

Abstract
Poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) are promising targeted therapeutics for breast and ovarian cancers bearing a germline BRCA1/2 mutation (BRCA m), and several have already received regulatory approval in the United States. In patients with a BRCA m cancer, PARPi can increase the burden of unrepaired DNA double-strand breaks by blocking PARP activity and trapping PARP1 onto damaged DNA. Resistance to PARP inhibitors can block the formation of DNA double-strand breaks through BRCA-related DNA repair pathway. MET is a hyper-activated receptor tyrosine kinase expressed in multiple cancer types and the activation contributes to resistance to DNA damage-inducing therapeutic drugs. Our previous study showed that MET inhibition by pan-kinase inhibitors has synergism with PARPi in suppressing growth of breast cancer in vitro and in xenograft tumor models. In this study, we validated the inhibitory effect of novel inhibitors, HS10241 (selective MET inhibitor) and HS10160 (PARPi), to their target respectively in triple-negative breast cancer (TNBC) and high-grade serous ovarian cancer (HGSOC) cells. We further demonstrated that these two inhibitors function synergistically in eliminating TNBC and HGSOC cells; combining with HS10241 increased DNA double-strand breaks induced by HS10160 in cancer cells; and PARP1 tyrosine (Y)-907 phosphorylation (PARP1 p-Y907) can be an effective biomarker as an indicator of MET-mediated PARPi in HGSOC. Our results suggest that the combination of HS10241 and HS10160 may benefit patients bearing tumors overexpressing MET as well as those resistant to single-agent PARPi treatment.
Keywords: Targeted therapy, PARP, MET, triple-negative breast cancer, high-grade serous ovarian cancer, resistance, SHR-3162
Introduction
Breast cancer has the highest incidence in women worldwide and leads in incidence in 154 countries in 2018 [1]. Triple-negative breast cancer (TNBC) is an aggressive subtype of breast cancer lacking estrogen and progesterone receptors and expressing low levels of HER2. The lack of these hormone receptors makes TNBC unresponsive to standard hormone receptor-targeted therapies for breast cancer. TNBC accounts for 15-20% of breast cancer cases and 25% of breast cancer-associated deaths [2]. Compared to other breast cancer subtypes, TNBC is more likely to metastasis and relapse at early stage, and the median survival time is shorter for TNBC patients than those who bears other breast cancer subtypes (4.2 vs. 6 years) [2,3]. Another female cancer with high incidence is ovarian cancer [4]. Ovarian cancer is the most lethal gynecologic malignancy. High-grade serous ovarian cancer (HGSOC), which comprises the majority of epithelial ovarian cancer, is characterized by p53 mutation, initial sensitivity to platinum agents, and overall chromosomal instability, and because of these features, HGSOC is much more aggressive than other types of ovarian cancer and carries a poorer prognosis [5]. It is reported that primary HGSOC tumor is highly heterogeneous based on its various epigenetic RNA and miRNA expression and promoter methylation regulations [6,7]. To date, aside from the BRCA mutation (BRCA m), none of the molecular specific traits has been referenced in HGSOC patient treatment in the clinic [5,8,9]. The heterogeneity of TNBC and HGSOC is the key challenge in treating the disease in the clinic [2]. Thus, the strategies identifying targetable biomarker in TNBC and HGSOC personalized treatment are urgently needed.
Among emerging targeted therapies for breast and ovarian cancer, PARP inhibitors (PARPi) exhibit promising efficacies in treating a subpopulation of cancer patients who carries BRCA m [10,11]. PARP1, the major target of PARPi, can be activated by cellular stress induced DNA damage adducts [12,13]. PARP1 mediates DNA damage repair pathways to remove damaged base from DNA by post-translationally adding ADP-ribose chain onto DNA repair proteins (PARylation) [14]. The use of PARPi blocks PARP-mediated repair of single-stranded DNA damages and traps PARP1 onto damaged DNA [15-18]. These working mechanisms of PARPi increase the burden of unrepaired double-stranded DNA, which should be repaired by BRCA-mediated homologous recombination pathway [19]. Therefore, PARPi induce synthetic lethality in BRCA m cancer cells [20-23]. In ovarian cancer, 9% of HGSOC harbor BRCA m [24]. Most of the BRCA m HGSOC respond well to PARPi, and thus PARPi are approved by the U.S. Food and Drug Administration (FDA) as a standard targeted therapy for BRCA m ovarian cancer [25]. In addition, a subpopulation of TNBC patients (10-20%) harbors BRCA m or shows similar phenotype as BRCA m, making these patients more sensitive PARPi [26,27]. Although most PARPi inhibit PARP1 enzymatic activity at similar efficacy, the ability of trapping PARP onto damaged DNA varies among different PARPi [16]. While the major cytotoxicity effect in BRCA m cells comes from PARP trapping [28], it is necessary to investigate each PARPi for potential combination treatment individually because the resistant mechanisms may vary based on the different designs of PARPi. In this study, we focused on a novel PARPi, HS-10160 (fluzoparib, SHR-3162), which is currently under clinical trial study in multiple solid tumor types [ClinicalTrials.gov Identifier: NCT03509636, NCT03075462, NCT03026881].
In both clinical trial data and cancer cell models, PARPi sensitivity and resistance are not fully restricted to BRCA m [29]. Previous studies showed that altered receptor tyrosine kinases (RTK) activation contributes to resistance to multiple DNA damage-inducing therapeutic drugs, including PARPi [30-35]. Among the RTKs, MET is a suitable target for targeted therapy in TNBC and HGSOC treatment because MET is one of the RTKs that is overexpressed in these cancer types [36]. Activated MET induces PARPi resistance in TNBC model by interacting with and phosphorylating PARP1 at Y-907 residue [34]. The PARP1 p-Y907 has lower binding affinity to PARPi and also facilitates DNA damage repair efficiency [15]. It has been reported that pan-tyrosine kinase inhibitors that target various RTKs, including MET (foretinib and crizotinib), demonstrated synergistic effect with PARPi in BRCA wild-type TNBC and a population of liver cancer cells [34,35]. In TNBC, the synergism is attributed to the increased double-strand DNA breaks as indicated by serine 139 phosphorylated H2AX (γH2AX) [34]. Currently, the therapeutic efficacy of selective MET inhibitor (METi), HS-10241, is being investigated as a single agent in solid tumor in clinical trials globally [ClinicalTrials.gov Identifier: NCT02759640, NCT02977364, NCT02981108, NCT03243643], but the combination of METi and PARPi has not yet been examined in clinical trials.
In this study, we asked whether the combination of PARPi and selective METi show synergism in TNBC and HGSOC. We on purpose selected two drugs that are developed by the same company in order to facilitate future clinical trials if the results turn positive. To this end, we chose PARPi HS-10160 and METi HS-10241, and focused on two TNBC and two HGSOC cell lines that express high levels of MET protein. By treating the cell lines with HS-10160 (PARPi) and HS-10241 (METi), we demonstrated that HS-10160 and HS-10241 inhibited PARylation and MET activation, respectively, under H2O2-treatment and that the combination of these inhibitors induced more γH2AX formation and reduce growth of cancer cells synergistically. Our findings suggested that MET also contributes to PARP1 Y-907 phosphorylation in HGSOC similar to that in TNBC. Therefore, PARP1 p-Y907 has the potential to serve as a biomarker to stratify TNBC and HGSOC patients for METi and PARPi combination treatment.
Methods
Chemicals and antibodies
Olaparib, was purchased from Selleck Chemical (Houston, TX) and crizotinib was from LC Laboratories (Woburn, MA). Fluzoparib (HS10160) and HS10241 were kindly provided by Jiangsu Hengrui Medicine Co. Ltd (Shanghai, China). All small molecule inhibitors were dissolved in dimethyl sulfoxide (DMSO). Hydrogen peroxide and antibody detecting actin (#A2066) was from Sigma-Aldrich (St. Louis, MO). FITC-conjugated antibody detecting Ser139 phosphorylated-H2AX (#613404) was purchased from BioLegend (San Diego, CA). Antibody against phosphotyrosine (#05-321, clone 4G10) was obtained from EMD Millipore (Billerica, MA). Antibodies against PARP (#9532), MET (#8198) and phosphorylated MET (Tyrosine 1234/1235) (#3077) were purchased from Cell Signaling Technology (Danvers, MA). Antibody against PARP1 p-Y907 was kindly provided by China Medical University (Taichung, Taiwan) [34]. Mounting buffer for immunofluorescence imaging containing DAPI was purchased from Electron Microscopy Science (Hatfield, PA).
Cell culture
All cells lines, except SUM149, were purchased from ATCC (Manassas, VA) and were incubated in Dulbecco modified Eagle medium (DMEM)/F12 medium supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 mg/mL streptomycin, or in Hyclone DMEM/high glucose medium with 15% FBS, 100 units/mL penicillin, and 100 mg/mL streptomycin. SUM149 cell line was purchased from Asterand Biosciences (Detroit, MI) and maintained in F12K medium supplied with 5% FBS, 10 mM HEPES, 1 mg/ml hydrocortisone, 5 µg/ml insulin, 100 units/mL penicillin, and 100 mg/mL streptomycin. Cell lines were validated by STR DNA fingerprinting using the AmpF_STR Identifiler kit according to manufacturer’s instructions (Applied Biosystems cat 4322288). The STR profiles were compared to known ATCC fingerprints (ATCC.org), and to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/) [37] and matched known DNA fingerprints.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin) containing protease inhibitors (bimake.com) and phosphatase inhibitors (biotool.com). Protein concentrations of the lysates were determined by using Pierce BCA protein assay kit (Fisher PI-23227) following manufactory’s protocol. Total protein (30 µg) was electrophoresed in a 10% Bis-Tris SDS PAGE gel and transferred to a polyvinylidene difluoride (PVDF) membrane (Life Technologies). The PVDF membranes were hybridized with primary antibodies overnight at 4°C after blocking in either 5% non-fat milk or 4% BSA. Excess antibodies were washed off with TBST buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.05% Tween-20). The membranes were then subjected to hybridization with secondary antibodies, either anti-mouse-HRP or anti-rabbit-HRP (e Bioscience), for one hour at room temperature, and imaged by using ECL reagents (Bio-Rad Laboratories) and ImageQuant LAS 4010 (GE Healthcare).
Confocal microscopy analysis of γ-H2AX foci
Cells were incubated on chamber slides (Labtek, Scotts Valley, CA) overnight before treated with indicated chemicals for 18 h. After washing with ice-cold phosphate-buffered saline solution (PBS), cells were fixed with 4% paraformaldehyde in PBS (pH 7.4) containing 0.03 M sucrose for 20 min on ice. The samples were then permeabilized with 0.2% triton X-100/PBS, washed with PBS, and blocked for 15 min in 5% bovine serum albumin in PBS. Samples were then incubated with FITC-conjugated γ-H2AX antibodies on shaker overnight in 4°C and washed three times with PBS for 15 min on shaker each time. Samples were mounted in DAPI-containing mounting buffer for 30 min and then subjected to confocal microscopy analysis (LSM 710 laser-scanning microscope; Carl Zeiss, Thornwood, NY). The ZEN and ImageJ software (NIH, Bethesda, MD) were used for data analysis. Cells were counted by using DAPI signal, and cells with more than two γH2AX foci were counted as γH2AX positive.
Colony formation assay
Cells were seeded in 24-well plates at a density of 4,000 cells/well for BT-549, 3,000 cells/well for MDA-MB-231, 4,000 cells/well for DOV13, and 5,000 cells/well for OVCA433. Cells were incubated overnight to allow attachment before treatment. Inhibitors were diluted into the indicated concentrations in each experiment with cell culture medium. Cells were then treated with inhibitor-containing medium for 7 days before washing with PBS and staining with 0.5% crystal violet for 4 hours. Excess crystal violet was washed off under running tap water and images were scanned by using HP Scanjet 5590. Cell survival was determined by using 33% acetic acid to extract the color and optical density measured at 565 nm.
MTT assay and calculation of combination index (CI) calculation of drug synergy
For MTT assay, MDA-MB-231 cells (3,000 cells per well), BT-549 (3,500 cells per well), DOV13 (4,000 cells per well), and OVCA433 (5,000 cells per well) were seeded in 96-well plate and incubated overnight before treatment. Cells were treated with inhibitors either alone or in combination as indicated for 3 days. MTT was added into each well at a final concentration of 0.5 µg/ml and incubated with cells for 30 minutes before formazan was extracted by using DMSO. Cell survival was calculated by measuring the optical density at 590 nm of each well and normalized to that of the untreated wells. The half maximal inhibitory concentration (IC50) was calculated from MTT assay using the GraphPad Prism 8.0 software. The Chou-Talalay combination index (CI) was calculated by using Compusyn software (www.combosyn.com) with cell survival data from MTT assay. Drug combinations were designed using constant ratio of two inhibitors following the suggestions from the Compusyn program [38,39].
Statistical analysis
All bar graphs were illustrated and analyzed by using GraphPad Prism 8.0 and statistics analyzed using the Student’s t test. A p value < 0.05 was considered statically significant.
Results
HS-10160 inhibits H2O2-induced PARylation in TNBC and HGSOC cells
The major function of PARPi is to inhibit PARylation of PARP or its recruitment of substrate proteins [40]. Since the capacity of HS10160 to inhibit DNA damages-induced PARylation has not been well documented in the literature, we tested this activity in two TNBC cell lines (MDA-MB-231 and BT549) and two HGSOC cell lines (DOV13 and OVCA433). Because PARP is activated by oxidative stress-induced DNA damages, we induced the DNA damage by treating cancer cells with H2O2 and assessed the effect on PARylation of cellular proteins by Western blot analysis. As expected, H2O2 treatment induced PARylation in the four cell lines tested, and the level of PARylation was reduced by HS10160 treatment in a dose-dependent manner (Figure 1). In TNBC cells, HS-10160 substantially inhibited PARylation at a concentration of 2.5 µM in MDA-MB-231 and 625 nM in BT-549 (Figure 1A, 1B). HS-10160 also significantly inhibited PARylation in HGSOC at a concentration of 5 µM in Ovca433 and 625 nM in DOV13 (Figure 1C, 1D). In addition, we found that 2.5 µM of the well-studied PARPi, olaparib, markedly inhibited PARylation under the same H2O2 stimulation condition in MDA-MB-231 cell (Figure S1). Our data suggested that HS-10160 can inhibit PARP1 activity efficiently in both TNBC and HGSOC cells and that its PARylation inhibition efficacy is comparable to that of olaparib.
Figure 1.
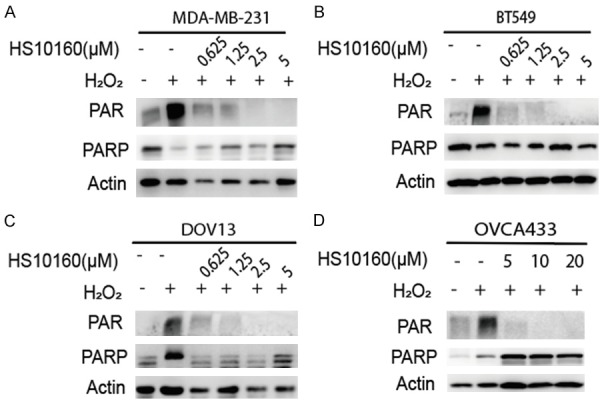
HS10160 inhibits H2O2-induced PARylation in TNBC and HGSOC cell lines. Human TNBC cells (BT549 and MDA-MB-231) and HGSOC cells (DOV13 and OVCA433) were treated with indicated concentrations of PARP inhibitor HS10160 for 3 h prior to 30 min 20 mM H2O2 treatment. Cells were then subjected to Western blot analysis to determine PARylation (PAR) and PARP1 expression. Actin were used as protein quantity loading control among different samples.
HS-10241 inhibits H2O2-induced MET activation in both TNBC and HGSOC cells
MET can be activated by cellular oxidative stress such as H2O2, and MET activation is indicated by its Y1230/1234/1235 phosphorylation [41]. To investigate whether HS-10241 inhibits H2O2-induced MET activation as it has been demonstrated with crizotinib and selective METi SU11274 in previous publications [34,41], we treated the TNBC and HGSOC cell lines with HS-10160 and H2O2 followed by detection of MET activation (Y1234/1235 phosphorylated MET, p-MET) by Western blot analysis. As predicted, H2O2 increased the levels of p-MET, which decreased gradually with increasing concentrations of HS10241 (Figure 2). In TNBC cells, HS-10241 substantially inhibited p-MET at concentrations lower than 2.5 µM in two cell lines tested (Figure 2A, 2B). Crizotinib, at a 2.5 µM is enough to inhibit MET activation in MDA-MB-231 cell (Figure S2). HS-10241 (5 µM) sufficiently inhibited more than 90% of p-MET in OVCA433 cell whereas a much higher concentration of 40 µM was required in DOV13 for the same effect (Figure 2C, 2D). These data suggested that HS-10241 inhibits oxidative stress-induced MET activation effectively in TNBC with concentration around 2.5-5 µM and the dose may vary by a larger range in HGSOC. It is likely that DOV13 may associate with unknown mechanism that is insensitive to HS-10241. By comparing the p-MET inhibitory effect of HS-10241 and crizotinib, we concluded that HS10241 exhibits inhibition efficacy similar to that of crizotinib in MDA-MB-231 cells.
Figure 2.
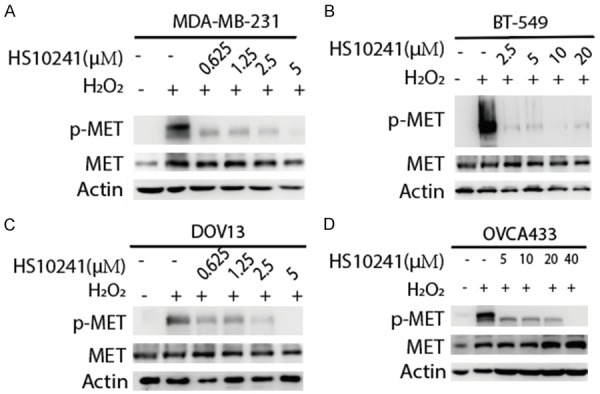
HS10241 suppresses H2O2-induced MET activation in TNBC and HGSOC cells. Indicated cells were treated with HS10241 at different concentrations for 3 h before exposed to 20 mM H2O2 for 20 min. Cells were then harvested for Western blot analysis with indicated antibodies. Activated MET was indicated by the phospho-Y1234/Y1235 MET (p-MET). Actin were used as protein quantity loading control among different samples.
Combination of HS-10160 and HS-10241 increases DNA double-strand breaks and induces cytotoxicity synergistically in TNBC and HGSOC cells
In theory, the use of PARPi increases the burden of unrepaired DNA double-strand breaks in cancer cells and specifically eliminates cells that cannot repair DNA breaks efficiently. Serine 139-phosphorylated histone H2AX (γH2AX) is a crucial component of DNA damage repair signaling cascade that is mediated by DNA double-strand break repair kinases, such as DNA-PK, ATM, and ATR [42]. Therefore, γH2AX is used as an indicator of cells bearing un-repaired DNA breaks [43]. To investigate whether the combination of HS-10241 and HS-10160 enhances the burden of un-repaired double-strand breaks in TNBC and HGSOC cells, we examined γH2AX foci by immunofluorescence staining in cells treated with HS-10160, HS-10241, or the combination. In these experiments, TNBC and HGSOC cells were treated with 20 mM H2O2 to verify their capability of γH2AX induction in response to severe DNA damages. As expected, all cells treated with H2O2 showed positive H2AX foci signaling in a majority of the cells (Figure 3A-D, bottom rows and white histograms). γH2AX was detected only in a small population of cells treated with either HS-10160 or HS-10241 alone (around 5% in TNBC cells and 2% in HGSOC cells), indicating that both inhibitors cannot induce significant double-strand breaks in a majority of the treated cells (Figure 3A-D, second and third rows, blue and green histograms). However, the combination of HS-10160 and HS-10241 induced substantially more γH2AX formation in TNBC cells (by more than 50%) and higher γH2AX positive population in HGSOC cells (around 10%) (Figure 3A-D, fourth row and red histograms).
Figure 3.

Combination of HS10241 and HS10160 induces formation of double strand DNA breaks and eliminate cancer cell synergistically. A-D. Double strand DNA breaks are indicated by measuring γ-H2AX foci through the use of immunofluorescence staining and confocal microscope. Cells indicated were treated with HS10160 (10 µM) and HS-10241 (10 µM) alone or in combination for 18 h before immunofluorescence staining. Representative images of γ-H2AX are shown. Percentages of treated cells containing γ-H2AX foci (γH2AX positive) were summarized from counting 100 cells in each samples. Cells treated with 20 mM H2O2 for 20 min were used as positive control of γH2AX induction. Histograms shown represent mean and standard deviations among 3 repeats. E-H. Synergism was assessed by the Chou-Talalay method. Cells were treated with various concentrations of HS10241 and HS10160 either alone or in combination. Cell survival is measured by using MTT assay. The combination index (CI) for each pair of agents in each cell line were calculated and plotted by using the Compusyn software.
We further determined the synergism of HS-10241 and HS10160 in both TNBC and HGSOC cell lines using the Chou-Talalay combination index (CI) method to. In general, a CI in the range of 0.8-1.2 indicates additive effect between inhibitors; < 0.8 indicates synergism between inhibitors; and > 1.2 represents antagonistic effect between inhibitors [44]. Our data showed that in MDA-MB-231 and DOV13 cells, the CI was lower than 0.3 at concentrations that killed more than 40% of the cells (Fa > 0.4) (Figure 3E, 3G). In BT-549 cells, the CI was generally between 0.5-1 with different cell killing effects (Fa 0.05-0.95; Figure 3F). In OVCA433 cell, CI ranged from 0.7 to 0.3 with increasing cell killing effects (Fa 0.5-0.95; Figure 3H). Taken together, our data demonstrated that the combination of HS10241 and HS10160 has synergistic cell killing effects in the TNBC and HGSOC cells tested.
PARP1 p-Y907 as biomarker for HS-10160 and HS-10241 combination treatment in TNBC and HGSOC cells
Previous publication indicates that PARP1 Y907 is phosphorylated by p-MET, and the PARP1 p-Y907 has lower affinity to PARPi than Y907 un-phosphorylated PARP1 and has higher efficiency in mediating DNA damage repair [34]. Therefore, PARP1 p-Y907 is a suitable biomarker for indicating p-MET induced PARPi resistance in TNBC [34]. To examine whether PARP1 p-Y907 can also be a biomarker of MET-mediated PARPi resistance in HGSOC and the potential of using HS-10241 to overcome it, we measured H2O2 induced PARP1 p-Y907 formation and its reduction caused by HS-10241 treatment in TNBC and HGSOC cell lines. By using Western blotting, we found that H2O2 can induce PARP1 p-Y907 in both HGSOC lines tested (Figure 4A-D). As expected, the use of HS-10241 effectively inhibits p-MET as well as decreases PARP1 p-Y907 in both TNBC and HGSOC cells (Figure 4A-D). These data suggest that PARP1 p-Y907 is also mediated by MET in HGSOC. Further, our data suggest that HS-10241 is potent for overcoming MET-induced PARPi resistance in both TNBC and HGSOC.
Figure 4.
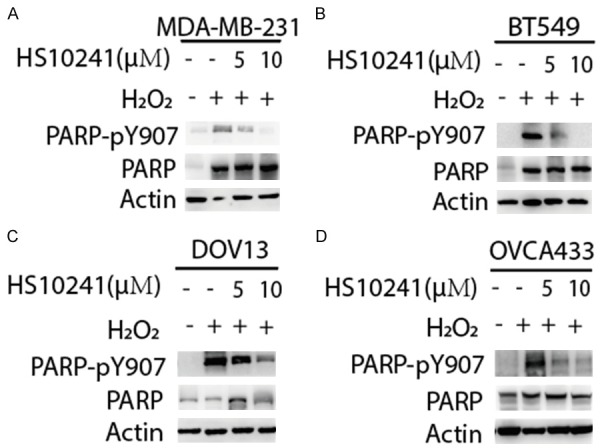
HS10241 suppresses PARP1 p-Y907 in TNBC and HGSOC cells. Cells were treated with either 5 µM or 10 µM HS10241 (as indicated) for 3 h before 20 mM H2O2 treatment. Cells were then harvested for detecting PARP p-Y907 and PARP1 expression by using Western blotting analysis. Actin were used as protein quantity loading control among different samples.
Discussion
In the current study, we specifically chose PARPi and METi developed by the same pharmaceutical company for this study as this strategy may facilitate the translation process from research institute to clinical trial studies. We confirmed that, compared to BRCA mutated, PARPi-sensitive-SUM149 cell, the cell lines we chose for this study are resistant to HS10160 (Figure S3). However, the sensitivity of the cells to HS10241 are similar (Figure S4). In this study, we confirmed that HS-10160 inhibited PARylation of PARP1 and HS-10241 inhibited MET activation effectively in both non-BRCA m TNBC and HGSOC cells (Figures 1 and 2). By treating the cancer cells with the inhibitor alone or in combination, we concluded that combination of HS-10160 and HS-10241 is suitable to overcome MET-induced intrinsic PARPi resistance by demonstrating the synergistic effect between these two drugs (Figure 3). Moreover, our data indicated that PARP1 p-Y907 has the potential to serve as biomarker to stratify patients with non-BRCA m TNBC and HGSOC for this combination treatment (Figure 4).
In preclinical studies, BRCA m is not the only factor affecting PARPi sensitivity. The sensitivity to PARPi can be independent of BRCA m status [45]. The observed synergism in BRCA wild-type cancer cells reported in this study is consistent with our previous findings that MET-mediated resistance to PARPi is independent of BRCA protein expression [34,35]. Expanding from our previous findings on the use of METi to overcome PARPi resistance in TNBC, we demonstrated in this study that PARP1 p-Y907 can be a biomarker to indicate MET-mediated PARPi resistance in both cancer types tested. Taking into consideration of the reported PARP1 p-Y907 as a biomarker in indicating PARPi resistance in liver cancer [35], we proposed that PARP1 p-Y907 can serve a biomarker that can be applied to multiple cancer types as well as a reference guide for precision METi/PARPi combination therapy.
Using METi as an example, our findings suggested that PARPi can be applied to treat TNBC and HGSOC in combination with kinase inhibitor to increase double-strand DNA damage burdens in cancer cells and thus enhance the therapeutic efficacy. Since MET is not the only oncogenic kinase in human cancers [46], the combination of PARPi with other kinase inhibitors may be potential therapeutic strategies in both preclinical and clinical studies [47]. However, only a few PARPi and kinase inhibitor combinations have moved into Phase II/III clinical trials swiftly [47]. Therefore, biomarkers are needed to evaluate the contribution of specific kinase activation in PARPi resistance in order to better stratify patients. With the urgent need of precision targeted therapy, we demonstrated that MET and PARP1 p-Y907 as a model in which a single biomarker can be applied to multiple cancer types.
Acknowledgements
We thank Kathryn Hale from the Department of Scientific Publications at MD Anderson for editing the manuscript. STR DNA fingerprinting was done by the Cancer Center Support Grant-funded Characterized Cell Line core, NCI, CA016672. This research is supported by the following grants: National Institutes of Health (CCSG CA016672, CA211615, and CA201777); Breast Cancer Research Foundation (BCRF-17-069); National Breast Cancer Foundation, Inc.; Patel Memorial Breast Cancer Endowment Fund; The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund (to M-C.H.); Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW107-TDU-B-212-114024 and MOHW107-TDU-B-212-112015); Future Talents Grant from China Medical University affiliated Shengjing Hospital to Y. Han; The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences 2017-2018 Larry Deaven Ph.D. Fellowship in Biomedical Sciences to M.K. Chen; The NIH T32 training grant in Cancer Biology at The University of Texas MD Anderson Cancer Center to M.K. Chen.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Lee A, Djamgoz MBA. Triple negative breast cancer: emerging therapeutic modalities and novel combination therapies. Cancer Treat Rev. 2018;62:110–122. doi: 10.1016/j.ctrv.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Wu X, Baig A, Kasymjanova G, Kafi K, Holcroft C, Mekouar H, Carbonneau A, Bahoric B, Sultanem K, Muanza T. Pattern of local recurrence and distant metastasis in breast cancer by molecular subtype. Cureus. 2016;8:e924. doi: 10.7759/cureus.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, Gaudet MM, Jemal A, Siegel RL. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68:284–296. doi: 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez VD, Samusik N, Chen TJ, Savig ES, Aghaeepour N, Quigley DA, Huang YW, Giangarra V, Borowsky AD, Hubbard NE, Chen SY, Han G, Ashworth A, Kipps TJ, Berek JS, Nolan GP, Fantl WJ. Commonly occurring cell subsets in high-grade serous ovarian tumors identified by single-cell mass cytometry. Cell Rep. 2018;22:1875–1888. doi: 10.1016/j.celrep.2018.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V Cancer Genome Atlas Research Network. Hu H. An integrated TCGA Pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416. e411. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krzystyniak J, Ceppi L, Dizon DS, Birrer MJ. Epithelial ovarian cancer: the molecular genetics of epithelial ovarian cancer. Ann Oncol. 2016;27(Suppl 1):i4–i10. doi: 10.1093/annonc/mdw083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RD, Clamp AR, Evans DGR, Edmondson RJ, Jayson GC. PARP inhibitors in platinum-sensitive high-grade serous ovarian cancer. Cancer Chemother Pharmacol. 2018;81:647–658. doi: 10.1007/s00280-018-3532-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinzelmann-Schwarz V, Knipprath Meszaros A, Stadlmann S, Jacob F, Schoetzau A, Russell K, Friedlander M, Singer G, Vetter M. Letrozole may be a valuable maintenance treatment in high-grade serous ovarian cancer patients. Gynecol Oncol. 2018;148:79–85. doi: 10.1016/j.ygyno.2017.10.036. [DOI] [PubMed] [Google Scholar]
- 10.Gadducci A, Guerrieri ME. PARP inhibitors in epithelial ovarian cancer: state of art and perspectives of clinical research. Anticancer Res. 2016;36:2055–2064. [PubMed] [Google Scholar]
- 11.Turk AA, Wisinski KB. PARP inhibitors in breast cancer: bringing synthetic lethality to the bedside. Cancer. 2018;124:2498–2506. doi: 10.1002/cncr.31307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lonskaya I, Potaman VN, Shlyakhtenko LS, Oussatcheva EA, Lyubchenko YL, Soldatenkov VA. Regulation of poly(ADP-ribose) polymerase-1 by DNA structure-specific binding. J Biol Chem. 2005;280:17076–17083. doi: 10.1074/jbc.M413483200. [DOI] [PubMed] [Google Scholar]
- 13.Eustermann S, Wu WF, Langelier MF, Yang JC, Easton LE, Riccio AA, Pascal JM, Neuhaus D. Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Mol Cell. 2015;60:742–754. doi: 10.1016/j.molcel.2015.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7:20–37. doi: 10.1158/2159-8290.CD-16-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, Gao J, Boothman DA. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr. 2014;24:15–28. doi: 10.1615/critreveukaryotgeneexpr.2013006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michelena J, Lezaja A, Teloni F, Schmid T, Imhof R, Altmeyer M. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat Commun. 2018;9:2678. doi: 10.1038/s41467-018-05031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen Y, Aoyagi-Scharber M, Wang B. Trapping poly(ADP-Ribose) polymerase. J Pharmacol Exp Ther. 2015;353:446–457. doi: 10.1124/jpet.114.222448. [DOI] [PubMed] [Google Scholar]
- 19.Mylavarapu S, Das A, Roy M. Role of BRCA mutations in the modulation of response to platinum therapy. Front Oncol. 2018;8:16. doi: 10.3389/fonc.2018.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carey JPW, Karakas C, Bui T, Chen X, Vijayaraghavan S, Zhao Y, Wang J, Mikule K, Litton JK, Hunt KK, Keyomarsi K. Synthetic lethality of PARP inhibitors in combination with MYC blockade is independent of BRCA status in triple-negative breast cancer. Cancer Res. 2018;78:742–757. doi: 10.1158/0008-5472.CAN-17-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, Gil J, Leung AY, Ashworth A, So CW. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med. 2015;21:1481–1490. doi: 10.1038/nm.3993. [DOI] [PubMed] [Google Scholar]
- 22.Faraoni I, Graziani G. Role of BRCA mutations in cancer treatment with poly(ADP-ribose) polymerase (PARP) inhibitors. Cancers (Basel) 2018;10 doi: 10.3390/cancers10120487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mittica G, Ghisoni E, Giannone G, Genta S, Aglietta M, Sapino A, Valabrega G. PARP inhibitors in ovarian cancer. Recent Pat Anticancer Drug Discov. 2018;13:392–410. doi: 10.2174/1574892813666180305165256. [DOI] [PubMed] [Google Scholar]
- 24.Neff RT, Senter L, Salani R. BRCA mutation in ovarian cancer: testing, implications and treatment considerations. Ther Adv Med Oncol. 2017;9:519–531. doi: 10.1177/1758834017714993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colombo I, Lheureux S, Oza AM. Rucaparib: a novel PARP inhibitor for BRCA advanced ovarian cancer. Drug Des Devel Ther. 2018;12:605–617. doi: 10.2147/DDDT.S130809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Shi Y, Huang D, Guan X. Emerging therapeutic modalities of PARP inhibitors in breast cancer. Cancer Treat Rev. 2018;68:62–68. doi: 10.1016/j.ctrv.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, Manyam GC, Wu W, Luo Y, Basourakos S, Song JH, Gallick GE, Karantanos T, Korentzelos D, Azad AK, Kim J, Corn PG, Aparicio AM, Logothetis CJ, Troncoso P, Heffernan T, Toniatti C, Lee HS, Lee JS, Zuo X, Chang W, Yin J, Thompson TC. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal. 2017;10 doi: 10.1126/scisignal.aam7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsels LA, Karnak D, Parsels JD, Zhang Q, Velez-Padilla J, Reichert ZR, Wahl DR, Maybaum J, O’Connor MJ, Lawrence TS, Morgan MA. PARP1 Trapping and DNA replication stress enhance radiosensitization with combined WEE1 and PARP inhibitors. Mol Cancer Res. 2018;16:222–232. doi: 10.1158/1541-7786.MCR-17-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tung NM, Garber JE. BRCA1/2 testing: therapeutic implications for breast cancer management. Br J Cancer. 2018;119:141–152. doi: 10.1038/s41416-018-0127-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen MK, Hung MC. Regulation of therapeutic resistance in cancers by receptor tyrosine kinases. Am J Cancer Res. 2016;6:827–842. [PMC free article] [PubMed] [Google Scholar]
- 31.Balaji K, Vijayaraghavan S, Diao L, Tong P, Fan Y, Carey JP, Bui TN, Warner S, Heymach JV, Hunt KK, Wang J, Byers LA, Keyomarsi K. AXL inhibition suppresses the DNA damage response and sensitizes cells to PARP inhibition in multiple cancers. Mol Cancer Res. 2017;15:45–58. doi: 10.1158/1541-7786.MCR-16-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagan MP, Yacoub A, Dent P. Radiation-induced PARP activation is enhanced through EGFR-ERK signaling. J Cell Biochem. 2007;101:1384–1393. doi: 10.1002/jcb.21253. [DOI] [PubMed] [Google Scholar]
- 33.Ivy SP, Liu JF, Lee JM, Matulonis UA, Kohn EC. Cediranib, a pan-VEGFR inhibitor, and olaparib, a PARP inhibitor, in combination therapy for high grade serous ovarian cancer. Expert Opin Investig Drugs. 2016;25:597–611. doi: 10.1517/13543784.2016.1156857. [DOI] [PubMed] [Google Scholar]
- 34.Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GR 4th, Chen MK, Nakai K, Hsu MC, Chen CT, Sun Y, Wu Y, Chang WC, Huang WC, Liu CL, Chang YC, Chen CH, Park M, Jones P, Hortobagyi GN, Hung MC. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201. doi: 10.1038/nm.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong Q, Du Y, Li H, Liu C, Wei Y, Chen MK, Zhao X, Chu YY, Qiu Y, Qin L, Yamaguchi H, Hung MC. EGFR and c-MET cooperate to enhance resistance to PARP inhibitors in hepatocellular carcinoma. Cancer Res. 2019;79:819–829. doi: 10.1158/0008-5472.CAN-18-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mo HN, Liu P. Targeting MET in cancer therapy. Chronic Dis Transl Med. 2017;3:148–153. doi: 10.1016/j.cdtm.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romano P, Manniello A, Aresu O, Armento M, Cesaro M, Parodi B. Cell line data base: structure and recent improvements towards molecular authentication of human cell lines. Nucleic Acids Res. 2009;37:D925–932. doi: 10.1093/nar/gkn730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 39.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 40.Cortesi L, Toss A, Cucinotto I. Parp inhibitors for the treatment of ovarian cancer. Curr Cancer Drug Targets. 2018;18:877–893. doi: 10.2174/1568009618666180308104646. [DOI] [PubMed] [Google Scholar]
- 41.Jagadeeswaran R, Jagadeeswaran S, Bindokas VP, Salgia R. Activation of HGF/c-Met pathway contributes to the reactive oxygen species generation and motility of small cell lung cancer cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1488–1494. doi: 10.1152/ajplung.00147.2006. [DOI] [PubMed] [Google Scholar]
- 42.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 43.Enriquez-Rios V, Dumitrache LC, Downing SM, Li Y, Brown EJ, Russell HR, McKinnon PJ. DNA-PKcs, ATM, and ATR interplay maintains genome integrity during neurogenesis. J Neurosci. 2017;37:893–905. doi: 10.1523/JNEUROSCI.4213-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wildum S, Zimmermann H, Lischka P. In vitro drug combination studies of Letermovir (AIC246, MK-8228) with approved anti-human cytomegalovirus (HCMV) and anti-HIV compounds in inhibition of HCMV and HIV replication. Antimicrob Agents Chemother. 2015;59:3140–3148. doi: 10.1128/AAC.00114-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hassan SN, Esch A, Heiser L, Gray JW. Biological indicators of response and resistance to PARP inhibition in BRCA wild-type breast cancer. J. Clin. Oncol. 2015;33:125–125. [Google Scholar]
- 46.Tsatsanis C, Spandidos DA. The role of oncogenic kinases in human cancer (Review) Int J Mol Med. 2000;5:583–590. doi: 10.3892/ijmm.5.6.583. [DOI] [PubMed] [Google Scholar]
- 47.Bitler BG, Watson ZL, Wheeler LJ, Behbakht K. PARP inhibitors: clinical utility and possibilities of overcoming resistance. Gynecol Oncol. 2017;147:695–704. doi: 10.1016/j.ygyno.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.