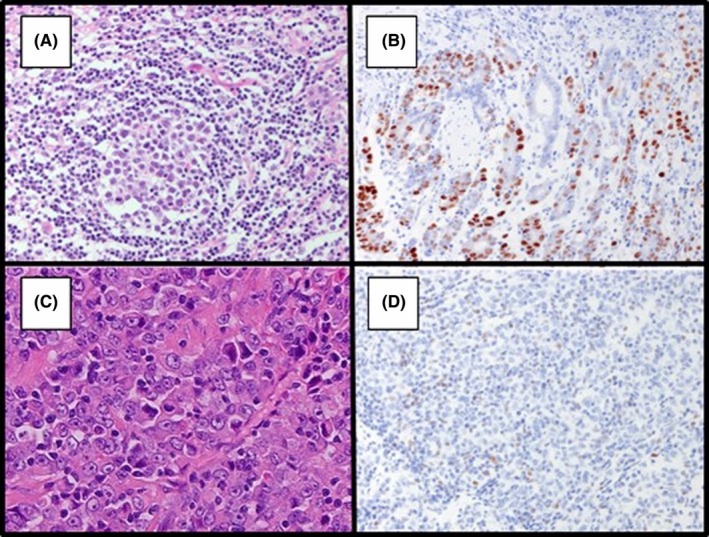
Representative cases of Epstein‐Barr virus (EBV)‐positive and microsatellite instability (MSI) tumor. A,B, EBV‐positive tumor revealed by H&E staining (A) and EBV‐encoded small RNA‐in situ hybridization (EBER‐ISH) analysis (B). C,D MSI tumor by H&E staining (C) and immunohistochemistry of mismatch repair proteins, including MLH1 (D), MSH2, MSH6, and PMS2
Figure 3.
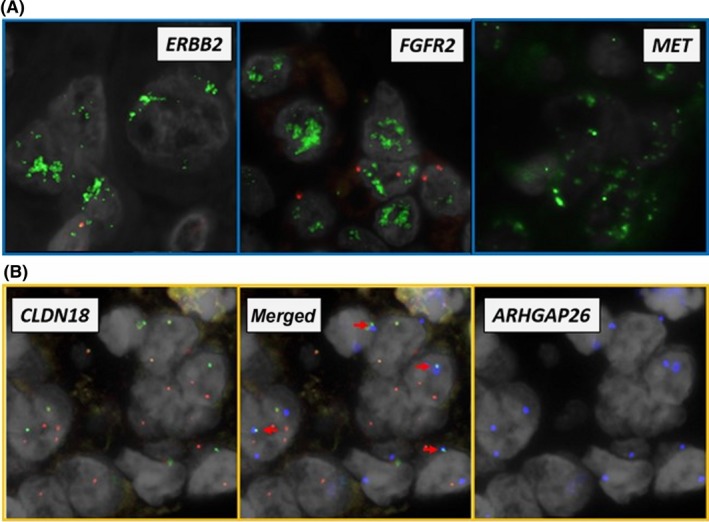
Examination of gene amplifications and rearrangements in FISH analysis. A, Representative images positive for gene amplification. DNA probes for indicated genes are labeled with green fluorophore. For internal controls, FISH probe to centromeres is labeled with red fluorophore. B, Representative images positive for the CLDN18‐ARHGAP26 fusion gene. The split FISH analysis indicates gene breakage when 2 different fluorophores labeled both sides of the gene fail to colocalize. Note that green (5′ side of CLDN18) and red (3′ side of CLDN18) signals were separated (left panel). The fusion FISH analysis indicates the presence of fusion genes when 2 different fluorophores labeled to fused pairs colocalize. Note that the blue signals from ARHGAP26 (right panel) overlap with green‐labeled CLDN18 in the merged panel (middle panel, arrows)
To illustrate the distinctive features of GCYAs, we compared the histological classification and molecular subtypes with TCGA cohort (Figure 4A). Consistent with previous reports,20 our GCYA cohort was characterized by diffuse‐type histology, GS subtype predominant, and rare MSI tumor. The detailed clinical, pathological, and molecular profile for each case is summarized in Figure 4B.
Figure 4.
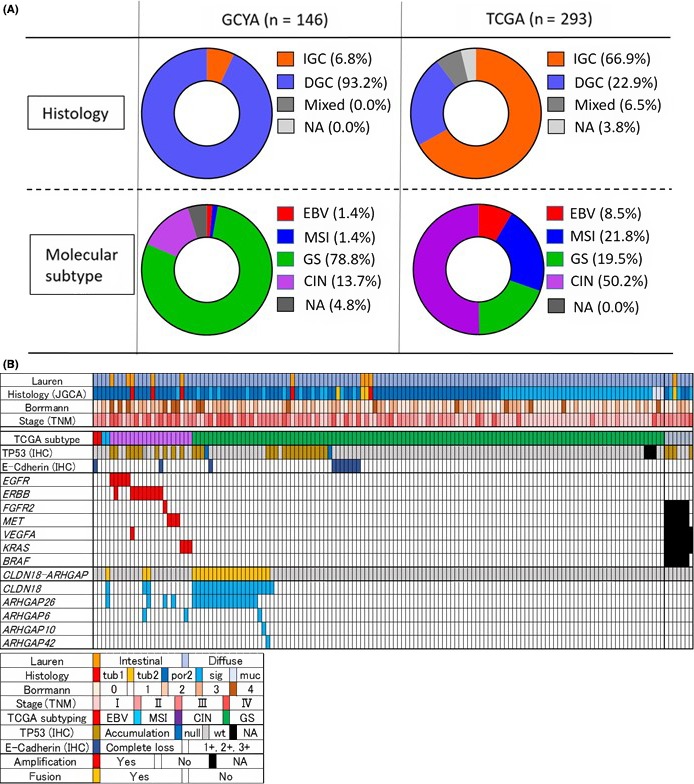
Histological and molecular landscape of gastric cancer in young adults (GCYA). A, Comparison of histology and molecular subtypes between GCYA and The Cancer Genome Atlas (TCGA) cohorts without 2 patients <40 years old. Note that the GCYA cohort is characterized by diffuse‐type histology, it is genomically stable (GS) subtype predominant, and microsatellite instability (MSI) tumors are rare. B, Clinicopathological features, Epstein‐Barr virus (EBV) positivity, MSI status, and genetic alterations detected by FISH or immunohistochemical (IHC) methods are summarized (n = 146) (upper panel), according to the classifications as indicated (bottom panel). CIN, xxxx; DGC, xxxx; IGC, xxxx; JGCA, Japanese Gastric Cancer Association; muc, xxxx; NA, xxxx; por, xxxx; sig, xxxx; tub, xxxx
The rearrangement of ALK, RET, ROS1, and NTRK1 are known to be driver fusions in lung cancer, and FISH analysis is a versatile method to screen for those fusions.31 Our FISH analysis did not find any fusions with ALK, RET, ROS1, or NTRK1; however, we repeatedly found fusion genes between CLDN18 and ARHGAP. Through the split‐FISH analysis, breakage of CLDN18, ARHGAP26, and ARHGAP6 genes were indicated in 23, 20, and 3 patients, respectively. Among them, CLDN18‐ARHGAP26 fusion was confirmed by fusion‐FISH in 18 patients (12.3%) and CLDN18‐ARHGAP6 in 2 patients (1.4%). For fusion‐FISH negative cases, we undertook RNA sequencing when fresh‐frozen samples were available, and identified 2 novel types of fusion, that is, CLDN18‐ARHGAP10 or CLDN18‐ARHGAP42 (Figure 5). In summary, we found 22 cases that were positive for CLDN18‐ARHGAP. This comprises 15.1% of our analysis, which is a major subgroup in our GCYA cohort (Figure 4B).
Figure 5.
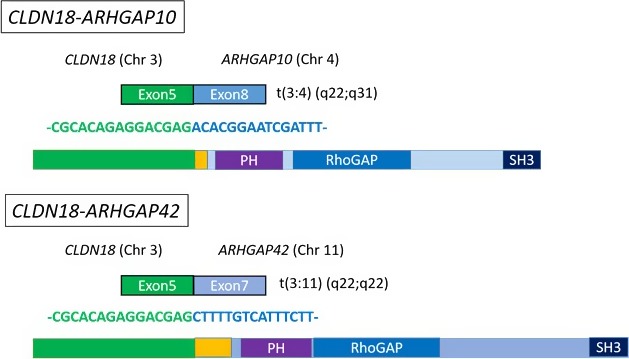
Novel fusion transcripts of CLDN18‐ARHGAP. RNA sequencing revealed the novel fusion transcripts, CLDN18‐ARHGAP10 and CLDN18‐ARHGAP42. The breakpoint of the CLDN18 side was identical to the known CLDN18 site. CLDN18 is fused to ARHGAP10 or ARHGAP42 in an in‐frame manner, suggesting that these chimeric proteins also contain the Rho‐GAP domain, similar to other types of fusion
3.4. Characterization of CLDN18‐ARHGAP fusion
The fusion genes between CLDN18 and ARHGAP26 reported in TCGA were found between exon 5 of CLDN18 and either exon 10 or 12 of ARHGAP26.19 The majority of the fusions occurred between exon 12, which we called variant 1 (and the other variant 2). To identify the nature of fusion genes found in our cohort, we extracted RNA from the FFPE specimens and their cDNA was sequenced for 18 cases positive for CLDN18‐ARHGAP26 fusion in the FISH analysis (Figure 6). Among them, 12 revealed either variant 1 (n = 11) or variant 2 (n = 1). Interestingly, in 6 cases, both of these types were detected, indicating heterogeneous origin of the fusion gene. In 2 cases of CLDN18‐ARHGAP6, the breakpoint was identical to that reported in TCGA cohort (exon 2 of ARHGAP6).19
Figure 6.
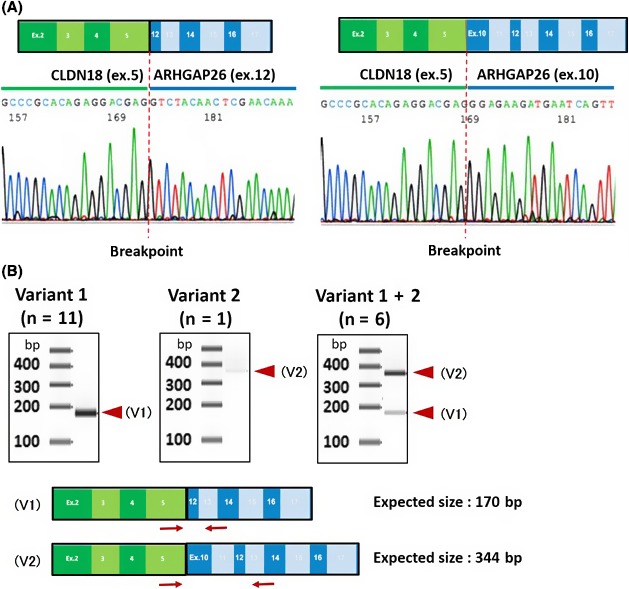
Structure of CLDN18‐ARHGAP fusion genes. A, Direct sequencing of the fused point between CLDN18 and ARHGAP26. Representative results showing that CLDN18 was fused to exon 10 (right panel) or exon12 (left panel) of ARHGAP26. The breakpoint for CLDN18 part was identical in all 22 cases, confirmed by sequencing DNA or RNA transcripts or both. B, Variants of CLDN18‐ARHGAP26. Primer sets for RT‐PCR analysis were designed as indicated in red arrows (forward primer, CLDN18 exon5; reverse primer, ARHGAP26 exon13). The estimated size of variant 1 (V1) is 170 bp and variant 2 (V2) is 344 bp. Among 18 CLDN18‐ARHGAP26 fusion‐positive cases, 12 revealed either V1 or V2 (left and middle panels), whereas in 6 cases both species were detected (right panel)
Furthermore, we could find unprecedented types of fusion of CLDN18 with ARHGAP, namely ARHGAP10 (exon 8) or ARHGAP42 (exon 7). All of these ARHGAP genes were found to be fused to CLDN18 in an in‐frame manner, implying an expression of chimeric protein with the GAP activity is preserved among these ARHGAP subtypes.
3.5. Clinical relevance of CLDN18‐ARHGAP
To address the significance of the CLDN18‐ ARHGAP fusion gene, we compared clinicopathological features between fusion‐positive and ‐negative cases. As summarized in Table 2, we found no statistically significant differences for gender, tumor location, histological type, gross appearance, or invasion depth of tumors between positives and negatives. However, patients with CLDN18‐ARHGAP fusion were diagnosed at earlier an age than those without (Figure 7A, left panel). The proportion of CLDN18‐ARHGAP fusion positive cases accounted for 27.6% in the younger population (35 years old or younger, n = 58) of GCYA (Figure 7A, right panel). Of note, positive cases often revealed larger tumor size, associated with LN metastases, and patients were diagnosed with advanced stage disease (Figure 7B,C and Table 2). We did not find any clinicopathological features that specify the newfound fusions, CLDN18‐ARHGAP10 and CLDN18‐ARHGAP42 (n = 2) from previously known fusions (n = 20; data not shown).
Table 2.
Characteristics of CLDN18‐ARHGAP‐positive and ‐negative tumors in patients with gastric cancer
| Negative (n = 124) | Positive (n = 22) | P value | |
|---|---|---|---|
| Sex, n (%) | |||
| Male | 63 (50.8) | 8 (36.4) | .252 |
| Female | 61 (49.2) | 14 (63.6) | |
| Location of primary tumor, n (%) | |||
| Upper third | 38 (30.6) | 3 (13.6) | .168 |
| Middle third | 55 (44.4) | 10 (45.5) | |
| Lower third | 31 (25.0) | 9 (40.9) | |
| Lauren classification, n (%) | |||
| Intestinal | 10 (8.1) | 0 (0.0) | .360 |
| Diffuse | 114 (91.9) | 22 (100.0) | |
| Gross appearance, n (%) | |||
| Early | 64 (51.6) | 8 (36.4) | .169 |
| Type 2 | 10 (8.1) | 0 (0.0) | |
| Type 3 | 30 (24.2) | 10 (45.5) | |
| Type 4 | 17 (13.7) | 3 (13.6) | |
| Others | 3 (2.4) | 1 (4.5) | |
| Tumor size (cm) | |||
| 10< | 20 (16.1) | 8 (36.4) | .038 |
| ≤10 | 104 (83.9) | 14 (63.6) | |
| Depth of invasion, n (%) | |||
| m/sm | 51 (41.1) | 5 (22.7) | .121 |
| mp | 17 (13.7) | 1 (4.5) | |
| ss | 16 (12.9) | 5 (22.7) | |
| se/si | 40 (32.3) | 11 (50.0) | |
| LN metastasis, n (%) | |||
| Absent | 72 (58.1) | 4 (18.2) | <.001 |
| Present | 52 (41.9) | 18 (81.8) | |
| Stage (JGCA 14th edition) | |||
| Stage I/II | 85 (68.5) | 6 (27.3) | <.001 |
| Stage III/IV | 39 (26.7) | 16 (72.7) | |
Patients with fusion had more association with large tumor, metastatic lymph nodes (LNs), and advanced stage.
JGCA, Japanese Gastric Cancer Association m, mucosa; mp, muscularis propria; se, tumor penetration of serosa; si, tumor invasion of adjacent structures; sm, submucosa; ss, subserosa.
Figure 7.
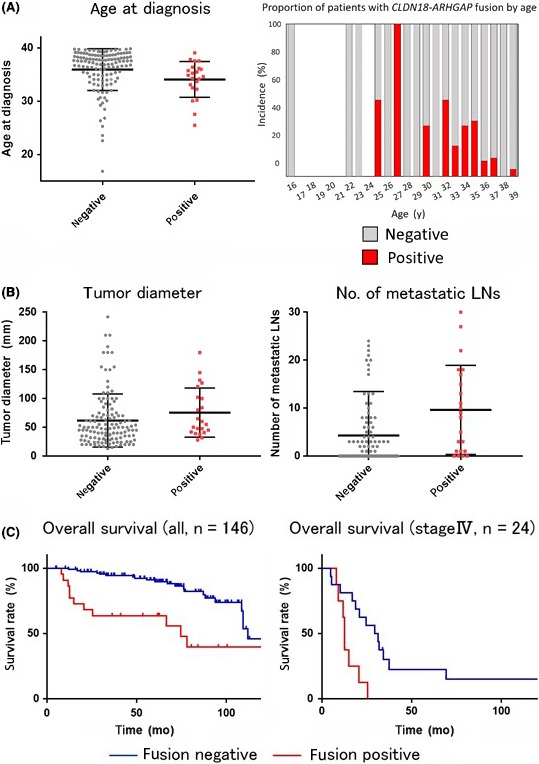
Clinical implications of the CLDN18‐ARHGAP fusion. A, Age at diagnosis for CLDHN18‐ARHGAP fusion‐positive and ‐negative subgroups (left panel). Bars indicate the median and 95% confidence interval. Incidence of CLDN18‐APHGAP positivity by age (right panel). Note the higher incidence of the fusion in younger patients within the gastric cancer in young adults cohort. B, Clinicopathological characteristics in the presence or absence of CLDN18‐ARHGAP fusion. Patients with CLDN18‐ARHGAP fusion had larger sized tumors and larger numbers of lymph node (LN) metastasis than patients without. Mann‐Whitney U test indicated their tendencies are statistically significant (P values). C, Kaplan‐Meier curve of overall survival in the whole cohort (left panel) and the stage IV population (right panel). Patients with CLDN18‐ARHGAP fusion revealed worse prognosis than patients without fusion in both analysis. Note that a significant difference was also indicated for the stage IV cohort
A Kaplan‐Meier analysis indicated that cases with the CLDN18‐ARHGAP fusion gene showed worse prognosis than those without (P < .001). The median follow‐up of patients who survived was 75.3 months (95% confidence interval, 70.0‐80.7) and 34 patients (23.3%) died by the cut‐off date. Importantly, when 24 patients diagnosed with stage IV disease were grouped, the OS rate turned out to be significantly different between the positives and negatives (P = .004) (Figure 7C).
To further address whether the age factor might affect the relevance of the fusion, we grouped our GCYA cohort into younger and older populations by median age. Interestingly, in 22 CLDN18‐ARHGAP fusion‐positive cases the OS rate was lower in younger patients (n = 11) than in older (n = 11), whereas in 124 fusion‐negative cases age revealed little effect on survival (Figure 8).
Figure 8.
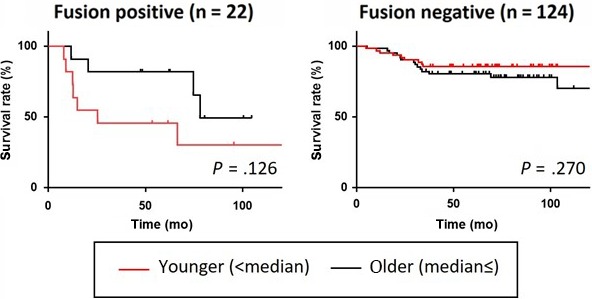
Impact of CLDN18‐ARHGAP fusion in younger age groups. CLDN18‐ARHGAP‐positive and ‐negative patients of our cohort were grouped into younger and older subgroups by the median age. Note that in CLDN18‐ARHGAP fusion‐positive cases the overall survival rate was lower in younger patients than in older, whereas in fusion‐negative cases age revealed little effect on survival
4. DISCUSSION
In our GCYA cohort, however, approximately 95% of patients were indicative for HP infection, which is the major environmental factor associated with tumorigenesis of GC, and classified as a class I carcinogen by WHO.32 Among these cases, 28 patients (19.2%) experienced peptic ulcer in the past, suggesting the history of severe HP infection. This implies that HP infection must also have a significant impact on the younger‐age onset,33 and that the genomic alterations toward carcinogenesis in GCYA could follow a process similar to that of the general GC population.
Cancers in young patients are often associated with inherited risk factors, as exemplified in Lynch syndrome34 or hereditary breast and ovarian cancer.35 The vast majority of GC arises sporadically, and GC linked to hereditary diffuse‐type GC or familial intestinal GC is rare (<3%).32, 36 Although a germline mutation analysis is required to assess the inherited factors, there was no remarkable family history for cancer or GC in our GCYA population, discounting the possibility that inherited factors are involved.37, 38 This would indicate that the tragedy of GCYA can be controllable.The meta‐analysis of EBV and MSI tumor estimated their incidence to be 8.7% and 13.1%, respectively.39, 40 The incidence of EBV and MSI tumor in our GCYA cohort was clearly lower than these (2.8%), as previously reported for GC in individuals younger than 45 years.20 Two lines of observation support the idea that GCYAs have a different group of etiologies from EBV and MSI GC subtypes. First, EBV‐positive tumors are often biased to men,19, 39, 41 and MSI‐positive GC typically affects elders.19 Second, longstanding accumulation of DNA methylations are thought to underlie MSI tumors, suppressing the expression of gene sets including MLH1.19, 42
Does any type of gene amplification characterize GCYA? The Cancer Genome Atlas analysis indicated that the amplification of genes related to the RTK signaling pathways are frequently observed in the CIN subtype.19 In our cohort, however, gene amplifications were barely detected: ERBB2 and others in RTK signaling pathways were seen only in a limited number of cases. Thus, it is unlikely that somatic copy‐number alternations, as seen in the upper gastrointestinal adenocarcinoma, widely underlie GCYA. The infrequent copy number variation is also consistent with our estimation that the GS subtype ranges over 75% of GCYA, as envisioned based on clinicopathological similarities between GCYA and GS type tumors.19
We identified the CLDN18‐ARHGAP fusion genes in as many as 15.1% of GCYA, which is in line with the TCGA data indicating that the frequency of this fusion is enriched up to 13.8% if grouped by the GS subtype.19 Notably, CLDN18‐ARHGAP fusions were mutually exclusive with CDH1 and RHOA somatic mutations.19 Thus, this fusion seems to constitute a significant genetic alteration in GS type tumors. CLDN18‐ARHGAP fusion‐positive cases tend to contain significantly larger tumors and greater numbers of metastatic LNs, thus classifying them to advanced stages more often than fusion‐negative cases. An appealing prediction is that cancers with the CLDN18‐ARHGAP fusion have higher malignant potential and accelerate development of GC. Together with a previous study,43 the finding that presence of CLDN18‐ARHGAP associates with aggressive disease in our younger patients would further underline the clinical relevance of CLDN18‐ARHGAP fusion in GCYA.
How might the chimeric protein CLDN18‐ARHGAP have pathogenic significance? Human claudin 18 (CLDN18) is one of the 26 claudin protein family member expressed specifically in stomach epithelial cells and has essential functions in tight junctions.45, 46 There is a transmembrane domain at its amino‐terminus which locates CLDN18 to the plasma membrane, and the carboxyl‐terminal domain is known to interact with actin regulatory proteins such as cadherin, integrin, and RhoA.47 Thus, claudin lacking the carboxyl‐terminal domain could disconnect all the actin regulatory proteins from the junctional complex and loosen cell‐to‐cell and/or cell‐to‐matrix connections.
The Rho‐GTPase activity of ARHGAP converts active RhoA to the inactive form.48 The existence of multiple patterns for ARHGAP that we and others have found strongly suggests that this fusion protein acquires a pathological role by recruiting GAP activity to CLDN18.49, 50 Thus, the activity of Rho‐GAP of ARHGAP26 will be ectopically enriched in the vicinity of the plasma membrane by virtue of CLDN18, and constitutively inhibit the RhoA activity at that site.19, 49, 50 As a constitutive inactive mutation of RhoA relates to the acquisition of oncogenic phenotype,48 this ectopic activity of ARHGAP26 must be involved in its pathogenicity.
Similar to other known fusions, the newfound CLDN18‐ARHGAP10 and CLDN18‐ARHGAP42 contained the catalytic RhoGAP domain. These new fusions were different from the others in that both also contained an intact PH domain. Although the relevance of the PH domain has been discussed,49 we consider it more likely that it is dispensable for the pathogenicity of the CLDN18‐ARHGAP fusion gene, given the small number of cases that contained the domain.
Therefore, in a worst scenario, both continuous RhoA inactivation and disruption of the junctional complex might take place simultaneously in the presence of the CLDN18‐ARHGAP fusion protein, contributing to the diffuse property of the tumors. Supporting this hypothesis, overexpression of CLDN18‐ARHGAP promoted cell motility in vitro.49 It will be interesting to address how its expression would affect cellular proliferation and if the fusion protein is druggable.
In conclusion, GCYAs arise based on the chronic inflammation of gastric mucosa, typically in association with HP infection, similarly to GC in older age groups. Among the characteristics of GCYA shared with the GS subtype, formation of the CLDN18‐ARHGAP fusion gene reveals a remarkable genetic feature, which might contribute to the aggressive nature of GC in younger patients.
CONFLICT OF INTEREST
Authors declare that they have no conflict of interest relating this work.
ACKNOWLEDGMENTS
We thank Drs. Akihito Dobashi, Yuki Togashi, and Satoko Baba for sharing their expertise on FISH analysis and sequencing. This work has been supported by research grants from the Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports and Technology of Japan.
Nakayama I, Shinozaki E, Sakata S, et al. Enrichment of CLDN18‐ARHGAP fusion gene in gastric cancers in young adults. Cancer Sci. 2019;110:1352–1363. 10.1111/cas.13967
REFERENCES
- 1. World Health Organization, International Agency for Research on Cancer . GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012.
- 2. Isobe Y, Nashimoto A, Akazawa K, et al. Gastric cancer treatment in Japan: 2008 annual report of the JGCA nationwide registry. Gastric Cancer. 2011;14:301‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. SEER Cancer Statistics Review, 1975‐2015. National Cancer Institute, Bethesda. https://seer.cancer.gov/csr/1975_2015. Accessed Xxxx 00, 0000.
- 4. Park JC, Lee YC, Kim JH, et al. Clinicopathological aspects and prognostic value with respect to age: an analysis of 3,362 consecutive gastric cancer patients. J Surg Oncol. 2009;99:395‐401. [DOI] [PubMed] [Google Scholar]
- 5. Qiu MZ, Wang ZQ, Zhang DS, et al. Clinicopathological characteristics and prognostic analysis of gastric cancer in the young adult in China. Tumour Biol. 2011;32:509‐514. [DOI] [PubMed] [Google Scholar]
- 6. Liu S, Feng F, Xu G, et al. Clinicopathological features and prognosis of gastric cancer in young patients. BMC Cancer. 2016;16:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anderson WF, Camargo MC, Fraumeni JF Jr, et al. Age‐specific trends in incidence of noncardia gastric cancer in US adults. JAMA. 2010;303:1723‐1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katai H, Sasako M, Sano T, Maruyama K. Gastric carcinoma in young adults. Jpn J Clin Oncol. 1996;26:139‐143. [DOI] [PubMed] [Google Scholar]
- 9. Maehara Y, Emi Y, Tomisaki S, et al. Age‐related characteristics of gastric carcinoma in young and elderly patients. Cancer. 1996;77:1774‐1780. [DOI] [PubMed] [Google Scholar]
- 10. Santoro R, Carboni F, Lepiane P, Ettorre GM, Santoro E. Clinicopathological features and prognosis of gastric cancer in young European adults. Br J Surg. 2007;94:737‐742. [DOI] [PubMed] [Google Scholar]
- 11. Matley PJ, Dent DM, Madden MV, Price SK. Gastric carcinoma in young adults. Ann Surg. 1988;208:593‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chung HW, Noh SH, Lim JB. Analysis of demographic characteristics in 3242 young age gastric cancer patients in Korea. World J Gastroenterol. 2010;16:256‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al‐Refaie WB, Hu CY, Pisters PW, Chang GJ. Gastric adenocarcinoma in young patients: a population‐based appraisal. Ann Surg Oncol. 2011;18:2800‐2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takatsu Y, Hiki N, Nunobe S, et al. Clinicopathological features of gastric cancer in young patients. Gastric Cancer. 2016;19:472‐478. [DOI] [PubMed] [Google Scholar]
- 15. Park HJ, Ahn JY, Jung HY, et al. Clinical characteristics and outcomes for gastric cancer patients aged 18‐30 years. Gastric Cancer. 2014;17:649‐660. [DOI] [PubMed] [Google Scholar]
- 16. Hsieh FJ, Wang YC, Hsu JT, Liu KH, Yeh CN. Clinicopathological features and prognostic factors of gastric cancer patients aged 40 years or younger. J Surg Oncol. 2012;105:304‐309. [DOI] [PubMed] [Google Scholar]
- 17. Kong X, Wang JL, Chen HM, Fang JY. Comparison of the clinicopathological characteristics of young and elderly patients with gastric carcinoma: a meta analysis. J Surg Oncol. 2012;106:346‐352. [DOI] [PubMed] [Google Scholar]
- 18. Bautista MC, Jiang SF, Armstrong MA. Impact of age on clinicopathological features and survival of patients with noncardia gastric adenocarcinoma. J Gastric Cancer. 2014;14:238‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cancer Genome Atlas Research Network . Nature. 2014;513:202‐209.25079317 [Google Scholar]
- 20. Cho SY, Park JW, Liu Y, et al. Sporadic early‐onset diffuse gastric cancers have high frequency of somatic CDH1 alterations, but low frequency of somatic RHOA mutations compared with late‐onset cancers. Gastroenterology. 2017;153:536‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Japanese Gastric Cancer Association . Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer. 2011;14:101‐112. [DOI] [PubMed] [Google Scholar]
- 22. Lauren P. The two histological main types of gastric carcinoma. Acta Pathol Microbiol Scand. 1965;64:31‐45. [DOI] [PubMed] [Google Scholar]
- 23. Hofmann M, Stoss O, Shi D, et al. Assessment of a HER2 scoring system for gastric cancer: results from a validation study. Histopathology. 2008;52:797‐805. [DOI] [PubMed] [Google Scholar]
- 24. Nicorici D, Satalan M, Edgren H, et al. FusionCatcher – a tool for finding somatic fusion genes in paired‐end RNA‐sequencing data. bioRxiv. 011650. 10.1101/011650 [DOI] [Google Scholar]
- 25. López‐Basave HN, Morales‐Vásquez F, Ruiz‐Molina JM, et al. Gastric cancer in young people under 30 years of age: worse prognosis, or delay in diagnosis? Cancer Manag Res. 2013;5:31‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449‐456. [DOI] [PubMed] [Google Scholar]
- 27. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro‐oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO‐4538‐12, ATTRACTION‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390:2461‐2471. [DOI] [PubMed] [Google Scholar]
- 29. Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD‐1 inhibition in metastatic gastric cancer. Nat Med. 2018;24:1449‐1458. [DOI] [PubMed] [Google Scholar]
- 30. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non‐small cell lung cancer. Nature. 2018;553:446‐454. [DOI] [PubMed] [Google Scholar]
- 31. Perner S, Wagner PL, Demichelis F, et al. EML4‐ALK fusion lung cancer: a rare acquired event. Neoplasia. 2008;10:298‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tan P, Yeoh KG. Genetics and molecular pathogenesis of gastric adenocarcinoma. Gastroenterology. 2015;149:1153‐1162. [DOI] [PubMed] [Google Scholar]
- 33. Pucułek M, Machlowska J, Wierzbicki R, Baj J, Maciejewski R, Sitarz R. Helicobacter pylori associated factors in the development of gastric cancer with special reference to the early‐onset subtype. Oncotarget. 2018;9:31146‐31162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hampel H, Bennett RL, Buchanan A, et al. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition. Genet Med. 2015;17:70‐87. [DOI] [PubMed] [Google Scholar]
- 36. McLean MH, El‐Omar EM. Genetics of gastric cancer. Nat Rev Gastroenterol Hepatol. 2014;11:664‐674. [DOI] [PubMed] [Google Scholar]
- 37. Bernini M, Barbi S, Roviello F, et al. Family history of gastric cancer: a correlation between epidemiologic findings and clinical data. Gastric Cancer. 2006;9:9‐13. [DOI] [PubMed] [Google Scholar]
- 38. Kawasaki K, Kanemitsu K, Yasuda T, Kamigaki T, Kuroda D, Kuroda Y. Family history of cancer in Japanese gastric cancer patients. Gastric Cancer. 2007;10:173‐175. [DOI] [PubMed] [Google Scholar]
- 39. Murphy G, Pfeiffer R, Camargo MC, Rabkin CS. Meta‐analysis shows that prevalence of Epstein‐Barr virus‐positive gastric cancer differs based on sex and anatomic location. Gastroenterology. 2009;137:824‐833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Choi YY, Bae JM, An JY, et al. Is microsatellite instability a prognostic marker in gastric cancer? A systematic review with meta‐analysis. J Surg Oncol. 2014;110:129‐135. [DOI] [PubMed] [Google Scholar]
- 41. Kim HS, Shin SJ, Beom SH, et al. Comprehensive expression profiles of gastric cancer molecular subtypes by immunohistochemistry: implications for individualized therapy. Oncotarget. 2016;7:44608‐44620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Y, Sethi NS, Hinoue T, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell. 2018;33:721‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tanaka A, Ishikawa S, Ushiku T, et al. Frequent CLDN18‐ARHGAP fusion in highly metastatic diffuse‐type gastric cancer with relatively early onset. Oncotarget. 2018;9:29336‐29350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. 2016;17:564‐580. [DOI] [PubMed] [Google Scholar]
- 45. Günzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013;93:525‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jovov B, Van Itallie CM, Shaheen NJ, et al. Claudin‐18: a dominant tight junction protein in Barrett's esophagus and likely contributor to its acid resistance. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1106‐G1113. [DOI] [PubMed] [Google Scholar]
- 47. Fukata M, Kaibuchi K. Rho‐family GTPases in cadherin‐mediated cell‐cell adhesion. Nat Rev Mol Cell Biol. 2001;2:887‐897. [DOI] [PubMed] [Google Scholar]
- 48. Hanna S, El‐Sibai M. Signaling networks of Rho GTPases in cell motility. Cell Signal. 2013;25:1955‐1961. [DOI] [PubMed] [Google Scholar]
- 49. Yao F, Kausalya JP, Sia YY, et al. Recurrent fusion genes in gastric cancer: CLDN18‐ARHGAP26 induces loss of epithelial integrity. Cell Rep. 2015;12:272‐285. [DOI] [PubMed] [Google Scholar]
- 50. Kataoka K, Ogawa S. Variegated RHOA mutations in human cancers. Exp Hematol. 2016;44:1123‐1129. [DOI] [PubMed] [Google Scholar]