

Abstract
Tumor molecular profiling is becoming a standard of care for patients with cancer, but the optimal platform for cancer sequencing remains undetermined. We established a comprehensive assay, the Todai OncoPanel (TOP), which consists of DNA and RNA hybridization capture‐based next‐generation sequencing panels. A novel method for target enrichment, named the junction capture method, was developed for the RNA panel to accurately and cost‐effectively detect 365 fusion genes as well as aberrantly spliced transcripts. The TOP RNA panel can also measure the expression profiles of an additional 109 genes. The TOP DNA panel was developed to detect single nucleotide variants and insertions/deletions for 464 genes, to calculate tumor mutation burden and microsatellite instability status, and to infer chromosomal copy number. Clinically relevant somatic mutations were identified in 32.2% (59/183) of patients by prospective TOP testing, signifying the clinical utility of TOP for providing personalized medicine to cancer patients.
Keywords: clinical sequencing, junction capture method, molecular profiling, personalized medicine, Todai OncoPanel
1. INTRODUCTION
The advent of large‐scale cancer genomic analyses has identified numerous driver genes1, 2 and germ line variants that increase cancer susceptibility.3 We increasingly understand the molecular determinants of oncogenesis, including genomic alterations that may affect proliferation‐associated pathways. Although next‐generation sequencing (NGS)‐based laboratory tests have been rapidly developed and several large‐scale clinical sequencing studies have proved their utility in cancer precision medicine,4 such tests are still available only at limited institutes. Most of these cancer gene panels are based on sequencing the gDNA of target genes isolated either by polymerase chain reaction (PCR)5, 6 or probe hybridization,7, 8, 9, 10 enabling the detection of single nucleotide variants (SNV), small insertions/deletions (indels) and copy number variation (CNV).
Fusion transcripts or aberrantly spliced transcripts are another important class of oncogenic somatic alterations. Examples such as BCR‐ABL1 fusion in chronic myeloid leukemia11 or EML4‐ALK fusion in non‐small‐cell lung cancer (NSCLC)12 have led to the development of novel first‐line therapies with corresponding tyrosine kinase inhibitors and have improved the outcome of patients harboring these fusions. Recent advances in sequencing technology have enabled the comprehensive detection of rearrangements in the cancer genome and transcriptome and have indeed resulted in the further discovery of novel oncogenic fusions, such as RET, ROS1, NTRK1/2/3, NRG1 and FGFR1/2/3 fusions in NSCLC.13, 14, 15, 16, 17, 18 Moreover, the precise detection of fusion genes in soft tissue sarcoma is essential for an accurate molecular diagnosis of this disorder.19 Because fusion oncogenes are relatively rare and diverse, the development of methods to detect multiple fusions from formalin‐fixed, paraffin‐embedded (FFPE) specimens is urgently needed for personalized medicine.
However, the detection of fusion genes and transcriptional abnormalities is still difficult in clinical settings where the available material is mainly FFPE samples. Capturing and sequencing intronic DNA to identify gene fusions may be problematic because genomic breakpoints are broadly distributed within corresponding introns that frequently contain multiple repeat sequences.20, 21, 22 Although NGS analysis of transcriptomes (RNA‐seq) is an alternative approach, RNA‐seq using FFPE samples is challenging because the RNA recovered from FFPE is severely fragmented and modified.23, 24, 25, 26 Although other approaches such as the cDNA capture method27 or anchored multiplex PCR‐based method28 were recently reported to be successful methods for RNA‐seq with FFPE samples, large‐scale validation for their clinical use is required.
Here, we describe the development of Todai OncoPanel (TOP), which is a twin‐panel system that enables the comprehensive characterization of cancer‐related genes from FFPE samples. The TOP DNA panel can detect SNV, indels and (CNV) in 464 genes and can infer the tumor mutation burden (TMB) and microsatellite instability (MSI) statuses, while the TOP RNA panel, which uses our original “junction capture method”, sensitively and accurately detects gene fusions and exon skipping and provides transcriptome profiling. Using the TOP panels, we launched a prospective clinical sequencing project at The University of Tokyo Hospital to examine the utility of this NGS‐based molecular cancer profiling for cancer diagnosis and treatment.
2. MATERIALS AND METHODS
2.1. Patient samples
Sixty‐one and 14 FFPE NSCLC specimens were obtained from the Department of General Thoracic Surgery, Graduate School of Medicine, Juntendo University, or the Department of Thoracic Surgery, Graduate School of Medicine, The University of Tokyo, respectively. Samples included resected specimens, endobronchial biopsies, computed tomography‐guided core needle biopsies, and fine‐needle aspiration specimens. Tumor tissue specimens were collected and analyzed using a protocol approved by institutional review boards (IRB) at The University of Tokyo (no. G3546) and Juntendo University (No. 2014176). Informed consent was obtained from all patients involved in this study. Prospective TOP testing for patients with advanced cancer was ordered by the treating physician to identify clinically relevant genomic alterations that could potentially inform treatment decisions. Patients undergoing TOP testing signed a clinical consent form and enrolled in an IRB‐approved research protocol (no. G10114) that permitted the return of results from clinical sequencing and broader genomic characterization of banked specimens for research. Following consent, archival samples were obtained, and blood was drawn as a source of matched normal (germ line) DNA. In total, TOP testing was requested for 183 unique patients between February 2017 and April 2018.
2.2. gDNA target sequencing
Genomic DNA was isolated from FFPE samples using GeneRead DNA FFPE Kits (Qiagen, Hilden, Germany); DNA quality was evaluated by TaqMan FFPE DNA QC Assay v2 (Thermo Fisher Scientific, Waltham, MA, USA), and 500 ng of each sample was subjected to target fragment enrichment using a SureSelectXT Custom kit (Agilent Technologies, Santa Clara, CA, USA). Custom‐made probes were designed to hybridize and capture the gDNA of the target genes listed in Table S1, intronic DNA of 4327 SNP within the targeted gene regions, 10 microsatellite regions including those designated in the Bethesda panel and 17 genes for the detection of genomic rearrangement. Massive parallel sequencing of the isolated fragments was performed using a HiSeq 2500 or Next‐seq platform (Illumina, San Diego, CA, USA) with the paired‐end option. BWA‐MEM (http://bio-bwa.sourceforge.net/) was used to align the paired‐end reads to the human reference genome GRCh38. After removing the PCR duplicates, SRMA29 was used to improve variant discovery through joint local realignments of tumor and matched normal bam files. Somatic mutations were called using Karkinos (https://github.com/genome-rcast/karkinos), which detects SNV, short indels, chromosomal CNV and tumor purity. As previously reported,30, 31 a heuristic algorithm and Fisher's exact tests were used for SNV detection by checking variant allelic frequency, reads cycle, strand bias, mismatch occurrence and presence in the 1000 genomes (http://www.1000genomes.org) and dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/) databases. Further, to optimize the capture deep sequencing, mutations were discarded if they had a read depth of less than 100 or a variant allele frequency (VAF) of less than 0.05. Annotation of the SNV was performed with ANNOVAR.32
2.3. Comparison of mutation rates for the TOP and whole exome sequence (WES)
Libraries for 37 tumor samples were recaptured using an Agilent Exome Kit (v6) and sequenced on an Illumina HiSeq 2500 platform at an average coverage of ×200. Sequence reads were processed using the same bioinformatics procedure described in the previous section. TMB was calculated as the total number of mutations divided by the length of the total genomic target region captured with the exome assay. Similarly, the TMB from the TOP was calculated by dividing the number of sequence mutations reported by the TOP assay by the total genomic area where the mutations were reported. The overall TMB distribution was used to identify a threshold for highly mutated tumors through the following formula: the third quartile (TMB) + 1.5 × IQR (TMB), where IQR is the interquartile range. Samples with a mutation burden of 8.47 or more mutations/Mb were considered hypermutated.
2.4. RNA‐seq using poly‐A selection
Total RNA was extracted from fresh frozen samples using RNA‐Bee (Tel‐Test, Gainesville, FL, USA) and was then treated with DNase I (Thermo Fisher Scientific) and subjected to poly(A)‐RNA selection prior to cDNA synthesis. The library used for RNA‐seq was prepared with an NEBNext Ultra Directional RNA Library Prep Kit (NEB, Ipswich, MA, USA), according to the manufacturer's protocol. NGS sequencing was conducted from both ends of each cluster using a HiSeq2500 or Next‐seq platform (Illumina).
2.5. RNA‐seq using cDNA capture
Total RNA was extracted from FFPE samples using an RNeasy FFPE Kit (Qiagen) and was then treated with DNase I (Thermo Fisher Scientific). RNA quality was evaluated on a 2200 TapeStation using the HighSensitivity RNA Screen Tape system to calculate RNA integrities (RIN) and DV200. cDNA synthesis and library preparation for coding exon capture were conducted using the TruSight RNA Pan‐Cancer Panel (Illumina), according to the manufacturer's protocol. cDNA synthesis and library preparation for junction capture were conducted using a SureSelect RNA Capture kit (Agilent Technologies), according to the manufacturer's protocol. Custom‐made probes for the junction capture method were designed to hybridize and capture the junctional sequences of the target genes listed in Table S1. NGS sequencing was conducted from both ends of each cluster using a HiSeq2500 or Next‐seq platform (Illumina).
2.6. Fusion detection
For the gDNA capture method, the first 30‐mers of paired‐end Read 1 and Read 2 were mapped to hg38 using the paired‐end alignment feature of BWA‐MEM. Reads mapped to the regions of different genes were selected as candidate fusion‐supporting reads. The putative fusion junction points were predicted by mapping those selected reads to hg38 using BLAT. The number of split reads supporting fusion junction sequences was counted using the single‐end alignment function of BWA‐MEM. For the cDNA capture method used to detect novel fusions, the read datasets of cDNA were mapped to Reference Sequence (RefSeq) built by the National Center for Biotechnology Information with the single‐end alignment function of BWA‐MEM. Single‐end reads that mapped to two different genes were predicted to be candidate fusion genes. All possible in‐frame exon‐exon combinations of each constituent candidate fusion gene were constructed to create a hypothetical fusion reference sequence. The number of split reads that supported 60‐mers of fusion junction sequences was counted. The reference sequence of the putative fusion junction was created, and the number of reads that supported 60‐mers of fusion junctions was counted to evaluate the 658 previously reported fusions. The number of reads that supported 60‐mers of the wild‐type constituents of the fusion gene was also counted to estimate the existence of fusion genes using the following criteria: positive, fusion‐supporting read number more than 0; negative, wild‐type read of 5′‐gene or 3′‐gene number of 50 or more and fusion‐supporting read number of 0; or N.D. (not determined), wild‐type read of 5′‐gene or 3′‐gene number of less than 50 and fusion‐supporting read number of 0. Using the data from housekeeping genes, we set the following criteria for good quality RNA‐seq experiments: the mean coverage of housekeeping genes was more than ×500, and the percentage of housekeeping gene regions with more than ×100 coverage was more than 70%.
2.7. Detection of exon skipping
To detect MET exon 14 skipping, we generated a reference sequence comprising the 3′ junction of MET exon 13 and the 5′ junction of exon 15. Similar to fusion detection, exon skipping was estimated by counting split reads that supported 60‐mers of the junction.
2.8. Clinical assessment and matching to clinical trials
For the annotation of somatic mutations, we checked OncoKB, a curated knowledge base of the oncogenic effects and treatment implications of gene alterations (http://oncokb.org); CIViC (Clinical Interpretations of Variants in Cancer), a community‐based curation database (https://civicdb.org); and ClinVar. In addition, we used COSMIC (Catalogue of Somatic Mutations in Cancer, https://cancer.sanger.ac.uk/cosmic) to assess mutation frequency in cancer. We also annotated some gene mutations using specific databases: BRCA Exchange (http://brcaexchange.org) for BRCA1/2, IARC TP53 (http://p53.iarc.fr) for TP53, and InSiGHT (https://www.insight-database.org/genes) for mismatch repair genes. We classified mutations in a tumor type‐specific manner according to the level of evidence that each gene alteration is a predictive biomarker of drug response, as indicated in the Results section. We annotated tumor samples according to the highest level of evidence for any mutation identified by TOP testing. We constructed a database of Pharmaceuticals and Medical Devices Agency‐approved drugs and biomarker targets using their websites. We also constructed a database of US Food and Drug Administration (FDA)‐approved drugs and biomarker targets using the FDA and National Cancer Institute websites. Our knowledge database also included some clinical trial databases. We collected datasets from ClinicalTrials.gov and the Japanese clinical trials databases UMIN, JAPIC and JMACCT. Our database uses natural language processing according to gene symbols, drug names and cancer types, enabling us to find clinical trials that target gene mutations. We also introduced English‐Japanese machine translation because these data sources are written in either English or Japanese. In the database, we normalized the clinical trial fields such as inclusion/exclusion criteria, title, intervention and status for cross‐over search.
2.9. RNA expression analysis
The TOP RNA panel probes were not designed to achieve uniform depth. To calculate gene expression using the TOP RNA panel, we determined weights for correcting the probe depth at each position. Weighted reads per kilobase million (RPKM) wrpkmg,n with gene g and specimen n was computed by adjusting the read depth using the weight. For comparisons among specimens, a mean value mn of the logarithm of the weighted RPKM for the housekeeping genes included in the TOP RNA panel (ACTB, GAPDH, H3F3A, HPRT1, HSP90AB1, NPM1, PPIA, RPLP0, TFRC and UBC) was defined by , where n = 1, …, N. G and N are the number of housekeeping genes included in the TOP RNA panel and the number of the identical tumors with FFPE and frozen specimens, respectively. In this case, G = 10 and N = 7. nf and nF are frozen and FFPE specimens. As in the trimmed mean of M‐value normalization method, a correction coefficient was defined by the mean of difference between the logarithm of the weighted RPKM of the housekeeping gene and the mn for each specimen. There were high correlations between normalized RPKM of 109 genes of FFPE and frozen specimen of the identical tumors. Moreover, a heatmap representation with dendrogram and partition was drawn using normalized RPKM values of 338 specimens that exceeded quality control criteria of TOP RNA panel.
2.10. Reverse transcription (RT)‐PCR analysis
Total RNA was extracted from FFPE samples using an RNeasy FFPE Kit (Qiagen) and was then treated with DNase I (Thermo Fisher Scientific). RT was performed with SuperScript™ IV VILO (Thermo Fisher Scientific). The resulting first‐strand cDNA were used as templates and amplified by PCR using PrimeSTAR polymerase (Takara Bio, Shiga, Japan). The following primer sets were used: AHRR‐NCOA2, 5′‐CGCGGATGCAAAAGTAAAAGC‐3′ (sense) and 5′‐TGCTGGGTTCCGAATCATACC‐3′ (antisense); and TAF15‐NR4A3, 5′‐ACCAGCAGTCAGGCTATGATC‐3′ (sense) and 5′‐TATTCCGAGCTGTATGTCTGC‐3′ (antisense). PCR products were subjected to capillary sequencing using a 3130xl Genetic Analyzer (Thermo Fisher Scientific).
3. RESULTS
3.1. Development of the TOP DNA panel
The capturing probes for the TOP DNA panel version (V)1 were designed to examine 464 cancer‐related genes chosen by the molecular tumor board at The University of Tokyo Hospital (Table S1A). The TOP DNA panel V1 assesses 410 genes tested by MSK‐IMPACT (a common NGS‐based panel in the USA)4 and the promoter region of TERT. For detecting allele‐specific CNV, capture probes for examining 4327 heterozygous SNP around 464 target genes were designed (Figure S1). The intronic DNA of 17 genes and 10 microsatellite regions including those designated in the Bethesda panel are covered for the detection of genomic rearrangement and MSI, respectively. Thus, the DNA panel is capable of detecting all SNV/indels/CNV of 464 genes as well as TERT promoter mutations, MSI and genomic rearrangement in the selected genes.
3.2. Junction capture method for RNA‐seq of the FFPE samples
Although gene fusions can be detected by capturing and sequencing the gDNA of intronic regions, the number of capture probes for such methods tends to become large because human gene introns can exceed more than 10 kbp. Conversely, fusion detection with RNA capture may be unreliable because RNA in FFPE samples are often severely degraded. Therefore, we first investigated which method is more sensitive for the detection of fusion genes from FFPE specimens.
For an RNA capture approach, we developed the “junction capture method” in which capture probes are designed only within 120 bases from the putative junction sites of fusion gene transcripts (Figure 1A,B). Because the size of RNA isolated from FFPE samples is approximately 200 bases, sequence reads captured by the probes at distances of more than 120 bp from the junction points generally do not support fusion events (hypothesizing 80‐bp overlapping of a probe with a target sequence is necessary for capturing). Therefore, we set capture probes at the junction site at a ×5‐10 tiling density to maximize the sensitivity. Furthermore, our computational pipeline was designed to compare the number of sequence reads encompassing each junction site with that of the reads supporting the corresponding wild‐type transcript (Figure 1C); thus, we could eliminate false‐negative results in which the fusion reads and wild‐type reads were both negative (Figure 1C).
Figure 1.
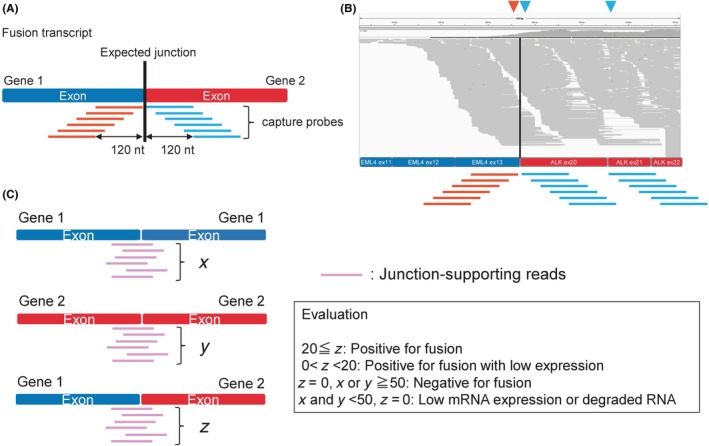
Exon junction capture method. A, Capture probes were designed to target the cDNA sequences within 120 bases from the estimated fusion junctions to efficiently and specifically obtain split reads that support the fusion transcript. B, Visualization of the sequence reads. The y‐axis represents the read coverage; the x‐axis represents the reference for the putative EML4‐ALK fusion transcript. Reads aligned to the reference sequence are shown in gray. The red arrowhead indicates the 3′ junction of EML4 exon 13, and the blue arrowheads indicate the 5′ junctions of ALK exons 20 and 21. The red and blue lines represent the capture probes for exon 13 of EML4 and exon 20 or 21 of ALK. C, The number of sequence reads that support the fusion transcript breakpoint (x) was counted and compared with wild‐type transcripts for the individual genes (y and z) to evaluate whether the fusion transcript is dominantly expressed. The existence of each wild‐type gene transcript and the absence of a fusion gene transcript confirm that the fusion transcript truly does not exist. Notably, low mRNA expression or mRNA degradation should be considered when a read number for each wild‐type gene transcript is not obtained
For a pilot experiment, we developed a target RNA‐seq panel based on the junction capture method (TOP RNA panel V1) that covered 67 fusion genes (Table S1B) and examined it and the TOP DNA panel V1. We also tested the TruSight RNA Pan‐Cancer Panel (Illumina), which captures coding exons of cancer‐related genes, and Archer FusionPlex Solid Tumor Kit (ArcherDX, Boulder, CO, USA) which is based on anchored multiplex PCR.
For the pilot study, 10 NSCLC FFPE specimens that were previously identified as positive for fusion genes were used. Interestingly, as shown in Table 1 and Figure 2A, the TOP RNA panel more accurately detected fusion genes than the TOP DNA panel, and the number of split reads for the corresponding fusion points was much larger for the TOP RNA panel than that obtained with the TOP DNA panel, suggesting that junction capture RNA‐seq is the preferable method for detecting fusion genes.
Table 1.
Summary of a pilot study to compare the capability of the junction capture method to detect fusion genes with that of a gDNA capture method
| Case # | Diagnosis | Sample year | RIN score | Fusion gene | TOP RNA panel (junction capture) | gDNA capture | Anchored multiplex PCR | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Split read number | Raw read number | TOP version | Split read number | Raw read number | Split read number | Raw read number | |||||
| #1 | NSCLC | 2012 | N.D. | EML4‐ALK | 232 | 28 873 868 | V1 | 12 | 53 647 702 | 68 | 1 941 324 |
| #2 | NSCLC | 2012 | N.D. | EML4‐ALK | 284 | 29 302 268 | V1 | 108 | 61 299 256 | 22 | 2 354 508 |
| #3 | NSCLC | 2013 | N.D. | EML4‐ALK | 180 | 29 903 016 | V1 | 44 | 58 450 392 | 10 | 2 639 006 |
| #4 | NSCLC | 2014 | N.D. | EML4‐ALK | 4179 | 30 546 206 | V1 | 0 | 66 769 928 | 278 | 3 019 450 |
| #5 | NSCLC | 2012 | N.D. | KIF5B‐RET | 514 | 27 969 576 | V1 | 0 | 57 830 964 | 0 | 54 170 756 |
| #6 | NSCLC | 2013 | N.D. | KIF5B‐RET | 2898 | 27 396 626 | V1 | 150 | 69 819 612 | 0 | 58 501 738 |
| #7 | NSCLC | 2014 | 1.3 | KIF5B‐RET | 1189 | 27 750 096 | V1 | 53 | 62 606 194 | 5 | 65 226 738 |
| #8 | NSCLC | 2011 | N.D. | SLC34A2‐ROS1 | 1633 | 30 614 778 | V1 | 17 | 64 415 016 | 13 | 45 395 450 |
| #9 | NSCLC | 2012 | 1.4 | CD74‐ROS1 | 5268 | 29 494 774 | V1 | 0 | 60 541 260 | 0 | 53 252 448 |
| #10 | NSCLC | 2013 | N.D. | CD74‐ROS1 | 2491 | 28 970 804 | V1 | 77 | 69 241 262 | 0 | 56 481 374 |
N.D., not detected; NSCLC, non‐small‐cell lung cancer; PCR, polymerase chain reaction; RIN, RNA integrity number.
Figure 2.
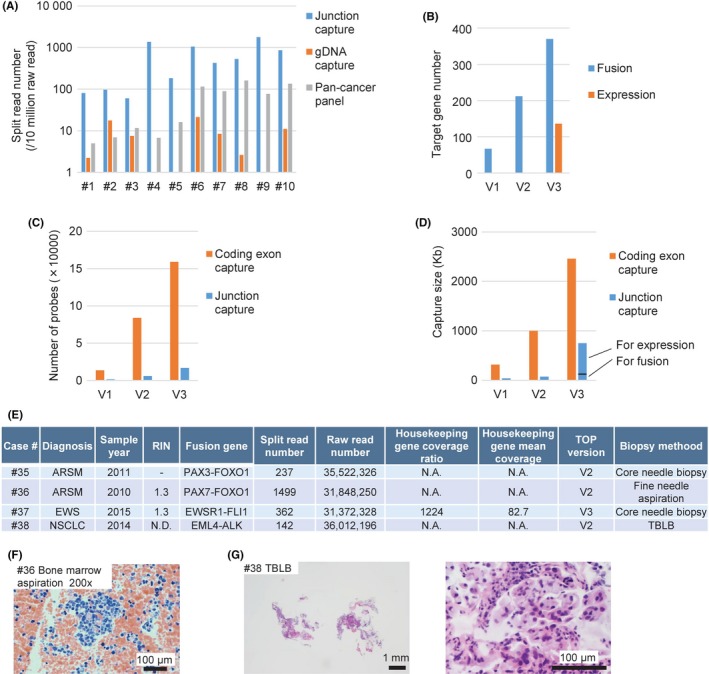
Efficiency of the junction capture method and its application to biopsy specimens. A, The split read number per 10 million raw reads obtained using each indicated method. B, The target gene number for fusion gene detection or expression analysis using each version of the TOP RNA panel. The estimated probe number (C) and target capture size (D) using the junction capture method or the coding exon capture method for each TOP RNA panel. E, A summary of the pilot study used to assess the capability of TOP RNA panel to detect fusion genes using non‐small cell lung cancer and sarcoma formalin‐fixed, paraffin‐embedded biopsy samples that were previously identified as positive for fusion genes. ARMS, alveolar rhabdomyosarcoma; EWS, Ewing sarcoma; N.A., not available; N.D., not detected; NSCLC, non‐small‐cell lung cancer; RIN, RNA integrity number; TBLB, transbronchial lung biopsy. F, Representative photograph of a bone marrow aspiration specimen stained with hematoxylin‐eosin (original magnification ×200; scale bar, 100 μm). G, Representative photographs of TBLB specimens stained with hematoxylin‐eosin (left: ×40 magnification; scale bar, 1 mm; right: ×400; scale bars, 100 μm)
3.3. Development of the TOP RNA panel
Therefore, we decided to use a twin‐panel system, with both DNA‐ and RNA‐based panels, for clinical sequencing. The TOP RNA panel was revised several times. V2 was designed to cover sarcoma fusion genes (Table S1C). V3 was developed to examine all fusion genes reported in the COSMIC database (http://cancer.sanger.ac.uk/cosmic) (Table S1D) and to measure the expression levels of 10 housekeeping genes as well as a total of 109 oncogenes and tumor suppressor genes reported in a previous study (Table S1E).33 In contrast to the junction capture method, the capture probes for the gene expression analyses were designed for all coding exons at a ×2 tiling density (Figure 2B). A minor version up was performed to cover two additional fusion genes in V4. In total, V4 can detect 365 fusion transcripts as well as MET exon skipping, and it can evaluate the expression levels of 109 cancer‐associated genes (Table S1D,E).
To investigate the analytical validity of the TOP RNA panel, 24 specimens of NSCLC and sarcoma already known to be positive for fusion genes were examined. Although the RIN score of the RNA extracted from such FFPE specimens was 1.1‐2.3, suggesting severe degradation, all fusion transcripts were successfully detected (Table 2). Notably, both the probe number and total capture size of the TOP RNA panel V1 were 14‐times smaller than those of panels that capture total coding exons such as the TruSight RNA Pan‐Cancer Panel (Figure 2C,D and Table S2), suggesting that our junction capture method is very cost‐effective.
Table 2.
Capability of the junction capture method to detect fusion genes in non‐small cell lung cancer and sarcoma FFPE
| Case # | Diagnosis | Sample year | RIN score | Fusion gene | TOP RNA panel (junction capture) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Split read number | Housekeeping gene coverage ratio | Housekeeping gene mean coverage | Raw read number | TOP version | |||||
| #11 | SS | 2011 | N.D. | SS18‐SSX1 | 4410 | N.A. | N.A. | 38 679 304 | V2 |
| #12 | SS | 2012 | 1.1 | SS18‐SSX2 | 492 | N.A. | N.A. | 36 669 700 | V2 |
| #13 | SS | 2012 | N.D. | SS18‐SSX1 | 629 | N.A. | N.A. | 42 335 766 | V2 |
| #14 | SS | 2013 | 2 | NAB2‐STAT6 | 4232 | N.A. | N.A. | 37 190 310 | V2 |
| #15 | SS | 2015 | 2 | EWSR1‐FLI1 | 110 | N.A. | N.A. | 37 234 264 | V2 |
| #16 | SS | 2016 | 2.2 | SYT‐SSX1 | 1446 | N.A. | N.A. | 35 024 302 | V2 |
| #17 | LGFS | 2014 | 1.4 | FUS‐CREB3L2 | 213 | N.A. | N.A. | 35 995 548 | V2 |
| #18 | NSCLC | 2014 | 1 | EML4‐ALK | 109 | 90.8 | 1180 | 33 405 300 | V3 |
| #19 | NSCLC | 2014 | N.D. | EML4‐ALK | 119 | 90 | 1171 | 32 722 906 | V3 |
| #20 | NSCLC | 2014 | 1.9 | EML4‐ALK | 175 | 90.1 | 1158 | 41 014 170 | V3 |
| #21 | NSCLC | 2014 | 1.8 | EML4‐ALK | 156 | 90.8 | 1184 | 40 714 948 | V3 |
| #22 | NSCLC | 2011 | 1.7 | EML4‐ALK | 396 | 88.5 | 1130 | 31 226 146 | V3 |
| #23 | NSCLC | 2012 | N.D. | EML4‐ALK | 107 | 89.8 | 1172 | 37 998 262 | V3 |
| #24 | NSCLC | 2012 | N.D. | KIF5B‐RET | 369 | 89.6 | 1175 | 28 702 718 | V3 |
| #25 | NSCLC | 2013 | N.D. | KIF5B‐RET | 423 | 89.6 | 1175 | 31 530 656 | V3 |
| #26 | NSCLC | 2012 | N.D. | CD74‐ROS1 | 13 | 87.9 | 1147 | 28 263 560 | V3 |
| #27 | NSCLC | 2013 | N.D. | CD74‐ROS1 | 28 | 88.8 | 1160 | 31 899 126 | V3 |
| #28 | NSCLC | 2014 | 1.9 | TPM3‐ROS1 | 32 | 89.7 | 1186 | 29 557 566 | V3 |
| #29 | NSCLC | 2014 | 1.3 | CD74‐NRG1 | 59 | 88.6 | 1174 | 34 571 210 | V3 |
| #30 | ARSM | 2005 | N.D. | EWSR1‐FLI1 | 35 | 89.9 | 1115 | 29 546 176 | V3 |
| #31 | EWS | 2012 | 1 | EWSR1‐FLI1 | 412 | 92.3 | 1145 | 29 850 060 | V3 |
| #32 | EWS | 2013 | 1.6 | EWSR1‐FLI1 | 544 | 93.2 | 1195 | 30 060 100 | V3 |
| #33 | EWS | 2015 | 2.2 | EWSR1‐FLI1 | 394 | 92.3 | 1145 | 31 835 816 | V3 |
| #34 | EWS | 2015 | 1.3 | EWSR1‐FLI1 | 384 | 90.7 | 1090 | 34 319 676 | V3 |
ARMS, alveolar rhabdomyosarcoma; EWS, Ewing sarcoma; FFPE, formalin‐fixed, paraffin‐embedded; LGFS, low‐grade fibromyxoid sarcoma; RIN, RNA integrity number; SS, synovial sarcoma. ; N.A., not available; N.D., not detected; NSCLC, non‐small‐cell lung cancer
Using our computational pipeline for the TOP RNA panel V3, 658 putative fusion junctions were investigated. As an example, the evaluation of 31 putative fusion genes was demonstrated for case 23, which was positive for EML4‐ALK (Figure S2). Supporting reads for the EML4‐ALK(E20;A20) fusion were successfully detected as expected. Another 27 candidate fusion genes in case 23 were filtered out as fusion‐negative by our pipeline because their fusion transcript read numbers were 0 while those of the corresponding wild‐type transcripts were more than 50. Evaluations of the other three putative fusion genes could not be performed because the read numbers of the wild‐type transcripts of the genes were too small (<50) to exclude the possibility of pseudonegative results.
We further evaluated whether the TOP RNA panel was applicable for small biopsy specimens. RNA was prepared from the FFPE sections of fusion‐positive core‐needle biopsy, fine‐needle aspiration or transbronchial lung biopsy specimens. Surprisingly, the TOP RNA panel detected a large number of split reads, successfully supporting the presence of fusion transcripts in each specimen (Figure 2E‐G).
3.4. TOP RNA panel can detect exon skipping
We further evaluated the capability of junction capture RNA‐seq to detect aberrant transcripts, such as MET exon 14 skipping, which was previously reported to be oncogenic.34, 35 RNA was extracted from FFPE sections of surgically resected tissues from five lung adenocarcinoma cases that had been shown to harbor MET exon 14 skipping by RNA‐seq. We counted the number of split reads that supported the skipping of MET exon 13 to exon 15. The TOP RNA panel successfully identified MET exon 14 skipping in all five FFPE samples, whereas no split reads were detected in the other skipping‐negative 34 cases (Figure 3 and Figure S3, Table S3).
Figure 3.
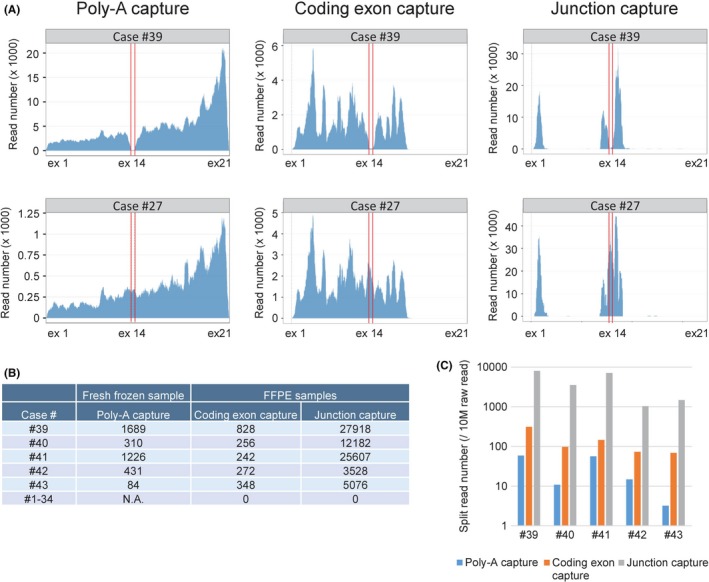
Detection of MET exon 14 skipping. A, RNA‐seq reads for a MET exon 14 skipping‐positive case (#39) and a MET exon 14 skipping‐negative case (#27) mapped to virtual MET cDNA constructed on the x‐axis that corresponds to the transcript of NM_000245. RNA‐seq was performed using three different methods: poly(A) selection of RNA (NEBNext Ultra Directional RNA Library Prep Kit, NEB) extracted fresh frozen samples and coding exon capture (TruSight RNA Pan‐Cancer Panel, Illumina) or junction capture of cDNA synthesized from RNA extracted from formalin‐fixed, paraffin‐embedded (FFPE) samples. Regions between red lines indicate MET exon 14. B, The number of split reads that support the transcript of MET exon 13 connected to exon 15 was counted in five cases positive for MET exon 14 skipping or in 34 negative cases by RNA‐seq using three different methods, as described in (A). C, The number of split reads that support the junction of MET exon 13 to exon 15 per 10 million raw reads of each indicated method is shown
3.5. TOP RNA panel can measure gene expression profiles
Expression analysis was conducted for seven tumors to compare the performance of the TOP RNA panel using FFPE specimens with poly(A)‐RNA‐seq using frozen specimens. The mRNA expression values of the 109 genes in the TOP RNA Panel were highly concordant with those determined by poly(A)‐RNA sequencing, even though the former data were obtained using FFPE specimens (Figure S4).
3.6. Retrospective clinical sequencing of NSCLC cases with the TOP
We next applied our junction capture RNA‐seq method to FFPE specimens from 35 cases of surgically resected NSCLC at stage II/III that were negative for mutations in KRAS and EGFR. MET exon 14 skipping, EML4‐ALK fusion and CCDC6‐RET fusion were detected in 17.1% of the total cases by the TOP RNA panel V3, whereas NF1, ERBB2 or BRAF mutation was found in 14.3% of the total cases by the TOP DNA panel V1 (Figure S5 and Table S4).
3.7. Development of a TOP clinical sequence workflow
In our Clinical Laboratory Improvement Amendments (CLIA) compliant laboratory at The University of Tokyo Hospital, a prospective research study for clinical sequencing using the TOP twin‐panel (TOP DNA panel V1 and RNA panel V4) was initiated in February 2017 (Figure 4). For every patient, the TOP DNA panel was used to examine both tumor and paired‐normal specimens, and the RNA panel was used to analyze only tumors. We could thus assess the somatic and germ line mutations independently. The resultant mutation information was referred to a cancer knowledge database developed in house and was used to produce clinical reports.
Figure 4.
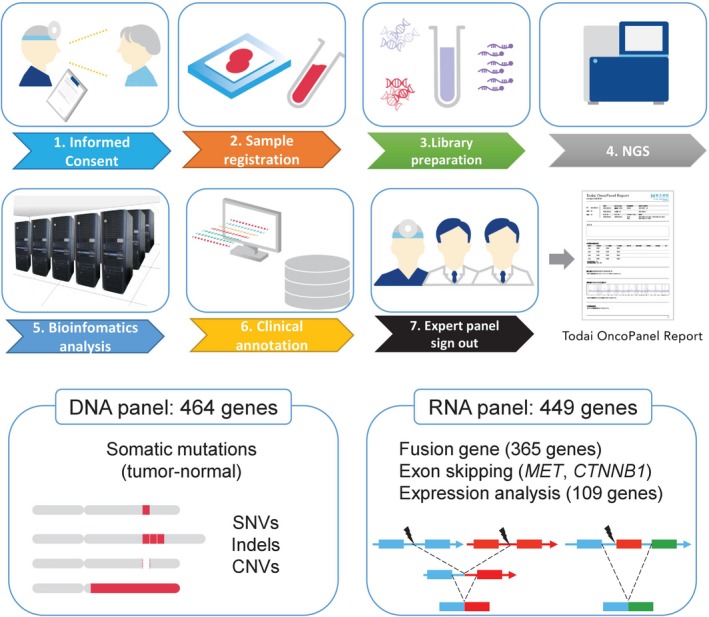
Overview of the TOP workflow. Patients provide informed consent for paired tumor‐normal sequence analysis, and a blood sample is collected as a source of normal DNA. DNA is extracted from the tumor and blood samples, and sequence libraries are prepared and captured using hybridization probes targeting all coding exons of 463 genes and the TERT promoter region. RNA is extracted from tumor samples and reverse transcribed. cDNA sequence libraries are prepared and captured using hybridization probes targeting 365 fusion genes, MET and CTNNB1 exon skipping, and 109 genes for expression analysis. Following sequencing, paired reads are analyzed through a custom bioinformatics pipeline that detects multiple classes of genomic and transcriptional alterations. The results are loaded into a genomic variants database developed in house, where they are manually reviewed for quality and accuracy. After expert panel (molecular tumor board) review of each case, the final version of each TOP report is signed out and transmitted to the electronic medical record. CNV, copy number variant; NGS, next‐generation sequencing; SNV, single nucleotide variant
We built an expert panel (a molecular tumor board) consisting of physicians, pathologists, genetic counselors, molecular biologists, cancer genome researchers, bioinformaticians and bioethics researchers. The expert panel met biweekly to review every case and to sign out the clinical reports that were automatically sent to the electronic medical record server (Figure 4).
3.8. Prospective TOP clinical sequencing
Between February 2017 and May 2018, we obtained 315 specimens (210 primary tumors) from 183 individuals for prospective TOP sequencing (Figure 5A and Figure S6) in our CLIA‐conformed laboratory. We achieved an average throughput of 50 specimens per month over the last 2 months of this study (Figure S6).
Figure 5.
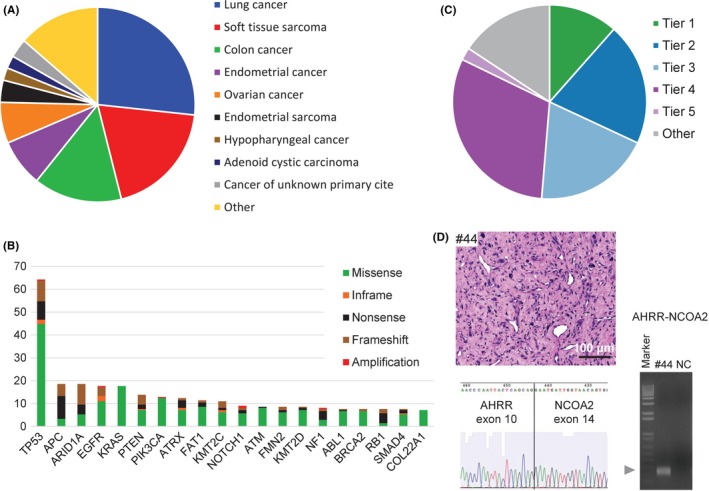
Overview of the TOP prospective cohort. A, Distribution of tumor types among the cases successfully sequenced from 183 patients. Cases represented more than 10 principal tumor types. B, The 20 most recurrent somatic alterations. Bars indicate the percentage of cases harboring the mutations, and the types of genomic alterations are color‐coded. C, Clinical actionability of somatic alterations revealed by the TOP. Alterations were annotated based on their clinical actionability according to TOP classification (Figure S10), and samples were assigned to the level with the most actionable alteration. Briefly, the levels of evidence vary according to whether the mutations are a Pharmaceuticals and Medical Devices Agency‐recognized biomarker (Tier 1), a Japanese clinical trial‐targeted biomarker or a US Food and Drug Administration‐recognized biomarker (Tier 2), an investigational agent sensitizing gene alteration or an oncogenic alteration supported by a knowledge database (Tier 3), an oncogenic alteration supported by a research paper (Tier 4) or a recurrently reported alteration in a knowledge database. D, A representative photograph of tumor #44 stained with hematoxylin‐eosin (scale bar, 100 μm; upper left panel). The proliferation of spindle cells with atypical nuclei and surrounding myxoid interstitial tissue were observed. A fresh frozen sample from case #44 was subjected to reverse transcription (RT) polymerase chain reaction with the fusion‐RT primer set to detect AHRR‐NCOA mRNA (lower left panel). The arrowhead indicates the estimated sizes of the fusion transcript. The band was extracted from the gel and subjected to Sanger sequencing. The electrophoretogram obtained from the band supported the junction sequences of AHRR‐NCOA2 (right panel). Marker, 1‐kb DNA ladder; NC, negative control
Given the diversity of the cases and specimen types submitted, the samples exhibited a wide range of nucleic acid quality metrics. Tissues with insufficient DNA yield (DNA < 200 ng; n = 9, 2.9%) were reported as inadequate, and we excluded samples (n = 7, 2.2%) that did not meet strict postsequencing quality control criteria (Figure S7). DNA input and sample age influenced sequencing performance (Figure S7). Altogether, we successfully sequenced 298 tumor specimens (94.9%) from 181 individuals (94.8%).
For RNA‐seq, we excluded samples that did not meet strict postsequencing quality control criteria using the coverage of housekeeping genes. RNA input and DV200 (the percentage of RNA fragments >200 nucleotides) influenced the sequencing performance (Figure S8). We successfully sequenced 269 tumor specimens (85.4%) from 167 individuals (87.4%) and achieved a success rate of 100% when we analyzed samples with an RNA input of more than 200 ng and a DV200 of more than 40%.
Tumors were sequenced with deep coverage (mean = 913) to ensure a high sensitivity for detecting genomic alterations in heterogeneous and low‐purity specimens (Figure S9A). Altogether, we detected 2337 non‐synonymous mutations, with a median VAF of 0.21, as well as 145 CNV, 53 fusion transcripts and one instance of MET exon 14 skipping. Among the 210 primary tumors, the most frequently altered gene was TP53, followed by APC, ARID1A EGFR, KRAS, PTEN, PIK3CA, ATRX, FAT1 and KMT2C (Figure 5B). To investigate how our results compared with those from one of the largest clinical sequencing cohorts (at Memorial Sloan Kettering Cancer Center), we examined MSK‐IMPACT data from cBioPortal (http://www.cbioportal.org/). Overall, the TOP results were highly consistent with the MSK‐IMPACT findings, exhibiting strong concordance in terms of the identities and frequencies of the mutations detected (Figure S9B).
We systematically evaluated the clinical utility of prospective molecular profiling to guide treatment decisions. We established a TOP classification (Figure S10), using databases for clinical trials as well as several curated cancer knowledge databases such as OncoKB (http://oncokb.org/) and COSMIC (https://cancer.sanger.ac.uk/cosmic), to group all mutations into tiers of clinical actionability. Mutations were classified in a tumor type‐specific manner according to the level of evidence that each mutation is a predictive biomarker of drug response. Altogether, 32.2% of cases (n = 59) harbored at least one actionable alteration (Tier 1 or Tier 2), while 85.8% of cases (n = 157) harbored at least one clinically annotated alteration (Tier 1 to Tier 5) (Figure 5C). The proportion of actionable mutations in lung cancer, sarcoma, gynecological cancer and colorectal cancer were 43.1%, 13.6%, 52.6% and 13.8%, respectively (Figure S11).
Tumor mutation burden may help predict the response to immune checkpoint inhibitors. To determine whether TMB could be inferred from the targeted capture data, we first calculated the distribution of the TOP sequencing mutation rates and then identified a threshold of 8.5 mutations/Mb as indicative of a high mutation burden (Figure S12A).
Comparisons with matched WES data from 37 tumors in this cohort revealed a high correlation in TMB when the mean depth of TOP sequencing was more than 300 (r = 0.87; Figure S12B). We further assessed the mutation signature of high‐TMB specimens (n = 13, 4.1%) and found that tumors with mutations in mismatch‐repair (MMR) genes such as MSH2, MSH6 or MLH1 exhibited Signature 6, which is associated with defective DNA mismatch repair and is found in microsatellite unstable tumors36 (Figure S13).
Germ line mutations in hereditary cancer genes can predict good response to therapeutic reagents, as shown in the response of cases with BRCA1/2 mutations to PARP inhibitors.37 We have established an IRB‐approved process for prospective germ line analysis whereby inherited pathogenic variants of 10 genes (TP53, BRCA1, BRCA2, MLH1, MSH2, MSH6, PMS2, APC, RB1 and PTEN) detected by the TOP DNA panel are reported to patients who wish to receive this information. Clinical sequencing of the 187 individuals in this study revealed seven pathogenic germ line mutations (four BRCA1, two BRCA2 and one MSH2 mutation) (Figure S14), three of which were reported to the corresponding patients in the presence of a certified genetic counselor.
Using the TOP RNA panel, 54 fusion transcripts and one instance of MET exon 14 skipping were identified, and 27.8% of them were clinically relevant (Tier 1 to Tier 5) (Figure S15A). Among 33 tumors without any clinically annotated DNA mutations, nine (27.2%) tumors harbored aberrant transcripts identified by the TOP RNA panel. Among 41 patients with sarcoma analyzed prospectively, one patient harbored a TAF15‐NR4A3 fusion, consistent with the pathological diagnosis of extraskeletal myxoid chondrosarcoma (Figure S15B).38 Another patient was diagnosed with myxofibrosarcoma due to the proliferation of spindle cells with atypical nuclei and surrounding myxoid interstitial tissue (Figure 5D). However, the pathological diagnosis was changed after detecting the AHRR‐NCOA2 fusion gene, which is known to be specific for angiofibroma of soft tissues (Figure 5D).39 The presence of TAF15‐NR4A3 and AHRR‐NCOA2 fusions was further confirmed by RT‐PCR and sequencing of the PCR products (Figure 5D and Figure S15B).
3.9. Clinical utility of gene expression profiling by the TOP RNA panel
The clinical utility of the TOP RNA panel in predicting the primary organs of cancer was tested by clustering the 338 specimens based on mRNA expression profiles, which revealed that these specimens could be grouped into five branches (Figure 6A). Clusters 1, 2a, 3, 5a and 5b separately consisted of either sarcoma, lung cancer, gynecological cancer or colon adenocarcinoma, suggesting that expression profiling with the TOP RNA panel may reflect the primary organ sites of tumors.
Figure 6.
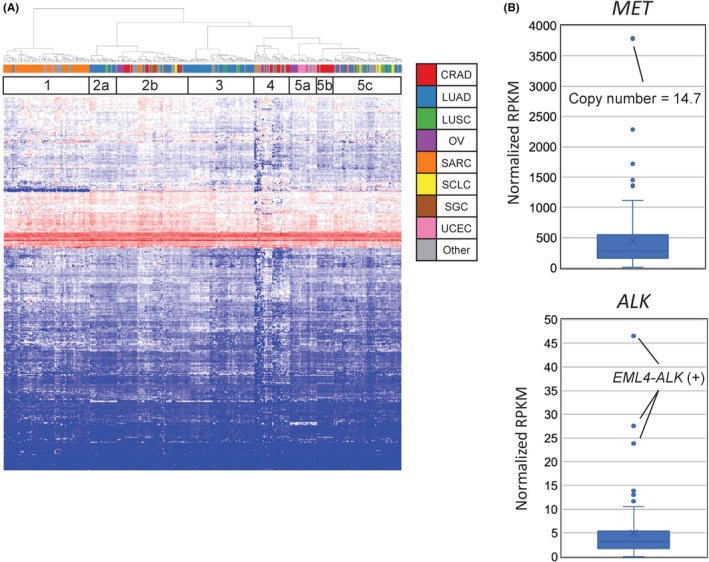
mRNA expression analysis using the TOP RNA panel. A, mRNA expression clustering analysis for the 338 specimens was performed to investigate the clinical utility of the TOP RNA panel in the prediction of the primary organs of cancers of unknown primary sites or the histopathological identities of unclassified tumors. The 338 specimens analyzed by the TOP RNA panel V4 were classified into five large categories. Clusters 1, 2a, 3, 5a and 5b dominantly consisted of either sarcomas, lung cancers, gynecological cancers or colon adenocarcinoma. The tumor types are color‐coded in the upper label. CRAD, colorectal adenocarcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian cancer; SARC, sarcoma; SCLC, small cell lung cancer; SGC, seminoma and germ cell tumor; UCEC, uterine corpus endometrial carcinoma. B, Expression of the MET gene in a case of copy number amplification and of the ALK gene in fusion‐positive cases were quantified and compared with those of the other 80 cases of lung adenocarcinoma
To examine the concordance between gene copy number/rearrangements and gene expression levels, as an example, we analyzed CNV and the expression of MET and ALK. As shown in Figure 6(B), among 80 lung adenocarcinoma specimens, the expression level of MET was highest in a tumor with gene amplification (MET copy number = 14.7). Similarly, abundant expression of ALK mRNA was observed only in the three cases with an EML4‐ALK fusion gene. Thus, the TOP twin panel system can accurately reveal how genetic anomalies affect the expression levels of cancer‐related genes.
3.10. Revision of the TOP DNA panel for MSI assessment
Because the MSI status of cancer cells is used as a response predictor for immune checkpoint inhibitors, we designed DNA capture probes for 10 microsatellite regions including those designated in the Bethesda panel.40 Moreover, additional probes were included in the TOP DNA panel V2 to interrogate 500 microsatellite loci that were commonly altered in our WES analysis of high‐MSI colorectal cancer.41 To detect MMR‐deficient tumors, MSIsensor scores42 were calculated using the TOP DNA sequence data. Among 36 specimens of colorectal cancer with Lynch syndrome, 34 had an MSI score of more than 3.5, the cut‐off value used to determine high MSI in a previous study.42 Furthermore, 31 of 34 specimens with such MSI scores had a TMB of more than 8.5. In contrast, five of 27 specimens of other cancer types with unknown MMR status had an MSI score of more than 3.5, and three of these cases had a TMB of more than 8.5 (Figure S16).
4. DISCUSSION
Although NGS‐based tumor genome analyses have proved their utility in molecular profiling for personalized medicine, the types of cancer gene panel systems that most reliably detect genetic alterations in tumors have not been fully validated. Here, we reveal that RNA‐seq has much better sensitivity and specificity in gene fusion detection than genomic DNA sequencing, even when using FFPE specimens. Furthermore, for the detection of fusion transcripts by RNA‐seq, targeting the sequences that support fusion (split reads) is more cost‐effective than targeting the whole coding regions of transcripts. We thus invented the junction capture method, which specifically targets breakpoint exons. The junction capture method detects novel fusion transcripts if one of the constituent genes is targeted by our capture panel. In addition, our pipeline counts not only fusion reads but also their corresponding wild‐type reads around the junction sites, which is highly useful in excluding false‐positive and false‐negative results. This aspect is particularly important in clinical sequencing because FFPE samples are the main source of diagnostic material and RNA from FFPE specimens is often severely degraded.
In our cohort of stage II or III NSCLC without EGFR/KRAS mutations, the RNA junction capture method successfully identified MET exon skipping and ALK or RET gene fusions in 17.1% of the cases. In contrast, TOP DNA panel analyses on the same cohort identified mutations in BRAF, ERBB2 or NF1 in 14.3% of cases. Therefore, by combining the two panels, one‐third of the cases were shown to harbor target able genetic changes. Because the 5‐year survival rates for patients with stage II and III NSCLC are approximately 55% and less than 40%, respectively,43 molecular profiling is beneficial for these patients.
Furthermore, the identification of fusion genes using junction capture RNA‐seq is useful in the molecular diagnosis of sarcoma, which is characterized by very diverse mutations with various (and often disease‐specific) fusion genes. In fact, the versatility of the TOP system was demonstrated in a case in which the detection of an AHRR‐NCOA2 fusion changed the diagnosis from myxofibrosarcoma, one of the most aggressive types of soft tissue neoplasms, to angiofibroma of soft tissues, a benign fibrovascular soft tissue tumor. However, caution is warranted because the morphological findings were atypical of angiofibroma of soft tissues, suggesting the possibility of an atypical AHRR‐NCOA2 fusion‐positive sarcoma.
The TOP DNA panel succeeded in obtaining homogenous coverage throughout the targeted genes and achieved similar sensitivity and specificity as those of WES using fresh frozen specimens. In addition to the detection of somatic mutations such as missense mutations, insertions, deletions and CNVs, the TOP DNA pipeline also implemented MSIsensor for the prediction of MSI status and the calculation of TMB to predict sensitivity to immunocheckpoint inhibitors.
With the TOP twin panels, we constructed an infrastructure for clinical sequencing at The University of Tokyo Hospital and assembled an expert team to properly assess the resultant sequencing data and provide final reports for the patients. These efforts to build a well‐organized system were the most essential factors for success when we implemented cutting‐edge technology in the clinic to accomplish personalized medicine.
Encouragingly, our results showed that 85.8% of all patients harbored at least one clinically annotated alteration and that 32.2% harbored some actionable alterations (Tier 1 or 2).
However, lower rates of clinical trial enrollment after clinical sequencing may be a big hurdle for precision medicine. Thus, cancer gene panel results should be seamlessly integrated into larger numbers of basket‐type (histologically independent) clinical trials. To accomplish this goal, molecular profiles accompanied by clinicopathological patient information should be registered in a nationwide database that pharmaceutical companies can access.
Two limitations of our study need to be considered. The first is regarding the threshold for hypermutation status. The cut‐off value of 8.47 mutations/Mb was relatively low compared with other studies.4, 44 This may reflect an overall low frequency of highly mutated cases in our dataset used to determine the cut‐off value. Therefore, the appropriate threshold should be further investigated in larger cohorts. The second is that the mRNA expression profiling to predict primary tumor origins is not accurate enough to really function as a clinical test. Addition of probes for tissue‐specific genes in the TOP RNA panel will probably improve the performance. Integration of other mutational profiles such as SNV and CNV may also increase the accuracy.
In this study, we developed the junction capture method for RNA‐seq and confirmed its validity, utility and feasibility in clinical practise using FFPE samples in combination with DNA‐seq. This methodology has the potential to replace several currently used more labor‐intensive methods, such as fluorescence in situ hybridization, immunohistochemistry and RT‐PCR, which lack scalability for multitarget testing and/or require diagnostic expertise. We believe that the TOP panel system accelerates personalized medicine and broadens cancer treatment options by precisely detecting fusion oncogenes, abnormal transcripts while evaluating gene expression as well as SNV, indels and CNV in cancer.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank F. Tanabe, A. Maruyama and H. Tomita for technical assistance. We thank our collaborating company, Xcoo (Tokyo, Japan), which made contributions to the knowledge database and reporting for the TOP. This study was financially supported in part through grants from the Program for Integrated Database of Clinical and Genomic Information under grant number 17kk0205003h0002, the Leading Advanced Projects for Medical Innovation (LEAP) under grant number JP17am0001001, the Practical Research for Innovative Cancer Control under grant number JP17ck0106252, and the Project for Cancer Research And Therapeutic Evolution (P‐CREATE) under grant number JP17cm0106502 from the Japan Agency for Medical Research and Development, AMED. Sequencing analysis of the clinical specimens was funded in part by the Sysmex. This work was supported in‐part by a grant from Eisai.
Kohsaka S, Tatsuno K, Ueno T, et al. Comprehensive assay for the molecular profiling of cancer by target enrichment from formalin‐fixed paraffin‐embedded specimens. Cancer Sci. 2019;110:1464–1479. 10.1111/cas.13968
Contributor Information
Shinji Kohsaka, Email: skohsaka@ncc.go.jp.
Hiroyuki Aburatani, Email: haburata-tky@umin.ac.jp.
Hiroyuki Mano, Email: hmano@ncc.go.jp.
2.11. Data availability
We have deposited the raw sequencing data in the Japanese Genotype‐Phenotype Archive (http://trace.ddbj.nig.ac.jp/jga), which is hosted by the DNA Data Bank of Japan, under the accession number JGAS00000000164.
REFERENCES
- 1. Ding L, Bailey MH, Porta‐Pardo E, et al. Perspective on oncogenic processes at the end of the beginning of cancer genomics. Cell. 2018;173:305‐320.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bailey MH, Tokheim C, Porta‐Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371‐385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang KL, Mashl RJ, Wu Y, et al. Pathogenic germline variants in 10,389 adult cancers. Cell. 2018;173:355‐370.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Porreca GJ, Zhang K, Li JB, et al. Multiplex amplification of large sets of human exons. Nat Methods. 2007;4:931‐936. [DOI] [PubMed] [Google Scholar]
- 6. Dahl F, Stenberg J, Fredriksson S, et al. Multigene amplification and massively parallel sequencing for cancer mutation discovery. Proc Natl Acad Sci USA. 2007;104:9387‐9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra‐long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okou DT, Steinberg KM, Middle C, Cutler DJ, Albert TJ, Zwick ME. Microarray‐based genomic selection for high‐throughput resequencing. Nat Methods. 2007;4:907‐909. [DOI] [PubMed] [Google Scholar]
- 9. Hodges E, Xuan Z, Balija V, et al. Genome‐wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522‐1527. [DOI] [PubMed] [Google Scholar]
- 10. Albert TJ, Molla MN, Muzny DM, et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903‐905. [DOI] [PubMed] [Google Scholar]
- 11. Erikson J, Griffin CA, ar‐Rushdi A, et al. Heterogeneity of chromosome 22 breakpoint in Philadelphia‐positive (Ph+) acute lymphocytic leukemia. Proc Natl Acad Sci USA. 1986;83:1807‐1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature. 2007;448:561‐566. [DOI] [PubMed] [Google Scholar]
- 13. Kohno T, Ichikawa H, Totoki Y, et al. KIF5B‐RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378‐381. [DOI] [PubMed] [Google Scholar]
- 15. Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fernandez‐Cuesta L, Plenker D, Osada H, et al. CD74‐NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014;4:415‐422. [DOI] [PubMed] [Google Scholar]
- 17. Vaishnavi A, Capelletti M, Le AT, et al. Oncogenic and drug‐sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19:1469‐1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang R, Wang L, Li Y, et al. FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non‐small cell lung cancer. Clin Cancer Res. 2014;20:4107‐4114. [DOI] [PubMed] [Google Scholar]
- 19. Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, Ladanyi M. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer. 2011;11:541‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang JG, Goldman JM, Cross NC. Characterization of genomic BCR‐ABL breakpoints in chronic myeloid leukaemia by PCR. Br J Haematol. 1995;90:138‐146. [DOI] [PubMed] [Google Scholar]
- 21. Obata K, Hiraga H, Nojima T, Yoshida MC, Abe S. Molecular characterization of the genomic breakpoint junction in a t(11;22) translocation in Ewing sarcoma. Genes Chromosom Cancer. 1999;25:6‐15. [PubMed] [Google Scholar]
- 22. Meyer C, Burmeister T, Groger D, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adiconis X, Borges‐Rivera D, Satija R, et al. Comparative analysis of RNA sequencing methods for degraded or low‐input samples. Nat Methods. 2013;10:623‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hester SD, Bhat V, Chorley BN, et al. Editor's highlight: dose‐response analysis of RNA‐Seq profiles in archival formalin‐fixed paraffin‐embedded samples. Toxicol Sci. 2016;154:202‐213. [DOI] [PubMed] [Google Scholar]
- 25. Hedegaard J, Thorsen K, Lund MK, et al. Next‐generation sequencing of RNA and DNA isolated from paired fresh‐frozen and formalin‐fixed paraffin‐embedded samples of human cancer and normal tissue. PLoS ONE. 2014;9:e98187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jovanovic B, Sheng Q, Seitz RS, et al. Comparison of triple‐negative breast cancer molecular subtyping using RNA from matched fresh‐frozen versus formalin‐fixed paraffin‐embedded tissue. BMC Cancer. 2017;17:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cieslik M, Chugh R, Wu YM, et al. The use of exome capture RNA‐seq for highly degraded RNA with application to clinical cancer sequencing. Genome Res. 2015;25:1372‐1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next‐generation sequencing. Nat Med. 2014;20:1479‐1484. [DOI] [PubMed] [Google Scholar]
- 29. Homer N, Nelson SF. Improved variant discovery through local re‐alignment of short‐read next‐generation sequencing data using SRMA. Genome Biol. 2010;11:R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kakiuchi M, Nishizawa T, Ueda H, et al. Recurrent gain‐of‐function mutations of RHOA in diffuse‐type gastric carcinoma. Nat Genet. 2014;46:583‐587. [DOI] [PubMed] [Google Scholar]
- 31. Totoki Y, Tatsuno K, Covington KR, et al. Trans‐ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267‐1273. [DOI] [PubMed] [Google Scholar]
- 32. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frampton GM, Ali SM, Rosenzweig M, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850‐859. [DOI] [PubMed] [Google Scholar]
- 35. Paik PK, Drilon A, Fan PD, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schrader KA, Cheng DT, Joseph V, et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016;2:104‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Domanski HA, Carlen B, Mertens F, Akerman M. Extraskeletal myxoid chondrosarcoma with neuroendocrine differentiation: a case report with fine‐needle aspiration biopsy, histopathology, electron microscopy, and cytogenetics. Ultrastruct Pathol. 2003;27:363‐368. [DOI] [PubMed] [Google Scholar]
- 39. Jin Y, Moller E, Nord KH, et al. Fusion of the AHRR and NCOA2 genes through a recurrent translocation t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation of aryl hydrocarbon receptor target genes. Genes Chromosom Cancer. 2012;51:510‐520. [DOI] [PubMed] [Google Scholar]
- 40. Gan C, Love C, Beshay V, et al. Applicability of next generation sequencing technology in microsatellite instability testing. Genes (Basel). 2015;6:46‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sato K, Kawazu M, Yamamoto Y, et al. Fusion kinases identified by genomic analyses of sporadic microsatellite instability‐high colorectal cancers. Clin Cancer Res. 2019;25:378‐389. [DOI] [PubMed] [Google Scholar]
- 42. Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor‐normal sequence data. Bioinformatics. 2014;30:1015‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldstraw P, Chansky K, Crowley J, et al. The IASLC lung cancer staging project: proposals for revision of the TNM Stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11:39‐51. [DOI] [PubMed] [Google Scholar]
- 44. Campbell BB, Light N, Fabrizio D, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017;171:1042‐1056.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have deposited the raw sequencing data in the Japanese Genotype‐Phenotype Archive (http://trace.ddbj.nig.ac.jp/jga), which is hosted by the DNA Data Bank of Japan, under the accession number JGAS00000000164.