

Abstract
Clear cell renal cell carcinoma (ccRCC) is the most common and lethal renal malignant tumor in adults. The aim of the present study was to identify the key genes involved in ccRCC metastasis. Expression profiling data for ccRCC patients with metastasis and without metastasis were obtained from The Cancer Genome Atlas database. The datasets were used to identify differentially expressed genes (DEGs) between the metastasis group and the non-metastasis group using the DESeq2 package. Function enrichment analyses of DEGs were performed. The protein-protein interaction (PPI) network was constructed and analyzed using the Search Tool for the Retrieval of Interacting Genes and Cytoscape for further analysis of the identified hub genes. A total of 472 DEGs were identified, including 247 that were upregulated and 225 that were downregulated in the metastasis group. Gene Ontology enrichment analysis revealed that DEGs were mainly enriched in cell transmembrane movement and mitotic cell cycle process. Kyoto Encyclopedia of Genes Genomes pathway analysis revealed that the DEGs were mainly involved in the ‘cell cycle’ (hsa04110), ‘collecting duct acid secretion’ (hsa04966), ‘complement and coagulation cascades’ (hsa04610) and ‘aldosterone-regulated sodium reabsorption’ (hsa04960) pathways. Using the PPI network, 35 hub genes were identified, and the majority of them were upregulated in ccRCC tissue compared with normal kidney tissue. The expression levels of certain hub genes (CDKN3, TPX2, BUB1B, CDCA8, UBE2C, NDC80, RRM2, NCAPG, NCAPH, PTTG1, FAM64A, ANLN, KIF4A, CEP55, CENPF, KIF20A, ASPM and HJURP) were significantly associated with overall survival and recurrence-free survival in ccRCC. The present study has identified key genes associated with the metastasis of ccRCC.
Keywords: clear cell renal cell carcinoma, metastasis, hub genes
Introduction
Renal cell carcinoma (RCC) is the most common and lethal renal malignant tumor in adults, and in contrast with stable or declining trends for the majority of malignant tumors, including lung cancer, prostate cancer, colorectal cancer and breast cancer, incidence rates of RCC have been indicated to be increased in both men and women (1,2). Clear cell RCC (ccRCC) accounts for >85% of RCC cases (1,2). Surgical excision is the standard treatment for localized ccRCC; however, 30% of ccRCC patients have metastatic disease at the time of diagnosis (3) and 30% of patients with localized disease eventually develop metastases (4,5). The number of available treatment for metastatic ccRCC has increased over the past decade, particularly, immunotherapy and targeted therapy have improved the outlook for metastatic ccRCC. However the 5-year survival and mortality rates remain poor (6,7).
The reduced treatment efficacy of metastatic ccRCC is largely attributed to an incomplete understanding of the molecular mechanisms that lead to ccRCC metastasis (8), therefore, it is crucial to discover novel therapeutic targets for metastasic ccRCC. High-throughput technology and bioinformatics methods have been widely used to analyze the gene expression data of various cancers, including hepatocellular carcinoma (9), lung cancer (10) and ccRCC (11). It has been reported as a promising method to identify potential biomarkers in tumor diagnosis and therapeutic targets (12,13). To the best of our knowledge, there is a limited number of studies that have used TCGA data regarding ccRCC to investigate the molecular mechanisms that lead to ccRCC metastasis. In the present study, bioinformatics methods were used to analyze ccRCC mRNA expression data obtained from The Cancer Genome Atlas (TCGA) database for ccRCC patients with metastasis and without metastasis to identify key genes of ccRCC metastasis and to further explore the molecular mechanisms of ccRCC metastasis.
Materials and methods
Data collection
Expression profiling and clinical records of patients with ccRCC in TCGA (https://cancergenome.nih.gov/) were obtained from UCSC Xena (http://xena.ucsc.edu/) (14). The gene expression profiles (dataset ID:TCGA-KIRC/Xena_Matrices/TCGA-KIRC.htseq_counts.tsv) were displayed as read counts based on the IlluminaHiSeq platform (Illumina Inc., San Diego, CA, USA), including 534 ccRCC and 72 healthy kidney tissues samples. Subsequent to removing healthy kidney tissue samples, the samples of ccRCC were included in the present study if the following criteria were met: i) Expression profile and clinical records were available; ii) the mRNAs with low abundances (i.e., all mRNAs with <50 read counts across all samples) were removed; iii) the patients could be classified in to metastasis and non-metastasis groups at the time of diagnosis according to the American Joint Committee on Cancer Tumor-Node-Metastasis system (https://cancerstaging.org/). A total of 416 ccRCCs met the criteria for the the metastasis group and 78 for the non-metastasis group. Approval by a local ethics committee was not required as the study adhered to TCGA publication guidelines and data access policies.
Analysis of differentially expressed genes (DEGs) for ccRCC with and without metastases
Expression levels of genes were compared between the metastasis group and the non-metastasis group to identify differentially expressed genes (DEGs) using the unpaired Student's t-test, within the DESeq2 3.8 software using R (15). |log2FoldChange|>0.585 and adjusted P-value <0.0001 were considered as threshold values for the DEGs.
Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analyses of DEGs
The R package within the 3.8 clusterProfiler software (16) was used to analyze and visualize functional profiles of gene and gene clusters from GO (17) and KEGG (18). GO and KEGG pathway enrichment analysis for DEGs was also performed using clusterProfiler package. A false discovery rate (FDR) of <0.05 was considered for a significant GO function and KEGG pathway.
Protein-protein interaction (PPI)
To obtain insights into the interactions among DEGs associated with ccRCC metastasis, a PPI network was constructed using the Search Tool for the Retrieval of Interacting Genes (STRING) (19), a database of known and predicted protein interactions. An interaction with a threshold combined score ≥0.4 was considered statistically significant. The PPI network was visualized by Cytoscape 3.6.1 software (http://www.cytoscape.org/) (20).
Hub genes selection and analysis
CytoHubba is a common tool for analyzing PPI networks (21). The hub genes were selected using the cytoHubba plugin of Cytoscape software. Any overlap in the top 50 list of genes, from the four ranking methods, were defined as hub genes. The four ranking methods include Degree (22), Maximum Neighborhood Component (23), Density of Maximum Neighborhood Component (23) and Maximal Clique Centrality (21). The expression levels of the hub genes in the ccRCC tissue were compared with the expression levels in normal kidney tissue from TCGA and Genotype-Tissue Expression (GTEx) projects (https://www.genome.gov/gtex/) using the Limma software (24) in Gene Expression Profiling Interactive Analysis (GEPIA) (25). In addition, the overall survival (OS) and recurrence-free survival (RFS) analyses of the hub genes were performed using the Kaplan-Meier survival method with log-rank test in GEPIA. GEPIA is a newly developed interactive web server for analyzing the RNA sequencing expression data of tumors and normal samples from TCGA and the GTEx projects. It should be noted that GEPIA does not provide specific P-values and confidence intervals.
Results
Identification of DEGs associated with ccRCC metastasis
Based on the aforementioned inclusion criteria, 494 ccRCCs in TCGA database were included and divided into the metastasis group, with 78 patients, and the non-metastasis group, with 416 patients (Table I). A total of 472 DEGs, including 247 upregulated DEGs and 225 downregulated DEGs, were identified in the metastasis group compared with the non-metastasis group. The results are presented as a volcano plot (Fig. 1). Red dots indicate high expression and blue dots indicate low expression. Gray dots represent the gene expression with the |log2FoldChange|<0.585 or adjusted P-value ≥0.0001.
Table I.
Features of clear cell renal cell carcinoma patients with metastasis (n=78) and without metastasis (n=416).
| Non-metastasis group | Metastasis group | |||
|---|---|---|---|---|
| Factors | n | % | n | % |
| Sex | ||||
| Male | 272 | 65.38 | 55 | 70.51 |
| Female | 144 | 34.62 | 23 | 29.49 |
| Age, years | ||||
| <65 | 253 | 60.82 | 55 | 70.51 |
| ≥65 | 163 | 39.18 | 23 | 29.49 |
| Histological grade | ||||
| G1 | 10 | 2.40 | 0 | 0.00 |
| G2 | 199 | 47.84 | 10 | 12.82 |
| G3 | 161 | 38.70 | 33 | 42.31 |
| G4 | 39 | 9.38 | 35 | 44.87 |
| Gx/Not known | 7 | 1.68 | 0 | 0.00 |
| Pathological T stage | ||||
| T1 | 241 | 57.93 | 4 | 5.13 |
| T2 | 54 | 12.98 | 10 | 12.82 |
| T3 | 118 | 28.37 | 56 | 71.79 |
| T4 | 3 | 0.72 | 8 | 10.26 |
| Pathological N stage | ||||
| N1 | 11 | 2.64 | 5 | 6.41 |
| N0 | 199 | 47.84 | 37 | 47.44 |
| Nx | 206 | 49.52 | 36 | 46.15 |
Figure 1.

A volcano plot of the differentially expressed genes of clear cell renal cell carcinoma between the metastasis group and the non-metastasis group. Red indicates upregulated genes and blue indicates downregulated genes. The gray area shows the gene expression below the threshold criteria (|log2Fold-Change|>0.585 or adjusted P-value <0.0001).
KEGG and GO enrichment analyses of DEGs
To analyze the biological classification of DEGs, functional and pathway enrichment analyses were performed using the clusterProfiler software. GO analysis results (Fig. 2A-C) showed that changes in the molecular function (MF) of DEGs were significantly enriched in ‘microtubule motor activity’ (GO:0003777), ‘active transmembrane transporter activity’ (GO:0022804), ‘inorganic anion transmembrane transporter activity’ (GO:0015103), ‘motor activity’ (GO:0003774) and ‘microtubule binding’ (GO:0008017). KEGG pathway analysis (Fig. 2D) revealed that the DEGs were mainly enriched in the ‘cell cycle’ (hsa04110), ‘collecting duct acid secretion’ (hsa04966), ‘complement and coagulation cascades’ (hsa04610) and ‘aldosterone-regulated sodium reabsorption’ (hsa04960) pathways.
Figure 2.

Annotation of the statistical significant enriched Gene Ontology and KEGG pathways of differentially expressed genes between the metastasis group and the non-metastasis group: (A) Cellular component, (B) biological process, (C) molecular function and (D) KEGG pathway analysis. KEGG, Kyoto Encyclopedia of Genes and Genomes.
PPI network construction
To obtain the interactions between the 472 DEGs in the metastasis group, a PPI network was constructed using the STRING database and visualized by cytoscape software. As shown in Fig. 3, the network included 264 nodes and 2,977 edges. Red nodes indicate upregulated genes and light blue nodes indicate downregulated genes in the metastasis group.
Figure 3.

A graphic representation of the protein-protein interaction network based on differentially expressed genes of clear cell renal cell carcinoma between the metastasis and non-metastasis group. Red nodes indicate upregulated genes and light blue nodes indicate downregulated genes in the metastasis group.
Hub gene selection and analysis
Among the lists of the top 50 genes selected respectively by the four ranking methods, 35 genes overlapped and were identified as hub genes (Figs. 4 and 5). All 35 genes were upregulated in the metastasis group and were subsequently analyzed using GEPIA. The results of the 35 hub genes analyzed by GEPIA are shown in Table II. Based on the same cutoffs (|log2FC|>0.585 and P<0.0001), 26 of the 35 hub genes were upregulated in ccRCC tissue compared with normal kidney tissue from TCGA and GTEx projects (Fig. 6). Subsequently, survival analysis of the hub genes was performed using a Kaplan-Meier curve in GEPIA. ccRCC patients with high expression (>median expression value) of some hub genes (CDKN3, TPX2, BUB1B, CDCA8, UBE2C, NDC80, RRM2, NCAPG, NCAPH, PTTG1, FAM64A, ANLN, KIF4A, CEP55, CENPF, KIF20A, ASPM and HJURP) showed worse OS time and RFS time (data not shown; P<0.05).
Figure 4.

Top 50 genes selected based on the four ranking methods. (A) Degree, (B) DMNC, (C) MCC and (D) MNC. The darker the color of the node, the higher the score. DMNC, density of maximum neighborhood component; MCC, maximal elique centrality; MNC, maximum neighborhood component.
Figure 5.
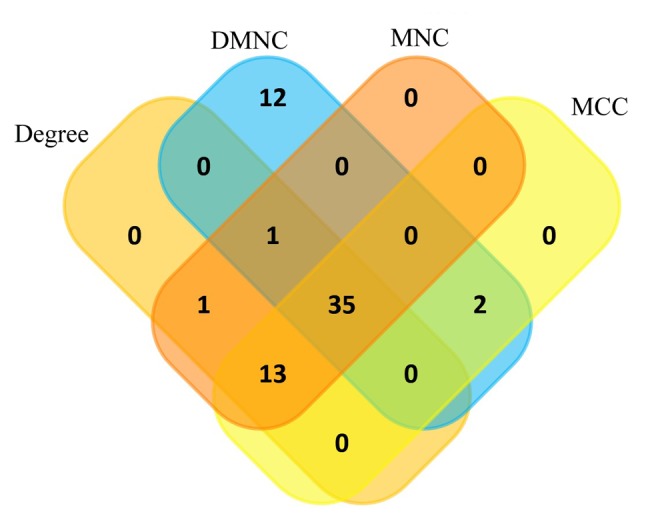
Venn plot of the overlapping genes for the top 50 genes selected based on the four ranking methods, Degree, DMNC, MNC, and MCC. DMNC, density of maximum neighborhood component; MCC, maximal elique centrality; MNC, maximum neighborhood component.
Table II.
Results of the 35 hub genes using the Gene Expression Profiling Interactive Analysis software.
| Hub gene | Overall survival associated | Recurrence-free survival associated | Clear cell renal cell carcinoma versus normal kidney tissue |
|---|---|---|---|
| KIF23 | √a | ||
| CDKN3 | √ | √ | Upregulated |
| TPX2 | √ | √ | Upregulated |
| BUB1B | √ | √ | Upregulated |
| CDCA8 | √ | √ | Upregulated |
| UBE2C | √ | √ | Upregulated |
| NDC80 | √ | √ | Upregulated |
| KIF15 | |||
| PBK | Upregulated | ||
| MELK | √ | Upregulated | |
| RRM2 | √ | √ | Upregulated |
| DLGAP5 | Upregulated | ||
| NCAPG | √ | √ | Upregulated |
| EXO1 | |||
| NUF2 | √ | Upregulated | |
| NCAPH | √ | √ | Upregulated |
| KIF18A | √ | ||
| PTTG1 | √ | √ | Upregulated |
| FAM64A | √ | √ | Upregulated |
| ANLN | √ | √ | Upregulated |
| SHCBP1 | |||
| CENPE | √ | ||
| MCM10 | |||
| MKI67 | Upregulated | ||
| KIF14 | √ | ||
| HMMR | √ | Upregulated | |
| KIF4A | √ | √ | Upregulated |
| CEP55 | √ | √ | Upregulated |
| CENPF | √ | √ | Upregulated |
| KIF20A | √ | √ | Upregulated |
| CDC45 | √ | Upregulated | |
| ASPM | √ | √ | Upregulated |
| NEK2 | √ | Upregulated | |
| TTK | √ | ||
| HJURP | √ | √ | Upregulated |
√. P<0.05; P<0.001 and |log2Fold-change|>0.585.
Figure 6.

Expression levels in clear cell renal cell carcinoma tissue for the 35 hub genes compared with normal kidney tissue in The Cancer Genome Atlas and Genotype-Tissue Expression projects. TPM, transcripts per kilobase million; (N), normal kidney tissue; (T), tumor tissue.
Discussion
Even with strict adherence to the National Comprehensive Cancer Network guidelines (NCCN Guidelines®), ~30% of ccRCC recurrences will be missed (26). According to the guidelines, patients with ccRCC would benefit from more aggressive treatment and monitoring management. To find potentially effective therapeutic targets, there is an urgent requirement to explore the molecular mechanisms that lead to ccRCC metastasis. The present study utilized a relatively large sample dataset obtained from TCGA. These data were analyzed to identify DEGs between ccRCC patients with metastasis and without metastasis. A total of 472 DEGs, including 247 upregulated genes and 225 downregulated genes, were identified in the metastasis group compared with the non-metastasis group. KEGG pathway analysis revealed that the DEGs were mainly involved in the ‘cell cycle’ (hsa04110), ‘collecting duct acid secretion’ (hsa04966), ‘complement and coagulation cascades’ (hsa04610) and ‘aldosterone-regulated sodium reabsorption’ (hsa04960) pathways. It is well known that the cell cycle pathway serves an important role in the development of different cancer types, including ccRCC (27). Askeland et al (28) showed that the cell cycle progression score can predict metastatic progression of ccRCC following resection. The present results suggest that the ‘collecting duct acid secretion’, ‘complement and coagulation cascades’ and ‘aldosterone-regulated sodium reabsorption’ pathways may also be associated with ccRCC metastasis. GO enrichment analysis revealed that DEGs were mainly associated with ‘cell transmembrane movement’ and ‘mitotic cell cycle process’. The present results provide bioinformatics evidence for further research.
The 35 overlapping genes among the top 50 genes in the PPI network found using four ranking methods were selected. All 35 genes were upregulated in the metastasis group, and 26 genes of them were upregulated in ccRCC tissues compared with normal kidney tissues. This result reveals that these genes may serve an important role in the progression of ccRCC. The expression level of CDKN3, TPX2, BUB1B, CDCA8, UBE2C, NDC80, RRM2, NCAPG, NCAPH, PTTG1, FAM64A, ANLN, KIF4A, CEP55, CENPF, KIF20A, ASPM and HJURP was significantly associated with overall survival and recurrence-free survival time (P<0.05). These findings may provide valuable prognostic biomarkers and therapeutic targets for ccRCC; however, further investigation is required.
Prior to the present study, few studies have addressed the gaps in the molecular mechanisms that lead to ccRCC metastases. Ho et al (29) identified and validated 7 genes that support ccRCC metastases by comparing gene expression profiles between metastatic tumors and their patient-matched primary tumor. The 7 genes (DCN, SLIT2, LUM, LAMA2, ADAMTS12, CEACAM6 and LMO3) were enriched for extracellular matrix (ECM) genes. Ghatalia et al (30) identified 9 overexpressed kinase genes (EPHB2, AURKA, GSG2, IKBKE, MELK, CSK, CHEK2, CDC7 and MAP3K8) (P<0.001) in metastatic ccRCC tumor tissue. In the present study, the aim was to focus on DEGs between the metastasis group and the non-metastasis group. However, due to lack of experimental validation, it is not clear whether these genes are causal or merely markers. Notably, the metastasis group was not only characterized by organ metastases, but also by more advanced tumors (stage T3 72 vs. 28%) and less differentiated tumors (grade 4 45 vs. 9%), when compared with the non-metastasis group, respectively. These results suggest DEGs between the groups may also be associated with locally advanced tumors. The main aim of the present study was to identify potential key genes for ccRCC with metastasis and without metastasis, considering that advanced ccRCC is just a relative definition that is likely to change as treatments improve (31). From a biological point of view, genes that promote tumor metastasis are likely to be genes that promote tumor progression. Therefore, it is reasonable that there were more T3/T4 or G3/G4 patients in the metastasis group as compared with the non-metastasis group, as the present study data shows. As few drugs have shown efficacy in the adjuvant treatment for preventing ccRCC metastasis or recurrence (32), more studies are required to identify biomarkers and explore the molecular mechanism of ccRCC metastasis.
There are a few important limitations to the present study. One limitation is that there were more patients within the non-metastatic group (n=416) compared with the metastatic group (n=78). Another limitation is the difference in the proportion of patients with T3/T4 or G3/G4 in the two groups. In addition, stratified differential expression gene analysis based on histological grade (or pathological T stage), was not performed. Although a powerful significance level (P<0.0001) was used, based on bioinformatic analysis, a study with a larger sample size and experimental validation is required.
In conclusion, the present study identified key DEGs in primary tumor tissues of ccRCC with metastasis compared with ccRCC without metastasis. The key genes involved in the metastasis of ccRCC may provide valuable prognostic biomarkers and therapeutic targets for ccRCC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during the present study are included in this published article.
Authors' contributions
XH and ZX designed the study. WW and YL analyzed the data and wrote the manuscript. ZG and YZ participated in analysis and interpretation of the data and reviewed the article. XH gave the final approval for publication. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med. 2005;353:2477–2490. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Clark PE. The role of VHL in clear-cell renal cell carcinoma and its relation to targeted therapy. Kidney Int. 2009;76:939–945. doi: 10.1038/ki.2009.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolff I, May M, Hoschke B, Zigeuner R, Cindolo L, Hutterer G, Schips L, De Cobelli O, Rocco B, De Nunzio C, et al. Do we need new high-risk criteria for surgically treated renal cancer patients to improve the outcome of future clinical trials in the adjuvant setting? Results of a comprehensive analysis based on the multicenter CORONA database. Eur J Surg Oncol. 2016;42:744–750. doi: 10.1016/j.ejso.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 5.Frank I, Blute ML, Cheville JC, Lohse CM, Weaver AL, Zincke H. An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: The SSIGN score. J Urol. 2002;168:2395–2400. doi: 10.1016/S0022-5347(05)64153-5. [DOI] [PubMed] [Google Scholar]
- 6.Pal SK, Williams S, Josephson DY, Carmichael C, Vogelzang NJ, Quinn DI. Novel therapies for metastatic renal cell carcinoma: Efforts to expand beyond the VEGF/mTOR signaling paradigm. Mol Cancer Ther. 2012;11:526–537. doi: 10.1158/1535-7163.MCT-11-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courtney KD, Choueiri TK. Updates on novel therapies for metastatic renal cell carcinoma. Ther Adv Med Oncol. 2010;2:209–219. doi: 10.1177/1758834010361470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009. doi: 10.1038/nrdp.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lv Y, Wei W, Huang Z, Chen Z, Fang Y, Pan L, Han X, Xu Z. Long non-coding RNA expression profile can predict early recurrence in hepatocellular carcinoma after curative resection. Hepatol Res. 2018;48:1140–1148. doi: 10.1111/hepr.13220. [DOI] [PubMed] [Google Scholar]
- 10.Lin Y, Lv Y, Liang R, Yuan C, Zhang J, He D, Zheng X, Zhang J. Four-miRNA signature as a prognostic tool for lung adenocarcinoma. Onco Targets Ther. 2018;11:29–36. doi: 10.2147/OTT.S155016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu L, Xiang L, Feng J, Li B, Zhou Z, Li J, Lin Y, Lv Y, Zou D, Lei Z, Zhang J. miRNA-21 and miRNA-223 expression signature as a predictor for lymph node metastasis, distant metastasis and survival in kidney renal clear cell carcinoma. J Cancer. 2018;9:3651–3659. doi: 10.7150/jca.27117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Idris SF, Ahmad SS, Scott MA, Vassiliou GS, Hadfield J. The role of high-throughput technologies in clinical cancer genomics. Expert Rev Mol Diagn. 2013;13:167–181. doi: 10.1586/erm.13.1. [DOI] [PubMed] [Google Scholar]
- 13.Le Gallo M, Lozy F, Bell DW. Next-generation sequencing. Adv Exp Med Biol. 2017;943:119–148. doi: 10.1007/978-3-319-43139-0_5. [DOI] [PubMed] [Google Scholar]
- 14.Goldman M, Craft B, Kamath A, Brooks AN, Zhu J, Haussler D. The UCSC Xena platform for cancer genomics data visualization and interpretation. BioRxiv. 2018 May 18; (Epub ahead of print). doi: https://doi.org/10.1101/326470. [Google Scholar]
- 15.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torto-Alalibo T, Purwantini E, Lomax J, Setubal JC, Mukhopadhyay B, Tyler BM. Genetic resources for advanced biofuel production described with the Gene Ontology. Front Microbiol. 2014;5:528. doi: 10.3389/fmicb.2014.00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanehisa M. The KEGG database. Novartis Found Symp. 2002;247:91–101. doi: 10.1002/0470857897.ch8. [DOI] [PubMed] [Google Scholar]
- 18.Yu G, Wang LG, Han Y, He QY. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;4:S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 23.Lin CY, Chin CH, Wu HH, Chen SH, Ho CW, Ko MT. Hubba: Hub objects analyzer-A framework of interactome hubs identification for network biology. Nucleic Acids Res. 2008;36:W438–W443. doi: 10.1093/nar/gkn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Champeris Tsaniras S, Kanellakis N, Symeonidou IE, Nikolopoulou P, Lygerou Z, Taraviras S. Licensing of DNA replication, cancer, pluripotency and differentiation: An interlinked world? Semin Cell Dev Biol. 2014;30:174–180. doi: 10.1016/j.semcdb.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Askeland EJ, Chehval VA, Askeland RW, Fosso PG, Sangale Z, Xu N, Rajamani S, Stone S, Brown JA. Cell cycle progression score predicts metastatic progression of clear cell renal cell carcinoma after resection. Cancer Biomark. 2015;15:861–867. doi: 10.3233/CBM-150530. [DOI] [PubMed] [Google Scholar]
- 29.Ho TH, Serie DJ, Parasramka M, Cheville JC, Bot BM, Tan W, Wang L, Joseph RW, Hilton T, Leibovich BC, et al. Differential gene expression profiling of matched primary renal cell carcinoma and metastases reveals upregulation of extracellular matrix genes. Ann Oncol. 2017;28:604–610. doi: 10.1093/annonc/mdw652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghatalia P, Yang ES, Lasseigne BN, Ramaker RC, Cooper SJ, Chen D, Sudarshan S, Wei S, Guru AS, Zhao A, et al. Kinase gene expression profiling of metastatic clear cell renal cell carcinoma tissue identifies potential new therapeutic targets. PLoS One. 2016;11:e0160924. doi: 10.1371/journal.pone.0160924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makhov P, Joshi S, Ghatalia P, Kutikov A, Uzzo RG, Kolenko VM. Resistance to systemic therapies in clear cell renal cell carcinoma: Mechanisms and management strategies. Mol Cancer Ther. 2018;17:1355–1364. doi: 10.1158/1535-7163.MCT-17-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Massari F, Di Nunno V, Ciccarese C, Graham J, Porta C, Comito F, Cubelli M, Iacovelli R, Heng DYC. Adjuvant therapy in renal cell carcinoma. Cancer Treat Rev. 2017;60:152–157. doi: 10.1016/j.ctrv.2017.09.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during the present study are included in this published article.