

Abstract
Programmed cell death ligand 1 (PD‐L1) on tumor cells suppresses anti‐tumor immunity and has an unfavorable prognostic impact in ovarian cancer patients. We herein report the pathophysiological and therapeutic impacts of PD‐L1 disruption in ovarian cancer. PD‐L1 was genetically disrupted in the murine ovarian cancer cell line ID8 using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9‐mediated genome editing. PD‐L1 knockout (KO) and control ovarian cancer cells were intraperitoneally inoculated into syngeneic mice, and survival and tumor dissemination were evaluated. Survival times were significantly longer in the PD‐L1‐KO ID8‐inoculated groups than in their control groups, and its therapeutic benefit was enhanced in combination with the cisplatin treatment. Tumor weights and ascites volumes were significantly lower in the PD‐L1‐KO ID8 groups than in their control groups. Immunohistochemical and immunofluorescence analyses showed that intratumoral CD4+ T cells, CD8+ T cells, NK cells and CD11c+ M1 macrophages were significantly increased, whereas regulatory T cells were significantly decreased in the PD‐L1‐KO ID8 groups compared with those in their control groups. The intratumoral mRNA expression of interferon‐γ, tumor‐necrosis factor‐α, interleukin (IL)‐2, IL‐12a, CXCL9 and CXCL10 was significantly stronger, while that of IL‐10, vascular endothelial growth factor, CXCL1 and CXCL2 was significantly weaker in the PD‐L1‐KO ID8 groups. These results indicate that CRISPR/Cas9‐mediated PD‐L1 disruption on tumor cells promotes anti‐tumor immunity by increasing tumor‐infiltrating lymphocytes and modulating cytokine/chemokine profiles within the tumor microenvironment, thereby suppressing ovarian cancer progression. These results suggest that PD‐L1‐targeted therapy by genome editing may be a novel therapeutic strategy for ovarian cancer.
Keywords: CRISPR/Cas9, genome editing, ovarian cancer, PD‐L1, tumor immunity
1. INTRODUCTION
Ovarian cancer remains the leading cause of cancer death among gynecological malignancies and is generally diagnosed at an advanced stage with peritoneal or distal metastasis. Treatments for advanced cases mainly involve debulking surgery and platinum/taxane‐based chemotherapy;1 however, the long‐term prognosis of patients has not satisfactorily improved over the past decade, and the 5‐year survival rate of advanced stage patients is approximately 25%‐30%.2 Therefore, novel therapeutic strategies are urgently needed to further prolong the survival of patients with this disease.
The immune system plays an important role in controlling cancer; however, tumors escape host immune surveillance using multiple mechanisms.3, 4, 5, 6 This acquisition of immune tolerance is essential for cancer growth and progression and may be the cause of resistance against conventional cancer immunotherapy. Thus, considerable attention has been paid to the development of treatment modalities to overcome these immune resistance mechanisms in many types of cancers, including ovarian cancer.7
Programmed cell death ligand 1 (PD‐L1) is a member of the B7 family (also known as CD274) and is expressed in a broad range of malignant tumors, as well as in the human tonsils, placental syncytiotrophoblasts, monocytes and lungs. PD‐L1 generates an inhibitory signal toward the receptor‐mediated activation of T lymphocytes (T cells) by binding to its receptor PD‐1 expressed on the surface of activated T cells.8, 9 Recent studies demonstrated that the PD‐L1 and PD‐1 signal attenuates local tumor immunity, and this immunosuppressive co‐signal (immune checkpoint) may be one of the most important components of the mechanism responsible for tumor immune tolerance.10, 11, 12 Several clinical trials on immune checkpoint inhibitors, such as the anti‐PD‐1 antibody and anti‐PD‐L1 antibody, have achieved significant anti‐tumor effects in patients with certain solid tumors and are already clinically applicable for patients with melanoma and non‐small‐cell lung cancer. These drugs are promising treatments for other solid tumors including ovarian cancer.13
In ovarian cancer, the expression of PD‐L1 on tumors is a poor prognostic factor.14, 15, 16 Furthermore, a recent study demonstrated that PD‐L1 on tumor cells promotes the peritoneal dissemination of ovarian cancer through a dysfunction in cytotoxic T cells in mouse models.15 Clinical trial data on ovarian cancer showed that the response rate of the anti‐PD‐1 or anti‐PD‐L1 antibody was 5.9%‐20.0%.12 The expression of PD‐L1 on tumors correlates with clinical responses to anti‐PD‐1 and anti‐PD‐L1 therapy in some types of cancers17 but not in ovarian cancer.18, 19 Thus, while the expression of PD‐L1 has an unfavorable prognostic impact, the pathophysiological and therapeutic roles of tumor‐derived PD‐L1 in the tumor microenvironment of ovarian cancer are still unclear.
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated protein 9 (Cas9) (CRISPR/Cas9) is a microbial adaptive immune system using RNA‐guide nucleases to cleave foreign genetic elements. This system has recently emerged as a powerful tool for genome engineering in various species and may be used for genome editing.20 The CRISPR/Cas9 system consists of the Cas9 protein and a site‐specific single guide RNA (sgRNA) and may target any genomic site in the form of 20 nucleotides and PAM domains. The Cas9 protein exhibits nuclease activity and induces double‐strand breaks (DSB) in any genomic DNA sequence including PAM domains guided by sgRNA. DSB are mainly ligated through the mutagenic non‐homologous end joining process, leading to the complete disruption of the targeted gene.21
The function of PD‐L1 on tumor cells has been evaluated in several types of cancer through the technique of RNA interference (RNAi).15, 22 However, the knockdown of target gene expression by RNAi is generally incomplete and temporary. Therefore, in the present study, we evaluated the pathophysiological and immunological roles of cancer cell‐derived PD‐L1 in the tumor microenvironment using PD‐L1 knockout (KO) ovarian cancer cells by the CRISPR/Cas9‐mediated genomic editing technique and attempted to clarify the therapeutic impact of PD‐L1 gene disruption on ovarian cancer progression using a mouse model.
2. MATERIAL AND METHODS
2.1. Regents and antibodies
Recombinant murine interferon (IFN)‐γ was purchased from PeproTech (Rocky Hill, NJ, USA). The following monoclonal antibodies (mAb) and polyclonal antibodies (pAb) were used. PE‐conjugated anti‐mouse PD‐L1 mAb (MIH1; BD Biosciences, San Jose, CA, USA) was used for flow cytometry. Goat anti‐mouse PD‐L1 pAb (AF1019; R&D Systems, Minneapolis, MN, USA) was used for western blotting. Rabbit anti‐mouse CD4 mAb (Abcam, Cambridge, MA, USA), rabbit anti‐mouse CD8α pAb (Abcam), rat anti‐mouse CD49b mAb (BD Pharmingen, Piscataway, NJ, USA), rabbit anti‐mouse Foxp3 pAb (BioLegend, San Diego, CA, USA), rat anti‐mouse F4/80 mAb (AbD Serotec, Oxford, UK), and rabbit anti‐mouse PD‐1 mAb (Abcam) were used for immunohistochemistry. Rabbit anti‐CD11c pAb (Bioss, Woburn, MA, USA), rabbit anti‐CD206 pAb (Abcam), rat anti‐F4/80 mAb (BMA Biomedicals, Rheinstrasse, Switzerland), rabbit anti‐CXCL9 pAb (Bioss) and rabbit anti‐CXCL10 pAb (Bioss) were used for double‐color immunofluorescence analyses.
2.2. Cell line and culture
The mouse ovarian carcinoma cell line, ID8, was kindly gifted from Dr Kathy Roby (University of Kansas Medical Center). The cell line was cultured in MEM‐alpha (Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS, penicillin, streptomycin and amphotericin B (Life Technologies).
2.3. Mice
Specific pathogen‐free 8‐week‐old female C57BL/6 (B6) mice were purchased from Japan SLC (Shizuoka, Japan). All animal experiments were approved (approval No. 792) by the Committee on Animal Care and Use at Wakayama Medical University.
2.4. Clustered regularly interspaced short palindromic repeats/Cas9‐mediated genome editing technique
The open‐access software programs, NCBI Gene (https://www.ncbi.nlm.nih.gov/gene) and CRISPR direct (https://crispr.dbcls.jp), were used to design sgRNAs targeted to Cd274 (Pdl1) exon 3 (NC_000085.6) (Figure 1A). The sgRNA targeting PD‐L1 (5′‐GGTCCAGCTCCCGTTCTACA‐3′) was cloned into pSpCas9(BB)‐2A‐GFP (pX458) (no 48138; Addgene, Cambridge, MA, USA). The plasmid vector (pX458‐sgPD‐L1) was sequenced to confirm successful ligation. ID8 was transiently transfected with pX458‐PD‐L1 or the pX458 empty vector using Lipofectamine 3000 (Life Technologies). Enhanced green fluorescent protein (EGFP)‐positive cells were sorted by FACS Aria II (BD Biosciences) and generated clones as single colonies by limiting dilutions. All isolated colonies were evaluated for the expression of PD‐L1 using flow cytometry and western blotting, and gene mutations were confirmed by sequencing for the presence of premature stop codons in all alleles.
Figure 1.

Generation of programmed cell death ligand 1 (PD‐L1) knockout ID8 cell lines using the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system. A, Schematic representation of PD‐L1 targeting guide RNA sequences. Single guide (sg) RNA target sites and the protospacer adjacent motif (PAM) are indicated by blue and red bars, respectively. B, PD‐L1‐KO ID8 cells and their control cells were cultured in vitro and treated with IFN‐γ (20 ng/mL) for 24 h. The expression of PD‐L1 was assessed by flow cytometry. PD‐L1‐KO ID8 cells without the IFN‐γ treatment (blue line histogram), PD‐L1‐KO ID8 cells with the interferon (IFN)‐γ treatment (red line histogram), and the matched isotype control (gray stained histogram). C, PD‐L1‐KO ID8 cells and their control cells were cultured in vitro and treated with or without IFN‐γ (20 ng/mL) for 24 h. PD‐L1 protein expression was analyzed by a western blot analysis. GAPDH was used as the loading control. Complete absence of PD‐L1 expression despite the IFN‐γ treatment in PD‐L1‐KO ID8 cells
In the detection of the genomic DNA of these clones, DNA fragments (998 bp) that included the target lesions were amplified using PCR (forward primer: 5′‐GTGTGGTTTAGTAGTAGAACACTTGGC‐3′, reverse primer: 5′‐GCTGAGTATTATCTGGTACTGAATCTCAAG‐3′) and sequenced to detect the mutation by Sanger sequencing. PCR products were subcloned into a pCR‐Blunt vector (Invitrogen, Carlsbad, CA, USA) and amplified in a bacterial host. Plasmid DNA was sequenced from individual colonies. All experiments with recombinant DNA technology were approved by the DNA Recombination Experiment Committee of Wakayama Medical University (approval no. 28‐12).
2.5. Proliferation assay
Cells (4 × 103/well) were cultured in 96‐well microplates for 24 to 72 hours. Cell viability was assayed using Cell Counting Kit‐8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's protocol.
2.6. Wound healing migration assay
Cells (1 × 105 cells/well) were seeded on 6‐well plates. Confluent monolayers of cells were wounded with a uniform scratch using a 200‐μL pipette tip. The wounds were observed 0, 8 and 24 hours after scratching and photographed at each time point using a 10× objective. The distance between the 2 edges of the wound was measured quantitatively at 10 random sites in each image.
2.7. Tumor formation in vivo
To evaluate the effects of PD‐L1 KO on tumor progression and survival, a mouse ovarian cancer peritoneal dissemination model was used as reported previously.23, 24 ID8 cells (2.5 × 106) were injected into the abdominal cavities of syngeneic mice, and the survival time, disseminated tumor weight and ascites volume were evaluated at the indicated time intervals after the inoculation. Mice were killed before reaching the moribund state. To evaluate the effects of PD‐L1 KO with a combination treatment by chemotherapy, mice were treated with an intraperitoneal injection of cisplatin (5 mg/kg) or physiological saline alone on days 20, 27 and 34 for ID8‐inoculated mice. Survival times were then evaluated.
In addition, to compare the effects of genetic and pharmacological PD‐L1 inhibition, ID8 cells (2.5 × 106) were intraperitoneally injected into the mice, and they were treated with an intraperitoneal injection of 200 μg of anti‐mouse PD‐L1 monoclonal antibody (clone 10F.9G2, BioLegend, San Diego, CA, USA) or isotype‐matched control IgG2bκ (clone RTK4530, BioLegend) on days 16, 23, 30, 37, 44 and 51 according to the protocol for their administration dose and schedule as previously reported.25 The disseminated tumor weight was measured at the indicated time and was compared with those in PD‐L1 KO ID8‐inoculated mice. Mice were killed before reaching the moribund state.
2.8. Flow cytometry
The expression of PD‐L1 was assessed by flow cytometry. Cells were incubated with or without a concentration of 20 ng/mL of recombinant mouse IFN‐γ for 24 hours. Cells were washed with PBS, and dead cells were excluded using the LIVE/DEAD Fixable Dead Cell Stain Kit (Invitrogen). Cells were then incubated with the PE‐conjugated anti‐mouse PD‐L1 antibody or matched isotype control at 4°C for 30 minutes, washed twice, and analyzed using FACS Verse (BD Biosciences). Results were analyzed using FlowJo software (LLC, Ashland, OR, USA).
2.9. Western blotting
Cells were incubated with or without 20 ng/mL of recombinant mouse IFN‐γ for 24 hours. Cells were then lysed in lysis buffer (20 mmol/L Tris‐HCl, pH 7.6, 150 mmol/L NaCl, 1% Triton, 1 mmol/L EDTA) containing Complete Protease Inhibitor Cocktails (Roche, Tokyo, Japan). Cells were centrifuged at 9 700 g for 20 minutes. A total of 7.5 μg of protein was electrophoresed in 10% SDS and transferred onto a nitrocellulose membrane. The membrane was incubated with the primary antibody against PD‐L1 (dilution 1:2000) or GAPDH (dilution 1:5000). After the incubation with the HRP‐conjugated secondary antibody, specific proteins were visualized using chemiluminescence detection (EZ West Lumi; ATTO, Tokyo, Japan).
2.10. Real‐time RT‐PCR
Total RNA was extracted using ISOGEN II (Nippon Gene, Tokyo, Japan). One microgram of total RNA was reverse transcribed into cDNA at 37°C for 15 minutes using the Prime‐Script RT Reagent Kit with gDNA Eraser (Takara Bio, Shiga, Japan). Generated cDNA was then subjected to a real‐time PCR analysis using the SYBR Premix Ex Taq II Kit (Takara Bio) with specific primer sets (Table 1). The amplification and detection of mRNA were performed using the Thermal Cycler Dice Real Time System (Takara Bio) according to the manufacturer's instructions. The relative quantity of target gene expression to the β‐actin gene was measured using the comparative Ct method as described previously.26
Table 1.
Sequences of primers used for real‐time RT‐PCR
| Transcript | Sequence |
|---|---|
| Ifng | (Fw.) 5′‐CGGCACAGTCATTGAAAGCCTA‐3′ |
| (Rv.) 5′‐GTTGCTGATGGCCTGATTGT‐3′ | |
| Tnfa | (Fw.) 5′‐AGCCTGTAGCCCACGTCGT‐3′ |
| (Rv.) 5′‐GGCACCACTAGTTGGTTGTCTTTG‐3′ | |
| Il2 | (Fw.) 5′‐CCCAGGATGCTCACCTTCA‐3′ |
| (Rv.) 5′‐CCGCAGAGGTCCAAGTTCA‐3′ | |
| Il12a | (Fw.) 5′‐TGTCTTAGCCAGTCCCGAAACC‐3′ |
| (Rv.) 5′‐TCTTCATGATCGATGTCTTCAGCAG‐3′ | |
| Il10 | (Fw.) 5′‐GCCAGAGCCACATGCTCCTA‐3′ |
| (Rv.) 5′‐GATAAGGCTTGGCAACCCAAGTAA‐3′ | |
| Vegfa | (Fw.) 5′‐ACATTGGCTCACTTCCAGAAACAC‐3′ |
| (Rv.) 5′‐GGTTGGAACCGGCATCTTTATC‐3′ | |
| Cxcl1 | (Fw.) 5′‐TGCACCCAAACCGAAGTC‐3′ |
| (Rv.) 5′‐GTCAGAAGCCAGCGTTCACC‐3′ | |
| Cxcl2 | (Fw.) 5′‐AAAGTTTGCCTTGACCCTGAA‐3′ |
| (Rv.) 5′‐CTCAGACAGCGAGGCACATC‐3′ | |
| Cxcl9 | (Fw.) 5′‐CCGAGGCACGATCCACTACA‐3′ |
| (Rv.) 5′‐AGTCCGGATCTAGGCAGGTTTG‐3′ | |
| Cxcl10 | (Fw.) 5′‐ATCATCCCTGCGAGCCTATCC‐3′ |
| (Rv.) 5′‐TGTCCATCCATCGCAGCAC‐3′ | |
| Actb | (Fw.) 5′‐CATCCGTAAAGACCTCTATGCCAAC‐3′ |
| (Rv.) 5′‐ATGGAGCCACCGATCCACA‐3′ |
2.11. ELISA
In the intraperitoneal dissemination model, ascites was collected 70 days after the tumor cell injection. Ascites was centrifuged at 9 00 g for 10 minutes, and the supernatant was subjected to ELISA. IFN‐γ, tumor‐necrosis factor‐α (TNF‐α), interleukin (IL)‐10, and vascular endothelial growth factor (VEGF) levels were measured with a commercially available ELISA Kit (R&D Systems) according to the manufacturer's instructions. The detection limits for each method were as follows: IFN‐γ > 9.4 pg/mL; TNF‐α > 10.9 pg/mL; IL‐10 > 15.6 pg/mL; VEGF > 7.8 pg/mL. Total protein in each supernatant was measured with a commercially available kit (BCA Protein Assay Kit; Pierce, MO, USA). Data were expressed as cytokine per protein (pg/mg) for each sample.
2.12. Immunohistochemical analyses
Tumor samples were fixed in 4% paraformaldehyde, and paraffin‐embedded specimens were cut into 4‐μm‐thick sections. Deparaffinized sections were immersed in 3% H2O2 to eliminate endogenous peroxidase activity. Antigen retrieval was performed by enzymatic digestion with trypsin‐EDTA at 37°C for 15 minutes or by boiling tissue sections in 10 mmol/L citrate buffer pH 6.0 or Tris/EDTA buffer pH 9.0. Sections were treated with PBS containing 1% normal serum corresponding to the secondary Abs and 1% BSA to reduce non‐specific reactions and incubated with the primary Abs at 37°C for 1 hour. After the incubation of the biotinylated secondary Abs, immune complexes were visualized using the Labeled Streptavidin Biotin Kit (Dako, Kyoto, Japan) or the Catalyzed Signal Amplification System (Dako). Cell nuclei were counterstained by hematoxylin. The number of CD4+ T cells, CD8+ T cells, NK cells, Treg cells and macrophages at the tumor site were counted on 15 randomly selected visual fields at 400× magnification, and the average of the 5 selected microscopic fields was calculated.
2.13. Double‐color immunofluorescence analyses
A double‐color immunofluorescence analysis was performed as previously reported.24, 27 Anti‐CD11c pAb or anti‐CD206 pAb and a rat anti‐F4/80 mAb were used to investigate the subtypes of macrophages infiltrating tumor tissues. Cy3 (Jackson Immuno Research, West Grove, PA, USA) was used to visualize CD11c‐poitive and CD206‐positive cells. FITC (Jackson Immuno Research) was used to visualize F4/80‐positive cells. DAPI staining was used for the counterstaining of nuclei. Similar immunofluorescence analysis was performed using anti‐CXCL9 pAb and anti‐CXCL10 pAb. Fluorescence immunostaining was observed using a fluorescence microscope, BZ‐X700 (Keyence, Osaka, Japan).
2.14. Statistical analyses
Means and SEM were calculated and presented for all parameters examined in the present study. The significance of differences was evaluated using Student's t test or Dunnett's test. The survival time was analyzed using Kaplan‐Meier curves, and the log‐rank test by JMP Pro (Ver. 13). P < .05 was accepted as being significant.
3. RESULTS
3.1. Clustered regularly interspaced short palindromic repeats/Cas9‐mediated programmed cell death ligand 1 gene editing
We used the CRISPR/Cas9 system to generate a stable ovarian cancer cell line exhibiting the deletion of the PD‐L1 gene. As previously reported,15 the expression of PD‐L1 was strongly induced by the IFN‐γ treatment at the protein level in control ID8 cells confirmed by FACS and western blotting (Figure 1B, C). We successfully established 3 clones in ID8 cells, labeled as PD‐L1‐KO1, PD‐L1‐KO2 and PD‐L1‐KO3, showing the complete absence of PD‐L1 expression from the unedited control despite the IFN‐γ treatment (Figure 1B, C). Plasmid DNA isolated from multiple colonies derived from each PCR product was sequenced. These plasmid DNAs had mutations. In PD‐L1‐KO1 and KO2, both alleles of exon 3 in the PD‐L1 gene had the same single base pair (bp) insertion (Figure 2). PD‐L1‐KO3 had 3 alleles with different 2‐bp deletions in 2 alleles and a single bp insertion in the other allele (Figure 2). These 3 PD‐L1 KO ID8 clones were used in subsequent experiments.
Figure 2.

Sequences of mutant alleles in programmed cell death ligand 1 (PD‐L1) KO ID8 cell clones. PCR products were subcloned and analyzed by DNA sequencing. sgRNA target sites and PAM sequences are labeled by blue and red bases. The number of sequences analyzed for each mutant allele is indicated behind the sequences. Green dashes, deleted bases (−); underlined green bases, insertions (+)
3.2. Effects of the programmed cell death ligand 1 deletion on in vitro cell behavior
No significant differences were observed in proliferative activity (Figure 3A), migratory potential (Figure 3B, C) or cell morphology among the control and 3 PD‐L1‐KO clones.
Figure 3.
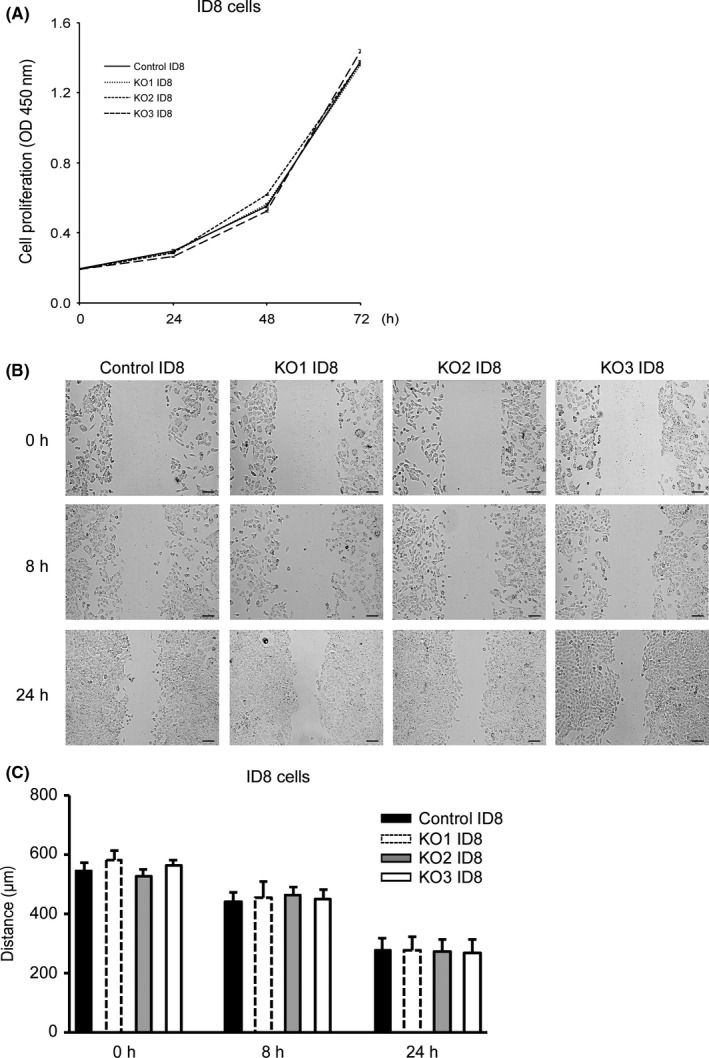
Comparison of the proliferative activity and migratory potential of programmed cell death ligand 1 (PD‐L1) KO ID8 cells and their control cells. A, In vitro cell proliferative activity of PD‐L1‐KO ID8 cells. The mean ± SE of 4 independent experiments is shown. O.D., optical density. Statistical significance determined by Dunnett's test. B, In vitro cell migratory potential of PD‐L1 KO ID8 cells evaluated 0, 8 and 24 h after wounding. Bar = 100 μm. C, Migration distance of PD‐L1‐KO ID8 cells versus their control cells for each time point and condition. All values represent the mean ± SE. Statistical significance determined by Dunnett's test
3.3. Effects of programmed cell death ligand 1 disruption on tumor progression and survival in a mouse peritoneal dissemination model
To evaluate the impact of the disruption of PD‐L1 on tumor progression and survival in vivo, PD‐L1‐KO1, 2, 3 or control ID8 cells were injected intraperitoneally into syngeneic mice. Survival times were significantly longer in the PD‐L1‐KO1, 2 and 3 ID8‐inoculated groups than in their control groups (Figure 4A). In addition, survival times were markedly longer in the PD‐L1‐KO2 ID8‐inoculated group when mice were treated with the intraperitoneal administration of cisplatin than in the control group with the cisplatin treatment (Figure 4B). We then evaluated intraperitoneal tumor dissemination and ascites volumes 70 days after the ID8 injection. Tumor weights and ascites volumes were significantly lower in the PD‐L1‐KO2 ID8‐inoculated group than in the control ID8‐inoculated group (Figure 4C, D). To compare the anti‐tumor effect of genetic PD‐L1 disruption with that of pharmacological inhibition, we treated tumor‐inoculated mice with anti‐PD‐L1 antibody on days 16, 23, 30, 37, 44 and 51 after intraperitoneal injection of control ID8 cells. The tumor weights on 70 days after tumor inoculation were significantly lower in the anti‐PD‐L1 antibody‐treated group compared to those in the control IgG‐treated group, although this tumor suppressive effect was significantly weaker than that in the PD‐L1 KO ID8‐inoculated group (Figure 4E, F). These results suggest that genetic disruption of tumor‐derived PD‐L1 using CRISPR/Cas9‐mediated genome editing may be more powerful for suppressing intraperitoneal tumor progression.
Figure 4.
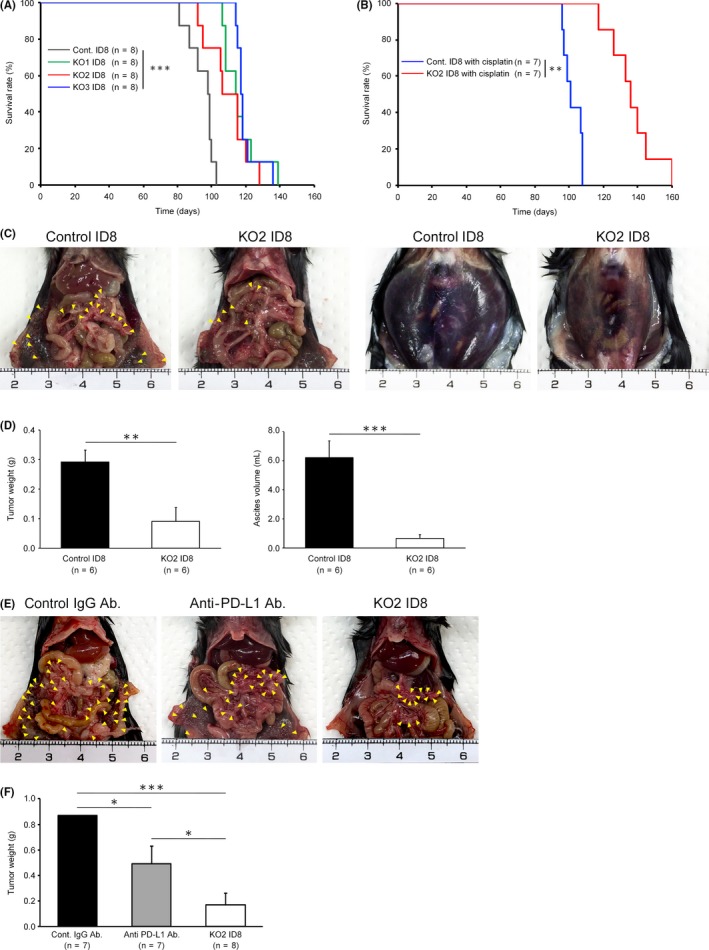
Effects of programmed cell death ligand 1 (PD‐L1) disruption on mouse survival and tumor dissemination. A, Kaplan–Meier curves and the log‐rank test in an overall survival analysis of PD‐L1‐KO1, KO2, KO3 ID8 and control ID8 cell‐inoculated mice (each group, n = 8); ***P < .001. B Kaplan–Meier curves and the log‐rank test in an overall survival analysis of PD‐L1‐KO2 ID8‐inoculated and control ID8‐inoculated mice treated with cisplatin (each group, n = 7); **P < .01. C, Photograph of peritoneal dissemination and ascites from PD‐L1‐KO2 ID8‐inoculated and control ID8‐inoculated mice on day 70. Yellow arrowheads: disseminated tumors on the peritoneum and omentum. Representative results from 6 independent mice in each group are shown. D, The weights of intraperitoneal tumors and ascites volumes were measured in PD‐L1‐KO2 ID8‐inoculated and control ID8 cell‐inoculated mice on day 70. All values represent the mean ± SE (n = 6). Statistical significance determined by Student's t test; **P < .01, ***P < .001. E, Photograph of peritoneal dissemination from control ID8‐inoculated mice treated with the control isotype‐matched IgG antibody, control ID8‐inoculated mice treated with the anti‐PD‐L1 antibody, and the PD‐L1 KO2 ID8‐inoculated mice on day 70. Yellow arrowheads: disseminated tumors on the peritoneum and omentum. F, The weights of intraperitoneal tumors were measured in the control ID8‐inoculated group treated with the control IgG antibody, the control ID8‐inoculated group treated with the anti‐PD‐L1 antibody, and the PD‐L1 KO2 ID8‐inoculated group on day 70. All values represent the mean ± SE (n = 7 to 8). Statistical significance determined by Student's t test; *P < .05, ***P < .001
3.4. Effects of programmed cell death ligand 1 disruption on intratumoral lymphocyte infiltration in a mouse peritoneal dissemination model
To elucidate the mechanism by which the disruption of PD‐L1 suppresses ovarian cancer progression, immunohistochemical analyses were performed, and tumor‐infiltrating lymphocyte profiling was performed. The numbers of intratumoral CD4+ T cells, CD8+ T cells and CD49+ NK cells were significantly higher in the PD‐L1‐KO ID8 groups than in their control groups (Figure 5A, B, C). In contrast, the number of Foxp3+ regulatory T cells was significantly lower in the PD‐L1‐KO ID8‐inoculated groups than in their control groups (Figure 5D). No significant differences were observed in the numbers of F4/80+ macrophages between the PD‐L1‐KO ID8‐inoculated groups and their control groups (Figure 5E). In addition, the number of PD‐1+ immune cells in the tumor microenvironment was significantly higher in the PD‐L1‐KO ID8 groups than in their control groups (Figure 5F).
Figure 5.
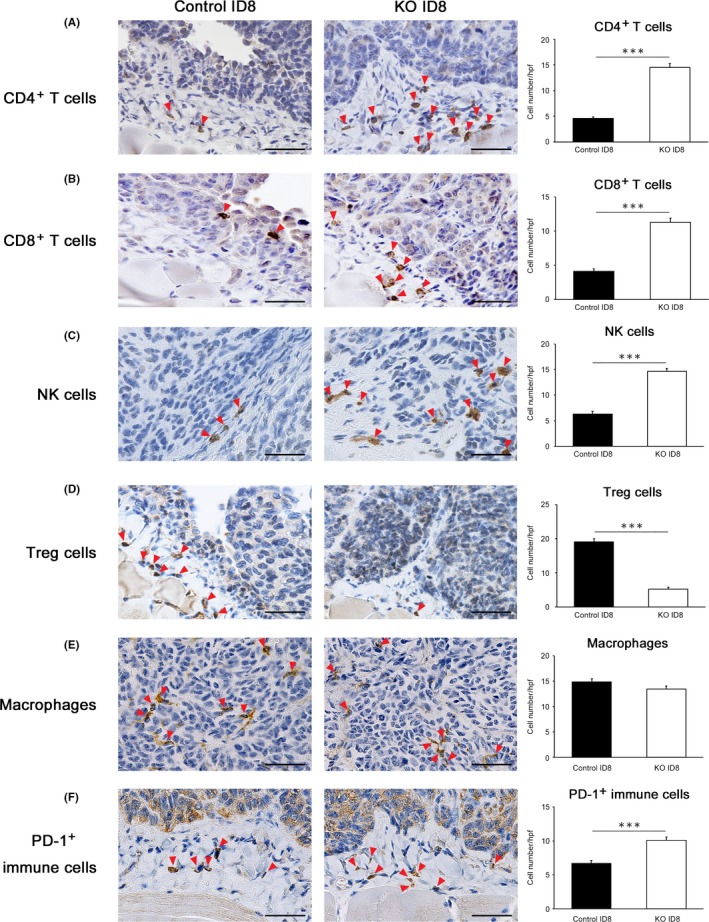
Programmed cell death ligand 1 (PD‐L1) disruption in tumors promotes anti‐tumor immunity by increasing tumor‐infiltrating lymphocytes. A‐F, CD4+ T cells, CD8+ T cells, natural killer (NK) cells (CD49), Treg cells (Foxp3), macrophages (F4/80) and PD‐1+ immune cells in the tumor microenvironment of mice transplanted with PD‐L1 KO ID8 and control ID8 on day 70. Representative results from 6 independent experiments are shown. Red arrowheads indicate positive cells. Original magnification, ×400. Bar = 50 μm. The number of CD4+ T cells, CD8+ T cells, NK cells (CD49), Treg cells (Foxp3), macrophages (F4/80) and PD‐1+ immune cells from peritoneally disseminated tumors of ID8. All values represent the mean ± SE (n = 6 in each group). Statistical significance determined by Student's t test; ***P < .001
3.5. The identification of macrophage subsets in tumor tissues
Double‐color immunofluorescence analyses were conducted to investigate whether the disruption of PD‐L1 affects the subsets of macrophages infiltrating tumor tissues. In the control ID8‐inoculated group, most of the intratumoral F4/80+ macrophages were CD206‐positive M2 macrophages (Figure 6A, C). In contrast, in the PD‐L1‐KO ID8‐inoculated groups, most of the intratumoral F4/80+ macrophages were CD11c‐positive M1 macrophages (Figure 6B, D). In addition, in the PD‐L1‐KO ID8‐inoculated groups, CXCL9 and CXCL10, which are chemokines that recruit CD8+ T cells, were detected on F4/80+ macrophages (Figure 6E, F).
Figure 6.
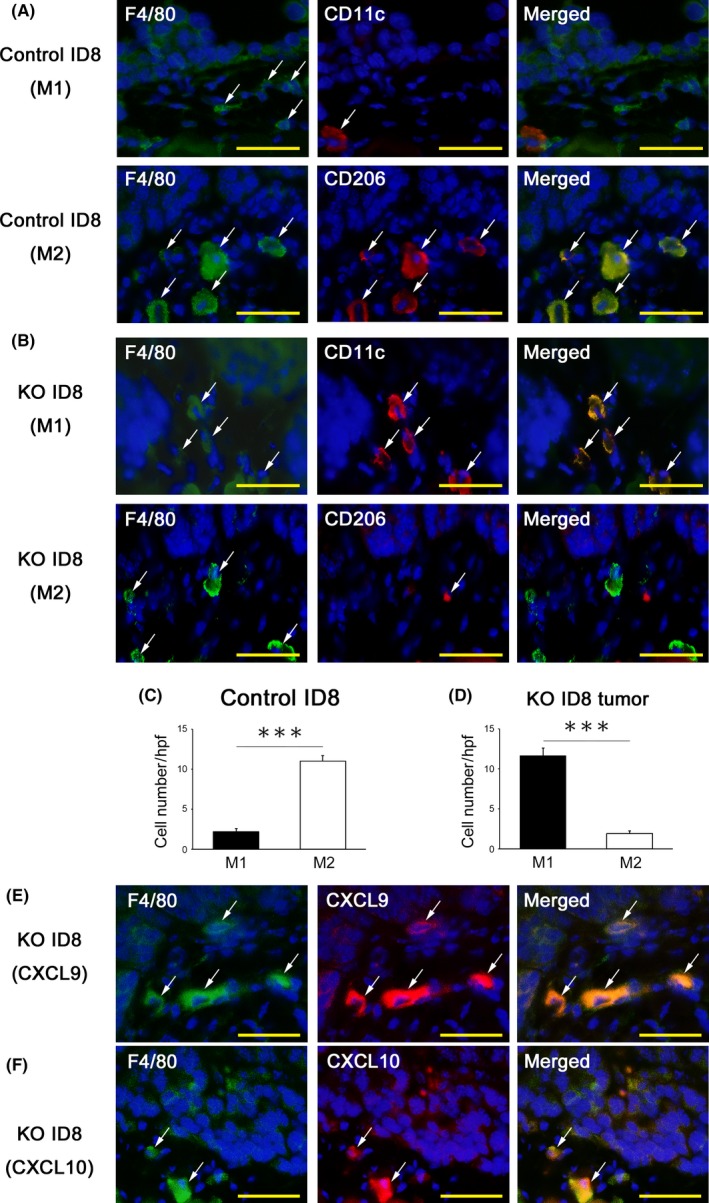
Identification of macrophage subsets and their chemokine expression in tumor tissues. A, B, The identification of macrophage subsets in tumor tissues. A double‐color immunofluorescence analysis was performed using the pair of anti‐F4/80 and CD11c for the M1 marker, and that of anti‐F4/80 and anti‐CD206 for the M2 marker. Fluorescent images were digitally merged. Nuclear staining is blue. Original magnification, ×400. Bar = 50 μm. C, D, The number of macrophage subsets (M1 and M2 macrophages) in the tumor microenvironment of mice transplanted with programmed cell death ligand 1 (PD‐L1) KO ID8 and control ID8. All values represent the mean ± SE (n = 6 in each group). Statistical significance determined by Student's t test; ***P < .001. E, F, Expression of CXCL9 and CXCL10 at disseminated tumors in PD‐L1‐KO ID8‐inoculated mice. A double‐color immunofluorescence analysis was performed using the pair of anti‐F4/80 and CXCL9 or CXCL10. Fluorescent images were digitally merged. Nuclear staining is blue. Original magnification, ×400. Bar = 50 μm
These results showed that the disruption of PD‐L1 on tumor cells induced the recruitment of anti‐tumor immune cells and reciprocally reduced that of immunosuppressive regulatory T cells. Furthermore, the disruption of PD‐L1 increased the number of M1, but not M2 macrophages, and upregulated the production of CXCL9 and CXCL10 by macrophages, resulting in the promoted infiltration of CD8+ killer T cells into the tumor microenvironment. These changes in immune cell profiles may contribute to the suppression of ovarian cancer progression.
3.6. Cytokine and chemokine levels in tumors and ascites of mice transplanted with programmed cell death ligand 1 deletion tumor cells
To investigate the mechanisms underlying PD‐L1 disruption‐mediated tumor suppression in more detail, intratumoral and intraascitic cytokine and chemokine profiles were evaluated. The intratumoral mRNA expression of IFNγ, TNFα, IL‐2, IL‐12a, CXCL9 and CXCL10 was significantly stronger in PD‐L1 KO ID8‐inoculated groups than in their control groups (Figure 7). In contrast, the mRNA expression of IL‐10, VEGF, CXCL1 and CXCL2 was significantly weaker in PD‐L1‐KO ID8‐inoculated groups than in their control groups (Figure 7). Moreover, the levels of IL‐10 and VEGF in ascites were significantly lower in the PD‐L1‐KO ID8‐inoculated groups than in their control groups (Figure 8). These results suggested that the disruption of PD‐L1 on tumor cells upregulates immune‐activating cytokines/chemokines, while downregulating immunosuppressive cytokines and tumor‐angiogenic factors.
Figure 7.
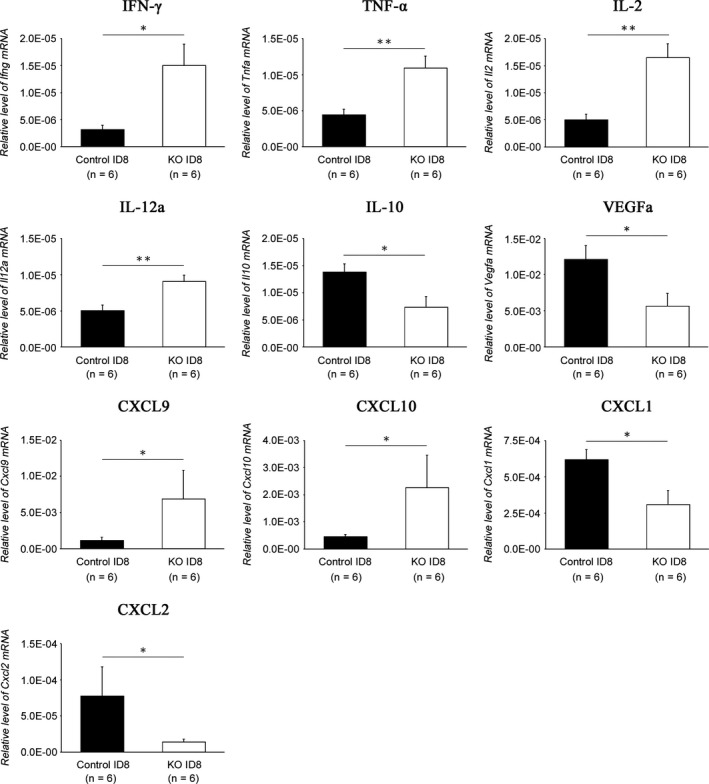
Cytokine and chemokine expression in disseminated tumors. The gene expression of cytokines and chemokines in peritoneally disseminated tumors from programmed cell death ligand 1 (PD‐L1) KO ID8‐inoculated and control ID8‐inoculated mice was examined by real‐time RT‐PCR. All values represent the mean ± SE (n = 6). Statistical significance determined by Student's t test; *P < .05, **P < .01, ***P < .001
Figure 8.

Cytokine levels in ascites from ovarian cancer‐inoculated mice. Cytokine protein levels in ascites from programmed cell death ligand 1 (PD‐L1) KO ID8‐ and control ID8‐inoculated mice were examined by ELISA. All values represent the mean ± SE (n = 6). Statistical significance determined by Student's t test; *P < .05, **P < .01
4. DISCUSSION
In the present study, we established a PD‐L1 KO ovarian cancer cell line using the CRISPR/Cas9 system and demonstrated that the complete disruption of PD‐L1 on tumor cells promoted anti‐tumor immunity by increasing tumor‐infiltrating lymphocytes and modulating cytokine/chemokine production within the tumor microenvironment, thereby suppressing ovarian cancer progression. This is the first study to show the therapeutic impact of PD‐L1 gene disruption in ovarian cancer cells using CRISPR/Cas9 genome editing.
Previous studies reported that PD‐L1 on tumor cells promotes peritoneal dissemination using a mouse ovarian cancer cell line, in which PD‐L1 was suppressed by an RNAi technique.15, 22 However, the knockdown of target gene expression by RNAi is generally incomplete and transient, and the function of PD‐L1 in ovarian cancer was not fully demonstrated in these in vitro or in vivo studies. Juneja et al28 recently reported that PD‐L1 on MC38 mouse colorectal adenocarcinoma cells plays an important role in suppressing antitumor immunity using PD‐L1 KO cells generated by the CRISPR/Cas9‐mediated genomic editing technique. They found that PD‐L1 on MC38 tumor cells reduced the CD8+ T cell/Treg cell ratio and CD8+ T‐cell cytotoxicity. In contrast, PD‐L1 on melanoma cells had no significant effect on antitumor immunity.28 In the present study, we clearly demonstrated that survival times were significantly longer, and tumor weights and ascites were significantly lower in PD‐L1‐KO ID8‐inoculated mice using PD‐L1 disrupted ovarian cancer cells by CRISPR/Cas9‐mediated genome editing. Furthermore, to compare the anti‐tumor effect of genetic PD‐L1 disruption with that of pharmacological PD‐L1 inhibition, control ID8 tumor‐inoculated mice were treated with anti‐PD‐L1 antibody. Our results showed that the tumor‐suppressive effect was significantly weaker in the anti‐PD‐L1 antibody‐treated group compared to those in the PD‐L1 KO ID8‐inoculated group. These findings suggest that complete genetic disruption of tumor‐derived PD‐L1 might be more powerful for suppressing intraperitoneal ovarian cancer progression, although it should be confirmed by further experiments of antibody therapy with another dose or schedule settings.
It is of interest that the survival times were markedly longer in PD‐L1‐KO ID8‐inoculated mice when combined with the cisplatin treatment. These results suggested that PD‐L1 disruption‐mediated anti‐tumor immunity may be promoted by chemotherapy. Although the underlying mechanism has not yet been elucidated in detail, studies demonstrated that immunogenic chemotherapy elicits antitumor immunity by promoting presentation of tumor antigens, increasing infiltration of CD8+ T cells into the tumor, and suppressing recruitment of regulatory T cells.25, 29, 30 These results suggest that the therapeutic effects of targeting PD‐L1 could be enhanced by combination chemotherapy.
Our results demonstrated that CRISPR/Cas9‐mediated PD‐L1 disruption on tumor cells induced the recruitment of tumor‐infiltrating CD4+ T cells, CD8+ T cells, NK cells and M1 macrophages, but reduced immunosuppressive Treg cells and M2 macrophages. In addition, we showed that the disruption of tumor‐derived PD‐L1 promoted intratumoral Th1‐type cytokines, including IFN‐γ, TNF‐α, IL‐2 and IL‐12a, but suppressed the Th2‐type cytokine IL‐10. These changes in immune cell profiles and subsequent changes in cytokine profiles within the tumor microenvironment may contribute to the suppression of ovarian cancer progression. Consistent with our results, the inhibition of the PD‐L1/PD‐1 pathway alters tumor‐infiltrating lymphocyte profiling. Previous studies demonstrated that the suppression or deletion of PD‐L1 on tumor cells increased the number of CD8+ T cells and decreased the number of Foxp3+ Treg cells.15, 28 In addition, recent studies reported that PD‐L1 on tumor cells inhibited the activated PI3K/AKT signaling of NK cells, thereby suppressing NK cell‐mediated anti‐tumor activity.31 Furthermore, PD‐1/PD‐L1 blockade was found to suppress tumor progression by promoting NK cell functions.31
Although PD‐1/PD‐L1 blockade activates T cells, the role for this pathway in tumor‐associated macrophages (TAM) remains unclear. Gordon et al32 showed that PD‐1‐positive TAM increased in a mouse model of colon cancer, and the expression of PD‐1 on TAM negatively correlated with phagocytic potency against tumor cells. They also showed that PD‐L1 KO tumor cell‐inoculated mice showed greater macrophage phagocytosis and less tumor progression than PD‐L1‐overexpressing tumor cell‐inoculated mice.32 However, PD‐L1 KO on tumors did not directly affect the number of PD‐1‐positive TAM. These findings may partly support the present results showing no significant difference in the number of F4/80+ macrophages between PD‐L1 KO ID8‐incoculated and control ID8‐inoculated mice. However, we revealed that CD11c+ M1, but not CD206+ M2 macrophages, increased in PD‐L1 KO ID8‐inoculated mice, suggesting that the disruption of PD‐L1 leads to a change in the macrophage subset from immunosuppressive M2 to immunoactivating M1.
In addition, our result demonstrated that CRISPR/Cas9‐mediated PD‐L1 disruption on tumor cells enhanced the recruitment of PD‐1+ immune cells. Recent studies have reported that PD‐1 positive tumor‐infiltrating lymphocytes are associated with a favorable prognostic factor for survival in ovarian cancer, head and neck cancer, and follicular lymphoma.16, 33, 34 Badoual et al33 showed that most (>95%) PD‐1 positive cells corresponded to CD4+ and CD8+ T cells. Although detailed significance of the higher number of PD‐1+ cells within the tumor microenvironment in the PD‐L1 KO tumors shown in our study remains to be clarified, these cells may contribute to enhanced anti‐tumor immunity.
It is of interest to investigate the impact of the disruption of PD‐L1 on the chemokine profile in the tumor microenvironment. Our results showed that the gene expression of CXCL9 and CXCL10 was enhanced, while that of CXCL1 and CXCL2 was suppressed in PD‐L1 KO ID8‐inoculated mice. Furthermore, the CXCL9 and CXCL10 proteins were exclusively detected in tumor‐infiltrating macrophages. The Th1‐type chemokines CXCL9 and CXCL10 have been shown to mediate the trafficking of CD8+ T cells into the tumor microenvironment.35, 36, 37 Furthermore, a positive correlation has been reported between high CD8+ T cells/CXCL9/CXCL10 and prolonged overall survival in patients with colon cancer.36 In human ovarian cancer and colon cancer, epigenetic silencing, including histone modifications and DNA methylation of the Th1‐type chemokines CXCL9 and CXCL10, caused the poor migration of effector T cells and NK cells into the tumor microenvironment.17, 35, 36 In addition, CXCL9 and CXCL10 production improved the therapeutic efficacy of the PD‐L1 checkpoint blockade in tumor‐bearing mice.17, 35 These findings support our results showing that the disruption of tumor‐derived PD‐L1 upregulated CXCL9 and CXCL10 production from macrophages and promoted the infiltration of CD8+ T cells into the tumor microenvironment. Our results also demonstrated that other chemokines, such as CXCL1 and CXCL2, were suppressed in PD‐L1 KO ID8‐inoculated mice. CXCL1 and CXCL2 are known to promote tumor growth, metastasis and angiogenesis in cancer.38 These chemokines are produced by M2 macrophages,39 and previous studies demonstrated that CXCL1 and CXCL2 were increased in ovarian cancer.38, 40 This mechanism may support tumor progression being suppressed in our study as a result of an altered macrophage subset from M2 macrophages producing CXCL1 and CXCL2 to M1 macrophages.
The clinical application of CRISPR/Cas9‐mediated genome editing targeting tumor cells themselves for cancer gene therapy is still limited because its delivery system into tumors in vivo is challenging. Gene delivery vectors based on adeno‐associated virus (AAV) have been utilized in cancer gene therapy because of their safety and efficacy.41, 42 Recently, some gene therapy models using the AAV of the CRISPR/Cas9 system were reported.43, 44 The administration of the AAV of CRISPR/Cas9 coupled with sgRNA to the mdx mouse model of Duchenne muscular dystrophy (DMD) with the mutated Dmd gene resulted in the excision of intervening DNA and restored the Dystrophin reading frame in myofibers, cardiomyocytes and muscle stem cells. An AAV‐CRISPR/Cas9‐mediated treatment partially recovered muscle functional deficiencies.43 Further studies are needed to create an AAV‐CRISPR/Cas9 vector targeting tumor‐cell derived PD‐L1 and to clarify its therapeutic efficacy in a preclinical mouse ovarian cancer peritoneal dissemination model for subsequent clinical trials.
In conclusion, we herein demonstrated that CRISPR/Cas9‐mediated PD‐L1 disruption on tumor cells promoted anti‐tumor immunity by increasing tumor‐infiltrating lymphocytes as well as modulating cytokine and chemokine production, and suppressed ovarian cancer progression. These results suggest that PD‐L1‐targeted therapy by genome editing may be a novel therapeutic strategy for ovarian cancer.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the Japan Society for the Promotion of Science Grants‐in‐Aid for the Scientific Research Grant JP17K16863 and the 2016 Wakayama Medical Award for Young Researchers from Wakayama Medical University, Japan.
Yahata T, Mizoguchi M, Kimura A, et al. Programmed cell death ligand 1 disruption by clustered regularly interspaced short palindromic repeats/Cas9‐genome editing promotes antitumor immunity and suppresses ovarian cancer progression. Cancer Sci. 2019;110:1279–1292. 10.1111/cas.13958
REFERENCES
- 1. Hardwick N, Frankel PH, Cristea M. New approaches for immune directed treatment for ovarian cancer. Curr Treat Options Oncol. 2016;17:14. [DOI] [PubMed] [Google Scholar]
- 2. American Cancer Society . Cancer Facts & Figures. Atlanta: American Cancer Society; 2016:17‐19. [Google Scholar]
- 3. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263‐274. [DOI] [PubMed] [Google Scholar]
- 4. Gajewski TF, Meng Y, Blank C, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131‐145. [DOI] [PubMed] [Google Scholar]
- 5. Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715‐727. [DOI] [PubMed] [Google Scholar]
- 6. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991‐998. [DOI] [PubMed] [Google Scholar]
- 7. Yigit R, Massuger LF, Figdor CG, Torensma R. Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol Oncol. 2010;117:366‐372. [DOI] [PubMed] [Google Scholar]
- 8. Dong H, Zhu G, Tamada K, Chen L. B7‐H1, a third member of the B7 family, co‐stimulates T‐cell proliferation and interleukin‐10 secretion. Nat Med. 1999;5:1365‐1369. [DOI] [PubMed] [Google Scholar]
- 9. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death‐1 ligand 1 interacts specifically with the B7‐1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Anti‐PD‐L1/PD‐1 immune therapies in ovarian cancer: basic mechanism and future clinical application. Int J Clin Oncol. 2016;21:456‐461. [DOI] [PubMed] [Google Scholar]
- 12. Hamanishi J, Mandai M, Konishi I. Immune checkpoint inhibition in ovarian cancer. Int Immunol. 2016;28:339‐348. [DOI] [PubMed] [Google Scholar]
- 13. Gaillard SL, Secord AA, Monk B. The role of immune checkpoint inhibition in the treatment of ovarian cancer. Gynecol Oncol Res Pract. 2016;3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamanishi J, Mandai M, Iwasaki M, et al. Programmed cell death 1 ligand 1 and tumor‐infiltrating CD8 + T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA. 2007;104:3360‐3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abiko K, Mandai M, Hamanishi J, et al. PD‐L1 on tumor cells is induced in ascites and promotes peritoneal dissemination of ovarian cancer through CTL dysfunction. Clin Cancer Res. 2013;19:1363‐1374. [DOI] [PubMed] [Google Scholar]
- 16. Darb‐Esfahani S, Kunze CA, Kulbe H, et al. Prognostic impact of programmed cell death‐1 (PD‐1) and PD‐ligand 1 (PD‐L1) expression in cancer cells and tumor‐infiltrating lymphocytes in ovarian high grade serous carcinoma. Oncotarget. 2016;7:1486‐1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zou W, Wolchok JD, Chen L. PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hamanishi J, Mandai M, Ikeda T, et al. Safety and antitumor activity of anti‐PD‐1 antibody, nivolumab, in patients with platinum‐resistant ovarian cancer. J Clin Oncol. 2015;33:4015‐4022. [DOI] [PubMed] [Google Scholar]
- 19. Disis ML, Patel MR, Pant S, et al. Avelumab (MSB0010718C), an anti‐PD‐L1 antibody, in patients with previously treated, recurrent or refractory ovarian cancer: a phase Ib, open‐label expansion trial. J Clin Oncol 2015;33:5509. [Google Scholar]
- 20. Valletta S, Dolatshad H, Bartenstein M, et al. ASXL1 mutation correction by CRISPR/Cas9 restores gene function in leukemia cells and increases survival in mouse xenografts. Oncotarget. 2015;6:44061‐44071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doudna JA, Charpentier E. Genome editing: the new frontier of genome engineering with CRISPR‐Cas9. Science 2014;346:1258096. [DOI] [PubMed] [Google Scholar]
- 22. Liu Y, Zeng B, Zhang Z, Zhang Y, Yang R. B7‐H1 on myeloid‐derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin Immunol. 2008;129:471‐481. [DOI] [PubMed] [Google Scholar]
- 23. Tanizaki Y, Kobayashi A, Toujima S, et al. Indoleamine 2,3‐dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci. 2014;105:966‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kobayashi A, Tanizaki Y, Kimura A, et al. AG490, a Jak2 inhibitor, suppressed the progression of murine ovarian cancer. Eur J Pharmacol. 2015;766:63‐75. [DOI] [PubMed] [Google Scholar]
- 25. Peng J, Hamanishi J, Matsumura N, et al. Chemotherapy induces programmed cell death‐ligand 1 overexpression via the nuclear factor‐κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015;75:5034‐5045. [DOI] [PubMed] [Google Scholar]
- 26. Caraguel CG, Stryhn H, Gagné N, Dohoo IR, Hammell KL. Selection of a cutoff value for real‐time polymerase chain reaction results to fit a diagnostic purpose: analytical and epidemiologic approaches. J Vet Diagn Invest. 2011;23:2‐15. [DOI] [PubMed] [Google Scholar]
- 27. Ishida Y, Kimura A, Kuninaka Y, et al. Pivotal role of the CCL5/CCR5 interaction for recruitment of endothelial progenitor cells in mouse wound healing. J Clin Invest. 2012;122:711‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Juneja VR, McGuire KA, Manguso RT, et al. PD‐L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214:895‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang P, Ma Y, Lv C, et al. Upregulation of programmed cell death ligand 1 promotes resistance response in non‐small‐cell lung cancer patients treated with neo‐adjuvant chemotherapy. Cancer Sci. 2016;107:1563‐1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu S, Tao Z, Hai B, et al. miR‐424(322) reverses chemoresistance via T‐cell immune response activation by blocking the PD‐L1 immune checkpoint. Nat Commun. 2016;7:11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Y, Cheng Y, Xu Y, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK‐cell‐mediated anti‐tumor function and indicates poor prognosis in digestive cancers. Oncogene. 2017;36:6143‐6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gordon SR, Maute RL, Dulken BW, et al. PD‐1 expression by tumour‐associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Badoual C, Hans S, Merillon N, et al. PD‐1‐expressing tumor‐infiltrating T cells are a favorable prognostic biomarker in HPV‐associated head and neck cancer. Cancer Res. 2013;73:128‐138. [DOI] [PubMed] [Google Scholar]
- 34. Carreras J, Lopez‐Guillermo A, Roncador G, et al. High numbers of tumor‐infiltrating programmed cell death 1‐positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J Clin Oncol. 2009;27:1470‐1476. [DOI] [PubMed] [Google Scholar]
- 35. Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1‐type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nagarsheth N, Peng D, Kryczek I, et al. PRC2 epigenetically silences Th1‐type chemokines to suppress effector T‐cell trafficking in colon cancer. Cancer Res. 2016;76:275‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Devaud C, John LB, Westwood JA, Darcy PK, Kershaw MH. Immune modulation of the tumor microenvironment for enhancing cancer immunotherapy. Oncoimmunology. 2013;2:e25961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kavandi L, Collier MA, Nguyen H, Syed V. Progesterone and calcitriol attenuate inflammatory cytokines CXCL1 and CXCL2 in ovarian and endometrial cancer cells. J Cell Biochem. 2012;113:3143‐3152. [DOI] [PubMed] [Google Scholar]
- 39. He C, Carter AB. The metabolic prospective and redox regulation of macrophage polarization. J Clin Cell Immunol. 2015;6:1279‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bolitho C, Hahn MA, Baxter RC, Marsh DJ. The chemokine CXCL1 induces proliferation in epithelial ovarian cancer cells by transactivation of the epidermal growth factor receptor. Endocr Relat Cancer. 2010;17:929‐940. [DOI] [PubMed] [Google Scholar]
- 41. Li C, Bowles DE, van Dyke T, Samulski RJ. Adeno‐associated virus vectors: potential applications for cancer gene therapy. Cancer Gene Ther. 2005;12:913‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Santiago‐Ortiz JL, Schaffer DV. Adeno‐associated virus (AAV) vectors in cancer gene therapy. J Control Release. 2016;240:287‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tabebordbar M, Zhu K, Cheng JKW, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ohmori T, Nagao Y, Mizukami H, et al. CRISPR/Cas9‐mediated genome editing via postnatal administration of AAV vector cures haemophilia B mice. Sci Rep. 2017;7:4159. [DOI] [PMC free article] [PubMed] [Google Scholar]