

Abstract
Capmatinib is a highly specific, potent and selective MET inhibitor. This was an open‐label, multicenter, dose‐escalation, phase I study conducted in Japanese patients with advanced solid tumors (not selected based on their MET status). The primary objective was to determine the maximum tolerated dose (MTD) and/or highest studied dose being safe. Secondary objectives included safety, pharmacokinetics and preliminary antitumor activity. Dose escalation was guided by a Bayesian Logistic Regression Model dependent on dose‐limiting toxicities (DLT) in cycle 1. Of 44 adult Japanese patients with confirmed advanced solid tumors enrolled, 29 received capmatinib capsules (doses ranging from 100 mg once daily [q.d.] to 600 mg twice daily [b.i.d.]) and 15 received tablets (200 mg b.i.d. and 400 mg b.i.d.). DLT occurred in two patients: grade 2 suicidal ideation (600 mg b.i.d. capsule) and grade 3 depression (400 mg b.i.d. tablet). MTD was not reached. The highest studied dose determined to be safe as tablet was 400 mg b.i.d., whereas it is not yet determined for capsules. Most common adverse events suspected to be drug‐related were increased blood creatinine, nausea, decreased appetite, vomiting and diarrhea. Following repeated daily dosing up to day 15 by q.d. or b.i.d. regimen using capsules, median time to reach maximum plasma drug concentration (T max) was 1.0‐4.0 hours; absorption was more rapid after dosing using tablets, with median T max of 1.0 hour on both days 1 and 15. Eight patients had a best overall response of stable disease. These data support further clinical development of capmatinib.
Keywords: capmatinib, INC280, Japan, MET, phase 1
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- AUC
area under the curve
- AUCinf
AUC from time zero to infinity
- AUClast
AUC from time zero to the last quantifiable concentration point
- AUCtau
AUC calculated to the end of the dosing interval
- BLRM
Bayesian Logistic Regression Model
- CI
confidence interval
- Cmax
maximum plasma drug concentration
- CNS
central nervous system
- CTCAE
Common Terminology Criteria for Adverse Events
- DDS
dose‐determining set
- DLT
dose‐limiting toxicity
- DOR
duration of response
- ECG
electrocardiogram
- ECOG PS
Eastern Cooperative Oncology Group performance status
- EGFR
epidermal growth factor receptor
- EWOC
escalation with overdose control
- FAS
full analysis set
- FISH
fluorescence in situ hybridization
- GCN
gene copy number
- GGT
γ‐glutamyltransferase
- HGF
hepatocyte growth factor
- IHC
immunohistochemistry
- IC50
half maximal inhibitory concentration
- MTD
maximum tolerated dose
- NGS
next‐generation sequencing
- NSCLC
non‐small‐cell lung cancer
- MAPK
mitogen‐activated protein kinase
- ORR
overall response rate
- PAS
PK analysis set
- PK
pharmacokinetics
- RECIST
Response Evaluation Criteria in Solid Tumors
- RP2D
recommended phase II dose
- SD
stable disease
- T1/2
terminal‐phase half‐life
- TKI
tyrosine kinase inhibitor
- Tmax
time to reach maximum plasma drug concentration
1. INTRODUCTION
MET, the proto‐oncogene, is a member of a distinct subfamily of heterodimeric receptor tyrosine kinases1, 2 and encodes the high‐affinity receptor for hepatocyte growth factor (HGF), which is the only known ligand for this receptor.1 Dysregulation of MET signaling results in activation of downstream pathways, including the RAS/MAPK, PI3K/AkT, and Rac/Rho pathways, which promote cell proliferation, survival and metastasis.3 Aberrant activation of the MET pathway may be a result of high‐level MET gene amplification, protein overexpression or gene mutations, and is associated with resistance to chemotherapy and radiotherapy and poor clinical outcomes in cancer patients.4, 5, 6 MET gene amplification typically occurs in approximately 3%‐5% of newly diagnosed patients with NSCLC7, 8 and is implicated in acquired resistance to EGFR TKI, reported in 5%‐22% of cases with EGFR‐inhibitor resistance, regardless of the presence of EGFR T790M mutation.4, 9, 10, 11 Overexpression of MET and/or elevated levels of HGF have been described in multiple tumor types, including lung, breast, colon and gastric cancers.3 MET protein overexpression has been reported in 20%‐37% of tumor tissues, normally through transcriptional upregulation,12, 13 whereas MET gene mutation in the splice site leading to exon 14 skipping14, 15, 16 has been reported in 2%‐4% of NSCLC adenocarcinoma and in 1%‐2% of other NSCLC subsets.17, 18 In addition, MET mutations have been identified in primary tumors as well as metastatic lesions of other cancers, including the head and neck, renal, liver and ovarian cancers.19, 20
Capmatinib (INC280) is a highly specific, potent and selective MET inhibitor in biochemical (IC50, 0.13 nM) and cellular assays across a range of tumor types including NSCLC, which also causes regression of MET‐dependent (amplified/autocrine) tumor models in animals at tolerable doses. Capmatinib has been demonstrated to be highly selective for MET kinase versus other kinases (10 000‐fold selectivity in panel of 57 human kinases) in large panels of biochemical and binding assays.21 Accordingly, a phase I, global, dose‐escalation study of capmatinib was conducted in patients with MET‐dependent solid tumors. The capsule formulation at 600 mg b.i.d. was initially selected as the RP2D.22 A tablet formulation was subsequently developed to improve patient compliance, as this allowed for higher dosage strengths and therefore fewer tablets compared with capsules. The protocol was amended accordingly to add tablet formulation based on the results of a bioavailability study. Thus, the RP2D of capmatinib in tablet formulation was stated as 400 mg tablet b.i.d. in the global study.23
This phase I, multicenter, open‐label, dose‐escalation study (ClinicalTrials.gov Identifier: NCT01546428) was conducted in Japan to determine the MTD and/or the highest studied dose determined to be safe, safety, PK and preliminary antitumor activity of capmatinib in patients with advanced solid tumors (not selected based on their MET dysregulation status) who had progressed despite standard therapy or for whom no effective therapy exists.
2. MATERIALS AND METHODS
2.1. Study population
Adult Japanese patients with histologically or cytologically confirmed advanced solid tumors who were refractory to standard therapies or had tumors for which no effective treatment was available were eligible. Patients were not selected based on the MET dysregulation status of their tumor. Other key inclusion criteria were an Eastern Cooperative Oncology Group performance status (ECOG PS) of 2 or less and adequate organ function and laboratory test results (required hematology and blood laboratory tests including neutrophil, platelet counts, and hemoglobin, creatinine, total bilirubin, AST, ALT, lipase and fasting serum triglyceride).
The key exclusion criteria were patients with symptomatic CNS metastases that were neurologically unstable or required increasing doses of steroids to control and patients with any CNS deficits; patients who had received previous or current treatment with a MET inhibitor or HGF‐targeting therapy; patients with significant cardiovascular disease or gastrointestinal dysfunction that might have impaired capmatinib absorption; patients who had undergone a bone marrow or solid organ transplant; previous cytotoxic chemotherapy, radiotherapy, biologic or investigational therapy, or major surgery 4 weeks or less prior to study treatment; and treatment with proton pump inhibitors within 3 days prior to study treatment.
This study was conducted in accordance with the principles of the Declaration of Helsinki and the Good Clinical Practice Guidelines of the International Conference on Harmonisation. The protocol and all the amendments were approved by the local institutional review board for each site that participated in the study and written consent was obtained from all patients before screening.
2.2. Study design and treatment plan
This was an open‐label, multicenter, dose‐escalation, phase I study conducted across two centers in Japan. Patients received at least one dose of capmatinib daily in a 28‐day cycle until disease progression, intolerable toxicity, withdrawal of consent or discontinuation for any other reason. Patients in subsequent cohorts received escalating doses of capmatinib (capsule or tablet formulation) until the MTD and/or highest studied dose determined to be safe was determined.
Dose escalation was guided by a 2‐parameter BLRM employing the EWOC principle.24, 25 The BLRM estimated the MTD by updating estimates of the probability of observing a DLT in the first cycle. A DLT was defined as a significant AE or abnormal laboratory parameter adjudged to be CTCAE, version 4.0, grade 3 or 4 in severity and considered unrelated to disease progression, intercurrent disease or concomitant medications, occurring 28 days or less following the first administration of the study treatment (cycle 1). For a given schedule, the MTD was defined as the highest drug dosage not expected to lead to DLT in more than 33% of patients in the first 28 days of capmatinib treatment under that schedule.
The primary objective was to determine the MTD and/or highest studied dose of single agent oral capmatinib determined to be safe. Secondary objectives included safety and tolerability, PK and preliminary antitumor activity of capmatinib. The exploratory objectives were to assess the effects on pharmacodynamic biomarkers for MET inhibition in tumor, to characterize the effects of capmatinib on circulating biomarkers relating to MET pathway, and to investigate the potential surrogate marker for capmatinib on the target and capmatinib drug action and/or to identify potential biomarkers that may correlate with efficacy, safety or resistance.
The expansion part was planned to assess the safety and explore the efficacy in additional patients with MET dysregulation; however, the expansion was not initiated as sufficient data for further development were already available from other clinical studies of capmatinib.22, 23
2.3. Study assessments
2.3.1. Safety analysis
Safety was assessed according to the CTCAE, version 4.0. Other safety data included regular laboratory evaluations, physical examinations, vital signs, weight and periodic ECG recordings. In order to be considered a DLT by the protocol, the toxicity must have occurred during the first 28‐day cycle of capmatinib treatment.
2.3.2. Pharmacokinetics
Blood samples (3 mL) were collected at each time point on day 1 and day 15 of cycle 1. Capmatinib concentrations in plasma were measured using a validated liquid chromatography‐mass spectrometry/mass spectrometry method with a lower limit of quantification of 1 ng/mL. The PK variables were classified into primary and secondary PK parameters. Primary PK parameters included AUC (AUClast, AUCinf, AUCtau), C max and T max. The secondary PK parameters were terminal elimination rate constant, T 1/2, apparent total body clearance of drug from the plasma, apparent volume of distribution during the terminal elimination phase, apparent volume of distribution during the terminal elimination phase at steady state and accumulation ratio. PK parameters were estimated using a non‐compartmental method with Phoenix WinNonlin (version 6.4; Pharsight, Mountain View, CA, USA). Dose proportionality of capmatinib was assessed by formulation and regimen by using a power model on a log‐transformed scale, that is, log (parameter) = log (α) + β × log (dose) + error. The results based on the power model were also presented graphically by PK parameters using the estimates of α and β.
2.3.3. Efficacy analysis
Computed tomography or magnetic resonance imaging‐based tumor assessments were performed according to RECIST version 1.1.
2.3.4. Biomarker analysis
Tumor tissues and blood samples were collected from all patients at different time points. Multiple biomarkers relevant to the mode of action of capmatinib such as MET mutation or amplification or overexpression, p‐c‐MET, p‐ERK, p‐AKT, p‐S6 and HGF levels were analyzed.
2.3.5. Statistical analyses
An adaptive BLRM guided by the EWOC principle was used in the dose‐escalation part for dose level selection and determination of MTD and/or the highest studied dose that was safe. In this study, four analysis sets were considered, FAS (all patients who received at least one dose of capmatinib), safety set (all patients who received at least one dose of capmatinib and had at least one valid postbaseline safety assessment), DDS (all patients from the safety set who either met the minimum exposure criterion [full planned daily dose of capmatinib administrated for ≥21 days out of 28 days] with sufficient safety evaluations [as determined by the investigators and Novartis] or who had experienced a DLT during the first cycle) and PAS (all patients who had at least one plasma sample providing evaluable PK). Patients who did not experience DLT during the first cycle were considered to have sufficient safety evaluations if they had been observed for 28 days or more following the first dose. Data were summarized using descriptive statistics (continuous data) and/or contingency tables (categorical data).
3. RESULTS
3.1. Patients
Between 10 February 2012 and 22 January 2016, 44 patients with advanced solid tumors who were not selected based on their MET dysregulation status were enrolled in this study. All patients were included in the FAS, safety set and PAS; however, five patients who did not meet the minimum exposure criterion were excluded from the DDS, which then consisted of 39 patients. Twenty‐nine patients (65.9%) received capsule formulation at eight dose levels (100 mg q.d., n = 3; 200 mg q.d., n = 4; 400 mg q.d., n = 3; 500 mg q.d., n = 4; 600 mg q.d., n = 4; 800 mg q.d., n = 4; 400 mg b.i.d., n = 4; and 600 mg b.i.d., n = 3) and 15 patients (34.1%) received tablet formulation at two dose levels (200 mg b.i.d., n = 3; and 400 mg b.i.d., n = 12). Twenty‐seven patients (61.4%) were male. The median age of patients was 62.0 years (range, 26‐74). A majority of the patients had an ECOG PS of 1 (56.8%). Overall, lung cancer was the most frequently reported primary site (15 patients [34.1%]), followed by stomach cancer (five patients [11.4%]). The most frequent histological type reported for all types of cancers was adenocarcinoma (20 patients [45.5%]). All patients received prior antineoplastic medication therapy, whereas 21 patients (47.7%) received prior radiotherapy (Table 1).
Table 1.
Baseline patient and disease characteristics
| Characteristic | Capmatinib capsule | Capmatinib tablet | All n = 44 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 mg q.d. n = 3 | 200 mg q.d. n = 4 | 400 mg q.d. n = 3 | 500 mg q.d. n = 4 | 600 mg q.d. n = 4 | 800 mg q.d. n = 4 | 400 mg b.i.d. n = 4 | 600 mg b.i.d. n = 3 | 200 mg b.i.d. n = 3 | 400 mg b.i.d. n = 12 | ||
| Age (median, y) | 74.0 | 57.0 | 65.0 | 60.5 | 60.0 | 62.5 | 61.0 | 64.0 | 51.0 | 66.0 | 62.0 |
| Age category (y), n (%) | |||||||||||
| <65 | 1 (33.3) | 4 (100) | 1 (33.3) | 3 (75.0) | 4 (100) | 3 (75.0) | 3 (75.0) | 2 (66.7) | 2 (66.7) | 5 (41.7) | 28 (63.6) |
| ≥65 | 2 (66.7) | 0 | 2 (66.7) | 1 (25.0) | 0 | 1 (25.0) | 1 (25.0) | 1 (33.3) | 1 (33.3) | 7 (58.3) | 16 (36.4) |
| Sex (male), n (%) | 1 (33.3) | 2 (50.0) | 2 (66.7) | 2 (50.0) | 2 (50.0) | 3 (75.0) | 3 (75.0) | 1 (33.3) | 3 (100) | 8 (66.7) | 27 (61.4) |
| ECOG PS, n (%) | |||||||||||
| 0 | 0 | 3 (75.0) | 2 (66.7) | 2 (50.0) | 2 (50.0) | 2 (50.0) | 1 (25.0) | 1 (33.3) | 2 (66.7) | 4 (33.3) | 19 (43.2) |
| 1 | 3 (100) | 1 (25.0) | 1 (33.3) | 2 (50.0) | 2 (50.0) | 2 (50.0) | 3 (75.0) | 2 (66.7) | 1 (33.3) | 8 (66.7) | 25 (56.8) |
| Key primary site of cancer | |||||||||||
| Lung | 1 (33.3) | 2 (50.0) | 1 (33.3) | 2 (50.0) | 0 | 2 (50.0) | 2 (50.0) | 0 | 0 | 5 (41.7) | 15 (34.1) |
| Stomach | 1 (33.3) | 0 | 1 (33.3) | 0 | 0 | 1 (25.0) | 0 | 0 | 0 | 2 (16.7) | 5 (11.4) |
| Esophagus | 0 | 0 | 0 | 0 | 0 | 1 (25.0) | 1 (25.0) | 2 (66.7) | 0 | 0 | 4 (9.1) |
| Pleura | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33.3) | 2 (16.7) | 3 (6.8) |
| Colon | 1 (33.3) | 1 (25.0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (4.5) |
| Rectum | 0 | 0 | 1 (33.3) | 0 | 1 (25.0) | 0 | 0 | 0 | 0 | 0 | 2 (4.5) |
| Time from most recent relapse/progression to first study drug (median, mo) | 1.40 | 2.25 | 1.50 | 1.35 | 1.65 | 0.90 | 1.40 | 3.00 | 0.40 | 1.05 | 1.40 |
| No. of prior antineoplastic medication regimens, n (%) | |||||||||||
| 1 | 0 | 1 (25.0) | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 1 (33.3) | 2 (16.7) | 5 (11.4) |
| 2 | 0 | 0 | 0 | 2 (50.0) | 2 (50.0) | 1 (25.0) | 0 | 2 (66.7) | 1 (33.3) | 1 (8.3) | 9 (20.5) |
| 3 | 1 (33.3) | 0 | 0 | 1 (25.0) | 0 | 1 (25.0) | 0 | 1 (33.3) | 1 (33.3) | 6 (50.0) | 11 (25.0) |
| 4 | 1 (33.3) | 1 (25.0) | 1 (33.3) | 0 | 1 (25.0) | 0 | 1 (25.0) | 0 | 0 | 2 (16.7) | 7 (15.9) |
| 5 | 0 | 0 | 1 (33.3) | 0 | 1 (25.0) | 0 | 2 (50.0) | 0 | 0 | 0 | 4 (9.1) |
| ≥6 | 1 (33.3) | 2 (50.0) | 0 | 1 (25.0) | 0 | 2 (50.0) | 1 (25.0) | 0 | 0 | 1 (8.3) | 8 (18.2) |
ECOG PS, Eastern Cooperative Oncology Group performance status.
All patients discontinued capmatinib treatment. The primary reason for discontinuation was disease progression, occurring in 38 patients (86.4%), followed by AE in four patients (9.1%) and loss to follow‐up and subject/guardian decision in one patient (2.3%) each. No on‐treatment deaths (within 28 days after discontinuation of the study treatment) were reported.
3.2. Safety
The median duration of treatment exposure was 7 weeks (range, 0.4‐32.3). The majority of the patients (72.7%) were in the relative dose intensity category of 90% to <110%. DLT were observed in two patients (5.1%): one patient treated with 600 mg b.i.d. capsule experienced grade 2 suicidal ideation, including grade 2 malaise and non‐compliance due to the size and number of capsules, causing the patient's refusal to continue treatment; and the second patient treated with 400 mg b.i.d. tablet reported grade 3 depression. Both the DLT events were suspected to be related to the study drug. The suicidal ideation was reported as recovered because it was resolved after 1 day, and depression was reported as not recovered/not resolved. In this study, the dose was escalated up to 400 mg b.i.d. for the tablet formulation and 600 mg b.i.d. for the capsule formulation; however, MTD was not reached. The highest studied dose determined to be safe as tablets was 400 mg b.i.d., but it is not yet determined for capsules.
Almost all patients (95.5%) experienced one or more AE during the study (Table 2). AE requiring dose adjustment and/or interruption were reported in 26 patients (59.1%). The most commonly occurring AE that required dose adjustment and/or interruption were increased blood creatinine (20.5%), nausea (13.6%), vomiting (6.8%) and decreased appetite (6.8%). Three patients (6.8%) experienced one dose reduction during the study, one each in the 800 mg q.d. capsule group, 400 mg b.i.d. capsule group and 400 mg b.i.d. tablet group. Twenty‐one patients (47.7%) experienced one or more dose interruptions during the study. Of these, the dose was interrupted only once in nine patients (20.5%). These dose interruptions were more frequent among patients in the 500 mg q.d. capsule group and 600 mg q.d. capsule group (n = 2, each). Two patients (5.1%) were permanently discontinued from the study treatment owing to the DLT, one (2.6%) each in the 600 mg b.i.d. capsule and 400 mg b.i.d. tablet groups. AE (all grades) leading to discontinuation were reported in four patients (9.1%): nausea (grade 2) in the 800 mg q.d. capsule group, suicidal ideation (grade 2) in the 600 mg b.i.d. capsule group, ulcerative colitis (grade 3) in the 200 mg b.i.d. tablet group and depression (grade 3) in the 400 mg b.i.d. tablet group. No on‐treatment deaths (within 28 days after discontinuation of study treatment) were reported.
Table 2.
Overall summary of AE
| Characteristic | Capmatinib capsule | Capmatinib tablet | All n = 44 n (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 mg q.d. n = 3 n (%) | 200 mg q.d. n = 4 n (%) | 400 mg q.d. n = 3 n (%) | 500 mg q.d. n = 4 n (%) | 600 mg q.d. n = 4 n (%) | 800 mg q.d. n = 4 n (%) | 400 mg b.i.d. n = 4 n (%) | 600 mg b.i.d. n = 3 n (%) | 200 mg b.i.d. n = 3 n (%) | 400 mg b.i.d. n = 12 n (%) | ||
| All deathsa | 0 | 0 | 0 | 1 (25.0) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.3) |
| On‐treatment deathsb | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total number of patients experiencing ≥1 AE | 3 (100) | 3 (75.0) | 2 (66.7) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 3 (100) | 3 (100) | 12 (100) | 42 (95.5) |
| Suspected to be drug‐related | 2 (66.7) | 2 (50.0) | 2 (66.7) | 3 (75.0) | 4 (100) | 3 (75.0) | 4 (100) | 2 (66.7) | 3 (100) | 12 (100) | 37 (84.1) |
| SAE | 0 | 1 (25.0) | 0 | 1 (25.0) | 1 (25.0) | 1 (25.0) | 1 (25.0) | 0 | 1 (33.3) | 1 (8.3) | 7 (15.9) |
| Suspected to be drug‐related | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 0 | 0 | 1 (33.3) | 0 | 2 (4.5) |
| AE leading to discontinuation | 0 | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 1 (33.3) | 1 (33.3) | 1 (8.3) | 4 (9.1) |
| AE requiring dose interruption and/or reduction | 2 (66.7) | 2 (50.0) | 1 (33.3) | 3 (75.0) | 4 (100) | 2 (50.0) | 2 (50.0) | 2 (66.7) | 0 | 8 (66.7) | 26 (59.1) |
| Suspected to be drug‐related | 1 (33.3) | 0 | 1 (33.3) | 2 (50.0) | 3 (75.0) | 1 (25.0) | 1 (25.0) | 1 (33.3) | 0 | 7 (58.3) | 17 (38.6) |
All deaths including those that occurred more than 28 days after discontinuation of study treatment are counted.
On‐treatment deaths occurring up to 28 d after discontinuation of study treatment are counted.
AE, adverse event; SAE, serious adverse event.
The most commonly (≥20%) reported AE (all grades) regardless of the study drug relationship were increased blood creatinine (52.3%), nausea (47.7%), decreased appetite (40.9%), vomiting (38.6%), diarrhea (25.0%), hypoalbuminemia (22.7%) and peripheral edema (20.5%) (Table 3). Grade 3/4 AE (regardless of study drug relationship) were reported in 15 patients (34.1%); hyponatremia (9.1%), decreased appetite (6.8%), ascites (4.5%), increased GGT (4.5%), hypoalbuminemia (4.5%) and hypophosphatemia (4.5%). AE suspected to be drug‐related (mainly grade 1/2) were reported in 37 patients (84.1%); the most common (≥20%) ones were increased blood creatinine (45.5%), nausea (31.8%), decreased appetite (29.5%), vomiting (29.5%) and diarrhea (20.5%). Grade 3/4 AE suspected to be drug‐related occurred in seven patients (15.9%): colitis ulcerative (n = 1), lung infection (n = 1), depression (n = 1), increased ALT and AST (n = 1), increased GGT (n = 1), hypophosphatemia (n = 1), decreased neutrophil count, decreased white blood cell count and hyponatremia (n = 1). There were no clinically meaningful changes or trends noted in vital signs and in ECG intervals. Treatment‐emergent prolongation of QTcB of more than 450 ms was observed in three patients, more than 480 ms was observed in one patient and QTcB increase from baseline of more than 30 ms was reported in three patients.
Table 3.
AE occurring in more than 15% patients at all grades or at grade 3/4 regardless of study treatment relationship
| Preferred term | Capmatinib capsule | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 mg q.d. n = 3 | 200 mg q.d. n = 4 | 400 mg q.d. n = 3 | 500 mg q.d. n = 4 | 600 mg q.d. n = 4 | 800 mg q.d. n = 4 | |||||||
| All grades n (%) | Grade 3/4 n (%) | All grades n (%) | Grade 3/4 n (%) | All grades n (%) | Grade 3/4 n (%) | All grades n (%) | Grade 3/4 n (%) | All grades n (%) | Grade 3/4 n (%) | All grades n (%) | Grade 3/4 n (%) | |
| Any AE | 3 (100) | 0 | 3 (75.0) | 1 (25.0) | 2 (66.7) | 0 | 4 (100) | 2 (50.0) | 4 (100) | 4 (100) | 4 (100) | 0 |
| Increased blood creatinine | 1 (33.3) | 0 | 1 (25.0) | 0 | 1 (33.3) | 0 | 3 (75.0) | 0 | 3 (75.0) | 0 | 1 (25.0) | 0 |
| Nausea | 1 (33.3) | 0 | 2 (50.0) | 0 | 1 (33.3) | 0 | 0 | 0 | 3 (75.0) | 0 | 2 (50.0) | 0 |
| Decreased appetite | 1 (33.3) | 0 | 2 (50.0) | 1 (25.0) | 0 | 0 | 0 | 0 | 3 (75.0) | 1 (25.0) | 2 (50.0) | 0 |
| Vomiting | 1 (33.3) | 0 | 3 (75.0) | 0 | 0 | 0 | 0 | 0 | 3 (75.0) | 0 | 1 (25.0) | 0 |
| Diarrhea | 0 | 0 | 0 | 0 | 1 (33.3) | 0 | 4 (100) | 0 | 1 (25.0) | 0 | 0 | 0 |
| Hypoalbuminemia | 1 (33.3) | 0 | 1 (25.0) | 0 | 0 | 0 | 1 (25.0) | 0 | 1 (25.0) | 1 (25.0) | 1 (25.0) | 0 |
| Edema peripheral | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 2 (50.0) | 0 | 0 | 0 |
| Malaise | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 0 | 0 |
| Pyrexia | 0 | 0 | 0 | 0 | 0 | 0 | 2 (50.0) | 0 | 1 (25.0) | 0 | 2 (50.0) | 0 |
| Anemia | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 1 (25.0) | 0 | 1 (25.0) | 0 |
| Constipation | 1 (33.3) | 0 | 1 (25.0) | 0 | 0 | 0 | 1 (25.0) | 0 | 0 | 0 | 2 (50.0) | 0 |
| Fatigue | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 1 (25.0) | 0 | 0 | 0 |
| Preferred term | Capmatinib capsule | Capmatinib tablet |
All n = 44 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
400 mg b.i.d. n = 4 |
600 mg b.i.d. n = 3 |
200 mg b.i.d. n = 3 |
400 mg b.i.d. n = 12 |
|||||||
|
All grades n (%) |
Grade 3/4 n (%) |
All grades n (%) |
Grade 3/4 n (%) |
All grades n (%) |
Grade 3/4 n (%) |
All grades n (%) |
Grade 3/4 n (%) |
All grades n (%) |
Grade 3/4 n (%) |
|
| Any AE | 4 (100) | 2 (50.0) | 3 (100) | 0 | 3 (100) | 1 (33.3) | 12 (100) | 5 (41.7) | 42 (95.5) | 15 (34.1) |
| Increased blood creatinine | 4 (100) | 0 | 1 (33.3) | 0 | 1 (33.3) | 0 | 7 (58.3) | 0 | 23 (52.3) | 0 |
| Nausea | 2 (50.0) | 0 | 2 (66.7) | 0 | 0 | 0 | 8 (66.7) | 0 | 21 (47.7) | 0 |
| Decreased appetite | 2 (50.0) | 1 (25.0) | 2 (66.7) | 0 | 1 (33.3) | 0 | 5 (41.7) | 0 | 18 (40.9) | 3 (6.8) |
| Vomiting | 0 | 0 | 2 (66.7) | 0 | 1 (33.3) | 0 | 6 (50.0) | 0 | 17 (38.6) | 0 |
| Diarrhea | 0 | 0 | 0 | 0 | 1 (33.3) | 0 | 4 (33.3) | 0 | 11 (25.0) | 0 |
| Hypoalbuminemia | 2 (50.0) | 1 (25.0) | 0 | 0 | 0 | 0 | 3 (25.0) | 0 | 10 (22.7) | 2 (4.5) |
| Edema peripheral | 1 (25.0) | 0 | 0 | 0 | 0 | 0 | 5 (41.7) | 1 (8.3) | 9 (20.5) | 1 (2.3) |
| Malaise | 2 (50.0) | 0 | 1 (33.3) | 0 | 2 (66.7) | 0 | 2 (16.7) | 0 | 8 (18.2) | 0 |
| Pyrexia | 1 (25.0) | 0 | 1 (33.3) | 0 | 0 | 0 | 1 (8.3) | 0 | 8 (18.2) | 0 |
| Anemia | 0 | 0 | 0 | 0 | 0 | 0 | 3 (25.0) | 0 | 7 (15.9) | 0 |
| Constipation | 0 | 0 | 0 | 0 | 0 | 0 | 2 (16.7) | 0 | 7 (15.9) | 0 |
| Fatigue | 1 (25.0) | 0 | 1 (33.3) | 0 | 0 | 0 | 3 (25.0) | 0 | 7 (15.9) | 0 |
AE, adverse event.
3.3. Pharmacokinetics
After single p.o. administration of capmatinib using capsule formulation on day 1, capmatinib was rapidly absorbed with median T max of 1.5‐2 hours, with the exception of 100 mg q.d. Following repeated daily dosing up to day 15 following the q.d. or b.i.d. regimen using capsules, median T max ranged 1.0‐4.0 hours (Table 4, Figures 1 and 2), whereas absorption was more rapid after dosing using a tablet with median T max of 1.0 hour on both day 1 and day 15.
Table 4.
Summary of PK parameters by treatment group
| PK parameter | Capmatinib capsule | Capmatinib tablet | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 100 mg q.d. n = 3 | 200 mg q.d. n = 4 | 400 mg q.d. n = 3 | 500 mg q.d. n = 4 | 600 mg q.d. n = 4 | 800 mg q.d. n = 4 | 400 mg b.i.d. n = 4 | 600 mg b.i.d. n = 3 | 200 mg b.i.d. n = 3 | 400 mg b.i.d. n = 12 | |
| Cycle 1 d 1 | ||||||||||
| C max, ng/mL | ||||||||||
| Geo‐mean | 227 | 1910 | 2620 | 7500 | 6370 | 3030 | 1900 | 7730 | 2190 | 3230 |
| CV% Geo‐mean | 60.9 | 50.4 | 360.2 | 54.6 | 80.1 | 107.6 | 158.9 | 53.4 | 118.0 | 80.8 |
|
T
max, h Median (min; max) |
3.98 (1.97; 4.00) |
1.50 (0.933; 2.00) |
1.98 (1.00; 3.98) |
1.97 (0.950; 2.02) |
1.48 (0.967; 2.00) |
2.02 (1.93; 2.12) |
1.96 (0.983; 2.00) |
1.90 (0.967; 1.93) |
0.950 (0.917; 0.967) |
1.00 (0.467; 3.95) |
| AUCtau, h × ng/mL | ||||||||||
| Geo‐mean | 1520 | 8740 | 14 600 | 37 600 | 29 900 | 17 500 | 7940 | 27 600 | 8170 | 12 500 |
| CV% Geo‐mean | 15.2 | 55.6 | 148.8 | 52.5 | 58.7 | 66.3 | 122.4 | 55.9 | 61.3 | 73.8 |
| AUClast, h × ng/mL | ||||||||||
| Geo‐mean | 1520 | 8730 | 14 600 | 37 600 | 29 900 | 17 500 | 7970 | 27 600 | 8200 | 12 500 |
| CV% Geo‐mean | 15.1 | 55.6 | 148.9 | 52.5 | 58.7 | 66.3 | 123.2 | 56.1 | 61.0 | 74.0 |
| Cycle 1 d 15 | ||||||||||
| C max, ng/mL | ||||||||||
| Geo‐mean | 393 | 2070 | 3550 | 7930 | 7140 | 5740 | 1540 | 16 400 | 2850 | 6450 |
| CV% Geo‐mean | 14.1 | 38.3 | 173.0 | 11.6 | 13.1 | 60.9 | 102.5 | 13.4 | 59.6 | 67.0 |
|
T
max, h Median (min; max) |
3.98 (1.02; 4.07) |
1.00 (0.983; 2.00) |
2.00 (1.95; 2.05) |
1.97 (1.03; 2.00) |
1.48 (0.967; 2.03) |
3.00 (2.00; 4.08) |
3.95 (2.00; 3.98) |
1.99 (1.97; 2.02) |
0.967 (0.967; 2.00) |
1.00 (0.500; 2.00) |
| AUCtau, h × ng/mL | ||||||||||
| Geo‐mean | 2640 | 9770 | 20 600 | 43 300 | 38 500 | 38 900 | 8960 | 62 500 | 11 000 | 26 300 |
| CV% Geo‐mean | 41.4 | 21.1 | 121.5 | 17.9 | 18.4 | 18.2 | 92.8 | 1.0 | 56.2 | 70.2 |
| AUClast, h × ng/mL | ||||||||||
| Geo‐mean | 2630 | 9760 | 20 600 | 43 300 | 38 500 | 38 900 | 8990 | 62 600 | 11 000 | 26 400 |
| CV% Geo‐mean | 41.6 | 21.2 | 121.5 | 17.9 | 18.4 | 18.2 | 93.4 | 1.0 | 56.2 | 70.4 |
Results are based on the power model: log (parameter) = log (α) + β × log (dose) + error.
AUC, area under the curve; AUClast, AUC from time zero to the last quantifiable concentration point; AUCtau, AUC calculated to the end of the dosing interval; C max, maximum plasma drug concentration; CV, coefficient of variation; PK, pharmacokinetic; T max, time to reach maximum plasma drug concentration.
Figure 1.
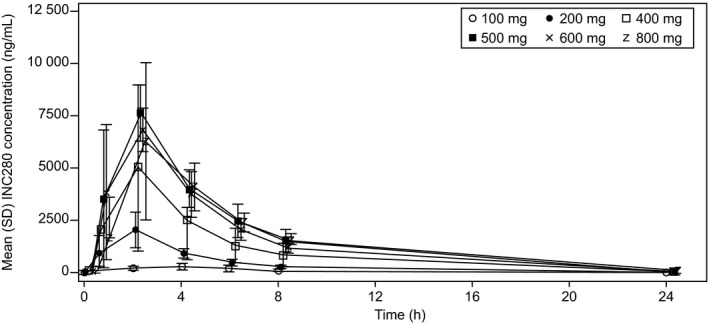
Plasma concentration profile over time on cycle 1, day 15 by treatment group (q.d. capsule group)
Figure 2.
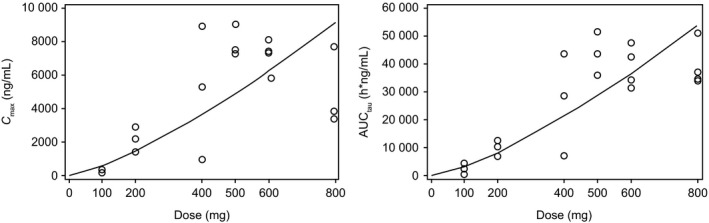
Relationship between dose and PK exposures (C max and AUC tau) on cycle 1, day 15 (q.d. capsule group). AUC tau, area under the curve calculated to the end of the dosing interval; C max, maximum plasma drug concentration; PK, pharmacokinetics
Pre‐dose concentrations were generally stable after day 15, indicating that a steady state was achieved by day 15. For the exposures after q.d. dosing using capsule formulations, a power model was used to assess dose proportionality for the dose range of 100‐800 mg. On day 1, estimated β for C max and AUClast was 1.33 (90% CI, 0.82‐1.85) and 1.28 (90% CI, 0.88‐1.68), respectively. Similar to day 1, β for day 15 was 1.34 (90% CI, 1.01‐1.67) and 1.35 (90% CI, 1.10‐1.60), respectively. As the estimated β was beyond 1 in addition to the lower limit of 90% CI being beyond 1 for day 15, C max and AUC seem to show an over‐proportional increase with dose. When capmatinib was administrated at the dose of 400 mg b.i.d., the mean AUCtau was 11 000 h × ng/mL (capsule group) or 31 400 h × ng/mL (tablet group). However, no clear conclusion about dose proportionality was obtained owing to the following: (a) lower exposures were observed at 800 mg compared with exposures observed at 600 mg; (b) generally high inter‐patient variability; and (c) limited sample number for each dose level.
Overall, the mean T 1/2 of capmatinib ranged 3.5‐6.4, 2.0‐3.1 and 2.5‐2.9 hours by q.d. capsule, b.i.d. capsule and b.i.d. tablet, respectively.
3.4. Efficacy
Overall, 37 patients (84.1%) had evaluable post‐treatment tumor assessments. Eight patients (18.2%) had a best overall response of SD as assessed by the investigator; of these, five patients (11.4%) received capsule formulation (one in 600 mg q.d., one each in 500 mg q.d., 800 mg q.d. and 600 mg b.i.d. groups) and three (6.8%) received tablet formulation (two in 400 mg b.i.d. and one in 200 mg b.i.d.) (Figure 3). In these eight patients, the primary sites of cancer were the lung (n = 2), Ewing sarcoma (n = 1), malignant peripheral nerve sheath tumor (n = 1), esophagus (n = 1), oral cavity (n = 1), osteosarcoma (n = 1) and small intestine (n = 1).
Figure 3.
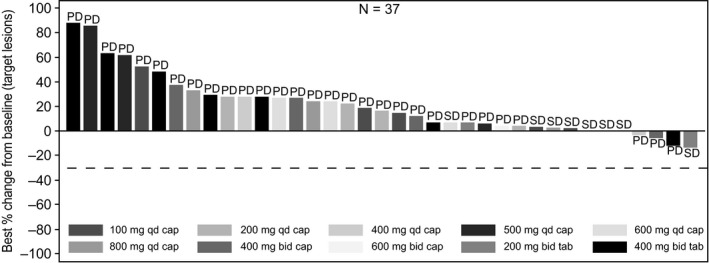
Waterfall plot for best percentage change from baseline in sum of diameters based on local radiology review in patients enrolled in the dose‐escalation part (n = 44). Patients with missing best percentage change from baseline and unknown overall response are not included. Primary site of cancer in the four patients who showed tumor decrease was stomach (PD, −3.33%; 400 mg q.d. capsule), adrenal (PD, −5.61%; 400 mg b.i.d. capsule), pleura (PD, −11.43%; 400 mg b.i.d. tablet) and oral cavity (SD, −13.16%; 200 mg b.i.d. tablet). cap, capsule; PD, progressive disease; SD, stable disease; tab, tablet
3.5. Biomarkers
Tumor samples were collected from all patients at the time of screening and were retrospectively analyzed by FISH, IHC and NGS. Of all patients, MET GCN with FISH analysis was high (9.4 and 8.3) in two patients. The patient in whom MET GCN was 8.3 had a best overall response of SD; however, MET protein expression was not detected by IHC in this patient. No patients with MET exon 14 skipping mutations were identified with NGS testing. MET copy number variant with NGS analysis was high (8) in one patient; however, this patient had a normal MET GCN (4.1) with FISH analysis likely because of tumor heterogeneity. MET mutations (L211W and P991S) were detected in two patients with NGS analysis; the patient with MET GCN 8.3 with FISH analysis had the L211W mutation.
Post‐treatment tumor sample was collected only from one patient; therefore, no formal analysis was performed for the levels of p‐c‐MET, p‐ERK, p‐AKT and p‐S6 in tumor samples.
The change in HGF concentration was not consistent over time. The overall mean decrease in serum HGF concentration from baseline (cycle 1, day 1) in cycle 1 was observed until day 8 (513.4 units at day 2; 347.3 units at day 8). Similarly, a decrease in HGF concentration from baseline (cycle 1, day 1) was observed at cycle 2, day 1 (454.6 units) and at the end of treatment (81.5 units). It was noted that the HGF concentrations in serum samples of the 800 mg q.d. capsule treatment group collected at cycle 1, day 1 (baseline) and of the 400 mg q.d. and 600 mg b.i.d. capsule treatment groups at cycle 1, day 15 were generally higher compared with the other treatment groups. Capmatinib administration did not consistently change the levels of HGF in the patient's blood sample.
4. DISCUSSION
This phase I, dose‐escalation study was conducted in Japanese patients with advanced solid tumors who had progressed despite standard therapy or for whom no effective therapy exists. The expansion was not initiated because sufficient data for further development were already available from other clinical studies of capmatinib.22, 23
Dose‐limiting toxicities were observed in two patients (5.1%; grade 2 suicidal ideation in the 600 mg b.i.d. capsule group and grade 3 depression in the 400 mg b.i.d. tablet group) and both events were suspected to be study drug‐related, prompting permanent discontinuation of the study treatment. However, the suicidal ideation event resolved after 1 day. The MTD for capmatinib was not determined in this study; however, the highest studied dose of capmatinib tablet formulation as single agent determined to be safe was 400 mg b.i.d.
The majority of the AE reported in this study were mild or moderate (CTCAE grade 1 or 2). The most commonly reported AE (all grades) were increased blood creatinine (52.3%), nausea (47.7%) and decreased appetite (40.9%). One patient (2.3%) treated with 500 mg q.d. capsule was suspected to have a drug‐related AST increase of grade 3 and ALT increase of grade 3. No on‐treatment deaths were reported. No clinically significant vital signs and 12‐lead ECG results were reported during the study. Overall, capmatinib was well tolerated in Japanese patients, and the safety profile was similar to that observed in the global study.23
After a single administration of capmatinib as a capsule or a tablet on day 1, the median T max ranged 1.5‐2.0 hours or 1.0 hour, respectively. On the contrary, the mean T 1/2 of capmatinib was independent of formulation. Similar T 1/2 but more rapid T max was observed with tablet formulation compared with capsule, indicating that the formulation seems to have a little impact on absorption but not on the elimination process of capmatinib. When capmatinib was administrated at a dose of 400 mg b.i.d., the mean C max on day 15 was 1980 ng/mL (capsule group) or 7570 ng/mL (tablet group) and the mean AUCtau was 11 000 h × ng/mL (capsule group) or 31 400 h × ng/mL (tablet group). These results are consistent with the data obtained from a relative global bioavailability study (CINC280X2103), which showed the C max geometric mean ratio (tablet vs capsule) as 3.0 and the AUC geometric mean ratios (tablet vs capsule) as 2.4‐2.6 (data on file).
In this study, where MET dysregulation was not an inclusion criterion, efficacy was marginal: SD was the best overall response reported, which occurred in eight (18.2%) patients. One of these patients with SD showed high MET GCN (8.3), whereas the H‐score of MET was 0. Therefore, the correlation between efficacy and MET amplification status in this patient was unclear. In the global phase I study (NCT01324479), capmatinib had demonstrated preliminary antitumor activity at the RP2D (400 mg b.i.d. tablet) in pretreated advanced NSCLC patients with either a high level of MET amplification and/or MET mutations leading to exon 14 deletion.23 Updated data from this study and the global phase II study (NCT02414139) are awaited for further confirmation of antitumor activity of capmatinib in patients with MET amplified/mutated NSCLC. Crizotinib, a multi‐kinase (ALK/ROS1/MET) inhibitor, showed clinically meaningful antitumor activity in patients with MET exon 14‐mutated NSCLC, with an ORR of 44% (95% CI, 22‐69).26 It has also demonstrated antitumor activity in patients with high MET amplification (MET/CEP7 ≥ 4) status, with an ORR of 40% (95% CI, 19.1‐63.9) and median DOR of 5.5 months.27 Tepotinib, another MET inhibitor, showed encouraging signs of activity with an ORR of 53.6% (95% CI, 33.9‐72.5) and median DOR of 12.39 months in patients with advanced NSCLC harboring MET exon 14 skipping mutations.28
A post‐treatment tumor sample was collected from only one patient in this study; therefore, no formal analysis was performed for the levels of biomarkers such as p‐c‐MET, p‐ERK, p‐AKT and p‐S6 in tumor samples. Failures of other MET inhibitors in phase III trials underscore the importance of identifying biomarkers that reliably predict benefit from MET inhibition. Issues have been raised regarding the use of MET as a biomarker based on IHC,29 highlighting the need to refine current assays and identify biomarkers to accurately predict patients likely to benefit from MET inhibition. Currently, MET mutations leading exon 14 deletion (MET exon 14 skipping mutations) and to some extent MET amplification appear to be the most relevant predictors of response to MET‐targeted therapies.
In summary, capmatinib administrated p.o. at the dose of 400 mg b.i.d. in tablet formulation was well tolerated, with a manageable safety profile similar to that in the non‐Japanese population. MTD was not reached, and the highest studied dose determined to be safe was 400 mg b.i.d. (tablet). PK exposures in Japanese patients were similar to those observed in non‐Japanese patients when capmatinib was administrated at the dose of 400 mg b.i.d. (tablet). Modest antitumor activity was observed in this dose‐escalation study of Japanese patients with heavily pretreated advanced solid tumors that were not selected by MET tumor status. These data support the further clinical development of capmatinib.
CONFLICTS OF INTEREST
Annual value of remuneration received: Kae Ishihara from Novartis Pharma K.K. (employment); Tomoyuki Kakizume from Novartis Pharma K.K. (employment); Kazuto Natsume from Novartis Pharma K.K. (employment); and Andrea Myers from Novartis Institute for Biomedical Research (employment). Annual profit from shares received: Andrea Myers has Novartis stock. Total annual value of daily allowances/honoraria received: Taito Esaki from Eli Lilly; Yoichi Naito from Chugai, Novartis and Pfizer; and Kiyotaka Yoh from Chugai, Eli Lilly and Astra Zeneca. Total annual value of research funds, endowments, endowed chairs and researcher‐employment costs received: Taito Esaki from Eli Lilly, Daiichi‐Sankyo, Merck Serono, Taiho, MSD, Novartis, Dainippon Sumitomo, Ono and Boehringer; Takashi Seto from Novartis Pharma; Hideaki Bando from Astra Zeneca and Sysmex; Yoichi Naito from Astra Zeneca, Pfizer and Eli Lilly; Kiyotaka Yoh from Bayer, Pfizer, Eli Lilly, Astra Zeneca, Taiho, Novartis and MSD; and Toshihiko Doi from Novartis. Total annual value of scholarship (incentive) endowments or research grants received: Taito Esaki from Ono.
ACKNOWLEDGMENTS
The study was sponsored by Novartis Pharmaceuticals Corporation. The authors thank the participating patients, their families, all study co‐investigators and research coordinators. Medical writing support was provided by Shilpa Garg and Pushkar Narvilkar (Novartis Health Care Pvt Ltd, Hyderabad).
Esaki T, Hirai F, Makiyama A, et al. Phase I dose‐escalation study of capmatinib (INC280) in Japanese patients with advanced solid tumors. Cancer Sci. 2019;110:1340–1351. 10.1111/cas.13956
REFERENCES
- 1. Birchmeier C, Birchmeier W, Gherardi E, et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915‐925. [DOI] [PubMed] [Google Scholar]
- 2. Christensen JG, Burrows J, Salgia R, et al. c‐MET as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225:1340‐26. [DOI] [PubMed] [Google Scholar]
- 3. Sierra JR, Tsao MS. c‐MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011;3:21‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sadiq AA, Salgia R. MET as a possible target for non‐small‐cell lung cancer. J Clin Oncol. 2013;31:1089‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lal B, Xia S, Abounader R, et al. Targeting the c‐Met pathway potentiates glioblastoma responses to gamma‐radiation. Clin Cancer Res. 2005;11:4479‐4486. [DOI] [PubMed] [Google Scholar]
- 6. Akervall J, Guo X, Qian CN, et al. Genetic and expression profiles of squamous cell carcinoma of the head and neck correlate with cisplatin sensitivity and resistance in cell lines and patients. Clin Cancer Res. 2004;10:8204‐8213. [DOI] [PubMed] [Google Scholar]
- 7. Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non‐small‐cell lung cancer patients. J Clin Oncol. 2009;27:1667‐1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawakami H, Okamoto I, Okamoto W, et al. Targeting MET amplification as a new oncogenic driver. Cancers (Basel). 2014;6:1540‐1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104:20932‐20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen HJ, Mok TS, Chen ZH, et al. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c‐MET amplification in tyrosine kinase inhibitor‐resistant Chinese non‐small cell lung cancer. Pathol Oncol Res. 2009;15:651‐658. [DOI] [PubMed] [Google Scholar]
- 11. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039‐1043. [DOI] [PubMed] [Google Scholar]
- 12. Tsao MS, Liu N, Chen JR, et al. Differential expression of Met/hepatocyte growth factor receptor in subtypes of non‐small cell lung cancers. Lung Cancer. 1998;20:1340‐16. [DOI] [PubMed] [Google Scholar]
- 13. Watermann I, Schmitt B, Stellmacher F, et al. Improved diagnostics targeting c‐MET in non‐small cell lung cancer: expression, amplification and activation? Diagn Pathol. 2015;10:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma PC, Kijima T, Maulik G, et al. c‐MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272‐6281. [PubMed] [Google Scholar]
- 15. Ma PC, Jagadeeswaran R, Jagadeesh S, et al. Functional expression and mutations of c‐Met and its therapeutic inhibition with SU11274 and small interfering RNA in non‐small cell lung cancer. Cancer Res. 2005;65:1479‐1488. [DOI] [PubMed] [Google Scholar]
- 16. Kong‐Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66:283‐289. [DOI] [PubMed] [Google Scholar]
- 17. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850‐859. [DOI] [PubMed] [Google Scholar]
- 18. Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lorenzato A, Olivero M, Patane S, et al. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res. 2002;62:7025‐7030. [PubMed] [Google Scholar]
- 20. Lee JH, Han SU, Cho H, et al. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19:4947‐4953. [DOI] [PubMed] [Google Scholar]
- 21. Liu X, Wang Q, Yang G, et al. A novel kinase inhibitor, INCB28060, blocks c‐MET‐dependent signaling, neoplastic activities, and cross‐talk with EGFR and HER‐3. Clin Cancer Res. 2011;17:7127‐7138. [DOI] [PubMed] [Google Scholar]
- 22. Bang Y‐J, Su W‐C, Nam D‐H, et al. Phase I study of the safety and efficacy of INC280 in patients with advanced MET‐dependent solid tumors. J Clin Oncol. 2014;32: abstract 2520. [Google Scholar]
- 23. Schuler MH, Berardi R, Lim W‐T, et al. Phase (Ph) I study of the safety and efficacy of the cMET inhibitor capmatinib (INC280) in patients (pts) with advanced cMET+ non‐small cell lung cancer (NSCLC). J Clin Oncol. 2016;34: abstract 9067. [Google Scholar]
- 24. Babb J, Rogatko A, Zacks S, et al. Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat Med. 1998;17:1103‐1120. [DOI] [PubMed] [Google Scholar]
- 25. Neuenschwander B, Branson M, Gsponer T, et al. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med. 2008;27:2420‐2439. [DOI] [PubMed] [Google Scholar]
- 26. Drilon AE, Camidge DR, Ou SH, et al. Antitumor activity and safety of crizotinib in patients (pts) with advanced MET exon 14‐altered non‐small cell lung cancer (NSCLC). J Clin Oncol. 2016;34: abstract 108. [Google Scholar]
- 27. Camidge DR, Otterson GA, Clark JW, et al. Crizotinib in patients (pts) with MET‐amplified non‐small cell lung cancer (NSCLC): updated safety and efficacy findings from a phase 1 trial. J Clin Oncol. 2018;36: abstract 9062. [Google Scholar]
- 28. Felip E, Horn L, Patel JD, et al. Tepotinib in patients with advanced non‐small cell lung cancer (NSCLC) harboring MET exon 14‐skipping mutations: phase II trial. J Clin Oncol. 2018;36: abstract 9016. [Google Scholar]
- 29. Rolfo C, Van Der Steen N, Pauwels P, et al. Onartuzumab in lung cancer: the fall of Icarus? Expert Rev Anticancer Ther. 2015;15:487‐489. [DOI] [PubMed] [Google Scholar]