

Abstract
Colorectal cancer (CRC) is currently the most common type of cancer in Japan, and its prognosis has improved because of development of diagnosis and advancement in treatments including surgery and chemotherapy. However, because of intratumor heterogeneity and clonal evolution, tumors often develop resistance to treatment. Genotyping tumor tissue in search of somatic genetic alterations for actionable information has become routine examination in clinical practice. However, the inherent molecular heterogeneity of metastatic tumors and the ability of cancer genomes to dynamically evolve are not properly captured by tissue specimens only. Circulating tumor DNA (ctDNA) carrying tumor‐specific genetic or epigenetic alterations is released into the circulation from tumor cells undergoing apoptosis or necrosis. Analysis of ctDNA has the potential to change clinical practice by exploiting blood rather than tissue, as a source of information. Here, we provide an overview of the characteristics of ctDNA and focus on detection methods for ctDNA, and the feasibility of use of ctDNA to monitor tumor dynamics for patients with colorectal cancer.
Keywords: circulating tumor DNA, colorectal cancer, liquid biopsy, next‐generation sequencing, tumor mutation burden
1. INTRODUCTION
Measurement of biomarkers before treatment is of importance in the optimum management of patients with colorectal cancer (CRC). For example, tissue‐based biomarkers such as RAS mutation, BRAF mutation, HER2 amplification, and microsatellite instability are used to determine prognosis and guide systemic therapy, especially in metastatic CRC.1, 2 Tissue‐based biomarkers have been extensively reviewed in recent years and remain the gold standard at present. However, sampling bias can readily occur in conventional sampling methods such as needle biopsies.3 This is due to the difficulty of obtaining sufficient material of adequate quality for cancer genome profiling4, 5 and sampling biases that arise from genetic heterogeneity (Table 1).6, 7, 8, 9 In contrast, a new diagnostic concept referred to as “liquid biopsy” has received considerable attention over the past few years.10, 11 Compared with a classic biopsy, liquid biopsies are more convenient, and present minimal procedural risk to the patient (Table 1).12 Over the past 10 years, large‐scale clinical studies have focused on the use of circulating tumor cell (CTC) counts as predictors for prognosis and response to therapy, particularly in breast and prostate cancer.10, 13 Furthermore, relevant molecular information may also be obtained by analyzing microRNA (miRNA) present in extracellular vesicles or exosomes.14 Recently, several reports have described the potential utility of management of patients with cancer as a result of advances in circulating tumor DNA (ctDNA) analysis.8, 10 Thus, in the present review, we focus on the potential clinical utility of ctDNA as key components of liquid biopsies in CRC.
Table 1.
Comparison of ctDNA vs tissue biopsy testing
| ctDNA assay | Tissue‐based assay |
|---|---|
| Advantage | |
| Quick | Substantial evidence for treatment selection in multiple malignancies for early and advanced cancers |
| Comprehensive tissue profile (reflection of inter‐ and intrasample heterogeneity) | High correlation with histology and cellular phenotype |
| Easy sampling | — |
| Minimally invasive | — |
| Disadvantage | |
| Limited evidence for treatment selection in advanced cancer | Time‐intensive procedure |
| Low correlation with histology or cellular phenotype | Localized sampling of tissue |
| — | Sampling is not easily carried out |
| — | Invasive |
ctDNA, circulating tumor DNA.
2. CIRCULATING CELL‐FREE DNA AND ctDNA BIOLOGY
Cell‐free DNA (cfDNA) was first reported in 1948 by Mandel and Métais who detected non−cell−bound nucleic acids in the bloodstream of individuals with cancer.15 cfDNA is thought to be released from cells mostly through apoptosis and necrosis.16, 17, 18, 19 Most studies have focused on cfDNA that is released into the blood of cancer patients. Notably, the categories of body fluids that can be profiled have recently expanded beyond blood and now include urine, cerebrospinal fluid, pleural fluid, and saliva.20, 21, 22 When tumor volume increases, cellular turnover also generally increases. Hence, patients with cancer have much higher levels of cfDNA than healthy individuals. cfDNA concentrations are also increased in other physiological conditions or clinical scenarios, such as acute trauma,23 cerebral infarction,24 exercise,25 transplantation,26 and infection.27 Most cfDNA fragments measure between 180 and 200 base pairs, suggesting that the majority of cfDNA is released from apoptotic cells. After release of cfDNA into circulation, clearance of cfDNA rapidly occurs in the kidneys, liver, and spleen. Observational studies have reported that the half‐life of cfDNA in circulation is between 16 minutes and several hours,28, 29, 30 which enables cfDNA analysis to be considered as a “real‐time” snapshot of disease burden. ctDNA is the fraction of cfDNA that originates from tumor cells, generally inferred by the detection of somatic variants, which are a small fraction of the total cfDNA.31 They are released from tumor cells into the bloodstream and, in principle, contain genetic defects identical to the tumor cells.32 Accordingly, molecular alterations that can be identified in tumors include point mutations, rearrangements, amplifications, and even gene copy number variations.32 For ctDNA analyses, plasma samples are preferable to serum samples,33 and the overall quantity of cfDNA is two‐ to 24‐fold higher in the serum,34 probably owing to extensive contamination of DNA released from immune cells that are lysed during the clotting process. Therefore, plasma is a superior source of ctDNA owing to the lower background levels of wild−type DNA.
3. DEVELOPMENT OF APPROACHES FOR DETECTION OF ctDNA
Analysis of ctDNA varies from analysis of a point mutation to whole‐genome analyses. Sensitivity of traditional approaches to DNA analysis (such as Sanger sequencing) is insufficient for detection of somatic mutations in plasma cfDNA in patients with cancer.11 To overcome this limitation, it is essential to develop highly sensitive and reproducible detection methods to identify ctDNA. Methodologies for detecting ctDNA are shown in Table 2.35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45
Table 2.
Comparison of technologies for ctDNA analysis
| Scale of analysis | Technology | LoD, % | Advantage | Disadvantage |
|---|---|---|---|---|
| Single‐locus or multiplexed assays | Microfluidic or allele‐specific PCR |
|
|
|
| Droplet digital PCR35, 36, 37 | 0.001 | |||
| Allele‐specific quantitative PCR38 | <0.01 | |||
| BEAMing39 | 0.01 | |||
| Targeted sequencing approaches | Amplicon sequencing |
|
|
|
| Safe‐SeqS40 | 0.10 | |||
| TAm‐Seq41 | >2 | |||
| Hybridization capture | ||||
| CAPP‐Seq42 | 0.01 | |||
| Genome‐wide analysis | Whole‐exome sequencing43, 44 | >1‐3 |
|
|
| Whole‐genome sequencing45 | 5‐10 |
BEAMing, beads, emulsions, amplification, magnetics; CAPP‐Seq, cancer personalized profiling by deep sequencing; cfDNA, cell‐free DNA; ctDNA, circulating tumor DNA; LoD, limit of detection; Safe‐SeqS, safe‐sequencing system; TAm‐Seq, tagged‐amplicon deep sequencing.
Digital droplet PCR uses a droplet generator to partition single pieces of DNA into droplets using an oil/water emulsion.46 Each molecule is individually analyzed for target sequences through an end‐point PCR, allowing the detection of mutant or wild‐type DNA fragments. This method can detect multiple mutations from the same sample with high sensitivity (from 0.05% to 0.001%), but its multiplexing capabilities are limited to the detection of five to 10 different target sequences. The BEAMing method, first described by Diehl et al,28 is based on single‐molecule PCR on microparticles in water‐in‐oil emulsions. Sensitivity is approximately 0.01%; however, this method is relatively complex and hardly usable for routine analysis. Thus, these methods are generally suited to investigate small numbers of mutations and are often applied to analysis of cancer hotspot mutations.
Targeted sequencing can allow the interrogation of multiple loci with high sensitivity using PCR amplicons or hybrid capture methods that suppress background noise. Regions for sequencing range from individual exons of interest (kilobases) to the entire exome (~50 megabases). By reducing the background error rates of sequencing, ctDNA can be detected at allele fractions below 0.1%.40, 47
Amplicon‐based assays that have been optimized for the purpose of ctDNA analysis can target dozens to hundreds of amplicons across multiple kilobases with high sensitivity.40 Capture‐based next generation sequencing (NGS) has the ability to enrich genomic regions of interest by hybridizing target genes/regions to antisense oligonucleotides before sequencing. This approach allows for the agnostic analysis of large portions of the genome and can identify multiple mutations with increased sensitivity.42, 47, 48 The sensitivity of ctDNA detection can be further enhanced, even with limited amounts of input material, by using multiplexed patient‐specific panels in combination with targeted sequencing methods.47 However, because the cost for detection of dozens of mutations in cfDNA is still expensive (>900 USD/test), technical development for genotyping and/or a NGS system for ctDNA detection is expected to reduce the cost in the near future.
4. RELATIONSHIP BETWEEN CLINICAL FACTORS AND ctDNA IN PATIENTS WITH CRC
To better understand the biology of ctDNA, it is important to consider the relationship between clinicopathological factors and ctDNA. Associations between clinicopathological factors and ctDNA are shown in Table 3.49, 50, 51 ctDNA is strongly correlated with initial tumor burden as estimated from the RECIST criteria,49, 51 suggesting that tumor volume is significantly related to the detection of ctDNA in plasma. Patients with liver metastasis showed significantly higher ctDNA compared to patients without metastasis at this site.50, 51 In contrast, peritoneal metastasis was not correlated with ctDNA.50, 51 The relationship between ctDNA and number of metastatic organs is controversial.49, 50, 51 These data suggest that intrinsic biological characteristics of tumors may impact ctDNA release as well as tumor burden. To further optimize the evaluation of ctDNA analysis, further studies exploring the external factors that may influence the results of ctDNA determination might be needed.
Table 3.
Correlation between ctDNA and clinicopathological characteristics
| Strong | Intermediate | No correlation |
|---|---|---|
| Liver metastasis50, 51 | Lung metastasis50, 51 | Peritoneal metastasis50, 51 |
| Tumor diameter49, 51 | CEA49, 51 | Primary tumor location49, 51 |
| CA19‐951 | ||
| LDH51 | ||
| Lymph node metastasis51 | ||
| Number of metastatic organs49, 50, 51 |
CA19‐9, carbohydrate antigen 19‐9; CEA, carcinoembryonic antigen; ctDNA, circulating tumor DNA; LDH, lactate dehydrogenase.
Strong: There are two or more reports suggesting association between clinical factors and ctDNA.
Intermediate: The association between clinical factors and ctDNA is controversial because there is only one report or there are two reports supporting and not supporting an association between clinical factors and ctDNA.
No correlation: There is no report suggesting an association between clinical factors and ctDNA.
5. CLINICAL RELEVANCE OF ctDNA FOR ADVANCED CRC
Circulating tumor DNA analysis can be applied to diagnose, identify and track tumor‐specific alterations during the course of the disease, and to guide therapeutic decisions (Figure 1). In this section, we focus on the significance of ctDNA as a prognostic biomarker, for minimal residual disease and recurrence monitoring and treatment response/resistance (Table 4).
Figure 1.
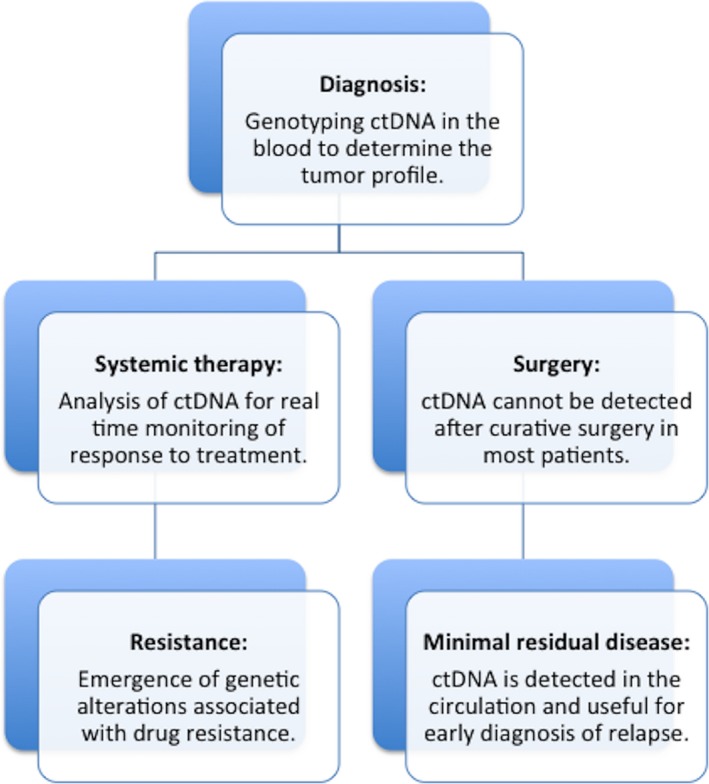
Clinical relevance of circulating tumor DNA (ctDNA) for advanced colorectal cancer. This figure shows key applications of liquid biopsies in the clinical setting. These include tumor genotyping in the diagnosis of cancer, assessing drug response, tracking minimal residual disease, and monitoring clonal evolution
Table 4.
Summary of studies on clinical relevance of ctDNA for colorectal cancer
| ctDNA application | Summary |
|---|---|
| Prognostic biomarker | |
| Minimal residual disease and recurrence monitoring | |
| Treatment response, clonal evolution, and resistance |
cfDNA, cell‐free DNA; ctDNA, circulating tumor DNA; mCRC, metastatic colorectal cancer.
5.1. Prognostic biomarkers
Patients with detectable ctDNA in their plasma were shown to have worse survival outcomes than those without.52, 53 Another study showed a strong association between ctDNA positivity and recurrence‐free survival (RFS), and overall survival (OS) in patients with CRC irrespective of tumor stage, study size, tumor markers, detection methods, and sample type.54 A meta‐analysis of 1076 patients with mCRC treated with chemotherapy confirmed that baseline total cfDNA levels correlate with OS.55 The utility of ctDNA and cfDNA as a prognostic parameter and alternative modality for mutation detection before treatment in metastatic CRC (mCRC) was demonstrated in the randomized CORRECT phase III trial.56 In this trial, 166 mCRC patients received a placebo and 337 were treated with regorafenib. Despite high interpatient variability, high cfDNA concentrations were associated with shorter median OS and progression‐free survival (PFS) in both placebo and regorafenib groups.56 PFS of patients with KRAS mutations in plasma in the regorafenib group was shorter than in those without mutations.57 The prognostic value of KRAS mutations in plasma but not in tumor tissue was confirmed.58 Thus, the detection of ctDNA and total cfDNA levels could have strong prognostic value in CRC and is directly related to disease burden.
5.2. Minimal residual disease and recurrence monitoring
Following surgery or treatment with curative intent, detection of ctDNA may signal the presence of a minimal residual disease (MRD) even in the absence of any other clinical evidence of disease. ctDNA‐based liquid biopsies could be optimized to capture and monitor MRD following curative resection, possibly preceding clinical or radiological recurrence.59, 60 A study reported the ability of ctDNA to detect MRD in 1046 plasma samples from a prospective cohort of 230 patients with resected stage II colon cancer.61 In patients without adjuvant chemotherapy, ctDNA was detected postoperatively in 14 of 178 patients (7.9%) and radiological recurrence was detected during follow up in 11 of these 14 patients (78.6%).61 In contrast, postoperative ctDNA was negative in the remaining 164 of 178 (92.1%) patients and disease recurrence was identified in only 16 (9.8%) patients.61 In patients treated with chemotherapy, presence of ctDNA after completion of chemotherapy was also associated with a shorter RFS (P = .001).61 Another study also reported the ability of ctDNA to detect MRD in patients with locally advanced rectal cancer (T3/T4 and/or nodal metastasis positive) planned for neoadjuvant chemoradiotherapy.62 Although there was no difference in RFS between patients with detectable ctDNA at baseline (pretreatment) and those without detectable ctDNA, patients with a ctDNA‐positive status after chemoradiotherapy or surgery had an increased risk of recurrence (chemoradiotherapy: hazard ratio [HR] 6.6, P < .001; surgery: HR 13.0, P < 0.001).62 Estimated 3‐year RFS was 33% for patients with postoperative ctDNA positivity and 87% for those with postoperative ctDNA negativity. These data suggest that postoperative ctDNA detection may be useful for the prediction of recurrence irrespective of the use of adjuvant chemotherapy. Thus, ctDNA‐based screening of patients with a higher risk of relapse may create opportunities for therapeutic intervention before the development of clinical metastasis.
5.3. Treatment response, clonal evolution, and resistance
Serial liquid biopsies may have particular utility in which resistant mutations could be prospectively identified and treatment could be changed in real time. For example, liquid biopsies have been successfully applied to identify mechanisms of resistance for epidermal growth factor (EGFR) inhibitor in mCRC patients.63 Significant proportions of CRC harbor mutations in KRAS, BRAF, or NRAS that could cause resistance to an EGFR inhibitor, and these hotspot mutations are candidates for mutations detectable in plasma.55, 58 Previous reports showed that patients with RAS wild‐type CRC in tissue, and plasma that is positive for KRAS and BRAF mutations can be resistant to the EGFR inhibitor.46, 64, 65, 66 Importantly, CRC presumably contain resistant mutant clones before treatment and the proportion of these resistant clones increase under therapeutic pressure.67 Time‐course analysis of ctDNA showed that several mutations rapidly emerge during EGFR blockades and can often be detected before radiological relapse (Figure 2).32, 64, 65, 66 For example, the emergence of resistant KRAS mutated clones could be detected for up to 10 months before radiographic confirmation of disease progression.64, 68 The presence of multiple KRAS mutations was detected in the circulation of patients with mCRC receiving EGFR inhibitor.66 Time‐course profiles of ctDNA in patients treated with EGFR inhibitor showed that KRAS mutant clones, which emerge during EGFR blockade, decline upon withdrawal of EGFR inhibitor, allowing for a rechallenge treatment of EGFR inhibitor that can again lead to a response.65
Figure 2.
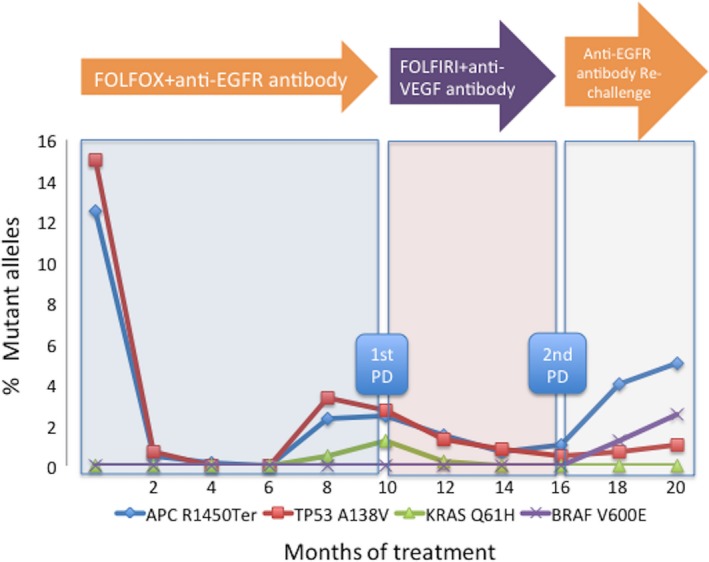
Liquid biopsies to monitor cancer evolution during target therapy. Time‐course analysis of tumor‐specific mutations in the blood of patients is useful to monitor a response and resistance to molecular targeted drugs. For example, we describe a patient with metastatic colorectal cancer treated with EGFR inhibitor. Circulating tumor DNA allows us to identify, track, and quantify clones bearing distinct alleles. Monitoring truncal mutations (TP53, APC) allow us to track tumor burden whereas lesion‐specific mutations (KRAS, BRAF) reflect clonal evolution during chemotherapy. Data of this figure are derived from a combination of our original data with other studies.32, 64, 65, 66 EGFR, epidermal growth factor receptor; PD, progressive disease; VEGF, vascular endothelial growth factor
6. FUTURE PERSPECTIVES AND CONCLUSION
Inhibitors of programmed death 1 (PD‐1) protein or its ligand (PD‐L1) have shown remarkable clinical benefits in many cancers.69 One emerging biomarker for response to anti‐PD‐1 or anti‐PD‐L1 therapy is the tumor mutational burden (TMB: total number of mutations per coding area of a tumor genome). Multiple recent studies have shown that TMB may be a surrogate for overall neoantigen load.70, 71 TMB, measured by whole‐exome sequencing, is associated with clinical benefits in multiple checkpoint inhibitors.72 This finding is supported by the clinical activity of anti‐PD‐1 therapy in CRC with mismatch repair deficiency, a tumor subtype with a high TMB, as compared to the CRC subtypes with mismatch repair proficiency.73 The ability to analyze tumor genomes through a simple blood draw has distinct advantages compared to tissue biopsy collection. Gandara et al74 reported the development, testing, and validation of a novel assay to measure TMB in blood (bTMB) in non‐small cell lung cancer patients treated with anti‐PD‐L1 antibody. This analysis showed that the result of bTMB was highly concordant compared to the results of tissue TMB (tTMB). Furthermore, there was a clear relationship between an increasing bTMB score and PFS.74 Kim et al75 also reported concordance of tumor mutational load between tumor tissue and plasma using a 73‐gene sequencing panel in metastatic gastric cancer. This analysis showed that there was moderate correlation between tumor plasma mutational load and tumor tissue mutational load (r 2 = 0.54).75 Furthermore, both overall response rate and PFS in patients treated with anti‐PD‐1 therapy with a high mutational load score of ctDNA significantly improved compared to patients with a low mutational load score of ctDNA.75 These data suggest that high bTMB could be a clinically actionable biomarker for anti‐PD‐1 or anti‐PD‐L1 therapy as well as high tTMB. As there are few reports on the relationship between tumor mutation burden (both tTMB and bTMB) and clinical benefit of anti‐PD‐1 or anti‐PD‐L1 therapy, especially in CRC patients with mismatch repair proficiency, further study will be needed. Moreover, although the number of patients with ctDNA detected in advanced cancers is likely to be high, that in early‐stage cancers is insufficient.76, 77 Further technical development for genotyping and/or genome sequencing to increase sensitivity and decrease the false‐positive rate is needed to make it possible to detect early cancer using liquid biopsy as a cancer screening test.
In conclusion, we believe that liquid biopsy is likely to be an alternative standard method for monitoring progressive genomic alterations during tumor evolution during exposure to molecular targeted drugs. Liquid biopsy has shown potential utility across a range of applications and will contribute to personalized oncology.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Osumi H, Shinozaki E, Yamaguchi K, Zembutsu H. Clinical utility of circulating tumor DNA for colorectal cancer. Cancer Sci. 2019;110:1148–1155. 10.1111/cas.13972
REFERENCES
- 1. Yoshino T, Arnold D, Taniguchi H, et al. Pan‐Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: a JSMO‐ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS. Ann Oncol. 2018;29:44‐70. [DOI] [PubMed] [Google Scholar]
- 2. Benson AB 3rd, Venook AP, Cederquist L, et al. Colon cancer, version 1.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15:370‐398. [DOI] [PubMed] [Google Scholar]
- 3. Overman MJ, Modak J, Kopetz S, et al. Use of research biopsies in clinical trials: are risks and benefits adequately discussed? J Clin Oncol. 2013;31:17‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vanderlaan PA, Yamaguchi N, Folch E, et al. Success and failure rates of tumor genotyping techniques in routine pathological samples with non‐small‐cell lung cancer. Lung Cancer. 2014;84:39‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ellis PM, Shepherd FA, Millward M, et al. Dacomitinib compared with placebo in pretreated patients with advanced or metastatic non‐small‐cell lung cancer (NCIC CTG BR.26): a double‐blind, randomised, phase 3 trial. Lancet Oncol. 2014;15:1379‐1388. [DOI] [PubMed] [Google Scholar]
- 6. Popper HH. Commentary on tumor heterogeneity. Transl Lung Cancer Res. 2016;5:433‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Mattos‐Arruda L, Weigelt B, Cortes J, et al. Capturing intra‐tumor genetic heterogeneity by de novo mutation profiling of circulating cell‐free tumor DNA: a proof‐of‐principle. Ann Oncol. 2014;25:1729‐1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lebofsky R, Decraene C, Bernard V, et al. Circulating tumor DNA as a non‐invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol Oncol. 2015;9:783‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murtaza M, Dawson SJ, Pogrebniak K, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015;6:8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alix‐Panabieres C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6:479‐491. [DOI] [PubMed] [Google Scholar]
- 11. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merker JD, Oxnard GR, Compton C, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol. 2018;36:1631‐1641. [DOI] [PubMed] [Google Scholar]
- 13. Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C. Molecular analysis of circulating tumour cells‐biology and biomarkers. Nat Rev Clin Oncol. 2014;11:129‐144. [DOI] [PubMed] [Google Scholar]
- 14. Best MG, Sol N, Kooi I, et al. RNA‐seq of tumor‐educated platelets enables blood‐based pan‐cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell. 2015;28:666‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mandel PM, Métais P. Les acides nucléiques du plasma sanguin chez l'Homme [French]. C R Seances Soc Biol Fil. 1948;142:241‐243. [PubMed] [Google Scholar]
- 16. Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35:347‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thakur BK, Zhang H, Becker A, et al. Double‐stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24:766‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kahlert C, Melo SA, Protopopov A, et al. Identification of double‐stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;289:3869‐3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stroun M, Lyautey J, Lederrey C, Olson‐Sand A, Anker P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta. 2001;313:139‐142. [DOI] [PubMed] [Google Scholar]
- 20. Melkonyan HS, Feaver WJ, Meyer E, et al. Transrenal nucleic acids: from proof of principle to clinical tests. Ann N Y Acad Sci. 2008;1137:73‐81. [DOI] [PubMed] [Google Scholar]
- 21. De Mattos‐Arruda L, Mayor R, Ng CK, et al. Cerebrospinal fluid‐derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Commun. 2015;6:8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y, Springer S, Mulvey CL, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci Transl Med. 2015;7:293ra104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodrigues FE, Simon D, Ikuta N, et al. Elevated cell‐free plasma DNA level as an independent predictor of mortality in patients with severe traumatic brain injury. J Neurotrauma. 2014;31:1639‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsai NW, Lin TK, Chen SD, et al. The value of serial plasma nuclear and mitochondrial DNA levels in patients with acute ischemic stroke. Clin Chim Acta. 2011;412:476‐479. [DOI] [PubMed] [Google Scholar]
- 25. Breitbach S, Sterzing B, Magallanes C, Tug S, Simon P. Direct measurement of cell‐free DNA from serially collected capillary plasma during incremental exercise. J Appl Physiol (1985). 2014;117:119‐130. [DOI] [PubMed] [Google Scholar]
- 26. De Vlaminck I, Valantine HA, Snyder TM, et al. Circulating cell‐free DNA enables noninvasive diagnosis of heart transplant rejection. Sci Transl Med. 2014;6:241ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Vlaminck I, Martin L, Kertesz M, et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc Natl Acad Sci USA. 2015;112:13336‐13341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. To EW, Chan KC, Leung SF, et al. Rapid clearance of plasma Epstein‐Barr virus DNA after surgical treatment of nasopharyngeal carcinoma. Clin Cancer Res. 2003;9:3254‐3259. [PubMed] [Google Scholar]
- 30. Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. 1999;64:218‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Crowley E, Di NF, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer‐genetics in the blood. Nat Rev Clin Oncol. 2013;10:472‐484. [DOI] [PubMed] [Google Scholar]
- 32. Bardelli A, Pantel K. Liquid biopsies, what we do not know (yet). Cancer Cell. 2017;31:172‐179. [DOI] [PubMed] [Google Scholar]
- 33. El Messaoudi S, Rolet F, Mouliere F, Thierry AR. Circulating cell free DNA: preanalytical considerations. Clin Chim Acta. 2013;424:222‐230. [DOI] [PubMed] [Google Scholar]
- 34. Jung M, Klotzek S, Lewandowski M, Fleischhacker M, Jung K. Changes in concentration of DNA in serum and plasma during storage of blood samples. Clin Chem. 2003;49:1028‐1029. [DOI] [PubMed] [Google Scholar]
- 35. Sanmamed MF, Fernandez‐Landazuri S, Rodriguez C, et al. Quantitative cell‐free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow‐up of patients with melanoma being treated with BRAF inhibitors. Clin Chem. 2015;61:297‐304. [DOI] [PubMed] [Google Scholar]
- 36. Hindson BJ, Ness KD, Masquelier DA, et al. High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604‐8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taly V, Pekin D, Benhaim L, et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem. 2013;59:1722‐1731. [DOI] [PubMed] [Google Scholar]
- 38. Mouliere F, El Messaoudi S, Pang D, Dritschilo A, Thierry AR. Multi‐marker analysis of circulating cell‐free DNA toward personalized medicine for colorectal cancer. Mol Oncol. 2014;8:927‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single‐molecule PCR on microparticles in water‐in‐oil emulsions. Nat Methods. 2006;3:551‐559. [DOI] [PubMed] [Google Scholar]
- 40. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:9530‐9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra68. [DOI] [PubMed] [Google Scholar]
- 42. Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Manier S, Park J, Capelletti M, et al. Whole‐exome sequencing of cell‐free DNA and circulating tumor cells in multiple myeloma. Nat Commun. 2018;9:1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Annala M, Vandekerkhove G, Khalaf D, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018;8:444‐457. [DOI] [PubMed] [Google Scholar]
- 45. Heitzer E, Ulz P, Belic J, et al. Tumor‐associated copy number changes in the circulation of patients with prostate cancer identified through whole‐genome sequencing. Genome Med. 2013;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med. 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schwaederle M, Husain H, Fanta PT, et al. Detection rate of actionable mutations in diverse cancers using a biopsy‐free (blood) circulating tumor cell DNA assay. Oncotarget. 2016;7:9707‐9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tie J, Kinde I, Wang Y, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol. 2015;26:1715‐1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vidal J, Muinelo L, Dalmases A, et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann Oncol. 2017;28:1325‐1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Osumi H, Shinozaki E, Takeda Y, et al. Clinical relevance of circulating tumor DNA assessed through deep sequencing in patients with metastatic colorectal cancer. Cancer Med. 2018;8:408‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Spindler KG, Appelt AL, Pallisgaard N, Andersen RF, Jakobsen A. KRAS‐mutated plasma DNA as predictor of outcome from irinotecan monotherapy in metastatic colorectal cancer. Br J Cancer. 2013;109:3067‐3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. El Messaoudi S, Mouliere F, Du MS, et al. Circulating DNA as a strong multimarker prognostic tool for metastatic colorectal cancer patient management care. Clin Cancer Res. 2016;22:3067‐3077. [DOI] [PubMed] [Google Scholar]
- 54. Basnet S, Zhang ZY, Liao WQ, Li SH, Li PS, Ge HY. The prognostic value of circulating cell‐free DNA in colorectal cancer: a meta‐analysis. J Cancer. 2016;7:1105‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spindler KG. Methodological, biological and clinical aspects of circulating free DNA in metastatic colorectal cancer. Acta Oncol. 2017;56:7‐16. [DOI] [PubMed] [Google Scholar]
- 56. Tabernero J, Lenz HJ, Siena S, et al. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 2015;16:937‐948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wong AL, Lim JS, Sinha A, et al. Tumour pharmacodynamics and circulating cell free DNA in patients with refractory colorectal carcinoma treated with regorafenib. J Transl Med. 2015;13:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Spindler KL, Pallisgaard N, Appelt AL, et al. Clinical utility of KRAS status in circulating plasma DNA compared to archival tumour tissue from patients with metastatic colorectal cancer treated with anti‐epidermal growth factor receptor therapy. Eur J Cancer. 2015;51:2678‐2685. [DOI] [PubMed] [Google Scholar]
- 59. Ryan BM, Lefort F, McManus R, et al. A prospective study of circulating mutant KRAS2 in the serum of patients with colorectal neoplasia: strong prognostic indicator in postoperative follow up. Gut. 2003;52:101‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang JY, Hsieh JS, Chang MY, et al. Molecular detection of APC, K‐ras, and p53 mutations in the serum of colorectal cancer patients as circulating biomarkers. World J Surg. 2004;28:721‐726. [DOI] [PubMed] [Google Scholar]
- 61. Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tie J, Cohen JD, Wang Y, et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut. 2018. 10.1136/gutjnl-2017-315852. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Parseghian CM, Loree JM, Morris VK, et al. Anti‐EGFR resistant clones decay exponentially after progression: implications for anti‐EGFR re‐challenge. Ann Oncol. 2018. 10.1093/annonc/mdy509. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature. 2012;486:532‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Diaz LA Jr, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Santini D, Vincenzi B, Addeo R, et al. Cetuximab rechallenge in metastatic colorectal cancer patients: how to come away from acquired resistance? Ann Oncol. 2012;23:2313‐2318. [DOI] [PubMed] [Google Scholar]
- 68. Misale S, Arena S, Lamba S, et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti‐EGFR therapies in colorectal cancer. Sci Transl Med. 2014;6:224ra26. [DOI] [PubMed] [Google Scholar]
- 69. Errico A. Immunotherapy: PD‐1‐PD‐L1 axis: efficient checkpoint blockade against cancer. Nat Rev Clin Oncol. 2015;12:63. [DOI] [PubMed] [Google Scholar]
- 70. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348:124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2018;30:44‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gandara DR, Paul SM, Kowanetz M, et al. Blood‐based tumor mutational burden as a predictor of clinical benefit in non‐small‐cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24:1441‐1448. [DOI] [PubMed] [Google Scholar]
- 75. Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD‐1 inhibition in metastatic gastric cancer. Nat Med. 2018;24:1449‐1458. [DOI] [PubMed] [Google Scholar]
- 76. Phallen J, Sausen M, Adleff V, et al. Direct detection of early‐stage cancers using circulating tumor DNA. Sci Transl Med 2017;9:eaan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fiala C, Diamandis EP. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018;16:166. [DOI] [PMC free article] [PubMed] [Google Scholar]