

Abstract
The correlation of genetic alterations with response to neoadjuvant chemotherapy (NAC) has not been fully revealed. In this study, we enrolled 247 breast cancer patients receiving anthracycline‐taxane‐based NAC treatment. A next generation sequencing (NGS) panel containing 36 hotspot breast cancer‐related genes was used in this study. Two different standards for the extent of pathologic complete response (pCR), ypT0/isypN0 and ypT0/is, were used as indicators for NAC treatment. TP53 mutation (n = 149, 60.3%), PIK3CA mutation (n = 109, 44.1%) and MYC amplification (n = 95, 38.5%) were frequently detected in enrolled cases. TP53 mutation (P = 0.019 for ypT0/isypN0 and P = 0.003 for ypT0/is) and ERBB2 amplification (P < 0.001 for both ypT0/isypN0 and ypT0/is) were related to higher pCR rates. PIK3CA mutation (P = 0.040 for ypT0/isypN0) and CCND2 amplification (P = 0.042 for ypT0/is) showed reduced sensitivity to NAC. Patients with MAPK pathway alteration had low pCR rates (P = 0.043 for ypT0/is). Patients with TP53 mutation (−) PIK3CA mutation (−) ERBB2 amplification (+) CCND1 amplification (−), TP53 mutation (+) PIK3CA mutation (−) ERBB2 amplification (+) CCND1 amplification (−) or TP53 mutation (+) PIK3CA mutation (+) ERBB2 amplification (+) CCND1 amplification (−)had significantly higher pCR rates (P < 0.05 for ypT0/isypN0 and ypT0/is) than wild type genotype tumors. Some cancer genetic alterations as well as pathway alterations were associated with chemosensitivity to NAC treatment. Our study may shed light on the molecular characteristics of breast cancer for prediction of NAC expectations when breast cancer is first diagnosed by biopsy.
Keywords: breast neoplasm, genetic variation, high‐throughput nucleotide sequencing, neoadjuvant therapy, pathologic complete response
1. INTRODUCTION
Neoadjuvant chemotherapy (NAC) is being increasingly used in locally advanced breast cancers to limit the extent of surgery in the breast and axilla. Some patients are sensitive to chemotherapy and reach pathologic complete response (pCR) after NAC. Those patients who reach pCR after NAC have more favorable prognosis than those who do not.1 Many biomarkers are reported to be associated with pCR, including negative ER expression,2 positive HER2 expression3 and high Ki67 expression4 Some studies have developed prediction models of pCR based on gene expression analysis with cDNA microarrays, such as Genomic Grade Index5 and DLDA‐30 Pharmacogenomic Predictor.6 These models had a predictive value in breast cancer patients receiving NAC treatment. Apart from those immunochemistry (IHC) biomarkers and prediction models, genetic polymorphism of a few genes, such as TP53,7 BRCA1/2 8 and FGFR4,9 was also reported to predict pCR. However, these studies based on conventional DNA sequencing only focused on limited DNA sites.
Next‐generation sequencing (NGS) is a high‐throughput, time‐effective and cost‐effective method for parallel analyses. Through NGS, whole‐exome sequencing was conducted for individual patients and portrayed mutation maps for breast cancers.10 For clinical application, many NGS gene panels containing mutational hotspots varied for different sequencing platforms. It is common to use NGS panels containing BRCA1/2 or other related genes for auxiliary diagnosis of hereditary breast cancers or to assess breast cancer risk.11, 12, 13, 14 Moreover, NGS also can be used for archived, formalin‐fixed and paraffin‐embedded tissue (FFPE) samples to analyze genetic mutation over time. Some designed NGS panels have also been used to depict gene mutation profiling for different purposes. For example, Park et al15, using FFPE samples and NGS panels, found that several actionable mutations were different in NAC patients who reached pCR compared with non‐pCR patients. However, the panel they used did not contain high frequency altered genes which were specific for breast cancers.
In the present study, we aimed to analyze the relationship between cancer alterations with chemosensitivity in stage II‐III breast cancer patients who received anthracycline‐taxane‐based NAC treatment. A commercial breast cancer‐specific gene panel, which contains hotspot genes for breast cancer, was used for deep sequencing on FFPE samples.
2. MATERIALS AND METHODS
2.1. Patients and samples
A total of 247 breast cancer patients who received anthracycline‐taxane‐based NAC from January 2015 to March 2017 were retrospectively enrolled in this study. No patients received neoadjuvant anti–HER2 targeted therapy or endocrine therapy before surgery. All patients were in clinical stages II or III and underwent a tumor core needle biopsy (CNB) under ultrasonographic guidance before NAC. Approval for this study was granted by the Ethics Committee of West China Hospital (IRS no.: 2017‐476) and the clinical trial was registered with the Chinese Clinical Trial Registry (http://www.chictr.org.cn/; registration no.: ChiCTR1800016763). The flowchart is shown in Figure S1.
In the present study, we used 2 different definitions of pCR: ypT0/isypN0 (no residual invasive disease in the breast and node) and ypT0/is (no residual invasive disease in the breast but possibly with nodal involvement). For each patient, estrogen receptor (ER) and progesterone receptor (PR) were evaluated using pre–NAC FFPE tissue blocks by IHC. For ER‐positive and PR‐positive disease, there was ≥1% positively stained nuclei in tumor tissues. ER and/or PR positive disease was regarded as hormone receptor (HR) positive disease. ER and PR negative disease was regarded as HR negative disease. Human epidermal growth factor receptor 2 (HER2) status was determined by IHC and FISH. Only HER2 IHC (3+) and/or FISH amplified disease was considered as HER2 positive disease. Ki67 expression was divided into a high expression group (>20%) and a low expression group (≤20%). Apart from those IHC biomarkers, age of patient at diagnosis, tumor size and lymph node status before NAC were collected from the patient medical histories.
2.2. DNA and library preparation
DNA was extracted from FFPE samples of CNB using a QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden). The concentration of extracted DNA was detected by Qubit 3.0 using a Qubit dsDNA HS Kit (Life Technologies). The quality of DNA was assessed using gel electrophoresis. Only samples with a total yield of more than 50 ng of DNA and no laddering effects (the most concentrated band above 1000 bp) were used for further analysis.
For library preparation, there were 2 main steps for quality control required for successful sequencing. After DNA extraction, DNA was purified using magnetic beads and then amplified using a PCR Cycler (Bio‐Rad Laboratories, Inc., CA, USA) with AmpliSeq HiFi Master Mix. The total yield of this pre–library of more than 500 ng was required for the next step. Then, this pre–library was processed for hybridization, capture and amplification using a PCR Cycler with HotStart Taq MasterMix. The final library should have concentration of no less than 4.5 ng/μL and fragment size between 280 and 500 bp, which was detected by Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., CA, USA). Only qualified library was accepted for NGS sequencing.
2.3. Next generation sequencing gene panel and gene alteration analysis
The panel (Beikang, Burning Rock Dx) consists of 36 breast cancer‐related genes, spanning 140 kb of the human genome. Details regarding detected regions of those selected genes are shown in Table S1. NGS sequencing was performed by MiSeq (Illumina Technologies) using Miseq Reagent Kit V3. Sequence data were mapped to the human genome (hg19) using BWA aligner 0.7.10. Local alignment optimization, variant calling and annotation were performed using GATK 3.2, MuTect and VarScan. Variants were filtered using the VarScan filter pipeline, with loci with depth less than 100 filtered out. For hotspot insertions and deletions (INDEL), a minimum of 5 supporting reads are needed at an allelic fraction (AF) >.5%; while 10 supporting reads are required for non‐hotspot INDEL at an AF >2%. A minimum of 8 supporting reads are needed for hotspot single nucleotide variants (SNV) to be called at an AF >1%, while 16 supporting reads are needed for non‐hotspot SNV at an AF >2%. According to the ExAC, 1000 Genomes, dbSNP, ESP6500SI‐V2 database, variants with population frequency over .1% were grouped as SNP and excluded from further analysis. Remaining variants were annotated with ANNOVAR and SnpEff v3.6. DNA translocation analysis was performed using Factera 1.4.3 as previously described. The limit of detection for SNV is 2% for hotspots and 5% for non‐hotspots. Copy number variation was detected by in‐house analysis scripts based on depth of coverage data of capture intervals. Coverage data were corrected against sequencing bias resulting from GC content and probe design. The average coverage of all captured regions was used to normalize the coverage of different samples to comparable scales. The copy number was calculated based on the ratio between the depth of coverage in tumor samples and the average coverage of an adequate number (n > 50) of samples without copy number variation as references for each capture interval. Copy number variation was confirmed if the coverage data of the gene region was quantitatively and statistically significantly different from its reference control. The limit of detection for CNVs is 1.5 for deletion and 2.64 for amplification.
2.4. Statistical analysis
Pearson's χ2 test and Fisher's exact test were used to set features, including clinical features and genetic alterations, associated with pCR. A multivariate analysis was performed using binary logistic regression analysis for genetic alterations to identify which were related to pCR. Nonsynonymous genetic alterations in at least 1 sample were mainly classified into 3 tumor‐associated pathways, including: (i) the retinoblastoma (RB) pathway (RB1, CCND1, CDK4); (ii) the mitogen‐activated protein kinase (MAPK) pathway (MAP2K4, MAP3K1, BRAF, KRAS, EGFR, FGFR1, FGFR2); (iii) and the phosphatidylinositol‐3‐kinase (PI3K) pathway (AKT1, AKT3, PTEN, PIK3CA, PIK3R1). Alterations of these 3 pathways were also included in the analysis. To further analyze the influence of genetic alterations on pCR, samples with complete records of HR and HER2 were classified into 4 subtypes: (i) HR−HER2− (n = 30); (ii) HR−HER2+ (n = 25); (iii) HR+HER2+ (n = 43); and (iv) HR+HER2− (n = 147). The associations between genetic alterations and pCR were analyzed in different subtypes, respectively. Statistical analysis was performed with SPSS 16.0 (SPSS, Chicago, IL, USA). The 2‐sided significance level was set at P < .05.
3. RESULTS
3.1. Patients’ characteristics
Patients’ characteristics are shown in Table 1. The median age of enrolled patients was 63 years (range from 26‐71 years). A total of 167 patients (67.6%) had tumor sizes of T1 or T2. A total of 189 patients (76.5%) had positive node status before treatment; 76.9% of all tumors were HR positive (ER and/or PR positive) and 27.5% of all tumors were HER2 positive. High Ki67 expression was detected in 187 patients (75.7%). After receiving anthracycline‐taxane‐based NAC, 30 patients (12.1%) reached ypT0/isypN0 and 42 patients (17.0%) had no residual invasive tumors in the breast (ypT0/is).
Table 1.
Patients’ characteristics
| Characteristics | Number (%) |
|---|---|
| Age, y | |
| <50 | 136 (52.6) |
| ≥50 | 111 (47.4) |
| Tumor size | |
| T1 | 30 (12.1) |
| T2 | 137 (55.5) |
| T3 | 32 (13.0) |
| T4 | 47 (19.0) |
| Unknown | 1 (.4) |
| Node status | |
| Negative | 58 (23.5) |
| Positive | 189 (76.5) |
| Hormone receptor status | |
| Negative | 55 (22.3) |
| Positive | 190 (76.9) |
| Unknown | 2 (.8) |
| Human epidermal growth factor receptor 2 status | |
| Negative | 177 (71.7) |
| Positive | 68 (27.5) |
| Unknown | 2 (.8) |
| Ki67 status | |
| ≤20% | 58 (23.5) |
| >20% | 187 (75.7) |
| Unknown | 2 (.8) |
| ypT0/isypN0 | |
| No | 217 (87.9) |
| Yes | 30 (12.1) |
| ypT0/is | |
| No | 205 (83.0) |
| Yes | 42 (17.0) |
3.2. Breast cancer genetic alterations and pathway involved
For sequencing depth, 234 cases (94.7%) had high median coverage depth (>500×) and 233 (94.3%) cases had high unique coverage depth (>100×). A total of 245 patients (99.2%) harbored at least 1 somatic alteration. The most frequently nonsynonymous alterations were detected on TP53 (n = 152, 61.5%), PIK3CA (n = 109, 44.1%), MYC (n = 99, 40.1%) and ERBB2 (n = 76, 30.8%). Gene amplifications were frequently detected on MYC (n = 95, 38.5%), ERBB2 (n = 67, 27.1%) and CCND1 (n = 37, 15.0%). All genetic alterations in this study are shown in Figure 1. The schematic structure of TP53 and PIK3CA is illustrated in Figure S2.
Figure 1.
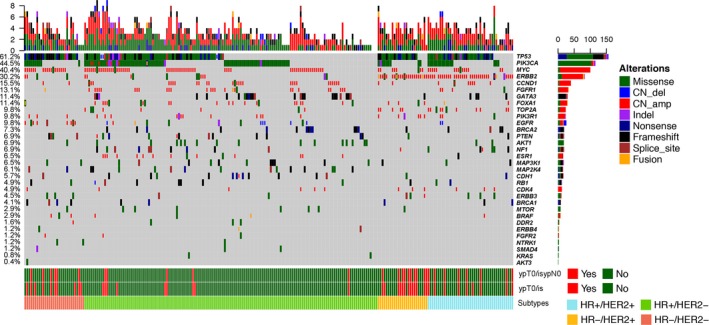
Heat map of gene alterations. Percentage of gene alterations is shown in the left row. Two cases with no records of hormone receptor (HR) and HER2 status were not included in this figure. The absolute count of altered genes in each case are shown in the upper bars. The absolute counts of cases which harbor genetic alterations of each detected gene are shown in the right bars
Based on the known function of genes, genetic alterations were mainly classified into 3 pathways (Figure 2). Sixty‐two cases (25.1%) had at least 1 related genetic alteration in the RB pathway. In this pathway, 49 of 62 cases (79.0%) harbored amplified genes (CCND1 and CDK4). RB1 was the most frequently (11 of 62 cases, 17.7%) mutated gene in this pathway. The second gene set was MAPK pathway related genes. Eighty cases (32.4%) had alterations in at least 1 gene related to MAPK pathway. FGFR1 was the most altered gene, and had 29 amplified cases and 3 mutated cases. The third gene set was associated with the PI3K pathway. Of 247 cases, 139 (56.3%) had a least 1 genetic alteration related to the PI3K pathway. In this pathway, PIK3CA was the most frequently (109 of 139 cases, 78.4%) altered gene, including 103 mutated cases and 6 amplified cases. Twenty‐six cases which harbored mutated PIK3CA also had other altered genes related to the PI3K pathway.
Figure 2.
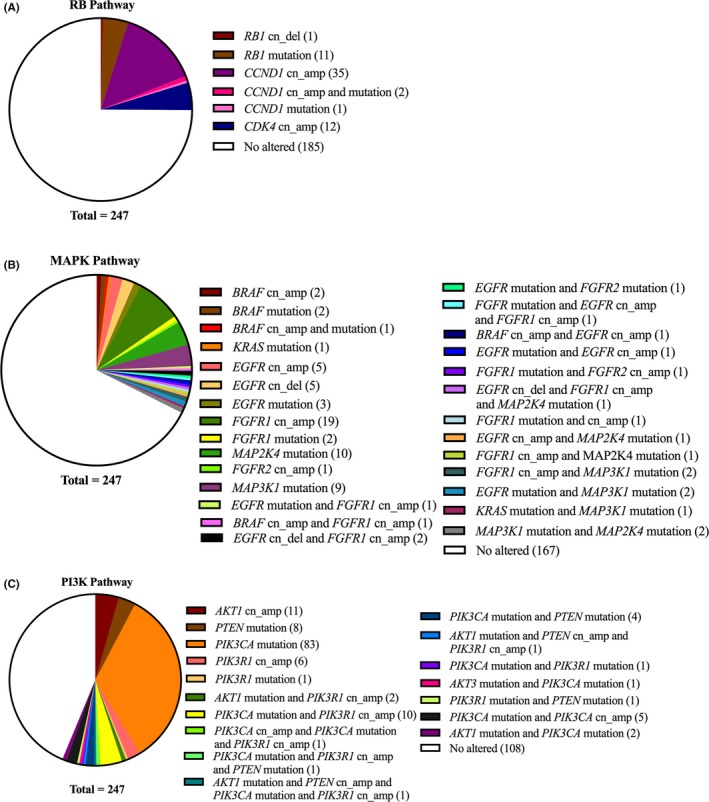
Distribution of gene alterations in 3 functional pathways. A, retinoblastoma (RB) pathway. B, mitogen activated protein kinase (MAPK) pathway. C, phosphatidylinositol‐3‐kinase (PI3K) pathway
In addition, we compared the frequency of genetic alterations among different subtypes of breast cancer (Table S2). The distribution of altered TP53 was statistically different among subtypes (P < .001). TP53 alterations were lower in HR+HER2− cancers (47.6%) than in the other 3 subtypes (76.7%‐90.0%). GATA3 alterations were only found in HR+ tumors (P = .013). TOP2A alterations were not detected in HR−HER2− tumors (P < .001). Altered MAPK pathway was detected in only 1 case (4.0%) in HR−HER2+ cancers; however, alterations of the MAPK pathway were found in 40.0% of HR−HER2− cancers. Moreover, alterations of the PI3K pathway were more frequent (63.9%) in HR+HER2− breast cancers than other subtypes (P = .048).
3.3. Clinical and genetic features associated with pathologic complete response
Clinical features and high‐frequency genetic alterations were included to analyze the relationship with ypT0/isypN0 or ypT0/is, respectively (Table 2). In this analysis, clinical features, such as positive node status (P = .005 for ypT0/isypN0; P < .001 for ypT0/is), negative HR expression (P < .001 for ypT0/isypN0 or ypT0/is) and positive HER2 status (P < .001 for ypT0/isypN0 or ypT0/is), were associated with higher possibility of pCR.
Table 2.
Clinical features and genetic alteration related to chemosensitivity
| Characteristics | N (%) | ypT0/isypN0 | χ2 | P | ypT0/is | χ2 | P | ||
|---|---|---|---|---|---|---|---|---|---|
| No | Yes | No | Yes | ||||||
| Age, y | |||||||||
| <50 | 136 (52.6) | 119 (87.5) | 17 (12.5) | .036 | .850 | 112 (82.4) | 24 (17.6) | .089 | .766 |
| ≥50 | 111 (47.4) | 98 (88.3) | 13 (11.7) | 93 (83.8) | 18 (16.2) | ||||
| Tumor stage | |||||||||
| T1‐T2 | 167 (67.6) | 145 (86.8) | 22 (13.2) | .465 | .495 | 136 (81.4) | 31 (18.6) | .815 | .367 |
| T3‐T4 | 79 (32.0) | 71 (89.9) | 8 (10.1) | 68 (86.1) | 11 (13.9) | ||||
| Unknown | 1 (.4) | – | – | – | – | ||||
| Node status | |||||||||
| Negative | 58 (23.5) | 57 (98.3) | 1 (1.7) | 7.715 | .005 | 57 (98.3) | 1 (1.7) | 12.540 | <.001 |
| Positive | 189 (76.5) | 160 (84.7) | 29 (15.3) | 148 (78.3) | 41 (21.7) | ||||
| Hormone receptor status | |||||||||
| Negative | 55 (22.3) | 39 (70.9) | 16 (29.1) | 20.232 | <.001 | 32 (58.2) | 23 (41.8) | 32.024 | <.001 |
| Positive | 190 (76.9) | 177 (93.2) | 13 (6.8) | 172 (90.5) | 18 (9.5) | ||||
| Unknown | 2 (.8) | – | – | – | – | ||||
| Human epidermal growth factor receptor 2 status | |||||||||
| Negative | 177 (71.7) | 166 (93.8) | 11 (6.2) | 19.315 | <.001 | 158 (89.3) | 19 (10.7) | 16.477 | <.001 |
| Positive | 68 (27.5) | 50 (73.5) | 18 (26.5) | 46 (67.6) | 22 (32.4) | ||||
| Unknown | 2 (.8) | – | – | – | – | ||||
| Ki67 status | |||||||||
| ≤20% | 58 (23.5) | 54 (93.1) | 4 (6.9) | 1.777 | .183 | 52 (89.7) | 6 (10.3) | 2.227 | .136 |
| >20% | 187 (75.7) | 162 (86.6) | 25 (13.4) | 152 (81.3) | 35 (18.7) | ||||
| Unknown | 2 (.8) | – | – | – | – | ||||
| TP53 mutation | |||||||||
| No | 98 (39.7) | 92 (93.9) | 6 (6.1) | 5.524 | .019 | 90 (91.8) | 8 (8.2) | 8.997 | .003 |
| Yes | 149 (60.3) | 128 (83.9) | 24 (16.1) | 115 (77.2) | 34 (22.8) | ||||
| PIK3CA mutation | |||||||||
| No | 138 (55.9) | 116 (84.1) | 22 (15.9) | 4.224 | .040 | 110 (79.7) | 28 (20.3) | 2.392 | .122 |
| Yes | 109 (44.1) | 101 (92.7) | 8 (7.3) | 95 (87.2) | 14 (12.8) | ||||
| ERBB2 amplification | |||||||||
| No | 180 (72.9) | 169 (93.9) | 11 (6.1) | 22.647 | <.001 | 162 (90.0) | 18 (10.0) | 23.067 | <.001 |
| Yes | 67 (27.1) | 48 (71.6) | 19 (28.4) | 43 (64.2) | 24 (35.8) | ||||
| MYC amplification | |||||||||
| No | 152 (61.5) | 135 (88.8) | 17 (11.2) | .342 | .558 | 128 (84.2) | 24 (15.8) | .413 | .520 |
| Yes | 95 (38.5) | 82 (86.3) | 13 (13.7) | 77 (81.1) | 18 (18.9) | ||||
| CCND1 amplification | |||||||||
| No | 210 (85.0) | 181 (86.2) | 29 (13.8) | 3.637 | .057 | 170 (81.0) | 40 (19.0) | 4.148 | .042 |
| Yes | 37 (15.0) | 36 (97.3) | 1 (2.7) | 35 (94.6) | 2 (5.4) | ||||
| Retinoblastoma pathway alteration | |||||||||
| No | 185 (74.9) | 161 (87.0) | 24 (13.0) | .473 | .654 | 152 (82.2) | 33 (17.8) | .363 | .696 |
| Yes | 62 (25.1) | 56 (90.3) | 6 (9.7) | 53 (85.5) | 9 (14.5) | ||||
| Mitogen‐activated protein kinase pathway alteration | |||||||||
| No | 167 (67.6) | 142 (85.0) | 25 (15.0) | 3.854 | .050 | 133 (79.6) | 34 (20.4) | 4.113 | .043 |
| Yes | 80 (32.4) | 75 (93.8) | 5 (6.2) | 72 (90.0) | 8 (10.0) | ||||
| Phosphatidylinositol‐3‐kinase pathway alteration | |||||||||
| No | 108 (43.7) | 91 (84.3) | 17 (15.7) | 2.324 | .127 | 85 (78.7) | 23 (21.3) | 2.505 | .113 |
| Yes | 139 (56.3) | 126 (90.6) | 13 (9.4) | 120 (86.3) | 19 (13.7) | ||||
For genetic alterations, patients with mutated TP53 status tended to have higher possibility of pCR (P = .019 for ypT0/isypN0; P = .003 for ypT0/is). ERBB2 amplification tested by NGS (P < .001 for both ypT0/isypN0 and ypT0/is) was also related to higher pCR rates. Mutated PIK3CA or amplified CCND1 was related to lower pCR rates with statistically significance (PIK3CA mutation, P = .040 for ypT0/isypN0; CCND1 amplification, P = .042 for ypT0/is). However, in analyzing the correlation between pathway alterations with pCR (Table 2), alterations of PI3K and RB pathways showed trends of lower pCR rates with no statistical significance. Altered MAPK pathway was significantly associated with pCR (P = .043 for ypT0/is). In multivariate analyses for genetic alterations (Table S3), ERBB2 amplification and CCND1 amplification demonstrated a relationship with pCR (ERBB2 amplification, P = .002 for ypT0/isypN0 and P = .003 for ypT0/is; CCND1 amplification, P = .032 for ypT0/is).
3.4. Different somatic mutation subtypes leading to diverse neoadjuvant chemotherapy effect
To further analyze the association between high frequency alterations and the NAC effect, we classified the patients with different status of 4 altered genes, which showed a relationship with pCR. We further analyzed these different somatic mutation subtypes with the NAC effect (pCR or non‐pCR). Sixteen different combinations of gene alterations among TP53 mutation, PIK3CA mutation, ERBB2 amplification and CCND1 amplification are listed in Figure 3, but only 6 combinations had more than 15 patients in each category. When compared with wild type genotype (TP53 mutation [−] PIK3CA mutation [−] ERBB2 amplification [−] CCND1 amplification [−]) tumors, patients with TP53 mutation (−) PIK3CA mutation (−) ERBB2 amplification (+) CCND1 amplification (−), TP53 mutation (+) PIK3CA mutation (−) ERBB2 amplification (+) CCND1 amplification (−) or TP53 mutation (+) PIK3CA mutation (+) ERBB2 amplification (+) CCND1 amplification (−) had significantly higher pCR rates (P < .05 for ypT0/isypN0 and ypT0/is, Figure 3A and B).
Figure 3.
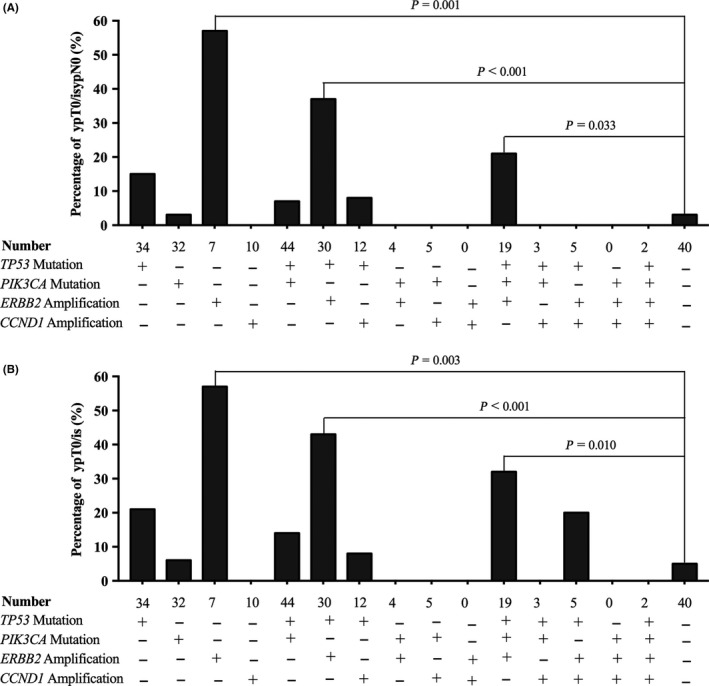
Different rates of pathologic complete response (pCR) (ypT0/isypN0 [A] or ypT0/is [B]) between patients with any combination of altered genes
3.5. Genetic alteration and related pathway that predispose to pathologic complete response in different subtypes
In addition, we analyzed the rates of ypT0/isypN0 (Table 3) and ypT0/is (Table S4) in genetic alterations within different subtypes to reveal relationships between genetic alterations and chemosensitivity. In HR−HER2+ tumors, PIK3CA mutation (P = .040 for ypT0/isypN0; P = .041 for ypT0/is) and MYC amplification (P = .020 for ypT0/isypN0; P = .002 for ypT0/is) were associated with lower pCR rates. However, in HR+HER2− tumors, MYC amplification was related to higher pCR rates (P = .074 for ypT0/isypN0; P = .028 for ypT0/is). For HR−HER2− and HR+HER2+ tumors, no genetic alterations had statistically significant association with pCR.
Table 3.
Association between genetic alterations and ypT0/isypN0 in different breast cancer subtypes
| Characteristics | N (%)a | HR−HER2− | χ2 | P | HR−HER2+ | χ2 | P | HR+HER2+ | χ2 | P | HR+HER2− | χ2 | P | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Yes | No | Yes | No | Yes | No | Yes | ||||||||||
| TP53 mutation | |||||||||||||||||
| No | 98 (40.0) | 2 (66.7) | 1 (33.3) | .370 | .501 | 3 (60.0) | 2 (40.0) | <.001 | 1.000 | 8 (80.0) | 2 (20.0) | .017 | .897 | 79 (98.8) | 1 (1.2) | 2.472 | .178 |
| Yes | 147 (60.0) | 22 (81.5) | 5 (18.5) | 12 (60.0) | 8 (40.0) | 27 (81.8) | 6 (18.2) | 63 (94.0) | 4 (6.0) | ||||||||
| PIK3CA mutation | |||||||||||||||||
| No | 136 (55.5) | 16 (80.0) | 4 (20.0) | <.001 | 1.000 | 7 (43.8) | 9 (56.2) | 4.890 | .040 | 22 (81.5) | 5 (18.5) | <.001 | 1.000 | 70 (95.9) | 3 (4.1) | .221 | .681 |
| Yes | 109 (44.5) | 8 (80.0) | 2 (20.0) | 8 (88.9) | 1 (11.1) | 13 (81.2) | 3 (18.8) | 72 (97.3) | 2 (2.7) | ||||||||
| ERBB2 amplification | |||||||||||||||||
| No | 180 (73.5) | 22 (78.6) | 6 (21.4) | .536 | 1.000 | 2 (66.7) | 1 (33.3) | .063 | 1.000 | 9 (100.0) | 0 (0) | 2.602 | .171 | 136 (97.1) | 4 (2.9) | 2.650 | .219 |
| Yes | 65 (26.5) | 2 (100.0) | 0 (0) | 13 (59.1) | 9 (40.9) | 26 (76.5) | 8 (23.5) | 6 (85.7) | 1 (14.3) | ||||||||
| MYC amplification | |||||||||||||||||
| No | 150 (61.2) | 12 (92.3) | 1 (7.7) | 2.172 | .196 | 8 (44.4) | 10 (55.6) | 6.481 | .020 | 25 (86.2) | 4 (13.8) | 1.362 | .404 | 89 (98.9) | 1 (1.1) | 3.705 | .074 |
| Yes | 95 (38.8) | 12 (70.6) | 5 (29.4) | 7 (100.0) | 0 (0) | 10 (71.4) | 4 (28.6) | 53 (53.0) | 4 (7.0) | ||||||||
| CCND1 amplification | |||||||||||||||||
| No | 208 (84.9) | 23 (82.1) | 5 (17.9) | 1.205 | .366 | 13 (56.5) | 10 (43.5) | 1.449 | .500 | 28 (77.8) | 8 (22.2) | 1.911 | .315 | 116 (95.9) | 5 (4.1) | 1.112 | .586 |
| Yes | 37 (15.1) | 1 (50.0) | 1 (50.0) | 2 (100.0) | 0 (0) | 7 (100.0) | 0 (0) | 26 (100.0) | 0 (0) | ||||||||
| Retinoblastoma pathway alteration | |||||||||||||||||
| No | 183 (74.7) | 21 (84.0) | 4 (16.0) | 1.500 | .254 | 12 (60.0) | 8 (40.0) | <.001 | 1.000 | 23 (74.2) | 8 (25.8) | 3.805 | .082 | 104 (97.2) | 3 (2.8) | .427 | .613 |
| Yes | 62 (23.7) | 3 (60.0) | 2 (40.0) | 3 (60.0) | 2 (40.0) | 12 (100.0) | 0 (0) | 38 (95.0) | 2 (5.0) | ||||||||
| Mitogen‐activated protein kinase pathway alteration | |||||||||||||||||
| No | 165 (67.3) | 12 (75.0) | 4 (25.0) | .536 | .657 | 15 (62.5) | 9 (37.5) | 1.563 | .400 | 28 (77.8) | 8 (22.2) | 1.911 | .315 | 86 (96.6) | 3 (3.4) | .001 | 1.000 |
| Yes | 80 (32.7) | 12 (85.7) | 2 (14.3) | 0 (0) | 1 (100.0) | 7 (100.0) | 0 (0) | 56 (96.6) | 2 (3.4) | ||||||||
| Phosphatidylinositol‐3‐kinase pathway alteration | |||||||||||||||||
| No | 106 (43.3) | 13 (81.2) | 3 (18.8) | .033 | 1.000 | 7 (53.8) | 6 (46.2) | .427 | .688 | 19 (79.2) | 5 (20.8) | .178 | 1.000 | 51 (96.2) | 2 (3.8) | .035 | 1.000 |
| Yes | 139 (56.7) | 11 (78.6) | 3 (21.4) | 8 (66.7) | 4 (33.3) | 16 (84.2) | 3 (15.8) | 91 (96.8) | 3 (2.2) | ||||||||
Two cases with no records of hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) status were not included in this table.
4. DISCUSSION
In this study, we detected cancer‐related genes using CNB tissues of breast cancer patients and the NGS method. To focus on the hotspot genes with high frequency in breast cancer samples and to obtain high quality data with deep sequence results, we used commercially available breast cancer‐specific NGS panel sequencing (Beikang, Burning Rock Biotech).
For 36 candidate genes, 32 genes had altered in at least 1 patient. TP53, PIK3CA and MYC had high frequency (>40%) of alterations, which was in accordance with previous studies.10, 16, 17 Most genes (24/32, 75.0%) had low‐frequency (<10%) of alterations.10 The alteration frequency of some genes was different among clinical subtypes. For example, TOP2A was closed to HER2 on chromosome 17 and was reported to be frequently co‐amplified in HER2 positive breast cancers.18, 19 We also found that TOP2A had higher frequency of alterations in HER2‐positive subtypes (Table S2). As for GATA3 and FOXA1, which have been proven to have functional effects on the ESR1 pathway20 and to have high expression levels in ER positive breast cancers,21, 22 they were also found to be altered frequently in HR‐positive tumors (Table S2).
There were 3 main definitions for pCR, which were ypT0/isypN0 (no residual invasive disease in the breast and node), ypT0ypN0 (no residual disease in the breast and node) and ypT0/is (no residual invasive disease in the breast but possibly with nodal involvement). For breast cancer patients who received NAC, pCR (no matter which definition was used) could be a favorable prognosis indicator.1, 23, 24 In particular, there was no statistically significant difference in the prognosis between ypT0/isypN0 and ypT0ypN0.23, 24 Although ypT0/is was not a better indicator for prognosis when compared with ypT0/isypN0 and ypT0ypN0,23 it could be used as a chemosensitivity indicator for breast cancer patients with NAC.1 Therefore, we used ypT0/isypN0 and ypT0/is simultaneously as definitions for pCR in this study.
Then we calculated associations between gene mutations or amplifications with pCR in all cases and different subtypes. Only genes with frequency of alteration over 15% were enrolled in this analysis. Therefore, high frequency gene mutation, such as for TP53 and PIK3CA, and high frequency gene amplification, such as for ERBB2, MYC and CCND1, were further analyzed.
It is known that TP53 is frequently altered in the majority of human cancers.25 Along with TP53 alterations, breast cancers were more sensitive to DNA‐damaging therapy because of loss of p53‐dependent transcriptional control.26 We found that TP53 was altered in 61.2% of cases and it had higher alteration rates in HR−HER2− tumors (Table S2). For breast cancers that harbored TP53 mutation, it was easier to reach pCR (Table 2). However, when analyzing the relationship between TP53 mutation and pCR in different subtypes, TP53 mutation had higher pCR rates in HR+HER2− tumors with no statistical significance (Table 3 and Table S4).
PIK3CA is the second most commonly mutated gene next to TP53 in many human cancers.25 In breast cancers, PIK3CA mutated frequently10 and was enriched in luminal tumors, especially in luminal A subtype.10, 25, 27 The same phenomenon was also found in our study (Table S2). We found that tumors with mutated PIK3CA were less sensitive to NAC (Table 2), as previously reported.28 Majewski et al29 reported that the pCR rate of HER2‐targeted therapy for PIK3CA wild‐type tumors was higher in ER−HER2+ subtypes than that in ER+HER2+ tumors. In our study, PIK3CA wild‐type tumors had higher pCR rates in HR‐HER2+ subtype than the other 3 subtypes and the association between PIK3CA mutation and pCR had statistical significance only in HR−HER2+ tumors (Table 3 and Table S4). Although patients with mutated PIK3CA were not sensitive to traditional chemotherapy, they may be benefit from dual inhibition of B cell lymphoma‐extra large (BCL‐XL) and mTOR/4E‐BP axes, which could sensitize these patients to traditional chemotherapy.30
In the present study, we investigated ERBB2 gene status using the NGS method. Among 65 cases (26.5%) with ERBB2 amplification, 56 (86.0%) were detected in HER2‐positive diseases. In this study, both HER2‐positive status (IHC‐3+ and/or amplified FISH results) and ERBB2 amplification tested by NGS were significantly related to higher pCR rates (Table 2). We also found 9 (3.6%) cases harboring mutated ERBB2 without ERBB2 amplification. Only 1 case (1/9, 11.1%) reached pCR after NAC treatment.
The MYC gene is overexpressed in many breast cancers. In our study, we found that its amplification rate was 38.5%. As for MYC amplification, it showed significant correlation with pCR in both HR−HER2+ and HR+HER2− tumors (Table 3 and Table S4). However, cases with MYC amplification had a higher pCR rate in HR+HER2− subtype and had a lower pCR rate in HR−HER2+ subtype. Yasojima et al31 also reported that c‐myc amplification was related to a higher pCR rate in ER‐positive but not ER‐negative breast cancers. Apart from sample size of HR+HER2− and HR−HER2+ breast cancers in our study, there may be different mechanisms of amplified MYC in responding to chemotherapy.
Overexpression of CCND1 may be related to gene amplification32 and result in tumor formation.33 CCND1 is a member of the cyclin D family and promotes G1‐S transition in cell proliferation.34 In the TransATAC substudy, breast cancers with CCND1 amplification were found to have a poor prognosis.32 In addition, overexpression of CCND1 demonstrates resistance to chemotherapy in vitro.34, 35, 36 In this study, CCND1 was amplified in 37 (15.0%) cases, with reported amplification rates from 8.7% to 30%.32, 37, 38 Cases with amplified CCND1 were negatively related to pCR (Table 2).
Moreover, we also classified different altered genes into 3 tumor‐related pathways. In our study, the RB pathway contained 3 major altered genes, which were RB1, CCND1 and CDK4. The distribution of RB pathway alteration was slightly more in HR‐positive disease. However, the RB pathway alteration showed lower sensitivity to chemotherapy with no statistical significance (Table 2). As for the MAPK pathway, which was altered significantly more in triple‐negative breast cancers in this study (Table S2), its aberrant activity is important for the progression of tumors. KRAS belonging to Ras families, which are small GTPases activated indirectly by external stimuli39 have low frequency of mutation in our study (.8%) and in other published studies.10, 40 BRAF are also an important component in the MAPK pathway. It mutates frequently in melanoma and papillary thyroid cancer.39 Targeted BRAF V600E treatment, such vemurafenib41, 42 and dacarbazine43, 44, is approved to be effective in melanoma. In breast cancers, BRAF is rarely reported to be mutated.45 The mutation rate was also under 3.0% in our study. In addition, MAPK pathway alteration was significantly related to a lower pCR rate (Table 2). Aberrant MAPK pathway is also reported to be involved in tamoxifen resistance.46 The third pathway analyzed in our study was the PI3K pathway, which was found altered in 139 (56.3%) cases. It is reported that alteration of the PI3K pathway is mainly observed in HR‐positive diseases.47 The highest alteration rate of the PI3K pathway was found in HR+HER2− breast cancers (Table S2). In addition, aberrant activation of the PI3K pathway is related to resistance to endocrine therapy and chemotherapy.48, 49 The resistance to endocrine therapy may be on account of inducing estrogen‐independent transcriptional activity.50 Activation of the PI3K pathway may overcome proapoptotic signals of anti–cancer drugs and leads to chemoresistance.51 Therefore, inhibition of the PI3K pathway is proved to promote cytotoxic effects in chemotherapy.52 In this study, cases with PI3K pathway alterations had lower pCR rates than non‐alteration cases with no statistical significance (Table 2).
With the development of targeted sequencing leading to lower cost and higher depth of coverage, more NGS panels are being applied in clinical practice. Multiple gene alterations can be detected at one time. Some gene alterations have been proven to be related to clinical outcomes, as discussed above. Moreover, patients with specific gene alterations may have the opportunity to receive targeted therapy, which may be approved or investigational. The targeted therapies of detected genes in this study are summarized in Table S5. It is a challenge in clinical practice to choose a suitable therapy for patients who harbor gene alterations.
In conclusion, we used NGS panels to detect breast cancer‐related gene alterations in FFPE CNB samples. Gene alterations, such as TP53 mutation, PIK3CA mutation, ERBB2 amplification and CCND1 amplification, and MAPK pathway alteration were associated with pCR in our study. Our study may shed light on the molecular characteristics of breast cancer for prediction of NAC expectations when breast cancer is first diagnosed by biopsy.
DISCLOSURE STATEMENT
Junjun Liu, Junyi Ye and Zhe Zhang are members of Burning Rock Biotech, Guangzhou, China.
Supporting information
ACKNOWLEDGMENTS
The authors wish to thank the research coordinators of the 7 institutions for data collection, and are grateful for the support from Burning Rock Biotech.
Yang L, Ye F, Bao L, et al. Somatic alterations of TP53, ERBB2, PIK3CA and CCND1 are associated with chemosensitivity for breast cancers. Cancer Sci. 2019;110:1389–1400. 10.1111/cas.13976
REFERENCES
- 1. Rastogi P, Anderson SJ, Bear HD, et al. Preoperative chemotherapy: updates of National Surgical Adjuvant Breast and Bowel Project Protocols B‐18 and B‐27. J Clin Oncol. 2008;26:778‐785. [DOI] [PubMed] [Google Scholar]
- 2. Li XR, Liu M, Zhang YJ, et al. ER, PgR, HER‐2, Ki‐67, topoisomerase IIalpha, and nm23‐H1 proteins expression as predictors of pathological complete response to neoadjuvant chemotherapy for locally advanced breast cancer. Med Oncol. 2011;28(Suppl 1):S48‐S54. [DOI] [PubMed] [Google Scholar]
- 3. Zhou B, Yang DQ, Xie F. Biological markers as predictive factors of response to neoadjuvant taxanes and anthracycline chemotherapy in breast carcinoma. Chin Med J (Engl). 2008;121:387‐391. [PubMed] [Google Scholar]
- 4. Petit T, Wilt M, Velten M, et al. Comparative value of tumour grade, hormonal receptors, Ki‐67, HER‐2 and topoisomerase II alpha status as predictive markers in breast cancer patients treated with neoadjuvant anthracycline‐based chemotherapy. Eur J Cancer. 2004;40:205‐211. [DOI] [PubMed] [Google Scholar]
- 5. Liedtke C, Hatzis C, Symmans WF, et al. Genomic grade index is associated with response to chemotherapy in patients with breast cancer. J Clin Oncol. 2009;27:3185‐3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hess KR, Anderson K, Symmans WF, et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J Clin Oncol. 2006;24:4236‐4244. [DOI] [PubMed] [Google Scholar]
- 7. van den Broek AJ, Broeks A, Horlings HM, et al. Association of the germline TP53 R72P and MDM2 SNP309 variants with breast cancer survival in specific breast tumor subgroups. Breast Cancer Res Treat. 2011;130:599‐608. [DOI] [PubMed] [Google Scholar]
- 8. Bayraktar S, Gutierrez‐Barrera AM, Liu D, et al. Outcome of triple‐negative breast cancer in patients with or without deleterious BRCA mutations. Breast Cancer Res Treat. 2011;130:145‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marme F, Werft W, Benner A, et al. FGFR4 Arg388 genotype is associated with pathological complete response to neoadjuvant chemotherapy for primary breast cancer. Ann Oncol. 2010;21:1636‐1642. [DOI] [PubMed] [Google Scholar]
- 10. Cancer Genome Atlas N . Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pilato B, Pinto R, De Summa S, et al. BRCA1‐2 diagnostic workflow from next‐generation sequencing technologies to variant identification and final report. Genes Chromosom Cancer. 2016;55:803‐813. [DOI] [PubMed] [Google Scholar]
- 12. Tung N, Battelli C, Allen B, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next‐generation sequencing with a 25‐gene panel. Cancer. 2015;121:25‐33. [DOI] [PubMed] [Google Scholar]
- 13. Lin PH, Kuo WH, Huang AC, et al. Multiple gene sequencing for risk assessment in patients with early‐onset or familial breast cancer. Oncotarget. 2016;7:8310‐8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lhota F, Zemankova P, Kleiblova P, et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2‐negatively tested breast cancer patients. Clin Genet. 2016;90:324‐333. [DOI] [PubMed] [Google Scholar]
- 15. Park K, Choi MK, Jung HH, et al. Molecular characterization of patients with pathologic complete response or early failure after neoadjuvant chemotherapy for locally advanced breast cancer using next generation sequencing and nCounter assay. Oncotarget. 2015;6:24499‐24510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ping Z, Xia Y, Shen T, et al. A microscopic landscape of the invasive breast cancer genome. Sci Rep. 2016;6:27545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jarvinen TA, Tanner M, Rantanen V, et al. Amplification and deletion of topoisomerase IIalpha associate with ErbB‐2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. Am J Pathol. 2000;156:839‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Konecny GE, Pauletti G, Untch M, et al. Association between HER2, TOP2A, and response to anthracycline‐based preoperative chemotherapy in high‐risk primary breast cancer. Breast Cancer Res Treat. 2010;120:481‐489. [DOI] [PubMed] [Google Scholar]
- 20. Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23:12‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418‐8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Badve S, Turbin D, Thorat MA, et al. FOXA1 expression in breast cancer–correlation with luminal subtype A and survival. Clin Cancer Res. 2007;13:4415‐4421. [DOI] [PubMed] [Google Scholar]
- 23. Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long‐term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384:164‐172. [DOI] [PubMed] [Google Scholar]
- 24. Mazouni C, Peintinger F, Wan‐Kau S, et al. Residual ductal carcinoma in situ in patients with complete eradication of invasive breast cancer after neoadjuvant chemotherapy does not adversely affect patient outcome. J Clin Oncol. 2007;25:2650‐2655. [DOI] [PubMed] [Google Scholar]
- 25. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ablain J, Poirot B, Esnault C, Lehmann‐Che J, de The H. p53 as an effector or inhibitor of therapy response. Cold Spring Harb Perspect Med. 2015;6:a026260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sabine VS, Crozier C, Brookes CL, et al. Mutational analysis of PI3K/AKT signaling pathway in tamoxifen exemestane adjuvant multinational pathology study. J Clin Oncol. 2014;32:2951‐2958. [DOI] [PubMed] [Google Scholar]
- 28. Yuan H, Chen J, Liu Y, et al. Association of PIK3CA mutation status before and after neoadjuvant chemotherapy with response to chemotherapy in women with breast cancer. Clin Cancer Res. 2015;21:4365‐4372. [DOI] [PubMed] [Google Scholar]
- 29. Majewski IJ, Nuciforo P, Mittempergher L, et al. PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2‐targeted therapies in breast cancer. J Clin Oncol. 2015;33:1334‐1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anderson GR, Wardell SE, Cakir M, et al. PIK3CA mutations enable targeting of a breast tumor dependency through mTOR‐mediated MCL‐1 translation. Sci Transl Med. 2016;8:369ra175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yasojima H, Shimomura A, Naoi Y, et al. Association between c‐myc amplification and pathological complete response to neoadjuvant chemotherapy in breast cancer. Eur J Cancer. 2011;47:1779‐1788. [DOI] [PubMed] [Google Scholar]
- 32. Lundgren K, Brown M, Pineda S, et al. Effects of cyclin D1 gene amplification and protein expression on time to recurrence in postmenopausal breast cancer patients treated with anastrozole or tamoxifen: a TransATAC study. Breast Cancer Res. 2012;14:R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV‐cyclin D1 transgenic mice. Nature. 1994;369:669‐671. [DOI] [PubMed] [Google Scholar]
- 34. Biliran H Jr, Wang Y, Banerjee S, et al. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin‐mediated apoptosis in an elastase‐myc transgene‐expressing pancreatic tumor cell line. Clin Cancer Res. 2005;11:6075‐6086. [DOI] [PubMed] [Google Scholar]
- 35. Tiemann K, Alluin JV, Honegger A, et al. Small interfering RNAs targeting cyclin D1 and cyclin D2 enhance the cytotoxicity of chemotherapeutic agents in mantle cell lymphoma cell lines. Leuk Lymphoma. 2011;52:2148‐2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kornmann M, Danenberg KD, Arber N, Beger HG, Danenberg PV, Korc M. Inhibition of cyclin D1 expression in human pancreatic cancer cells is associated with increased chemosensitivity and decreased expression of multiple chemoresistance genes. Cancer Res. 1999;59:3505‐3511. [PubMed] [Google Scholar]
- 37. Burandt E, Grunert M, Lebeau A, et al. Cyclin D1 gene amplification is highly homogeneous in breast cancer. Breast Cancer. 2016;23:111‐119. [DOI] [PubMed] [Google Scholar]
- 38. Roy PG, Pratt N, Purdie CA, et al. High CCND1 amplification identifies a group of poor prognosis women with estrogen receptor positive breast cancer. Int J Cancer. 2010;127:355‐360. [DOI] [PubMed] [Google Scholar]
- 39. Montagut C, Settleman J. Targeting the RAF‐MEK‐ERK pathway in cancer therapy. Cancer Lett. 2009;283:125‐134. [DOI] [PubMed] [Google Scholar]
- 40. Tilch E, Seidens T, Cocciardi S, et al. Mutations in EGFR, BRAF and RAS are rare in triple‐negative and basal‐like breast cancers from Caucasian women. Breast Cancer Res Treat. 2014;143:385‐392. [DOI] [PubMed] [Google Scholar]
- 41. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous‐cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oberholzer PA, Kee D, Dziunycz P, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30:316‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP‐74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989‐1000. [DOI] [PubMed] [Google Scholar]
- 44. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med. 2012;367:107‐114. [DOI] [PubMed] [Google Scholar]
- 45. Cejalvo JM, Perez‐Fidalgo JA, Ribas G, et al. Clinical implications of routine genomic mutation sequencing in PIK3CA/AKT1 and KRAS/NRAS/BRAF in metastatic breast cancer. Breast Cancer Res Treat. 2016;160:69‐77. [DOI] [PubMed] [Google Scholar]
- 46. Cui Y, Parra I, Zhang M, et al. Elevated expression of mitogen‐activated protein kinase phosphatase 3 in breast tumors: a mechanism of tamoxifen resistance. Cancer Res. 2006;66:5950‐5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Estevez LG, Garcia E, Hidalgo M. Inhibiting the PI3K signaling pathway: buparlisib as a new targeted option in breast carcinoma. Clin Transl Oncol. 2016;18:541‐549. [DOI] [PubMed] [Google Scholar]
- 48. Huang WC, Hung MC. Induction of Akt activity by chemotherapy confers acquired resistance. J Formos Med Assoc. 2009;108:180‐194. [DOI] [PubMed] [Google Scholar]
- 49. Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3’‐kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002;62:1087‐1092. [PubMed] [Google Scholar]
- 50. Campbell RA, Bhat‐Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3‐kinase/AKT‐mediated activation of estrogen receptor alpha: a new model for anti‐estrogen resistance. J Biol Chem. 2001;276:9817‐9824. [DOI] [PubMed] [Google Scholar]
- 51. Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153‐164. [DOI] [PubMed] [Google Scholar]
- 52. Badinloo M, Esmaeili‐Mahani S. Phosphatidylinositol 3‐kinases inhibitor LY294002 potentiates the cytotoxic effects of doxorubicin, vincristine, and etoposide in a panel of cancer cell lines. Fundam Clin Pharmacol. 2014;28:414‐422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials