

Abstract
Emerging evidence indicates that the long noncoding RNA UCA1 is upregulated in multiple cancers, including pancreatic ductal adenocarcinoma (PDAC), and plays a critical role in various complex biological processes. However, the functional roles of UCA1 in PDAC remain to be clarified. In the current study, we showed that UCA1 significantly promoted cell proliferation and tumor growth both in vitro and in vivo, and enhanced stemness maintenance of PDAC cell lines. Moreover, we found that UCA1 overexpression increased the activity and expression of oncogenic KRAS. Mechanistically, upregulated UCA1 increased phospho-KRAS protein levels by interacting with hnRNPA2B1, and KRAS facilitated high cytoplasmic accumulation of hnRNPA2B1. Additionally, we identified that UCA1 functioned as a competing endogenous RNA (ceRNA) to increase the expression of KRAS via sponging miR-590-3p, and in turn, KRAS promoted UCA1 expression. Collectively, these findings suggest that the UCA1-KRAS axis plays a crucial role in PDAC progression and that UCA1 may serve as a target for new PDAC therapies.
Keywords: Pancreatic ductal adenocarcinoma, long noncoding RNA, UCA1, KRAS, KRAS phosphorylation
Introduction
Pancreatic ductal adenocarcinoma (PDAC), the main form of pancreatic cancer (90%) and one of the most fatal malignancies, is the fourth leading cause of cancer-associated death worldwide [1-3]. Despite recent advancements in the treatment and the understanding of the molecular biology of the disease, the 5-year overall survival rate of pancreatic cancer patients is less than 5% because of rapid metastasis, chemotherapy resistance and the lack of an effective early detection modality [4-6]. Thus, an explicit understanding of the pathogenesis of and improved strategies for targeting this challenging malignancy are desperately needed.
Long noncoding RNAs (lncRNAs), a class of noncoding transcripts with a length of greater than 200 nucleotides, play vital roles in diverse biological processes and diseases, including the occurrence and development of tumors [7,8]. LncRNA urothelial carcinoma-associated 1 (UCA1), located at 19p13.12, was initially identified in human bladder carcinoma [9]. This lncRNA plays an oncogenic role in multiple types of malignancies, such as bladder cancer, breast cancer, colorectal cancer, gastric cancer [9-13]. Furthermore, upregulated UCA1 is associated with clinicopathological features, tumor stage and poor prognosis of pancreatic cancer patients and promotes the proliferation, migration, invasion and tumorigenesis of pancreatic cancer cells via regulating the Hippo pathway or sponging miR-135a and miR-96 [14-17]. However, little is known about whether UCA1 is involved in the KRAS pathway.
KRAS is the most commonly mutated oncogene in tumors of endodermal origin, such as those of the pancreas (95%), colon (40%), and lung (35%) [18,19]. The most frequently observed oncogenic KRAS mutation in PDAC occurs at codon 12 and results in KRAS remaining constitutively active in the GTP-bound state [18,20,21]. KRAS mediates signaling through three major effector pathways, i.e., the RAF/MEK/ERK, PI3K/AKT/mTOR and RalA/B pathways, as has been shown in human pancreatic cancer tissues, cancer cell lines and mouse models of PDAC [22,23]. Activating KRAS mutations are necessary for the initiation, progression and metastasis of PDAC. In addition, these mutations play a role in stemness maintenance [18,24-27]. Although the results in preclinical and phase I trials targeting KRAS to treat PDAC are encouraging, no significant clinical benefits have been observed. Therefore, there is an urgent need for a better understanding of the pathogenesis of and improved strategies for treating this challenging malignancy.
Here, we found that UCA1 expression was upregulated in PDAC tissues and cell lines. UCA1 was critical for cell growth both in vitro and in vivo as well as for stemness maintenance in PDAC cell lines. Importantly, mechanistic analysis demonstrated for the first time that UCA1 not only enhances phospho-KRAS activity by increasing the binding of hnRNPA2B1 to KRAS but also affects KRAS expression through sponging miR-590-3p to modulate the progression of PDAC. Thus, these results provide evidence for the importance of the UCA1/KRAS regulatory network and suggest that this pathway is a novel target for exploring new therapeutic strategies to treat PDAC.
Materials and methods
Cells and cell culture
The human PDAC cell lines PaTu8902, Mpanc96 and HPAF-II; a human embryonic kidney cell line (HEK293T); and the immortalized pancreatic ductal epithelial cell line H6C7 were purchased from the American Type Culture Collection. The human PDAC cell lines PANC-1, PaTu8988, and SW1990 were kindly provided by the Second Military Medical University in Shanghai. All cells were tested and authenticated by short tandem repeat analysis. Mpanc96 and HPAF-II cells were cultured in RPMI-1640 medium (HyClone, Beijing, China); PANC-1, PaTu8988, SW1990, H6C7 and HEK293T cells were cultured in DMEM (HyClone, Beijing, China). All media were supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 100 units/ml penicillin and 100 mg/ml streptomycin, and the cells were cultured in a humidified 5% CO2 incubator at 37°C.
Western blotting
The cultured cells were rinsed with cold PBS before being lysed with RIPA buffer at 100°C for 10 min. Then, the mixture was centrifuged at 12000 r/min for 5 min. Approximately 10 μl of protein was loaded in each lane of a 10% SDS-PAGE gel. After separation, the protein was transferred to a PVDF membrane. The membrane was blocked with 5% BSA for 1 h at room temperature and was then incubated with primary antibodies at 4°C overnight, followed by the secondary antibody. The antibodies used were rabbit anti-CD133 (Cell Signaling, CAT 64326), rabbit anti-OCT4 (Cell Signaling, CAT 2750), rabbit anti-SOX2 (Cell Signaling, CAT 3579), rabbit anti-NANOG (Cell Signaling, CAT 4903), rabbit anti-hnRNPA2B1 (Abcam, ab31645), mouse anti-KRAS (Santa Cruz, sc30), mouse anti-GFP (Cell Signaling, CAT 2955), and mouse anti-β-Tubulin (Cell Signaling, CAT 6181).
qRT-PCR
Total RNA was isolated using RNAiso Plus (Takara). Reverse transcription was performed using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer’s recommendations. SYBR green-based real-time PCR was then performed in triplicate using a CFX96 sequence detection system (Bio-Rad), and U6 was used as the internal control. The primers for qRT-PCR were as follows: U6 primer forward, 5’-CTCGCTTCGGCAGCACA-3’ and reverse, 5’-AACGCTTCACGAA TTTGCGT-3’; UCA1 primer forward, 5’-TTTGCCAGCCTCAGCTTAAT-3’ and reverse, 5’-TTGTCCCCATTTTCCATCAT-3’; and KRAS primer forward, 5’-ATTGTGAATGTTGGTGT-3’ and reverse, 5’-GAAGGTCTCAACTGAAATT-3’. The relative fold change in RNA expression was calculated using the 2-ΔΔCt method.
Plasmid construction
The double strand duplexes of the UCA1 and EGFP shRNA oligos (Sangon Biotech Co. Ltd. Shanghai, China) were inserted into the EcoRI and AgeI sites of pLKO.1-TRC (Sigma-Aldrich, St. Louis, MO, USA), as described previously [15]. The UCA1 expression plasmid was kindly provided by professor Yin-Yuan Mo at the University of Mississippi Medical Center, Jackson, MS, USA. The sh-KRAS, Flag-Vector, and Flag-KRAS plasmids were previously constructed in our laboratory. pSL-MS2-12X (YouBio, Changsha, China) was double digested with XhoI and ApaI, and the MS2-12X fragment was subcloned into pcDNA3.1 and pcDNA3.1-UCA1; the resulting constructs were named pcDNA3.1-MS2 and pcDNA3.1-MS2-UCA1, respectively. Mutations in UCA1 were generated by site-directed mutagenesis, and the relevant fragments were cloned into the pcDNA3.1-MS2 plasmid. The sequences were confirmed by DNA sequencing.
Cell transfection and generation of stably transfected cell lines
Cells were transfected with plasmid DNA or shRNAs using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
The lentiviral packaging vectors used were psPAX2 and pMD2.G. Cells were infected with 1 × 106 recombinant lentivirus transduction units in the presence of 8 mg/ml polybrene (Sigma-Aldrich). Puromycin (1 μg/ml) was added to the cells until all cells in the blank group died. The surviving cells were stably infected cells and were used for subsequent experiments.
Cell counting kit-8 (CCK-8) proliferation assay
Cell proliferation and viability were assessed by using a CCK-8. Cells (2 × 103) were seeded into each well of a 96-well culture plate and incubated in humidified air at 37°C for 24, 48, 72, 96, 120 and 144 h. After removal of the supernatant, 100 μl of medium was added to every well, and the plate was incubated for 1 h. The absorbance was read at 490 nm on a microplate reader.
Colony formation assay
Stable cell lines were harvested, resuspended in medium, transferred to six-well plates (500 cells per well) and cultured for 10-14 days until large colonies were visible. Cells were fixed in 4% paraformaldehyde for 15 min and were then stained with 0.05% crystal violet for 30 min. The number of colonies was counted.
Sphere formation assay
The sphere formation assay was performed by plating dissociated single cells from stable cell lines at a density of 1 cell/µl into 6-well plates in stem cell medium: serum-free DMEM/F12 medium supplemented with L-glutamine, sodium pyruvate, 100 U/ml penicillin/streptomycin, 20 ng/ml basic fibroblast growth factor (PeproTech, London, UK), 20 ng/ml epidermal growth factor (Cell Signaling Tech, Denver, MA, USA), and B27 (1:50, Gibco, Grand Island, NY, USA). The number of spheres was counted after 14 days of culture.
Xenograft mouse model
This study was carried out in accordance with the recommendations of NIH animal use guidelines. The protocol was approved by the Institutional Animal Care and Use Committee of Jiangsu University, Zhenjiang, China. HPAF-II cells (2.0 × 106 cells/site) stably transfected with sh-EGFP or sh-UCA1 were subcutaneously injected into 4-week-old nude mice to generate xenografts. The tumor volume was measured every week after injection and calculated using the following formula: length × (width2)/2.
RNA immunoprecipitation (RIP) assay
To confirm the interaction of hnRNPA2B1 and miR-590-3p with UCA1, we used an anti-GFP antibody to pull down UCA1. RIP assays were performed using the Magna RIP Kit (Millipore, Bedford, MA, USA) according to the manufacturer’s instructions. After the antibody was recovered by protein A/G beads, hnRNPA2B1 and miR-590-3p were detected in the precipitates by western blotting and qRT-PCR, respectively.
Immunoprecipitation (IP) assay
To determine the interaction between hnRNPA2B1 and KRAS, we performed an IP assay using an anti-Flag antibody. The IP assay was performed by using the Thermo Scientific™ Pierce Classic IP Kit (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. Briefly, cells were lysed in IP lysis/wash buffer containing protease inhibitors, and the cleared lysates were immunoprecipitated with the anti-Flag antibody. The input and immunocomplexes were analyzed by western blotting.
Immunofluorescence analysis
Cells were grown on coverslips in a 24-well plate for 48 h and were then washed twice with PBS, fixed with ice-cold 3% paraformaldehyde at room temperature for 15 min, and blocked with 3% BSA in PBS for 1 h. Cells were incubated with the primary antibody overnight at 4°C. The following antibodies were used: anti-hnRNPA2B1 and anti-KRAS. After being rewarmed for 1 h, the cells were incubated with Cy3-labeled goat anti-rabbit IgG (Beyotime Biotechnology) and Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) secondary antibodies for 2 h at 37°C in the dark. DAPI (1 μg/ml, Pierce, IL, USA) was used to counterstain nuclei. After extensive washing, the samples were mounted on glass slides, and fluorescence images were captured with a confocal microscope (DeltaVision Elite, GE Healthcare).
Phos-tag SDS-PAGE
Cells were lysed with RIPA buffer supplemented with a protease inhibitor mixture at 100°C for 10 min after rinsing with cold PBS and were then centrifuged at 12000 r/min for 5 min. Approximately 10 μl of protein was loaded on a 12% SDS-PAGE gel supplemented with 100 µM Phos-tag (Wako, 304-93521) and 100 µM MnCl2. The gel was run overnight at 5 mA/gel and soaked in a general transfer buffer containing 1 mM EDTA for 20 min, followed by a 10 min incubation with a transfer buffer without EDTA. Then, the gel was transferred to a PVDF membrane at 50 V overnight. After being blocked with 5% BSA for 1 h at room temperature, the membranes were blotted with the anti-KRAS antibody.
Luciferase reporter assay
The fragments of the 3’ untranslated region (UTR) of UCA1 and KRAS containing the predicted miR-590-3p binding site were amplified by chemical synthesis and inserted into a luciferase reporter vector to form the reporter vectors UCA1-wild-type (UCA1-WT) and KRAS-wild-type (KRAS-3’UTR-WT), respectively. The putative binding sites of miR-590-3p in the UCA1 or KRAS 3’ UTR were replaced to generate the UCA1-mutant-type (UCA1-MUT1) or KRAS-mutant-type (KRAS-3’UTR-MUT) constructs. The reporter vectors were cotransfected into HEK293T cells along with miR-590-3p or miRNA negative control. Luciferase activity was detected by a dual luciferase reporter assay system according to the manufacturer’s instructions.
Statistical analysis
All grouped data are presented as the means ± SDs from at least three independent experiments. Comparisons between groups were analyzed by ANOVA or Student’s t-test using GraphPad Prism 5 software. Kaplan-Meier curves were analyzed using log-rank analysis. P<0.05 was considered statistically significant.
Results
UCA1 expression is upregulated in PDAC tissues and cell lines
To determine the clinical relevance of UCA1 expression, we first used The Cancer Genome Atlas (TCGA) database to analyze the mRNA levels of UCA1 and found that UCA1 was highly expressed in PDAC tumor specimens compared to UCA1 expression in normal tissue (Figure 1A). Furthermore, we found from the TCGA database Kaplan-Meier survival curves that UCA1 was a negative prognostic factor for overall survival (Figure 1B). UCA1 transcript levels in 6 PDAC cell lines and the immortalized human pancreatic ductal epithelial cell line H6C7 were assessed by qRT-PCR. The results indicated that the UCA1 levels were significantly higher in the PDAC cell lines than in H6C7 cells and that although UCA1 mRNA remained highly abundant in Mpanc96 and HPAF-II cells, UCA1 was weakly expressed in PaTu8988 and PANC-1 cells (Figure 1C).
Figure 1.
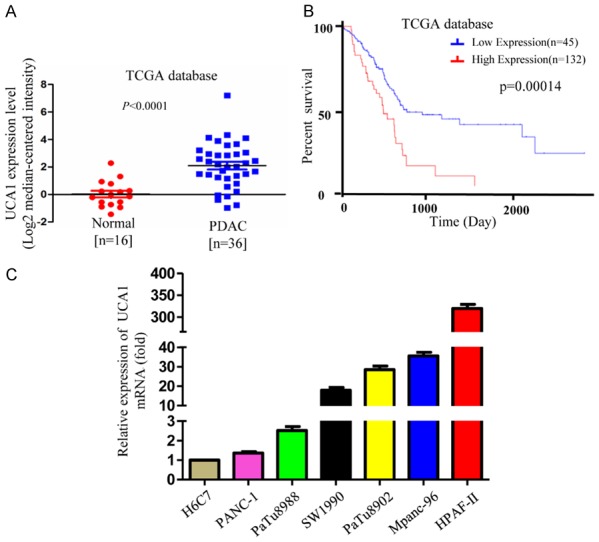
UCA1 is highly expressed in PDAC cells and tissues and is associated with overall survival. A. TCGA database analysis indicated that UCA1 expression was upregulated in PDAC tissues compared with that in normal pancreatic tissues (normal pancreas n=16; PDAC n=36; P<0.0001). B. The Kaplan-Meier survival curves showed a significant correlation between UCA1 overexpression and poor overall survival in PDAC patients (P<0.001). C. The relative expression of UCA1 in HPDE6-C7 cells and in 6 PDAC cell lines was assessed by qRT-PCR.
UCA1 promotes cell proliferation and tumor growth
To investigate whether UCA1 expression is required for proliferation in PDAC, we knocked down UCA1 with sh-UCA1. In Mpanc96 and HPAF-II cells, the levels of UCA1 were significantly reduced in the sh-UCA1 group compared to those in the sh-EGFP group (vector control) (Figure S1A). Similarly, UCA1 expression was effectively upregulated by the overexpression of UCA1 in PaTu8988 and PANC-1 cells (Figure S1B). Moreover, the CCK-8 and colony formation assays revealed that UCA1 downregulation largely suppressed the proliferative activity and colony formation of Mpanc96 and HPAF-II cells (Figure 2A and 2C). In contrast, overexpression of UCA1 in PaTu8988 and PANC-1 cells promoted proliferation and colony formation (Figure 2B and 2D). Furthermore, experiments in the xenograft mouse model revealed that sh-UCA1 also significantly reduced tumor growth (Figure 2E and 2F).
Figure 2.

UCA1 promotes proliferation and tumorigenesis in PDAC cell lines in vitro and in vivo. A, B. The effect of UCA1 on cell growth was assessed by a CCK-8 assay in Mpanc96/HPAF-II cells with UCA1 knockdown and PaTu8988/Panc-1 cells with UCA1 overexpression. C, D. Colony formation assay to examine the effect of UCA1 knockdown and overexpression on the colony formation capacity of PDAC cell lines. E, F. HPAF-II cells with UCA1 downregulation were injected (2.0 × 106 cells/site) subcutaneously into a mice, and the tumor volume was measured weekly (n=5 mice). *P<0.05.
UCA1 enhances the capability for stemness maintenance
To further determine the role of UCA1 in PDAC, we examined sphere formation by a tumorsphere formation assay in stem cell medium. In Mpanc96 and HPAF-II cells, sphere formation capability was decreased in the sh-UCA1 group compared to that in the vector control group (Figure 3A), as was the expression of the stemness markers CD133, OCT4, NANOG and SOX2 (Figure 3C). In contrast, overexpression of UCA1 in PaTu8988 and PANC-1 cells resulted in the opposite effects (Figure 3B and 3D). These results suggested that UCA1 is involved in stemness maintenance in PDAC cell lines.
Figure 3.
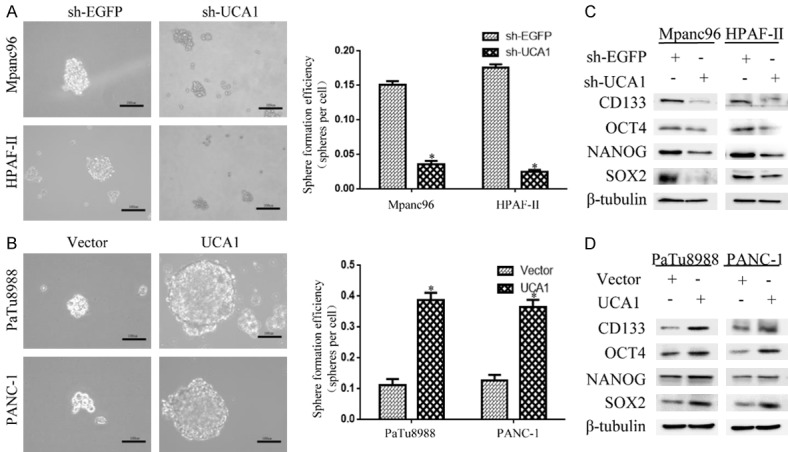
Elevated UCA1 expression induces stemness in PDAC cell lines. A, B. The effect of UCA1 on the sphere formation capability was assessed through a tumorsphere formation assay in stem cell medium and imaged by inverted microscopy. Representative images of PDAC cell line spheroid cultures after 10 days. Scale bar, 100 µm. *P<0.05. C, D. The effect of UCA1 on stemness was measured by a western blot analysis of the stemness markers CD133, OCT4, NANOG and SOX2. *P<0.05.
Identification of UCA1 as an hnRNPA2B1 binding partner
Various mechanisms have been proposed to explain lncRNA-mediated gene expression; an important mechanism is the ability to form ribonucleoprotein complexes through interactions with various proteins to regulate a large number of genes. As an RNA-binding protein, hnRNPA2B1 has multiple fundamental biological functions, such as RNA splicing, mRNA processing, telomere synthesis, gene expression modulation, mRNA translation and tumorigenesis [28-34]. To identify the means by which UCA1 is involved in the proliferation and stemness of PDAC cell lines, we further performed RIP assays using an anti-GFP antibody to determine whether hnRNPA2B1 can interact with UCA1. As expected, UCA1 interacted with hnRNPA2B1 (Figure 4A).
Figure 4.
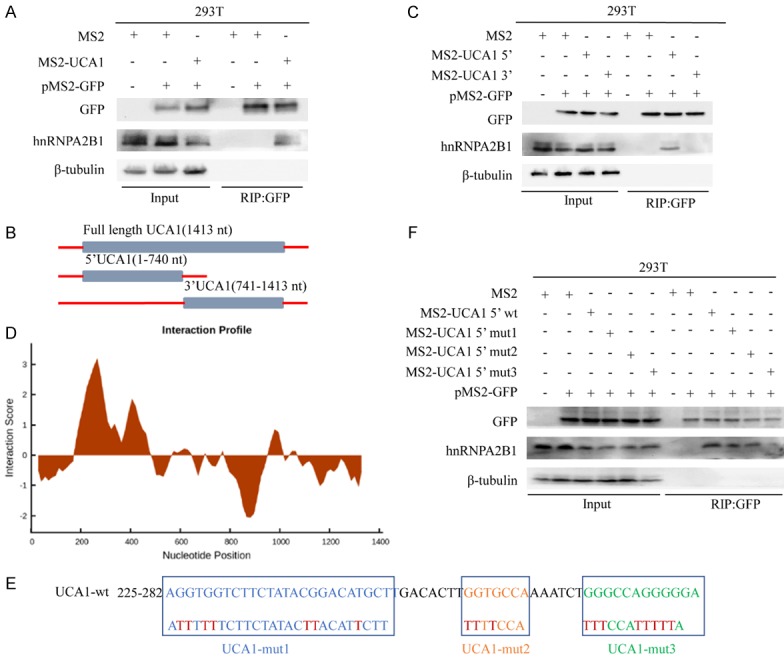
Identification of UCA1 as an hnRNPA2B1 binding partner. A. The RIP assay with an anti-GFP antibody showed that UCA1 interacts with hnRNPA2B1. B, C. The 5’ end of UCA1 is essential for its interaction with hnRNPA2B1, as confirmed by the RIP assay. D-F. A putative hnRNPA2B1-binding motif (nucleotides 271-282) in UCA1 is critical to its interaction with hnRNPA2B1.
To determine which fragment of UCA1 is involved in the interaction with hnRNPA2B1, we constructed two distinct plasmids, pcDNA3.1-MS2-12X-UCA1 5’ and pcDNA3.1-MS2-12X-UCA1 3’ (covering the 5’ end and the 3’ end of UCA1, respectively) and found that the UCA1 5’ end structure was sufficient for the interaction between UCA1 and hnRNPA2B1 (Figure 4B and 4C).
Furthermore, we used the catRAPID online algorithm (http://service.tartaglialab.com/page/catrapid group), which is based on secondary structure, hydrogen bonding and van der Waals contributions, to predict the RNA-protein interaction of UCA1 with hnRNPA2B1 (Figures 4D and S1C) [35]. To investigate whether UCA1 interacts with hnRNPA2B1 at the predicted RNA sequences, we hypothesized that the 3’ stem-loop structure of UCA1 was sufficient for the interaction between UCA1 and hnRNPA2B1, and we performed RIP on wild-type and point mutation-containing (Δ225-247, Δ258-264 or Δ271-282) UCA1 5’ end structures (Figure 4E and 4F). We found that the interaction was drastically reduced when UCA1 (Δ271-282) was mutated, suggesting that this hnRNPA2B1-binding motif is critical to UCA1-hnRNPA2B1 binding.
UCA1 enhances the interaction between hnRNPA2B1 and KRAS
A previous study showed that the interaction between hnRNPA2B1 and KRAS depends on the KRAS Ser181 phosphorylation status and that this interaction plays a key role in KRAS-dependent PDAC cell lines through the PI3K/AKT signaling pathway [36]. We confirmed the interaction of hnRNPA2B1 with KRAS by coimmunoprecipitation assays in HEK293T cells (Figure 5A and 5B). To determine whether UCA1 impacts the interaction between hnRNPA2B1 and KRAS, we performed a coimmunoprecipitation analysis in HEK293T cells cotransfected with UCA1 and Flag-KRAS. Clearly, UCA1 upregulation increased hnRNPA2B1 expression, as observed in the western blot analysis of the precipitated protein (Figure 5C). In addition, we validated the colocalization of hnRNPA2B1 and KRAS in KRAS-dependent PDAC cell lines (Mpanc96 and HPAF-II) with UCA1 downregulation by immunofluorescence microscopy. Interestingly, we observed a reduction in cytoplasmic hnRNPA2B1 and decreased colocalization of hnRNPA2B1 with the cytoplasm-localized KRAS in KRAS-dependent PDAC cell lines with UCA1 knockdown (Figure 5D). These data further confirmed that UCA1 is involved in regulating the nuclear/cytoplasmic distribution of hnRNPA2B1 and the interaction of hnRNPA2B1 with KRAS.
Figure 5.

Effect of UCA1 on the interaction between hnRNPA2B1 and KRAS. A. Samples were harvested from HEK293T cells transfected with Flag-hnRNPA2B1 for IP with antibodies against Flag. B. Samples were collected from HEK293T cells with Flag-KRAS overexpression for IP with the anti-Flag antibody. C. UCA1 facilitates the interaction of hnRNPA2B1 with KRAS, as detected by IP in HEK293T cells overexpressing UCA1. D. Representative immunofluorescence images showing hnRNPA2B1 (labeled in red) and KRAS (green) expression in Mpanc96/HPAF-II cells transfected with sh-EGFP or sh-UCA1.
UCA1 enhances phospho-KRAS expression by interacting with hnRNPA2B1
Having demonstrated that UCA1 upregulation significantly increased the physical interaction of the hnRNPA2B1-KRAS complex, we speculated that UCA1 might also be involved in regulating the expression of KRAS by interacting with hnRNPA2B1. Thus, we further examined the protein levels of hnRNPA2B1 and phospho-KRAS and assessed KRAS expression at both the protein and mRNA levels in KRAS-dependent PDAC cell lines (Mpanc96 and HPAF-II) with UCA1 knockdown. As expected, the expression of hnRNPA2B1, KRAS and phospho-KRAS, as well as the KRAS mRNA levels, were reduced after UCA1 downregulation (Figures 6A, 6B and 7C). To confirm the mechanism of action, we overexpressed full-length UCA1 (UCA1-wt) and a UCA1 construct with a mutation in the hnRNPA2B1-binding region (Δ271-282, UCA1-mut) and examined the expression of hnRNPA2B1, KRAS and phospho-KRAS, as well as the KRAS mRNA levels. Most importantly, the UCA1-mut group exhibited increased KRAS expression at both the protein and mRNA levels, but the expression of hnRNPA2B1 and phospho-KRAS was not affected, suggesting that UCA1 impacts phospho-KRAS expression by interacting with hnRNPA2B1 (Figure 6C and 6D). We also observed the cytoplasmic expression of hnRNPA2B1 and colocalization of hnRNPA2B1 and KRAS. Notably, immunofluorescence analysis showed that UCA1-wt promoted cytoplasmic expression of hnRNPA2B1 and increased the colocalization of hnRNPA2B1 and KRAS compared with these characteristics in the control group, while no change was observed in the UCA1-mut group (Figure 6E).
Figure 6.
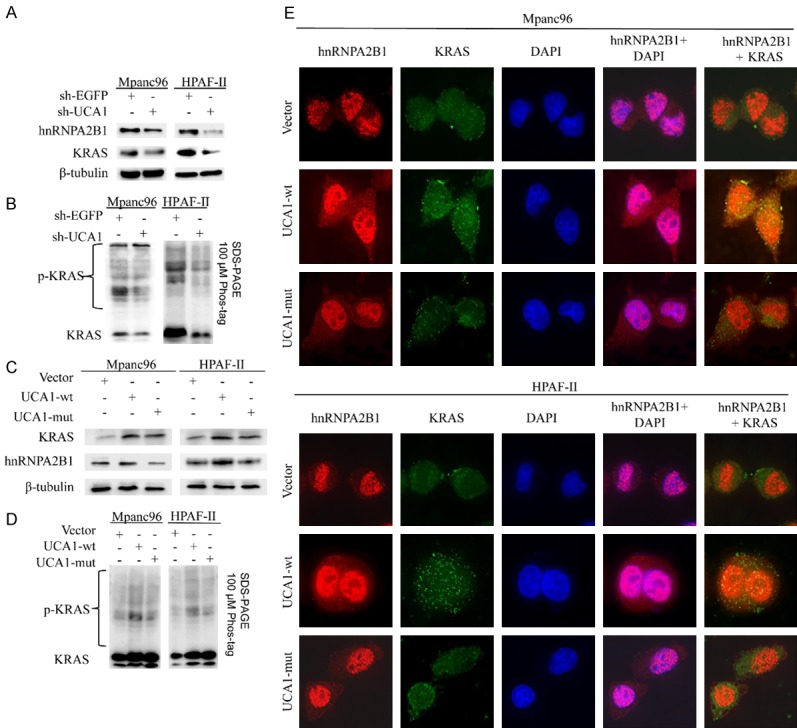
UCA1 increases phospho-KRAS expression by interaction with hnRNPA2B1 in KRAS-dependent PDAC cell lines. A. Immunoblotting of hnRNPA2B1 and KRAS in Mpanc96 and HPAF-II cells treated with sh-EGFP or sh-UCA1. β-tubulin was used as the loading control. B. Samples separated by conventional SDS-PAGE were loaded on the Phos-tag SDS-PAGE gel and assayed by immunoblotting using an anti-KRAS antibody. P-KRAS, phosphorylated KRAS. C. Immunoblotting of hnRNPA2B1 and KRAS in Mpanc96 and HPAF-II cells transfected with MS2, MS2-UCA1-wt or MS2-UCA1-mut. β-tubulin was used as the loading control. D. Samples separated by conventional SDS-PAGE were loaded on the Phos-tag SDS-PAGE gel and assayed by immunoblotting using an anti-KRAS antibody. P-KRAS, phosphorylated KRAS. E. Representative immunofluorescence images showing hnRNPA2B1 (labeled in red) and KRAS (green) expression in Mpanc96 and HPAF-II cells transfected with MS2, MS2-UCA1-wt or MS2-UCA1-mut.
Figure 7.
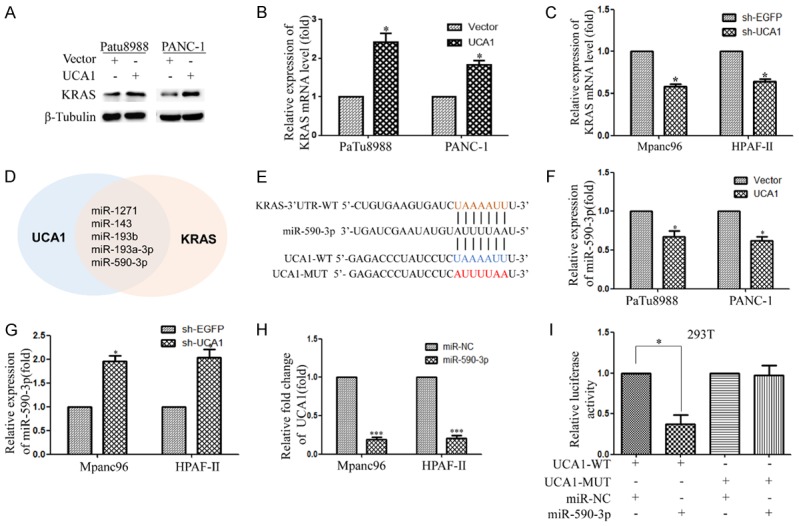
UCA1 acts as a miR-590-3p sponge. A, B. Overexpression of UCA1 in KRAS-independent PDAC cell lines (PaTu8988 and PANC-1 cells) promotes the expression of KRAS protein and mRNA. C. qRT-PCR analysis of KRAS mRNA levels in Mpanc96 and HPAF-II cells with UCA1 downregulation. D. Prediction of potential miRNAs targeting UCA1 and binding to the KRAS 3’UTR by bioinformatics analysis (miRanda; http://www.micro-RNA.org/). E. The bioinformatics prediction of the miR-590-3p binding sequence in UCA1 is shown. F, G. qRT-PCR analysis of miR-590-3p levels in PDAC cell lines with UCA1 silencing or overexpression. H. Relative expression of UCA1 after miR-590-3p upregulation in Mpanc96 and HPAF-II cells. I. The relative luciferase activity was measured by a dual luciferase reporter assay after the cotransfection of wild-type or mutant UCA1 and miR-590-3p.
UCA1 is a direct target of miR-590-3p
The above results first indicated that UCA1 regulates the expression of phospho-KRAS by interacting with hnRNPA2B1. However, the mechanism by which UCA1 modulates KRAS expression requires further investigation. Thus, we transfected KRAS-independent PDAC cell lines (PaTu8988 and PANC-1) with UCA1 or the vector control to examine KRAS expression. Interestingly, we found that UCA1 overexpression resulted in significant upregulation of KRAS expression and mRNA levels in PaTu8988 and PANC-1 cells (Figure 7A and 7B). These data further confirmed that UCA1 impacted the expression of KRAS in both KRAS-dependent and KRAS-independent PDAC cell lines. To seek insight into the potential molecular mechanism connecting UCA1 with KRAS, bioinformatics analysis (miRanda; http://www.micro-RNA.org/) was applied to predict potential miRNAs targeting UCA1 and binding to the KRAS 3’UTR (Figure 7D). Next, RIP was performed to identify which candidate miRNAs were involved in the interaction with UCA1. The involvement of miR-1271, miR-143, miR-193a-3p, and miR-590-3p was confirmed, as shown in Figure S2B. Next, we found that UCA1 and KRAS have a potential miR-590-3p binding site by bioinformatics prediction (Figure 7E).
In addition, miR-590-3p expression was lower in UCA1--overexpressing cells than in control cells (Figure 7F). In contrast, UCA1 knockdown resulted in the opposite effects in Mpanc96 and HPAF-II cells (Figure 7G). Moreover, miR-590-3p overexpression resulted in decreased expression of UCA1, suggesting that UCA1 negatively regulates miR-590-3p expression (Figure 7H). To clarify that the mechanism of miR-590-3p action on UCA1 is specific, we performed a luciferase activity assay to detect the potential association between UCA1 and miR-590-3p. The luciferase reporter assay revealed that the luciferase activity was significantly lower in the wild-type UCA1 group than in the vector control group, whereas the mutated UCA1 group did not show a significant response to miR-590-3p, indicating that UCA1 can directly bind to miR-590-3p (Figure 7I).
UCA1 acts as a competing endogenous RNA (ceRNA) of miR-590-3p to modulate KRAS expression
Furthermore, we observed that miR-590-3p suppressed KRAS protein expression and mRNA levels, as shown in Figure 8A and 8B. Moreover, the bioinformatics analysis and luciferase activity assay showed that miR-590-3p significantly reduced the luciferase activity of the 3’UTR-KRAS-WT reporter gene vector, but no significant change was observed for the 3’UTR- KRAS-MUT vector (Figure 8C and 8D). UCA1-WT increased the mRNA and protein levels of KRAS in PaTu8988 and PANC-1 cells compared with these levels in the control group but UCA1-MUT1 had no significant effect (Figure 8E and 8F). Taken together, these data suggested that UCA1 modulates KRAS expression by sponging miR-590-3p.
Figure 8.
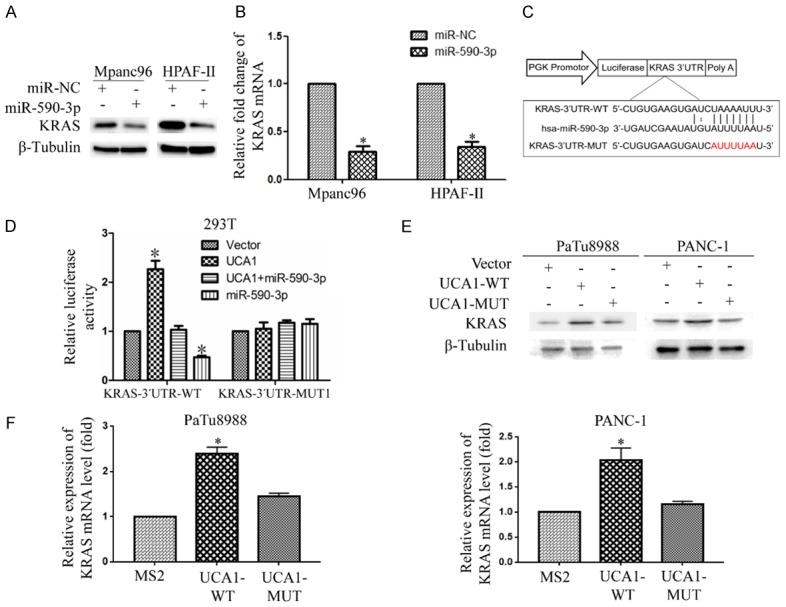
UCA1 promotes KRAS expression by sponging miR-590-3p. A, B. The protein and mRNA levels of KRAS were suppressed in Mpanc96 and HPAF-II cells after transfection with miR-590-3p compared to these levels in the corresponding cells transfected with miR-NC. C. The miR-590-3p binding sequence in the KRAS 3’UTR was predicted. D. The decrease in luciferase activity after the cotransfection of the 3’UTR-KRAS-WT construct and miR-590-3p into HEK293T cells was analyzed by a dual luciferase reporter assay. E, F. KRAS expression was assessed in PaTu8988 and PANC-1 cells after transfection with pSL-MS2-12X, UCA1-WT or UCA1-MUT. (*P<0.05, **P<0.01).
Discussion
Increasing evidence suggests that UCA1 is an oncogene in tumor growth and metastasis and may be a biomarker and therapeutic target in multiple human cancers. Our previous study suggested that UCA1 overexpression was found in human PDAC tissues and was associated with poorer patient outcomes. Here, we showed that UCA1 is also upregulated in PDAC cell lines and that its expression is positively correlated with oncogenic KRAS expression. The complex UCA1-KRAS regulatory network leads to cell growth in vitro and tumor growth in vivo, and this network is critical for stemness maintenance in PDAC cells (Figure S3). Thus, our results indicated that UCA1 acts as an oncogenic factor in PDAC.
LncRNAs, which lack protein-coding potential, have conserved secondary structures that can directly interact with proteins [37]. An important finding in our study is that hnRNPA2B1, as a binding partner of UCA1, plays an important role in the UCA1-mediated KRAS regulatory network. Activating KRAS mutations are known to be oncogenic in various types of cancer, are involved in the formation of pancreatic intraepithelial neoplasia and promote the development of PDAC. Moreover, these mutations are necessary for stemness maintenance. The expression of hnRNPA2B1 has been reported to be dysregulated in multiple cancers, including pancreatic cancer, lung cancer and hepatocellular carcinoma (HCC) [38-40]. As an RNA-binding protein, hnRNPA2B1 accelerates pre-mRNA processing via forming complexes with lncRNAs during the development of carcinoma. In addition, HCC patients with high cytoplasmic expression of hnRNPA2B1 exhibited a significantly higher incidence of poorly differentiated stages and lower survival rates than those with nuclear expression of hnRNPA2B1 [40]. Similarly, Barcelo et al. showed evident cytoplasmic hnRNPA2B1 staining in PDAC tissue and that hnRNPA2B1 is a novel interactor with oncogenic KRAS, which regulates the PI3K/AKT/mTOR pathway in KRAS-dependent PDAC [36]. Interestingly, these researchers proved that the interaction between hnRNPA2B1 and KRAS depends on the KRAS Ser181 phosphorylation status and that KRAS phosphorylation increases the recruitment of HNRNPA2B1 to the cytoplasm [36]. In this study, we demonstrated that UCA1 interacts with hnRNPA2B1 and identified the potential hnRNPA2B1-binding motif in UCA1. This motif was critical to UCA1-hnRNPA2B1 binding because the ability for this interaction was drastically reduced when it was mutated. In addition, UCA1 upregulation promoted the interaction of hnRNPA2B1 and KRAS. UCA1 knockdown reduced the protein levels of hnRNPA2B1, total KRAS and phospho-KRAS; the level of cytoplasmic hnRNPA2B1; and the colocalization of hnRNPA2B1 and KRAS in KRAS-dependent PDAC cell lines (Figure 6). However, although UCA1 overexpression enhanced the protein levels of hnRNPA2B1, total KRAS and phospho-KRAS; the level of cytoplasmic hnRNPA2B1; and the colocalization of hnRNPA2B1 and KRAS, only total KRAS expression was altered when the UCA1-hnRNPA2B1 binding motif was mutated (Figure 7). These results suggested that UCA1 promotes phospho-KRAS protein expression through interaction with hnRNPA2B1 and that the higher cytoplasmic accumulation of hnRNPA2B1 was a consequence of the increased hnRNPA2B1 recruitment by KRAS phosphorylation. These results might explain why hnRNPA2B1 expression was expressed higher in the cell cytoplasm with UCA1 overexpression.
Studies have shown the phosphorylation of KRAS at serine 181, which is located within the polybasic region [41,42]. Recent evidence has revealed that KRAS requires S181 phosphorylation to manifest its oncogenic properties, implying that KRAS phosphorylation is essential for cell survival and tumorigenic activity [43]. Furthermore, KRAS phosphorylation could modulate oncogenic KRAS activity, and it is necessary to activate the mitogen-activated protein kinase and PI3K/AKT pathways [44,45]. We showed that UCA1 upregulates the levels of KRAS phosphorylation for its involvement in the development of PDAC via hnRNPA2B1 binding; however, the molecular mechanism connecting UCA1 to KRAS has not yet been completely elucidated.
A recent study reported that lncRNAs can act as ceRNAs of miRNAs to regulate target mRNA levels [46]. UCA1 has been shown to contain binding sites for many miRNAs involved in multiple tumor types. Moreover, UCA1 acts as a ceRNA and is widely reported in several types of tumors. UCA1 plays an oncogenic role in inducing tumorigenesis in breast cancer via acting as a ‘sponge’ to bind miR-143 [47]. In addition, UCA1 functions as a ceRNA to increase the expression of ZEB1 via miR-204-5p and regulate glioma metastasis [48]. UCA1 activates CREB1 expression by sponging miR-590-3p to be involved in gastric cancer progression [49]. These examples piqued our interest in exploring whether a ceRNA mechanism is involved in the interaction between UCA1 and KRAS. Here, we showed by a bioinformatics analysis and RIP assay that miR-590-3p can directly bind UCA1. Moreover, we observed that UCA1 negatively regulates miR-590-3p expression in PDAC cell lines. The luciferase reporter assay provided further evidence of the specific interaction between UCA1 and miR-590-3p. Furthermore, we confirmed that KRAS was a direct downstream target of miR-590-3p by using bioinformatics analysis and a luciferase activity assay. Most importantly, we found that UCA1 downregulation suppressed the expression of the miR-590-3p target gene KRAS, whereas UCA1 upregulation inhibited miR-590-3p function, resulting in increased KRAS expression. These results showed that UCA1 functions as a molecular sponge by directly binding to miR-590-3p and up-regulating KRAS expression. Consistent with this finding, KRAS also significantly promoted UCA1 expression (Figure S2F and S2G), thus forming a positive feedback loop. However, the mechanism by which KRAS affects UCA1 expression remains to be further explored. Our observations demonstrated that the UCA1/KRAS regulatory mechanism is involved in proliferation and stemness maintenance in PDAC cell lines and that the effect of UCA1 in PDAC cell lines can be explained.
Taken together, the data from the present study revealed that UCA1 plays an oncogenic role in PDAC in part by enhancing the activity and expression of KRAS. Mechanistically, we provided the first evidence that UCA1 increases KRAS phosphorylation by interacting with hnRNPA2B1 and that UCA1 functions as a molecular sponge for miR-590-3p to promote KRAS expression. These findings provide a better understanding of the underlying mechanism of UCA1 in PDAC progression. The inhibition of the UCA1/KRAS regulatory network might be a target for new PDAC therapies.
Acknowledgements
We are grateful to professor Yin-Yuan Mo at University of Mississippi Medical Center for his support and comments. This study was supported by the National Natural Science Foundation of China (81672402 and 81472333), Provincial Natural Science Foundation of Jiangsu, China (BK20171305), Jiangsu key R&D program social development project, China (BE2018689), Research Programs of Jiangsu Provincial Commission of Health and Family Planning, China (H201434), “Six one Project” Research Projects of High-level Medical Personnel of Jiangsu Province (LGY2016054), “Six Talents Peak” High-level Talent Selection and Training Project of Jiangsu Province (2014-WSW-038), Science and Technology Support Program Project of Zhenjiang, China (SH2014024) and Qing Lan Project Funding of Jiangsu Colleges and Universities.
Disclosure of conflict of interest
None.
Abbreviations
- PDAC
pancreatic ductal adenocarcinoma
- lncRNAs
long noncoding RNAs
- UCA1
urothelial carcinoma-associated 1
- IP
immunoprecipitation
- RIP
RNA immunoprecipitation
Supporting Information
References
- 1.Pelosi E, Castelli G, Testa U. Pancreatic cancer: molecular characterization, clonal evolution and cancer stem cells. Biomedicines. 2017;5 doi: 10.3390/biomedicines5040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73–85. doi: 10.1016/S0140-6736(16)00141-0. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 4.Majumder K, Gupta A, Arora N, Singh PP, Singh S. Premorbid obesity and mortality in patients with pancreatic cancer: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2016;14:355–368. doi: 10.1016/j.cgh.2015.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanda M, Fujii T, Takami H, Suenaga M, Inokawa Y, Yamada S, Nakayama G, Sugimoto H, Koike M, Nomoto S, Kodera Y. Combination of the serum carbohydrate antigen 19-9 and carcinoembryonic antigen is a simple and accurate predictor of mortality in pancreatic cancer patients. Surg Today. 2014;44:1692–701. doi: 10.1007/s00595-013-0752-9. [DOI] [PubMed] [Google Scholar]
- 6.Hashimoto D, Chikamoto A, Ohmuraya M, Sakata K, Miyake K, Kuroki H, Watanabe M, Beppu T, Hirota M, Baba H. Pancreatic cancer in the remnant pancreas following primary pancreatic resection. Surg Today. 2014;44:1313–20. doi: 10.1007/s00595-013-0708-0. [DOI] [PubMed] [Google Scholar]
- 7.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Zhang H, Chen Z, Wang X, Huang Z, He Z, Chen Y. Long non-coding RNA: a new player in cancer. J Hematol Oncol. 2013;6:37. doi: 10.1186/1756-8722-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang XS, Zhang Z, Wang HC, Cai JL, Xu QW, Li MQ, Chen YC, Qian XP, Lu TJ, Yu LZ, Zhang Y, Xin DQ, Na YQ, Chen WF. Rapid identification of UCA1 as a very sensitive and specific unique marker for human bladder carcinoma. Clin Cancer Res. 2006;12:4851–8. doi: 10.1158/1078-0432.CCR-06-0134. [DOI] [PubMed] [Google Scholar]
- 10.Huang J, Zhou N, Watabe K, Lu Z, Wu F, Xu M, Mo YY. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1) Cell Death Dis. 2014;5:e1008. doi: 10.1038/cddis.2013.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang X, Liu W, Xu X, Zhu J, Wu Y, Zhao K, He S, Li M, Wu Y, Zhang S, Cao J, Ye Z, Xing C. Downregulation of long noncoding RNA UCA1 enhances the radiosensitivity and inhibits migration via suppression of epithelialmesenchymal transition in colorectal cancer cells. Oncol Rep. 2018;40:1554–1564. doi: 10.3892/or.2018.6573. [DOI] [PubMed] [Google Scholar]
- 12.Han Y, Yang YN, Yuan HH, Zhang TT, Sui H, Wei XL, Liu L, Huang P, Zhang WJ, Bai YX. UCA1, a long non-coding RNA up-regulated in colorectal cancer influences cell proliferation, apoptosis and cell cycle distribution. Pathology. 2014;46:396–401. doi: 10.1097/PAT.0000000000000125. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Q, Wu F, Dai WY, Zheng DC, Zheng C, Ye H, Zhou B, Chen JJ, Chen P. Aberrant expression of UCA1 in gastric cancer and its clinical significance. Clin Transl Oncol. 2015;17:640–6. doi: 10.1007/s12094-015-1290-2. [DOI] [PubMed] [Google Scholar]
- 14.Chen P, Wan D, Zheng D, Zheng Q, Wu F, Zhi Q. Long non-coding RNA UCA1 promotes the tumorigenesis in pancreatic cancer. Biomed Pharmacother. 2016;83:1220–1226. doi: 10.1016/j.biopha.2016.08.041. [DOI] [PubMed] [Google Scholar]
- 15.Zhang M, Zhao Y, Zhang Y, Wang D, Gu S, Feng W, Peng W, Gong A, Xu M. LncRNA UCA1 promotes migration and invasion in pancreatic cancer cells via the Hippo pathway. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1770–1782. doi: 10.1016/j.bbadis.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Gao F, Zhou L, Wang H, Shi G, Tan X. UCA1 regulates the growth and metastasis of pancreatic cancer by sponging miR-135a. Oncol Res. 2017;25:1529–1541. doi: 10.3727/096504017X14888987683152. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Zhou Y, Chen Y, Ding W, Hua Z, Wang L, Zhu Y, Qian H, Dai T. LncRNA UCA1 impacts cell proliferation, invasion, and migration of pancreatic cancer through regulating miR-96/FOXO3. IUBMB Life. 2018;70:276–290. doi: 10.1002/iub.1699. [DOI] [PubMed] [Google Scholar]
- 18.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 20.Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res. 2007;67:2098–106. doi: 10.1158/0008-5472.CAN-06-3752. [DOI] [PubMed] [Google Scholar]
- 21.Rachagani S, Senapati S, Chakraborty S, Ponnusamy MP, Kumar S, Smith LM, Jain M, Batra SK. Activated KrasG(1)(2)D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br J Cancer. 2011;104:1038–48. doi: 10.1038/bjc.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, Pham L, Wang MM, Karachaliou N, Cao MG, Manzano JL, Ramirez JL, Torres JM, Buttitta F, Rudin CM, Collisson EA, Algazi A, Robinson E, Osman I, Muñoz-Couselo E, Cortes J, Frederick DT, Cooper ZA, McMahon M, Marchetti A, Rosell R, Flaherty KT, Wargo JA, Bivona TG. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015;47:250–6. doi: 10.1038/ng.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D, Spellman PT, Phillips WA, Gray JW, McMahon M. A central role for RAF→MEK→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2:685–93. doi: 10.1158/2159-8290.CD-11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okada M, Shibuya K, Sato A, Seino S, Suzuki S, Seino M, Kitanaka C. Targeting the K-Ras--JNK axis eliminates cancer stem-like cells and prevents pancreatic tumor formation. Oncotarget. 2014;5:5100–5112. doi: 10.18632/oncotarget.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang DK, Grimmond SM, Biankin AV. Pancreatic cancer genomics. Current Opinion in Genetics & Development. 2014;24:74–81. doi: 10.1016/j.gde.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 26.Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–53. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsubara S, Ding Q, Miyazaki Y, Kuwahata T, Tsukasa K, Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci Rep. 2013;3:3230. doi: 10.1038/srep03230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–86. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen M, Zhang J, Manley JL. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010;70:8977–80. doi: 10.1158/0008-5472.CAN-10-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemieux B, Blanchette M, Monette A, Mouland AJ, Wellinger RJ, Chabot B. A function for the hnRNP A1/A2 proteins in transcription elongation. PLoS One. 2015;10:e0126654. doi: 10.1371/journal.pone.0126654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.El-Mazny A, Sayed M, Sharaf S. Human telomerase reverse transcriptase messenger RNA (TERT mRNA) as a tumour marker for early detection of hepatocellular carcinoma. Arab J Gastroenterol. 2014;15:68–71. doi: 10.1016/j.ajg.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Wang K, Ling T, Wu H, Zhang J. Screening of candidate tumor-suppressor genes in 3p21.3 and investigation of the methylation of gene promoters in oral squamous cell carcinoma. Oncol Rep. 2013;29:1175–82. doi: 10.3892/or.2012.2213. [DOI] [PubMed] [Google Scholar]
- 33.Chen T, Cui P, Chen H, Ali S, Zhang S, Xiong L. A KH-domain RNA-binding protein interacts with FIERY2/CTD phosphatase-like 1 and splicing factors and is important for pre-mRNA splicing in Arabidopsis. PLoS Genet. 2013;9:e1003875. doi: 10.1371/journal.pgen.1003875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lan X, Yan J, Ren J, Zhong B, Li J, Li Y, Liu L, Yi J, Sun Q, Yang X, Sun J, Meng L, Zhu W, Holmdahl R, Li D, Lu S. A novel long noncoding RNA Lnc-HC binds hnRNPA2B1 to regulate expressions of Cyp7a1 and Abca1 in hepatocytic cholesterol metabolism. Hepatology. 2016;64:58–72. doi: 10.1002/hep.28391. [DOI] [PubMed] [Google Scholar]
- 35.Bellucci M, Agostini F, Masin M, Tartaglia GG. Predicting protein associations with long noncoding RNAs. Nat Methods. 2011;8:444–5. doi: 10.1038/nmeth.1611. [DOI] [PubMed] [Google Scholar]
- 36.Barceló C, Etchin J, Mansour MR, Sanda T, Ginesta MM, Sanchez-Arévalo Lobo VJ, Real FX, Capellà G, Estanyol JM, Jaumot M, Look AT, Agell N. Ribonucleoprotein HNRNPA2B1 interacts with and regulates oncogenic KRAS in pancreatic ductal adenocarcinoma cells. Gastroenterology. 2014;147:882–892. e888. doi: 10.1053/j.gastro.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 37.Rivas E, Clements J, Eddy SR. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat Methods. 2017;14:45–48. doi: 10.1038/nmeth.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Liu HL, Li Y, Yuan P. Proteomic analysis of pancreatic intraepithelial neoplasia and pancreatic carcinoma in rat models. World J Gastroenterol. 2011;17:1434–41. doi: 10.3748/wjg.v17.i11.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dowling P, Pollard D, Larkin A, Henry M, Meleady P, Gately K, O’Byrne K, Barr MP, Lynch V, Ballot J, Crown J, Moriarty M, O’Brien E, Morgan R, Clynes M. Abnormal levels of heterogeneous nuclear ribonucleoprotein A2B1 (hnRNPA2B1) in tumour tissue and blood samples from patients diagnosed with lung cancer. Mol Biosyst. 2015;11:743–52. doi: 10.1039/c4mb00384e. [DOI] [PubMed] [Google Scholar]
- 40.Zhou ZJ, Dai Z, Zhou SL, Hu ZQ, Chen Q, Zhao YM, Shi YH, Gao Q, Wu WZ, Qiu SJ, Zhou J, Fan J. HNRNPAB induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by transcriptionally activating SNAIL. Cancer Res. 2014;74:2750–62. doi: 10.1158/0008-5472.CAN-13-2509. [DOI] [PubMed] [Google Scholar]
- 41.Ballester R, Furth ME, Rosen OM. Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J Biol Chem. 1987;262:2688–95. [PubMed] [Google Scholar]
- 42.Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, Miura J, Wiener HH, Wright L, Saba SG, Yim D, Fein A, Pérez de Castro I, Li C, Thompson CB, Cox AD, Philips MR. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–93. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Barceló C, Paco N, Morell M, Alvarez-Moya B, Bota-Rabassedas N, Jaumot M, Vilardell F, Capella G, Agell N. Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014;74:1190–9. doi: 10.1158/0008-5472.CAN-13-1750. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez-Moya B, López-Alcalá C, Drosten M, Bachs O, Agell N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene. 2010;29:5911–22. doi: 10.1038/onc.2010.298. [DOI] [PubMed] [Google Scholar]
- 45.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2011;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuo YL, Li XM, Luo J. Long noncoding RNA UCA1 modulates breast cancer cell growth and apoptosis through decreasing tumor suppressive miR-143. Eur Rev Med Pharmacol Sci. 2015;19:3403–11. [PubMed] [Google Scholar]
- 48.Liang C, Yang Y, Guan J, Lv T, Qu S, Fu Q, Zhao H. LncRNA UCA1 sponges miR-204-5p to promote migration, invasion and epithelial-mesenchymal transition of glioma cells via upregulation of ZEB1. Pathol Res Pract. 2018;214:1474–1481. doi: 10.1016/j.prp.2018.07.036. [DOI] [PubMed] [Google Scholar]
- 49.Gu L, Lu LS, Zhou DL, Liu ZC. UCA1 promotes cell proliferation and invasion of gastric cancer by targeting CREB1 sponging to miR-590-3p. Cancer Med. 2018;7:1253–1263. doi: 10.1002/cam4.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.