

Abstract
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide. The most important reason for the occurrence of HCC is hepatitis C or B infection. Moreover, genetic factors play an important role in the tumorigenesis of HCC. Here, we demonstrated that Krüppel‐like factor 2 (KLF2) expression was downregulated in HCC samples compared with adjacent tissues. Additionally, KLF2 was shown to inhibit the growth, migration and colony‐formation ability of liver cancer cells. Further mechanistic studies revealed that KLF2 can compete with Gli1 for interaction with HDAC1 and restrains Hedgehog signal activation. Together, our results suggest that KLF2 has potential as a diagnostic biomarker and therapeutic target for the treatment of HCC.
Keywords: HDAC1, Hedgehog signaling, hepatocellular carcinoma, intrahepatic metastasis, Krüppel‐like factor 2
1. INTRODUCTION
Hepatocellular carcinoma (HCC) is a serious threat to human health. The World Cancer Report issued in 2016 showed that HCC has become the third most dangerous disease in men and the sixth most dangerous disease in women.1 More than 600 000 individuals are newly diagnosed with HCC each year, and of these, approximately 360 000 patients are in China.2
Hepatocellular carcinoma features family aggregation, which suggests that genetic factors play a significant role in its occurrence. Oncogenes (e.g. c‐Myc, β‐catenin, Ras) contain amplification or activating mutations at the DNA level in liver cancer,3, 4, 5 while tumor suppressor genes (e.g. P53) often have inactivating mutations in HCC.6 Presently, it is widely believed that activation of the Ras‐ERK, Wnt/beta‐catenin and Hedgehog/Gli1 signaling pathways is closely associated with HCC occurrence.7
The Hedgehog (Hh) signaling pathway was first discovered in Drosophila. In mammals, three homologs of the Hedgehog gene have been reported: Sonic (Shh), Desert (Dhh) and Indian (Ihh). These genes code for secretory proteins that can function as ligands.8 The hh ligands can undergo various post‐translational modifications, including catalytic shearing and cholesterol modifications at their amino terminus, and thus, Hedgehog ligands can exert their maximal biological activity. The secreted Hh ligand binds to Hip1, Patched1 (Ptch1) and Patched2 (Ptch2) transmembrane receptors by diffusion.9 The transmembrane receptors Ptch1 and Ptch2 are expressed in most cells. When the amount of ligand is insufficient, the Ptch receptor can block the function of another transmembrane protein (Smoothened, Smo); the overall signaling pathway is static, but otherwise, Smo is activated. Activated Smo can promote Gli1 expression in the cytoplasm, and the N‐terminus of Gli1 enters the cell nucleus where it functions as a transcription factor. In vertebrate cells, three Gli genes have been reported, namely Gli1, Gli2 and Gli3, which act as transcriptional activators or transcriptional suppressors depending on the cell type.10 Generally, Gli1 protein acts as a transcriptional activator, and Gli3 protein acts as a transcriptional suppressor. After translation and shear, the N‐terminus of Gli1 finally becomes a transcriptional activator and enters the cell nucleus where it activates the transcription of downstream genes. Hedgehog signaling pathway activation can promote tumor cell growth, migration, metastasis and in vivo tumorigenesis.11 In hepatic stellate cells, the Hedgehog signaling pathway is activated, which promotes hepatoma cell angiogenesis and vasculogenic mimicry.12 Previous studies have indicated that activated Hedgehog signaling has important functions in HCC occurrence and development.
The Krüppel‐like factor (KLF) family comprises transcription factors with a zinc finger domain and includes 17 members.13 Studies showed that the KLF family can regulate multiple biological processes, such as cell proliferation, differentiation and migration, among others. The KLF family has important functions in the occurrence of multiple tumor types. For example, KLF2 participates in the genesis and development of intestinal cancer and breast cancer,14 KLF5 plays an important role in the progression of esophageal cancer and lung cancer,15, 16 and KLF10 expression is downregulated in pancreatic cancer and kidney cancer.17, 18 KLF4 can inhibit the growth, migration and infiltration of the liver cancer cell line MM189, while KLF6 can inhibit intestinal cancer growth and infiltration.19 Finally, KLF8 expression can promote the proliferation, infiltration and epithelial‐mesenchymal transition (EMT) of liver cancer cells.20 In liver cancer cells, KLF9 binds to the promoter region of the P53 gene and upregulates P53 expression to promote cell apoptosis and inhibit tumor progression.21 In HepG2 cells, the downregulation of KLF17 can promote EMT in cells and can enhance the ability of tumor cell invasion.22
In recent years, the role of KLF2 in tumor occurrence and development has been more frequently reported. KLF2 is downregulated in gastric cancer, liver cancer, non‐small‐cell lung cancer and pancreatic ductal adenocarcinoma.23, 24, 25 Multiple long‐chain non‐coding RNA (TUG1, ANRIL, XIST and ZFAS1) and miRNA (miR‐9) can regulate KLF2 expression23, 24, 25, 26, 27, 28. Although the function of KLF2 in liver cancer has not been directly investigated and reported, relevant studies have indicated that long‐chain non‐coding RNA such as TUG1 and ANRIL can promote the occurrence of liver cancer and development by apparent silencing of KLF2 expression.27, 28 Such studies all indicate that KLF2 can negatively regulate liver cancer occurrence and development.
In this study, we found that in the HCC tissues of patients and in a mouse model of liver cancer, KLF2 expression was downregulated. We discovered that KLF2 can inhibit the growth, migration and colony‐formation ability of liver cancer cells in vitro. Further studies have demonstrated that KLF2 can compete with Gli1 for interaction with HDAC1 through which it restrains Hedgehog signal activation. This study demonstrated the function of KLF2 in liver cancer occurrence and development, and the molecular mechanism by which it is regulated, which provides a new strategy for HCC therapy.
2. MATERIALS AND METHODS
2.1. HCC sample collection
Liver tumor samples from HCC patients were obtained from The Longgang Central Hospital of Shenzhen, Shenzhen, China. All samples collected were obtained from patients who were admitted to the hospital for liver cancer from 2010 to 2016. None of the patients received any form of treatment (including chemotherapy and radiotherapy) before surgery. The organization collected permission from the hospital's biomedical ethics committee and informed consent from patients. All patients provided signed, informed consent.
2.2. Mice
Alb‐Cre (016832), P53fl/fl (008462) and LSL‐KrasG12D (019104) mice all had a C57BL/6 background and were purchased from Nanjing Biomedical Research Institute of Nanjing University and were originally obtained from The Jackson Laboratory. All mice were maintained under specific pathogen‐free conditions. Genotyping was performed using the following primers: Alb‐Cre: forward, 5′‐ AGGTGTAGAGAAGGCACTCAGC‐3′, reverse, 5′‐CTAATCGCCATCTTCCAGCAGG‐3′; P53: forward, 5′‐CACAAAAACAGGTTAAACCCAG‐3′; reverse, 5′‐AGCACATAGGAGGCAGAGAC‐3′; and Kras: forward, 5′‐CTAGCCACCATGGCTTGAGT‐3′; reverse, 5′‐TCCGAATTCAGTGACTACAGATG‐3′.
2.3. Cells and reagents
Cells were maintained at 37°C under 5% CO2. L02, Chang, 7404 and Huh‐7 cell lines were purchased from the Chinese Academy of Sciences and were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), l‐glutamine (2 mmol/L) and penicillin‐streptomycin (100 U/mL).
Anti‐KLF2, anti‐GAPDH, anti‐FOXM, anti‐Patch, anti‐Gli1, anti‐Flag, anti‐HA, anti‐Myc, anti‐GST and DAPI were all purchased from Cell Signaling Technology (Danvers, MA, USA). Recombinant Human Sonic Hedgehog/Shh Protein (8908‐SH) was purchased from R&D Systems (Minneapolis, MN, USA).
2.4. Histological analysis
Paraffin‐embedded samples were prepared from the livers of the experimental mice and HCC patients. Hematoxylin‐eosin (HE) and immunohistochemistry (IHC) were used to stain the liver sections. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays were performed using the ApoAlert DNA Assay Kit (Clontech, Mountain View, CA, USA).
2.5. Plasmids and shRNA
CDNA for human KLF2 (NM_016270.3), HDAC1 (NM_004964.3) and Gli1 (NM_005269.3) were amplified from reverse‐transcribed cDNA from HEK293T cells (KLF2: forward, 5′‐ATGGCGCTGAGTGAACCCATCC‐3′; reverse, 5′‐CTACATGTGCCGTTTCATGT‐3′; HDAC1: forward, 5′‐ATGGCGCAGACGCAGGGCACCCG‐3′; reverse, 5′‐TCAGGCCAACTTGACCTCCT‐3′; and Gli1: forward, 5′‐ATGTTCAACTCGATGACCCC‐3′; reverse, 5′‐TTAGGCACTAGAGTTGAGGAA‐3′) and then were cloned into P23, pcDNA3.1 and pCMV‐HA vectors, respectively, to obtain the Flag, myc and HA tag. Plasmids were sequenced at Sangon Biotech (Shanghai, China) to confirm the sequence accuracy.
These genes were cloned into the p2329/pLKO.130 vectors or for transient (using the Lipofectamine 2000 according to the manufacturer's instructions; Thermo Fisher Scientific, Waltham, MA, USA) or stable expression in 7404 or Huh‐7 cells. The lentivirus was used to produce the stable transformants. Briefly, P23 or pLKO.1 plasmids were transfected into 293T cells with the psPAX and PMD2G plasmids using the Lipofectamine 2000 according to the manufacturer's instructions. Twenty‐four hours later, the supernatant was collected and used to infect the 7404 and Huh7 cells. Cells were selected with puromycin (1 μg/mL) for 1 week. The resistant cells were pooled and the expressions of KLF2 were examined using western blot analysis.
The plasmid pLKO.1 was also used as the carrier to construct the shRNA. The two target sequences are as follows: shKLF2 1#, 5′‐AACCCGAGTCCGGCGGCACCG‐3; and shKLF2 2#, 5′‐GAAGCGCGGCCGCCGCTCTTG‐3′.
2.6. Quantitative reverse transcription polymerase chain reaction (qRT‐PCR) analysis
Total RNA was isolated by RNAiso Plus reagent according to the manufacturer's instructions (Takara Bio, Mountain View, CA, USA). First‐strand cDNA was generated using M‐MLV reverse transcriptase (RNase H‐) (Takara Bio). Samples were amplified by a CFX‐96 machine (Bio‐Rad, Hercules, CA, USA) using SYBR Green master mix (DBI Bioscience, Shanghai, China) according to the manufacturer's instructions, and data were normalized to the 18S rRNA control. The qRT‐PCR primers were as follows: human KLF2: forward, 5′‐TGCGTTCTATTACCCCGAAC‐3′; reverse, 5′‐CAGCAGAAAGCGGCCGTGCA‐3′; and human 18S: forward, 5′‐GAGAAACGGCTACCACATCC‐3′; and reverse, 5′‐CACCAGACTTGCCCTCCA‐3′.
2.7. MTT
In all, 2000 cells were inoculated into each well of a 96‐well plate in a total volume of 200 μL. On days 2, 4 and 6 of the experiment, 20 μL MTT was added. At the end of the experiment, 200 μL dimethylsulfoxide was used to dissolve the MTT crystals, and the precipitate was added into each well; the optical density (OD) value was then measured at 540 nm.
2.8. Cell migration
Each Boyden chamber was divided into upper and lower layers so that film with holes could be placed in the middle. Collagen was first diluted 1:70 to coat the rough surface of the film, which was then incubated overnight at 4°C on a table. The film was then removed and air‐dried for later use. Before the cell migration experiment was performed, cell morphology was assessed under the microscope, and cell fusion of approximately 70% was deemed appropriate. The cells were digested and counted using a cell counter. Then, 3 × 105 cells were suspended in the 150 μL serum‐free DMEM culture medium. A total of 150 μL DMEM supplemented with 10% serum was added to each well of the lower Boyden chamber. The film, with an 8‐μm diameter hole in the middle, was placed with the rough side facing downward. Then, 1 × 105 cells (suspended in 50 μL serum‐free DMEM) were plated into the upper Boyden chamber. The upper and lower Boyden chambers were fixed with screws and were placed into a humidified box, which was maintained in a cell culture incubator. The box was kept in the incubator for 8 hours. After the experiment, the screws were loosened, and the upper and lower sections of the Boyden chambers were separated. Cells on the smooth surface of the film were removed with a cotton swab and then rinsed with clean water. The film was fixed in Eosin A solution for 5 minutes, which was followed by staining in Eosin B solution for 5 minutes. The film was then rinsed with clean water, and then images were obtained under the microscope.
2.9. Colony‐formation assay
First, 1.25% low melting‐point agar was placed in a 42°C water bath for more than 1 hour, and a lower‐layer gel was prepared with 20% FBS, 40% 2 × DMEM and 40% 1.25% agar. This layer was mixed well and placed on a 24‐well plate with 200 μL/well, coagulated and then placed into a 37°C incubator. After the cells were 70% confluent, they were digested, suspended and diluted to 2.5 × 104 cells/mL. The upper‐layer gel comprised the following: 25% FBS, 37.5% 2 × DMEM, 2 mmol/L l‐glutamine, 0.01 mmol/L β‐mercaptoethanol and 37.5% 1% agar. This gel was mixed with cells at a ratio of 4:1 and was then rapidly added to the lower‐layer gel at 200 μL/well. The mixture was maintained in a 37°C incubator for 2 weeks, after which images were obtained and the colony numbers were counted. Each cell type was plated in four parallel wells, and all experiments were performed three times.
2.10. Intrahepatic metastasis model
In all, 1 × 105 cells were injected into each mouse. Male nude mice aged 5 weeks were selected and each group comprised nine mice. After anesthesia, the dermal and muscular layers were cut along the abdominal midline to expose the liver. Cells were injected into the largest lobe of the liver, and then tweezers were used for extrusion and hemostasis. Then, the mouse's muscular and dermal layers were sutured. After the experiment, the mice were killed by cervical dislocation, and the lobes that were injected with cells and those that were not injected were fixed and embedded. After HE staining, tumor cell metastasis was determined. When the survival time was analyzed, the injection time was determined to be the starting point, and the duration until death for all mice in each group was the primary end‐point, according to which the survival curve was plotted.
2.11. Dual‐luciferase reporter assay
7404 or Huh‐7 cells were transfected with the Gli1 luciferase reporter, TK‐renilla and the indicated plasmids by Polyethylenimine (Polysciences, Warrington, PA, USA) according to the manufacturer's instructions. After 24 hours, the cells were harvested, and the luciferase activity was measured using a dual‐luciferase reporter assay system according to the manufacturer's instructions (Promega, Madison, WI, USA).
2.12. Immunostaining
7404 cells were mounted on coverslips, fixed, permeabilized and then stained with the indicated antibodies and Hoechst. Images were obtained using an Olympus IX81 microscope (Tokyo, Japan) or a Leica confocal microscope (Wetzlar, Germany). Fluorescence imaging analysis was performed in a blinded manner, and densitometry quantification or Pearson's correlation was determined by Image‐Pro 6.0 software.
2.13. Expression and purification of recombinant proteins
Full‐length human KLF2 was constructed as part of the pGEX4T‐1‐GST vector (GE Healthcare, Chicago, IL, USA) to generate GST‐fusion proteins. Recombinant proteins were expressed in Escherichia coli BL21 (DE3) Codon‐Plus strain (Novagen, Madison, WI, USA). BL21 cells were transformed with the above plasmids and grown in lysogeny broth supplemented with ampicillin (50 μg/mL). Expression of the recombinant proteins was induced by 0.1 mmol/L isopropyl β‐D‐1‐thiogalactopyranoside at 16°C for 20 hours. For purification, GST‐KLF2 or GST was purified by glutathione‐agarose beads according to the manufacturer's instructions (GE Healthcare). Purified KLF2 protein was identified by western blotting without boiling.
2.14. GST pull‐down
GST‐KLF2 or GST proteins at equimolar concentrations were incubated with 7404 and Huh‐7 cell lysates at 4°C for 2 hours in 100 μL pull‐down buffer (20 mmol/L Tris‐Cl, 100 mmol/L NaCl, 5 mmol/L MgCl2, 1 mmol/L ethylenediaminetetraacetic acid [EDTA], 1 mmol/L dithiothreitol, 0.5% (v/v) NP‐40 and 10 μg/mL bovine serum albumin, pH 7.5) followed by three washes. Samples were combined with sodium dodecylsulfate (SDS) loading buffer and were subjected to SDS polyacrylamide gel electrophoresis (PAGE) without boiling.
2.15. Immunoprecipitation analysis
7404 and Huh‐7 cells overexpressing the indicated proteins were washed with cold phosphate‐buffered saline before lysis in cold lysis buffer (25 mmol/L Tris‐Cl, 150 mmol/L NaCl, 1% [v/v] NP‐40, 5 mmol/L EDTA, 0.5% sodium deoxycholate and protease inhibitor cocktail, pH 7.2). Cell lysates were then centrifuged at 12 000 g for 15 minutes at 4°C. Following incubation of cell lysates with protein G Sepharose beads coated with the indicated antibodies and rotation at 4°C for 2 hours, the beads were then washed five times in lysis buffer and resuspended in SDS‐PAGE loading buffer for western blot analysis.
2.16. Statistical analysis
All sample sizes were sufficient to ensure proper statistical analysis. Data are represented as the means ± standard error of the mean of at least three experiments. Statistical analyses were performed using GraphPad Prism 6 software, version 6. Statistical significance was calculated using Student's two‐tailed unpaired t‐test. The log‐rank (Mantel‐Cox) test was used for survival comparisons (P > 0.05, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
3. RESULTS
3.1. KLF2 expression is reduced in HCC
First, to study the expression pattern of KLF2 in HCC tissue, we used fluorescence quantitative PCR to detect the mRNA level of KLF2 in tissue from 38 cases of liver cancer and in the corresponding paracarcinomatous tissues. The results showed that the expression of KLF2 RNA in paracarcinomatous tissue was significantly higher than that in tumor tissue (Figure 1A). Second, we used immunohistochemistry and western blot to detect the expression of KLF2 in paracarcinomatous and liver cancer tissues. KLF2 expression in HCC was significantly downregulated (Figure 1B‐D), which was consistent with previous results. Finally, we detected the KLF2 protein level in normal liver cells (L02 and Chang) and liver cancer cells (7404, Hep3B, Huh‐7 and HepG2). Experimental results showed that KLF2 expression in normal liver cells (L02 and Chang) was higher than in liver cancer cells (see Figure S1).
Figure 1.
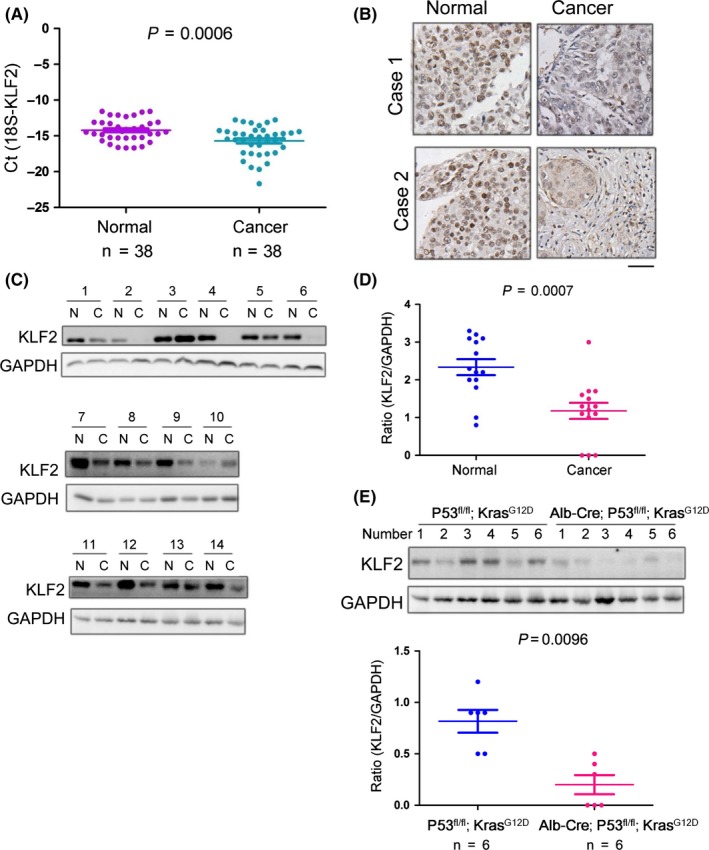
The expression of KLF2 is downregulated in liver cancer. A, Fluorescence quantitative polymerase chain reaction was used to detect the expression of Krüppel‐like factor 2 (KLF2) mRNA in liver tissues of 38 cases of liver cancer and in corresponding paracarcinomatous tissues; 18S rRNA served as the internal reference gene. B, Immunohistochemistry was used to detect the expression of KLF2 in liver cancer tissues and corresponding paracarcinomatous tissues of two random cases (scale bar: 50 μm, representative images). C‐D, Western blot was used to detect the protein level of KLF2 in hepatocellular carcinoma tissues and corresponding paracarcinomatous tissues of 14 cases. E, Western blot was used to detect the protein level of KLF2 in the mouse liver cancer model (Alb‐Cre; P53fl/fl; KrasG12D) and in control tissues, and the quantification was performed
In liver cancer, P53 deletion and the KrasG12D activating mutation are very common. Based on this, we established a model of spontaneous HCC (Alb‐Cre; P53fl/fl; KrasG12D) by crossing Alb‐Cre mice with mice expressing LSL‐KrasG12D and P53fl/fl 30, 31. Simply, the Cre enzyme expressed by the mice is regulated by the Alb gene promoter. Cre enzyme expression causes the deletion of P53 and the stop codon before the coding sequences of KrasG12D, which activates KrasG12Dexpression and drives the development of liver cancer. To determine the expression of KLF2 in the mouse liver cancer model, we selected six mice (Alb‐Cre; P53fl/fl; LSL‐KrasG12D) with liver cancer and six controls. Their liver tissues were separated and western blot analysis was performed. According to the results, KLF2 was downregulated in mice with liver cancer (Alb‐Cre; P53fl/fl; LSL‐KrasG12D) (Figure 1F). These studies showed that KLF2 is downregulated in liver cancer.
3.2. KLF2 inhibits liver cancer cell growth, migration and colony formation
Krüppel‐like factor 2 expression in HCC tissues and in liver cancer animal models is downregulated, which indicates that KLF2 may act as a tumor suppressor gene in liver cancer occurrence and development. To demonstrate this hypothesis, we first used a virus that overexpressed Flag‐KLF2 to infect the liver cancer cells 7404 and Huh‐7. After 72 hours, we sorted the GFP‐positive cells and performed a western blot to detect the expression of Flag‐KLF2 (see Figure S2a). After obtaining 7404 cells and Huh‐7 cells with stably expressed Flag‐KLF2, we used an MTT assay to detect the effect of KLF2 on the growth of 7404 cells and Huh‐7 cells. Experimental results showed that, on the 8th day, the OD value of the control cells was significantly higher than that of 7404 cells and Huh‐7 cells that overexpressed KLF2 (Figure 2A). The experimental results showed that the upregulation of KLF2 in liver cancer cells inhibited the growth of tumor cells. Tumor progression can also be embodied by the migration capacity of tumor cells, which can be detected by a Boyden chamber experiment. After 8 hours, for both 7404 cells and Huh‐7 cells, after KLF2 overexpression, cell migration capacity was weakened, and the number of cells that passed through the middle hole to reach the other side of the film was approximately 50% of the number in the control group (Figure 2B,C). According to the results, upregulated KLF2 in liver cancer cells could inhibit the migration capacity of cells. The other feature of tumor cells that can be used to distinguish them from normal cells is the ability for anchorage‐independent growth. The soft agar colony experiment is an important method for detecting the anchorage‐independent growth of tumor cells. We utilized the soft agar colony experiment to detect the effect of upregulated KLF2 on the anchorage‐independent growth of 7404 cells. The experimental results showed that after overexpression of KLF2, the number of colonies of 7404 cells on soft agar was reduced and that the size of the colonies was also smaller (Figure 2D,E). Such results showed that upregulated KLF2 could inhibit the colony‐formation ability of liver cancer cells.
Figure 2.
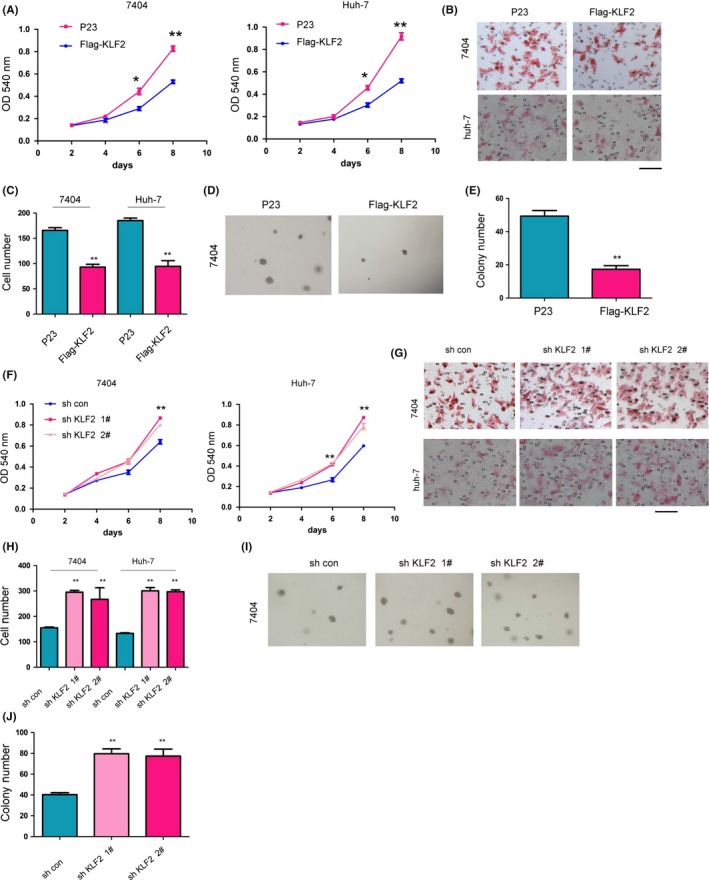
Krüppel‐like factor 2 (KLF2) inhibits liver cancer cell growth, migration and colony‐formation ability. A, MTT assay was used to study the effect of overexpressed KLF2 on liver cancer cell growth. B,C, Borden chamber experiment was used to study the effect of KLF2 overexpression on liver cancer cell migration (scale bar: 100 μm, representative images). D,E, Soft agar colony experiment was used to study the effect of KLF2 overexpression on the anchorage‐independent growth of cancer cells. F, MTT assay was used to study the effect of downregulating KLF2 on liver cancer cell growth. (G,H) Borden chamber experiment was used to study the effect of KLF2 downregulation on liver cancer cell migration (scale bar: 100 μm, representative images). I,J, Soft agar colony experiment was used to study the effect of KLF2 overexpression on the anchorage‐independent growth of cancer cells. *P < 0.05; **P < 0.01
To exclude false‐positive results from the overexpressed exogenous proteins in liver cancer cells, we utilized shRNA interference to downregulate the expression of KLF2 in 7404 cells and Huh‐7 cells. To exclude off‐target effects, we designed two independent sequences to downregulate the expression of KLF2. The two independent shRNA sequences (sh KLF2 1# and sh KLF2 2#) could effectively downregulate the level of KLF2 protein with an inhibition efficiency of approximately 50% (see Figure S2b). We used an MTT assay to detect the effect of KLF2 downregulation on cell growth. The experimental results showed that after KLF2 expression was downregulated, on days 6 and 8 of the experiment, the OD value of the experimental group was significantly higher than that of the control group, which indicated that interference of KLF2 expression promoted the growth of liver cancer cells (Figure 2F). Next, we performed a Boyden chamber assay to detect the effect of KLF2 downregulation on liver cancer cell migration. The results showed that after KLF2 expression was downregulated, the number of migrated cells was significantly higher than that of the control group (Figure 2G,H), which indicated that interference in KLF2 expression promoted the migration of liver cancer cells. In addition, the soft agar colony‐formation experiment demonstrated that after KLF2 expression was downregulated in 7404 cells, the colony‐formation ability of those cells on soft agar was enhanced (Figure 2I,J). In conclusion, KLF2 suppresses the growth, migration and colony‐formation ability of liver cancer cells.
3.3. Downregulated KLF2 expression promotes hepatic metastasis
Previous studies have shown that upregulation of KLF2 expression in liver cancer cells can inhibit the growth, migration and colony‐formation ability of liver cancer cells, while downregulation of KLF2 can promote the growth, migration and colony‐formation ability of liver cancer cells. However, the effect of KLF2 on liver cancer occurrence in vivo has not been clearly demonstrated. Thus, we utilized an intrahepatic metastasis model of liver cancer to investigate the effect of KLF2 on intrahepatic metastasis of liver cancer cells. Parental 7404 control (7404/sh con) tumor cells or those in which KLF2 expression was downregulated (7404/sh KLF2) were injected into the liver at the edge of the largest liver lobe. Due to the presence of abundant vessels, the tumor cells in this location could metastasize into the blood stream. Tumors that formed in the lobe that was injected with cells were considered tumors in situ, while tumors in the other lobe were considered metastatic tumors. In the 5th week of the experiment, the mice were killed and their livers were imaged (Figure 3A). Experimental results showed that the control cells were primarily in the lobe that was injected with cells, but that the 7404/sh‐KLF2 cells were not only in the injected lobe, but were also in the other liver lobes. This indicated that after KLF2 expression was downregulated, the metastasis and migration ability of 7404 cells was enhanced, which was consistent with the finding that downregulated KLF2 expression could promote liver cancer cell migration, as observed in the in vitro experiment. Later, we performed HE staining on liver tissue that was not injected with cells (Figure 3B). In the mice injected with 7404 control cells, the tumor area in the non‐injected liver lobe was smaller and was approximately 20% of the visual field. In the mice injected with 7404/sh‐KLF2 cells, the tumor area in the non‐injected liver lobe was larger and was approximately 90% of the visual field. This indicated that downregulated KLF2 expression could induce 7404 cells to form tumors in the non‐injected lobe of the liver. Next, we analyzed the survival of the mice. The median survival time of mice injected with 7404 control cells was approximately 6.8 weeks, but the survival time of mice injected with 7404/sh KLF2 cells was only 4 weeks. In addition, the mice injected with 7404 control cells all died during the 12th week of the experiment, whereas those injected with 7404/sh KLF2 cells died during the 6th week (Figure 3C). Finally, we utilized TUNEL staining to detect apoptosis of cells in the non‐injected lobe of the liver in the two groups of mice (Figure 3D‐E). The liver tissue of mice injected with 7404 control cells contained more TUNEL‐positive signals. This indicated that KLF2 downregulation inhibited apoptosis of tumor cells, which was consistent with the finding that KLF2 downregulation can promote the growth of liver cancer cells in vitro. These findings indicated that KLF2 downregulation in liver cancer cells promotes intrahepatic metastasis of liver cancer cells.
Figure 3.
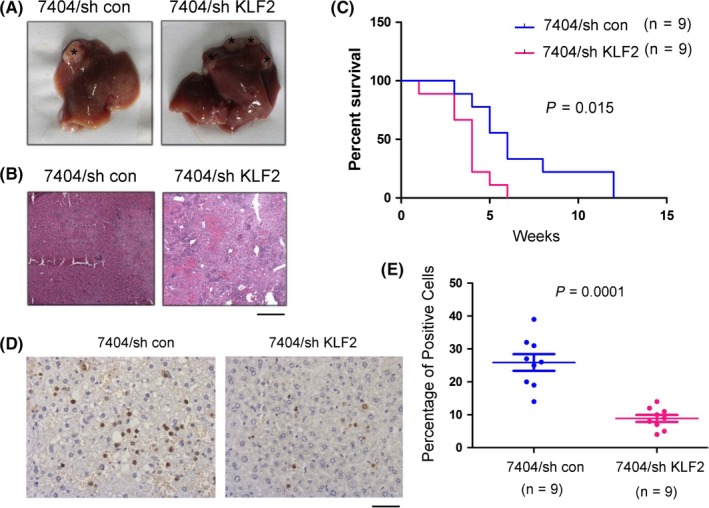
Downregulated Krüppel‐like factor 2 (KLF2) expression promotes hepatic metastasis. A, Photograph of a liver injected with 7404/sh con cells and 7404/sh KLF2 cells. B, Hematoxylin‐eosin staining was performed on the non‐injected lobe of liver and in the lobe that was injected with 7404/sh con cells or 7404/sh KLF2 cells (scale bar: 100 μm, representative images). C, Survival time of mice injected with 7404/sh con cells or 7404/sh KLF2 cells. D,E, Terminal deoxynucleotidyl transferase dUTP nick end labeling staining was performed on the non‐injected lobe of liver injected with 7404/sh con cells or 7404/sh KLF2 cells, and a statistical analysis was performed (scale bar: 20 μm, representative images). *P < 0.05; **P < 0.01. Lentivirus was used to produce stable KLF2 transformants in 7404 cells
3.4. KLF2 inhibits the Hedgehog/Gli1 signaling pathways in liver cancer cells
In previous studies, we demonstrated the potential biological function of KLF2 in liver cancer occurrence by analyzing the expression of KLF2 in liver cancer tissue and detecting the effect of KLF2 expression on the growth, migration and metastasis of liver cancer cells. However, the mechanism of KLF2 in the regulation of liver cancer cell growth, migration and metastasis has not been clearly demonstrated. First, using a luciferase reporter system, we screened the signaling pathways of KLF2 that possibly affect liver cancer occurrence. After the ligand protein Shh was added or the Gli1 expression vectors were co‐transfected, the Hedgehog/Gli1 signaling pathway was activated and KLF2 expression inhibited the activation of the Hedgehog/Gli1 signaling pathway by the Shh ligand and Gli1 expression (Figure 4A). This indicates that KLF2 can inhibit the Hedgehog/Gli1 signaling pathway in liver cancer cells. The Hedgehog signaling pathway can regulate the expression of multiple downstream target genes (FOXM, Patch and Gli1). Therefore, we detected the effect of KLF2 expression on downstream target genes of the Hedgehog/Gli1 signaling pathway (Figure 4B). The results indicated that upregulated KLF2 expression in 7404 and Huh‐7 liver cancer cells can result in downregulated expression of the downstream genes FOXM, Patch and Gli1. These experimental results showed that KLF2 can inhibit the Hedgehog and Gli1 signaling pathways in liver cancer cells. In previous studies, we discovered that overexpression of KLF2 can inhibit the colony‐formation ability of 7404 liver cancer cells. Next, we examined whether the transcription factor Gli1 could restore the biological function of KLF2. The expression of KLF2 inhibited the colony‐formation ability of 7404 cells on soft agar, and overexpression of Gli1 fully rescued the inhibition of KLF2 (Figure 4C,D). Moreover, the expression of KLF2 was negatively correlated with the expression of Gli1 and Patch (Figure 4E). This demonstrated that KLF2 can actually inhibit liver cancer occurrence and development through inhibition of Hedgehog/Gli1 signaling pathway.
Figure 4.
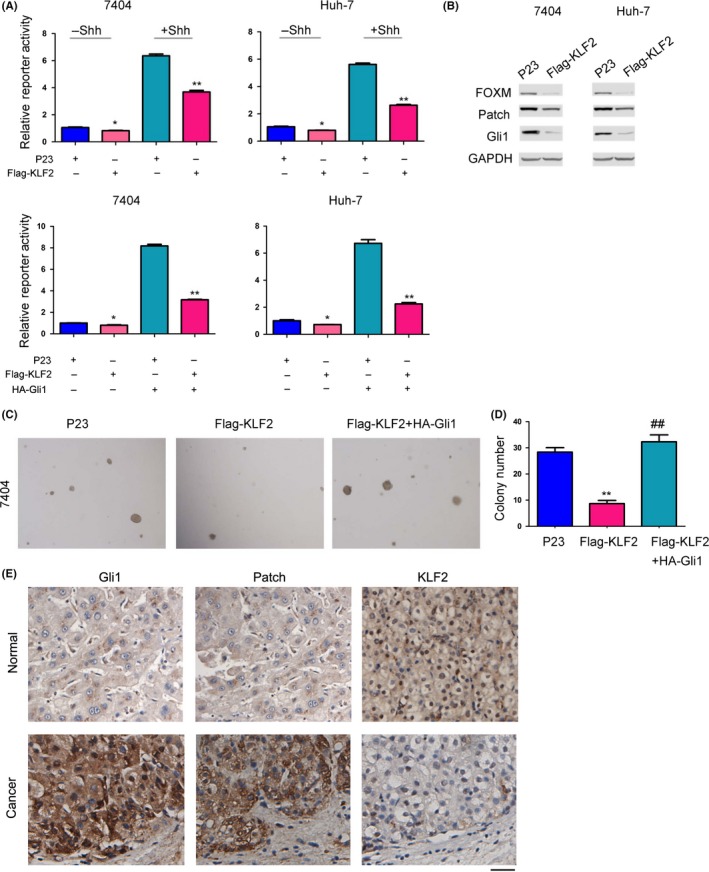
Krüppel‐like factor 2 (KLF2) inhibits the Hedgehog/Gli1 signaling pathways in liver cancer cells. A, Luciferase reporter assay shows the effect of KLF2 expression on the Hedgehog/Gli1 signaling pathway. Cells were treated with Shh protein (100 ng/mL) for 8 h or co‐transfected (transient transfection, using Lipofectamine® 2000) with Gli1 expression vectors for 24 h before harvested. B, Western blot detected the effect of KLF2 expression on the Hedgehog/Gli1 signaling pathway and its downstream target genes. C,D, A soft agar colony‐formation experiment was used to detect the response of Gli1 expression to KLF2 biological function. E, Immunohistochemistry was performed to examine the expression of Gli, Patch and KLF2 in hepatocellular carcinoma tissues and non‐cancerous tissues (scale bar: 50 μm, representative images). *P < 0.05; **P < 0.01; ## P < 0.01. B‐D, Lentivirus was used to produce stable KLF2 and Gli1 transformants in 7404 and Huh7 cells
3.5. KLF2 interacts with Gli1 in HCC cells
Previous studies showed that KLF2 inhibits the Hedgehog/Gli1 signaling pathway, but the detailed mechanism by which KLF2 suppresses the Hedgehog/Gli1 signaling pathway has not been clearly shown. Considering the function of KLF2 as a transcription factor, we first investigated the interaction between KLF2 and the transcription machinery of the Hedgehog/Gli1 signaling pathway in the cell nucleus. Gli1 is one of the most important transcription factors in Hedgehog/Gli1 signaling. Thus, we first detected the interaction between KLF2 and Gli1. We purified the GST‐KLF2 recombinant protein, which was incubated with 7404 or Huh‐7 cells in lysis buffer overnight; GST protein served as the control (Figure 5A). The results showed that the endogenously expressed Gli1 protein in 7404 or Huh‐7 cells could interact with the GST‐KLF2 fusion protein. HA‐Gli1 and Flag‐KLF2 were expressed in 7404 cells by plasmid transfection. After 48 hours, the protein was extracted and co‐immunoprecipitation was performed with an anti‐Flag antibody. The experimental results showed that HA‐Gli1 protein and Flag‐KLF2 protein interacted in 7404 cells (Figure 5B). In a further study, we used a co‐immunoprecipitation assay to detect the interaction of endogenously expressed KLF2 and Gli1. The detection results were consistent with the results discussed above, namely that endogenously expressed Gli1 and KLF2 were found to interact (Figure 5C). On the contrary, we performed immunofluorescence staining to detect the co‐localization of endogenously expressed KLF2 and Gli1 proteins in 7404 cells. The results showed that KLF2 and Gli1 were co‐localized (Figure 5D). As a transcription factor located in the cell nucleus, Gli1 can interact with multiple proteins. We next determined that the C‐terminal of Gli1 was responsible for its binding with KLF2 (Figure 5E). It has been reported that HDAC1 can interact with and deacetylate the C‐terminal of Gli1, which results in Hedgehog/Gli1 signal pathway activation.32 Finally, we investigated whether KLF2 could negatively regulate the interaction between HDAC1 and Gli1. As shown in Figure 5(F), The activation of Hedgehog/Gli1 signal pathway by HDAC1 could be repressed by KLF2. In addition, in 7404 cells, overexpressed KLF2 inhibited the interaction between HDAC1 and Gli1 (Figure 5G). These studies showed that KLF2 may negatively regulate the Hedgehog/Gli1 signaling pathway by inhibiting the interaction between HDAC1 and Gli1.
Figure 5.
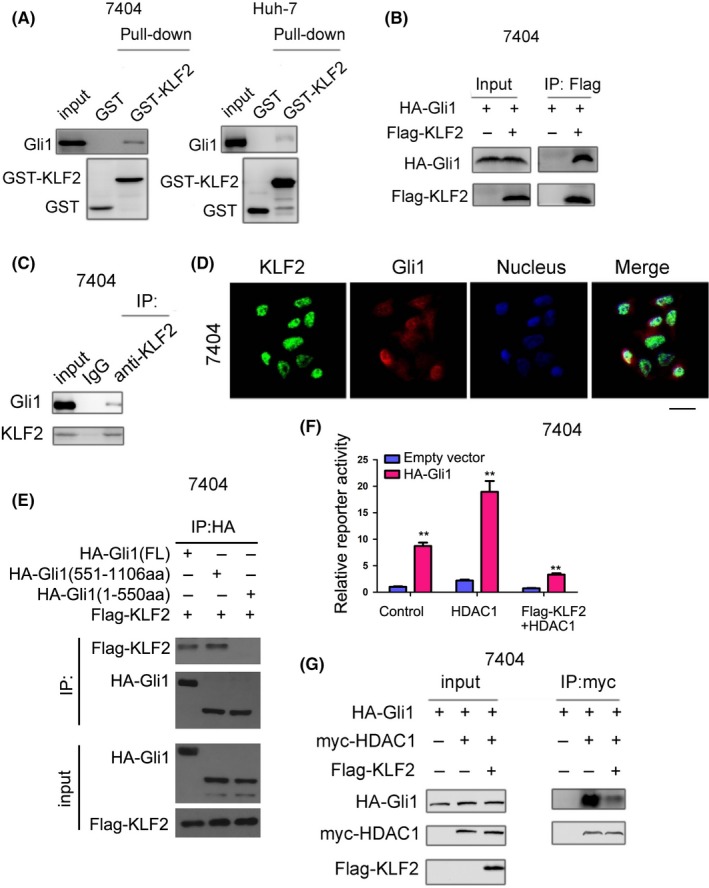
Krüppel‐like factor 2 (KLF2) interacts with Gli1 in hepatocellular carcinoma cells. A, A GST pull‐down assay was used to detect the interaction between recombinant protein GST‐KLF2 and Gli1 expressed by liver cancer cells. B, Co‐immunoprecipitation was used to detect the interaction between KLF2 (Flag‐KLF2) and Gli1 (HA‐Gli1). C, Co‐immunoprecipitation was used to detect the interaction between endogenously expressed KLF2 and Gli1. D, Immunofluorescence staining was used to detect the co‐localization between KLF2 and Gli1 (scale bar: 20 μm, representative images). E, Co‐immunoprecipitation was used to detect the domain of Gli1 for the binding of KLF2. F, Reporter assay was used to examine the effects KLF2 on the activation of Hedgehog/Gli1 signaling by HDAC1. G, Co‐immunoprecipitation was used to detect the effect of exogenously expressed KLF2 (Flag‐KLF2) on the interaction between HDAC1 and Gli1. Transient transfection was performed using the Lipofectamine 2000 according to the manufacturer's instructions
4. DISCUSSION
Hepatocellular carcinoma is one of the most common malignant tumors in China. Therefore, studies on the molecular mechanism of HCC occurrence and development are of great significance for the diagnosis and treatment of this disease. In this study, we discovered that in the process of HCC occurrence, KLF2 expression was downregulated. In an analysis of its cellular biological function, we discovered that upregulation of KLF2 in liver cancer cells can inhibit their growth, migration and colony‐formation ability. On the contrary, downregulation of KLF2 can promote tumor cell growth, migration and colony‐formation ability. In the mouse intrahepatic metastasis experiment, downregulated KLF2 in liver cancer cells can promote the intrahepatic metastasis of tumor cells and shorten the survival time of the mice. In the molecular mechanistic study, we revealed that KLF2 can interact with Gli1 to inhibit the interaction of Gli1 and HDAC1, which results in inhibition of the Hedgehog/Gli1 signaling pathway. Consistently, we observed that KLF2 can inhibit the reporter gene activity of the Hedgehog/Gli1 signaling pathway and the expression of downstream target genes and that overexpressed Gli1 can reverse the suppression of KLF2. These results indicated that KLF2 can inhibit liver cancer occurrence and development by negatively regulating the Hedgehog/Gli1 signaling pathway. In this study, we did not elucidate what type of molecules were used by KLF2 to affect the in vitro migration and in vivo metastasis of liver cancer cells. However, we observed that overexpressed KLF2 can result in the downregulation of the transcription factor Gli1, which can regulate the expression of multiple molecules related to cell migration. For example, Gli1 can induce EMT of intestinal cancer cells, which promotes the metastasis of intestinal cancer cells.33 Additionally, relevant studies have shown that through vascular endothelial growth factor upregulation, Gli1 can promote the infiltration and metastasis of skin squamous cell carcinoma.34 We also found that Gli1 can promote the metastasis of brain tumor cells.35 Such studies have shown that KLF2 may inhibit the migration capacity of liver cancer cells by inhibiting the expression of Gli1 target genes. In angiogenesis, KLF2 can mediate the inhibition of endothelial cell migration, which results from ERK5 inactivation, by inhibiting PAK1 expression.36 In tumorigenesis, KLF2 expression is downregulated in pancreatic ductal adenocarcinoma, as its overexpression inhibits the migration and metastasis of adenocarcinoma.32
This study demonstrated the important role of the interaction between KLF2 and Gli1 in the regulation of Hedgehog signaling activation. Gli1 protein can be modified, such as by phosphorylation, acetylation and ubiquitination. The acetyltransferase P300/CBP‐associated factor can acetylate Gli1, which inhibits Gli1 transcriptional activity.37, 38 The deacetylase HDAC1 promotes the deactivation of Gli1, which activates the Hedgehog/Gli1 signaling pathway.39 In this study, we revealed that KLF2 expression can inhibit the interaction between HDAC1 and Gli1, which demonstrates that HDAC1 and KLF2 may competitively bind to Gli1. In normal cells, the interaction of KLF2/Gli1 is balanced with that of HDAC1/Gli1. In tumor cells, changes in the expression of KLF2 and HDAC1 disrupt the balance between the interaction of KLF2/Gli1 and that of HDAC1/Gli1, and in this case, the Hedgehog/Gli1 signaling pathway is overactivated and tumors develop. Consistent with this hypothesis, some studies have reported on the upregulation of HDAC1 in liver cancer tissue,40 while our study also showed that KLF2 expression is downregulated in liver cancer tissue. These study results showed that the increased interaction between HDAC1 and Gli1 can activate the transcriptional activity of Gli1.
In conclusion, we observed that KLF2 expression in HCC tissue is downregulated and that KLF2 can inhibit the growth, migration and metastasis of liver cancer cells by inhibiting the Hedgehog/Gli1 signaling pathway. These findings propose KLF2 as a promising diagnostic marker for HCC occurrence, and the re‐expression of KLF2 in liver cancer cells may also be considered a new strategy for HCC treatment.
CONFLICTS OF INTEREST
Authors declare no conflict of interest for this article.
Supporting information
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported by grants from Science & Technology Project of Shenzhen Longgang District (20170403100420500), Guangdong Provincial Department of Science and Technology (2018A030313525) and talent grants from Guizhou Science and Technology Cooperation Platform (2017‐5634, 2018‐5801).
Lin J, Tan H, Nie Y, et al. Krüppel‐like factor 2 inhibits hepatocarcinogenesis through negative regulation of the Hedgehog pathway. Cancer Sci. 2019;110:1220–1231. 10.1111/cas.13961
JinBo Lin, Huifang Tan, Yingjie Nie and Dongwen Wu contributed equally to this study.
Contributor Information
Xiaoliang Chen, Email: 120497143@qq.com.
Tao Chen, Email: chen_tau@aliyun.com.
REFERENCES
- 1. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599‐616. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. Cancer J Clin 2016;66:7‐30. [DOI] [PubMed] [Google Scholar]
- 3. Ye H, Zhang C, Wang BJ, et al. Synergistic function of Kras mutation and HBx in initiation and progression of hepatocellular carcinoma in mice. Oncogene. 2014;33:5133‐5138. [DOI] [PubMed] [Google Scholar]
- 4. Chin R, Earnest‐Silveira L, Koeberlein B, et al. Modulation of MAPK pathways and cell cycle by replicating hepatitis B virus: factors contributing to hepatocarcinogenesis. J Hepatol. 2007;47:325‐337. [DOI] [PubMed] [Google Scholar]
- 5. Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16:313‐317. [DOI] [PubMed] [Google Scholar]
- 6. Lee YR, Park SY. P53 expression in hepatocellular carcinoma: influence on the radiotherapeutic response of the hepatocellular carcinoma. Clin Mol Hepatol. 2015;21:230‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zheng X, Zeng W, Gai X, et al. Role of the Hedgehog pathway in hepatocellular carcinoma (review). Oncol Rep. 2013;30:2020‐2026. [DOI] [PubMed] [Google Scholar]
- 8. Edeling M, Ragi G, Huang S, Pavenstadt H, Susztak K. Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat Rev Nephrol. 2016;12:426‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pasca dMM, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer 2003;3:903‐911. [DOI] [PubMed] [Google Scholar]
- 11. Shi Y, Sun X, He X. Overexpression of Aristaless‐Like Homeobox‐4 Inhibits Proliferation, Invasion, and EMT in Hepatocellular Carcinoma Cells. Oncol Res. 2017;25:11‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li W, Miao S, Miao M, et al. Hedgehog Signaling Activation in Hepatic Stellate Cells Promotes Angiogenesis and Vascular Mimicry in Hepatocellular Carcinoma. Cancer Invest. 2016;34:424‐430. [DOI] [PubMed] [Google Scholar]
- 13. O'Grady E, Mulcahy H, Adams C, Morrissey JP, O'Gara F. Manipulation of host Kruppel‐like factor (KLF) function by exotoxins from diverse bacterial pathogens. Nat Rev Microbiol. 2007;5:337‐341. [DOI] [PubMed] [Google Scholar]
- 14. Zhang W, Levi L, Banerjee P, Jain M, Noy N. Kruppel‐like factor 2 suppresses mammary carcinoma growth by regulating retinoic acid signaling. Oncotarget. 2015;6:35830‐35842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tarapore RS, Yang Y, Katz JP. Restoring KLF5 in esophageal squamous cell cancer cells activates the JNK pathway leading to apoptosis and reduced cell survival. Neoplasia. 2013;15:472‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang Y, Tetreault MP, Yermolina YA, Goldstein BG, Katz JP. Kruppel‐like factor 5 controls keratinocyte migration via the integrin‐linked kinase. J Biol Chem. 2008;283:18812‐18820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang VH, Tsai YC, Tsai YL, et al. Krupple‐like factor 10 regulates radio‐sensitivity of pancreatic cancer via UV radiation resistance‐associated gene. Radiother Oncol. 2017;122:476‐484. [DOI] [PubMed] [Google Scholar]
- 18. Venkov C, Plieth D, Ni T, et al. Transcriptional networks in epithelial‐mesenchymal transition. PLoS ONE. 2011;6:e25354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee UE, Ghiassi‐Nejad Z, Paris AJ, et al. Tumor suppressor activity of KLF6 mediated by downregulation of the PTTG1 oncogene. FEBS Lett. 2010;584:1006‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen YN, He HG, Shi Y, et al. Kruppel‐like factor 8 promotes cancer stem cell‐like traits in hepatocellular carcinoma through Wnt/beta‐catenin signaling. Mol Carcinog. 2017;56:751‐760. [DOI] [PubMed] [Google Scholar]
- 21. Sun J, Wang B, Liu Y, et al. Transcription factor KLF9 suppresses the growth of hepatocellular carcinoma cells in vivo and positively regulates p53 expression. Cancer Lett. 2014;355:25‐33. [DOI] [PubMed] [Google Scholar]
- 22. Liu FY, Deng YL, Li Y, et al. Down‐regulated KLF17 expression is associated with tumor invasion and poor prognosis in hepatocellular carcinoma. Med Oncol. 2013;30:425. [DOI] [PubMed] [Google Scholar]
- 23. Nie F, Yu X, Huang M, et al. Long noncoding RNA ZFAS1 promotes gastric cancer cells proliferation by epigenetically repressing KLF2 and NKD2 expression. Oncotarget. 2016;8(24):38227‐38238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li W, Sun M, Zang C, et al. Upregulated long non‐coding RNA AGAP2‐AS1 represses LATS2 and KLF2 expression through interacting with EZH2 and LSD1 in non‐small‐cell lung cancer cells. Cell Death Dis. 2016;7:e2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nie FQ, Sun M, Yang JS, et al. Long noncoding RNA ANRIL promotes non‐small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer Ther. 2015;14:268‐277. [DOI] [PubMed] [Google Scholar]
- 26. Fang J, Sun CC, Gong C. Long noncoding RNA XIST acts as an oncogene in non‐small cell lung cancer by epigenetically repressing KLF2 expression. Biochem Biophys Res Comm. 2016;478:811‐817. [DOI] [PubMed] [Google Scholar]
- 27. Huang MD, Chen WM, Qi FZ, et al. Long non‐coding RNA TUG1 is up‐regulated in hepatocellular carcinoma and promotes cell growth and apoptosis by epigenetically silencing of KLF2. Mol Cancer. 2015;14:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang MD, Chen WM, Qi FZ, et al. Long non‐coding RNA ANRIL is upregulated in hepatocellular carcinoma and regulates cell proliferation by epigenetic silencing of KLF2. J Hematol Oncol. 2015;8:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng YZ, Cai Z, Shi S, et al. Cilia loss sensitizes cells to transformation by activating the mevalonate pathway. J Exp Med. 2018;215:177‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cai Z, Qian ZY, Jiang H, et al. hPCL3s Promotes Hepatocellular Carcinoma Metastasis by Activating beta‐Catenin Signaling. Cancer Res. 2018;78:2536‐2549. [DOI] [PubMed] [Google Scholar]
- 31. Dow LE, O'Rourke KP, Simon J, et al. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell. 2015;161:1539‐1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang D, Dai Y, Cai Y, et al. KLF2 is downregulated in pancreatic ductal adenocarcinoma and inhibits the growth and migration of cancer cells. Tumour Biol. 2016;37:3425‐3431. [DOI] [PubMed] [Google Scholar]
- 33. Zhang C, Wang Y, Feng Y, et al. Gli1 promotes colorectal cancer metastasis in a Foxm1‐dependent manner by activating EMT and PI3K‐AKT signaling. Oncotarget. 2016;7:86134‐86147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun Q, Bai J, Lv R. Hedgehog/Gli1 signal pathway facilitates proliferation, invasion, and migration of cutaneous SCC through regulating VEGF. Tumour Biol. 2016;37:16215‐16225. [DOI] [PubMed] [Google Scholar]
- 35. Santoni M, Burattini L, Nabissi M, et al. Essential role of Gli proteins in glioblastoma multiforme. Curr Protein Pept Sci. 2013;14:133‐140. [DOI] [PubMed] [Google Scholar]
- 36. Komaravolu RK, Adam C, Moonen JR, Harmsen MC, Goebeler M, Schmidt M. Erk5 inhibits endothelial migration via KLF2‐dependent down‐regulation of PAK1. Cardiovasc Res. 2015;105:86‐95. [DOI] [PubMed] [Google Scholar]
- 37. Infante P, Canettieri G, Gulino A, Di Marcotullio L. Yin‐Yang strands of PCAF/Hedgehog axis in cancer control. Trends Mol Med. 2014;20:416‐418. [DOI] [PubMed] [Google Scholar]
- 38. Malatesta M, Steinhauer C, Mohammad F, Pandey DP, Squatrito M, Helin K. Histone acetyltransferase PCAF is required for Hedgehog‐Gli‐dependent transcription and cancer cell proliferation. Can Res. 2013;73:6323‐6333. [DOI] [PubMed] [Google Scholar]
- 39. Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3‐REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132‐142. [DOI] [PubMed] [Google Scholar]
- 40. Quint K, Agaimy A, Di Fazio P, et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011;459:129‐139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials