

Abstract
Transfer RNA is extensively modified by the actions of a variety of enzymes. The radical S-adenosyl-l-methionine enzyme TYW1 modifies tRNAPhe forming the characteristic tricyclic ring via the condensation of carbons 2 and 3 of pyruvate. This chapter details methods that are required for studies of TYW1.
1. Introduction
Transfer RNA (tRNA) in all kingdoms of life is extensively modified by introduction of either simple changes that are installed by a single enzyme, or hypermodifications that require the coordinated actions of multiple enzymes (Cantara et al., 2011; Machnucka et al., 2013). Many modifications are ubiquitous and are conserved in both structure and position across nearly all organisms. Position 37 of tRNAPhe in archaea and eukaryotes bears a fluorescent and acid labile tricyclic base, wyosine (Blobstein, Grunberger, Weinstein & Nakanishi, 1973). Wyosine derivatives were initially discovered in yeast, but have been found subsequently in other eukaryotes as well as in archaea (McCloskey et al., 1987; Kasai, Yamaizumi, Kuchino & Nishimura, 1979; Kasai et al., 1976; RajBhandary, Chang, Stuart, Faulkner, Hoskinson & Khorana, 1967; RajBhandary, Stuart, Hoskinson & Khorana, 1968; RajBhandary & Chang, 1968). The biosynthetic pathway to the tricyclic wyosine derivatives was established in Saccharomyces cerevisiae using deletion strains (Fig. 1) (Noma, Kirino, Ikeuchi & Suzuki, 2006; Waas, de Crécy-Lagard & Schimmel, 2005). Subsequent studies have identified additional enzymes that are responsible for the biosynthesis of structurally related derivatives (Noma et al, 2006; Noma, Ishitani, Kato, Nagao, Nureki & Suzuki, 2010; de Crécy-Lagard et al., 2010; Kato et al., 2011).
Figure 1. Biosynthesis of all known wyosine bases.

The biosynthetic pathway for wyosine derivatives starting from guanosine.
Wyosine bases are all derived from modification of a genetically encoded guanosine nucleotide (Li, Nakanishi, Grunberger & Weinstein, 1973) (Fig. 1), with the tricyclic base, 4-demethylwyosine (imG-14), serving as the precursor to all wyosine bases. ImG-14 is obtained in two steps that involve methylation of the G by TRM5 to form N-methylguanosine (m1G) (Björk, Jacobsson, Nilsson, Johansson. Byström & Persson, 2001; Droogmans & Grosjean, 1987), followed by formation of the imidazoline ring containing imG-14 base by the action of a radical S-adenosyl-l-methionine (SAM) enzyme, TYW1 (Noma et al, 2006; Waas et al, 2005). ImG-14 is further tailored by the actions of TYW2–5 and TRM5b (de Crécy-Lagard et al, 2010; Noma et al, 2006; Noma et al, 2010; Waas et al, 2005). While SAM plays a role in each of these steps, the reaction catalyzed by TYW1 is unique in that SAM is used to activate the substrate rather than as a group donor.
TYW1 is a member of the radical SAM (RS) superfamily, which was identified on the basis of a conserved CxxxCxxC motif (Sofia, Chen, Hetzler, Reyes-Spindola & Miller, 2001). The thiolates in this conserved sequence bind three atoms of a site-differentiated 4Fe-4S cluster, leaving the fourth iron with an open coordination site to bind SAM via it’s α-carboxyl and α-amino moieties (Berkovitch, Nicolet, Wan, Jarrett & Drennan, 2004; Walsby et al., 2002). In the catalytically active +1 oxidation state, the iron-sulfur cluster catalyzes reductive cleavage of the bond between the 5′ carbon of ribose and the sulfonium moiety in SAM forming methionine and 5′-deoxyadenosyl radical (dAdo•). The dAdo• initiates radical-mediated reactions by H-atom abstraction. While this paradigm is common to most RS enzymes, variations have been noted. For example, some members of the RS superfamily have been found to bind the cluster through a non-canonical sequence (Dowling, Bruender, Young, McCarty, Bandarian & Drennan, 2013; Kim et al., 2013; Zhang et al., 2010), others reductively cleave SAM to generate aminocarboxypropyl radical (Demick & Lanzilotta, 2011), and another group promote radical additions with dAdo• rather than H-atom abstraction (Mahanta, Fedoseyenko, Dairi & Begley, 2013). We note that while TYW2 catalyzes transfer of aminocarboxylpropyl group of SAM to 7-aminocarboxypropyl demethylwyosine, this enzyme is not a member of the RS superfamily and the transfer occurs by polar mechanisms (Noma et al, 2006; Umitsu, Nishimasu, Noma, Suzuki, Ishitani & Nureki, 2009).
While gene deletion experiments had established a role for TYW1 in the conversion of m1G to imG-14, the mechanism of this transformation had been enigmatic (Noma et al, 2006; Waas et al, 2005). The formation of the imidazoline ring from m1G requires two new carbon atoms, the source of which was not known. The breakthrough that has allowed mechanistic studies of TYW1 was the identification of pyruvate as the substrate (Young & Bandarian, 2011). Stable isotope labeling experiments have since tracked the fate of carbon and hydrogen atoms and show that C2 and C3 are incorporated into the imidazoline base (Fig. 2) (Young et al, 2011; Young & Bandarian 2015).
Figure 2. Source of atoms in imG-14.
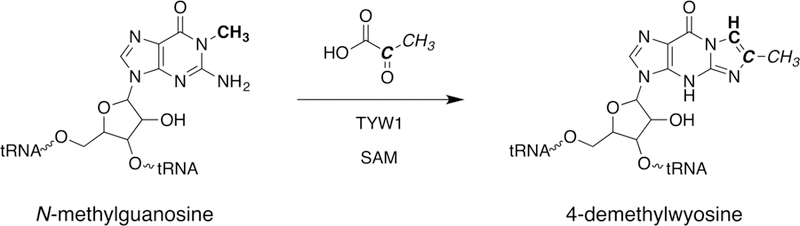
TYW1 incorporates C2 and C3 from pyruvate into m1G to form imG-14.
Two high resolution structures of TYW1 are available in the literature (Goto-Ito et al., 2007; Suzuki et al., 2007); however, neither structure reveals electron density for the anticipated RS cluster. Sequence alignments suggest that in addition to the CxxxCxxC motif, TYW1 has additional conserved Cys residues (Fig. 3), which appear to be clustered in the active site (Goto-Ito et al, 2007; Suzuki et al, 2007). Spectroscopic studies have revealed two magnetically distinct 4Fe-4S sites in TYW1, one of which is presumably the RS cluster and the other the second cluster, a so-called auxiliary cluster, bound to the additionally conserved Cys residues (Kathirvelu et al., 2017; Perche-Letuvée et al., 2012). There is a growing number of RS enzymes that catalyze complex transformations with the aid of both RS and auxiliary clusters. However, in most cases, the role of the auxiliary cluster remains unknown.
Figure 3. Sequence alignment of TYW1.

The alignment of TYW1 sequences from Methanocalococcus jannaschii (M. jannaschii), Pyrococcus horikoshii (P. horikoshii), Methanosarcina barkeri (M. barkeri), Homo sapiens (H. sapiens), and Saccharomyces cerevisiae (S. cerevisiae) is shown. The cysteine residues that coordinate the iron-sulfur clusters are highlighted in black boxes. The N-terminal sequence of both H. sapiens and S. cerevisiae are truncated and only the regions of the protein that are similar to the archaeal protein are shown.
A current mechanistic paradigm for TYW1 that is consistent with biochemical and spectroscopic studies is shown in Fig. 4. Pyruvate is proposed to be bound to the active site by forming a Schiff base to a conserved and catalytically essential Lys residue (Young et al, 2011; Young et al, 2015). Reductively cleaved SAM provides dAdo• to initiate catalysis by abstracting a hydrogen atom from m1G. The radical adds to the immobilized pyruvate, which subsequently undergoes decarboxylation. Transimination to form the imidazoline ring completes the catalytic cycle. The auxiliary cluster may active the pyruvate for decarboxylation by serving as either an electron sink or donor to the departing “COO-”.
Figure 4. Mechanistic paradigm for TYW1.
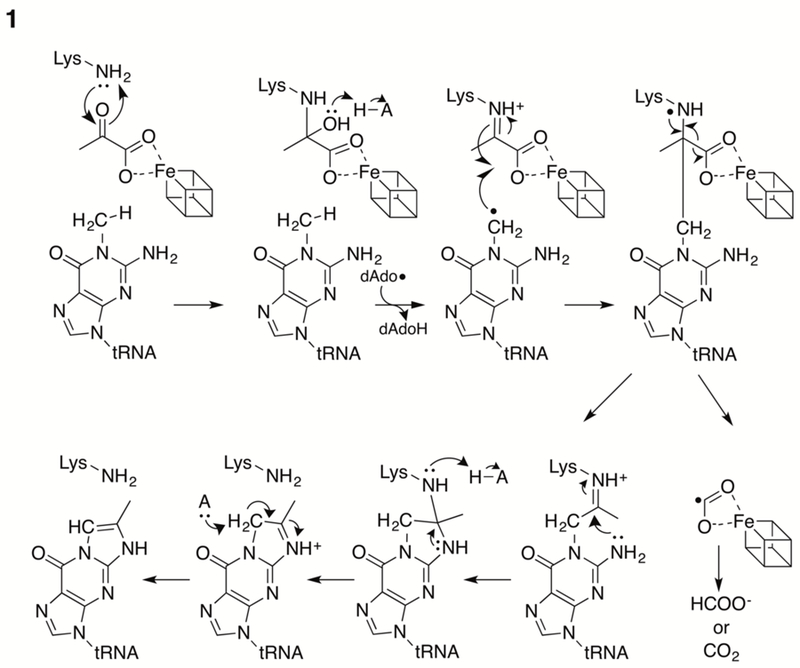
Reductive cleavage of SAM forms 5’-dAdo•, which is used to activate the substrate by H-atom transfer. Pyruvate is proposed to bind to TYW1 as a Schiff base to a conserved Lys residue. The auxiliary cluster is shown as a cube.
Studies of TYW1, in addition to cofactor-replete TYW1, also require production of m1G containing tRNA substrate. This chapter will outline our methods for preparation and characterization of TYW1.
2. Expression and Purification of TYW1
The extreme oxygen sensitivity of RS enzymes often necessitates use of purification tags that allow for rapid enrichment of the recombinant enzyme from crude mixtures. All in vitro studies with TYW1 have utilized homologs from thermophilic organisms (Goto-Ito et al, 2007; Kathirvelu et al, 2017; Perche-Letuvée et al, 2012; Suzuki et al, 2007; Young et al, 2011; Young et al, 2015), which permits the use of a heat step that denatures many of the proteins in the expression host. After this step, hydrophobic interactions and affinity chromatography using the genetically encoded purification tag allows pure TYW1 that is free nucleic acid contamination to be obtained. Methods for the expression and purification of TYW1 from Methanococcus janaschii are presented in this section.
2.1. Expression of TYW1 gene
To maximize the expression of TYW1, the gene for TYW1 from Methanococcus jannaschii was obtained in codon-optimized form (Genscript) (see Fig. 5 for codon-optimized sequence) and cloned between the NdeI and HindIII restriction sites of a modified pET28 vector, containing a Tobacco Etch Virus nuclear-inclusion-a endopeptidase (TEV) protease cleavage site (Thoden & Holden, 2005), creating pAY613. This vector contains a T7 promotor that is inducible with IPTG (isopropyl β-D-1-thiogalactopyranoside) and a kanamycin (Kan) resistance marker. We often co-transform the expression host with the plasmid pDB1282 to aid in assembly of the FeS clusters. This plasmid contains iscSUA-hscB-hscA-fdx-orf3 from Azobacter vinelandii (Frazon, Fick & Dean, 2002), which have been shown to be involved in biosynthesis of iron-sulfur clusters. The genes are maintained on pARA13 plasmid, which has an ampicillin (Amp) resistance marker and is inducible by arabinose. Co-transformation with pDB1282 leads to a higher level of iron-sulfur cluster biogenesis proteins, which supplement the endogenous E. coli iron sulfur cluster biogenesis machinery.
Figure 5. Codon optimized sequence of TYW1.

The top shows the codon optimized TYW1 sequence, expressed by pAY613 with the TEV protease site shown bolded and the restriction sites used to insert the gene in italics. The middle shows the translated protein sequence expressed by pAY613 with the TEV protease site shown bolded. The bottom shows the final sequence of TYW1 purified in this study with the extra amino acids not encoded by the original TYW1 gene are shown in bold italics.
Our general protocol entails overexpression of TYW1 in E. coli strain BL21(DE3) grown in lysogeny broth (LB) broth containing Kan and Amp to select for bacteria containing the plasmids for the expression of TYW1 and the isc operon. Protein expression is induced by the addition of IPTG (pAY613) and arabinose (pDB1282) and protein expression is carried out overnight at 37 °C with decreased shaking rate.
Protocol for Expression of TYW1 Gene
Optimal expression is achieved by always starting from a freshly transformed E. coli cells. The plates are discarded after 1 week.
Make LB (Lennox) agar plates by combining 5 g of tryptone, 2.5 g yeast extract, 2.5 g NaCl, and 7.5 g agar with 500 mL of water. Autoclave for 30 minutes and allow to cool to approximately 50 °C. Add 500 µL of 34 mg/mL Kan (in water) and 500 µL of 100 mg/mL Amp (in water) to the LB agar. Pour the plates and allow to solidify. This same recipe is used for preparation of LB liquid media, except that agar is excluded.
Co-transform E. coli cell line BL21(DE3) T1 phage resistant (New England Biolabs C2527) with pAY613 and pDB1282. Plate transformation mixture on a LB (Lennox) agar plate containing 34 µg/mL Kan and 100 µg/mL Amp and incubate at 37 °C overnight.
Make 1 L of LB broth as described in step 2. Transfer 125 mL to a autoclaved 250 mL Erlenmeyer flask aseptically.
Use a single colony from the transformation plate to inoculate a 125 mL culture of LB broth containing 34 µg/mL Kan and 100 µg/mL Amp in a 250 mL Erlenmeyer flask. Grow cells overnight at 37 °C with shaker set to 225 rpm.
Prepare 6 L of LB broth (as described in step 2 above) and distribute equally into six 2.8 L Fernbach flasks.
Inoculate 1 L of LB broth (in 2.8 L Fernbach flasks) containing 34 µg/mL Kan and 100 µg/mL Amp with 10 mL of the overnight culture.
Shake the flasks at 175 rpm at 37 °C to an optical density at 600 nm (OD600) of ~ 0.7.
Add 0.5 mL of 0.1 M iron(III)chloride (in water) to give a concentration of 50 µM along with 0.5 g solid arabinose (0.05% w/v) and induce expression by addition of 100 µL of 1 M IPTG (in water) to give a final concentration of 100 µM.
Immediately upon induction, the shaking rate is reduced to 125 rpm and the cultures are incubated at 37 °C for 18 h.
Harvest cells by centrifugation at 5,000 xg and flash freeze cell pellet in liquid nitrogen.
Typical yield from this procedure is 3 g wet cell paste from each L of media. Store cells at −80 °C.
2.2. Purification of TYW1
Purification of TYW1 should be performed in an anaerobic chamber under a nitrogen atmosphere. We use a Coy chamber, which typically has an atmosphere composed of 97% nitrogen and 3% hydrogen. Anaerobicity is achieved by allowing H2 to react with O2 on the surface of the palladium catalyst within the chamber. Excess moisture in the chamber is removed by both a dessicant pack within the chamber and a Coy dehumidifier. The anaerobicity of the chamber is monitored by a Coy cam-12 detector. The chamber is in a laboratory which is maintained at 22 °C. Samples are introduced into the glove box through an antechamber, which is automatically cycled through vacuum and gas filling cycles. We have modified the default cycle of the antechamber so that it is vacuumed to 19.9 inHg and refilled with N2 four times prior to final vacuum and fill cycle where a mixed gas (95%N2/5%H2) is used. To facilitate purification steps, we have placed an AKTAprime plus inside the chamber. All tube, tips, and other plasticware are placed in the glovebox at least 24 h prior to use. Water and concentrated buffer stocks are stirred in the glovebox for 12 h. Cell disruption is carried out on ice and the centrifugation steps are at 4 °C. Cells are disrupted by a Branson sonifier as indicated below. We maintain the horn of the sonifier in the glove box and attach it to a controller maintained outside the glove box.
The purification of TYW1 from M. jannaschii is greatly facilitated by a heat denaturation step, which removes the majority of the contaminating native E. coli proteins improving protein quality. Next, hydrophobic chromatography is utilized to remove E. coli nucleic acids that co-purify with TYW1 and potentially interfere with protein characterization. The final purification step is affinity chromatography utilizing the N-terminal His6. This step serves to purify the protein by removing a small number of contaminants, but it also concentrates the protein from 0.2 L to 0.05 L. At this point the purification of the His6-TYW1 is complete and protein may be frozen after desalting to remove imidazole and salt.
Protocol for Purification of His6-TYW1 protein
In an anaerobic chamber prepare the following buffers using water, solid salts, and concentrated buffer stocks that have been stirred in the chamber for 24 h: lysis buffer – 20 mM tris(hydroxymethyl)aminomethane hydrochloride (pH 8) (Tris-HCl); butyl column buffer – 20 mM Tris-HCl (pH 8) containing 1 M ammonium sulfate (AmSO4); HisTrap loading buffer – 50 mM potassium phosphate (pH 7.4) (Kpi) containing 0.5 M KCl, and 50 mM imidazole; HisTrap elution buffer – 50 mM Kpi (pH 7.4) containing 0.5 M KCl, and 0.5 M imidazole; desalting buffer – 50 mM piperazine-N,N’-bis(2-ethanesulfonic acid)-sodium hydroxide (pH 7.4) (PIPES-NaOH) containing 2 mM dithiothreitol (DTT).
Place 18 g of frozen cell pellet, a stir bar, and solid phenylmethane sulfonyl fluoride (PMSF) (to a final concentration of 1 mM) in a stainless steel beaker and transfer into the anaerobic chamber along with a crystallization dish packed with ice.
Add 0.15 L of anaerobic lysis buffer to the beaker and place beaker in the crystallization dish with the ice surrounding the beaker on all sides. Place crystallization dish on a stir plate and stir the suspension.
Disrupt cells using a Branson sonifier. Cell disruption is achieved with 48 bursts of 15 sec each, with 60 sec intervals to allow sample time remain cold. The amplitude of the bursts is kept at 50%.
Transfer lysate to a centrifuge tube that has a lid with an O-ring, cap the lid tightly, and centrifuge at 18,000 xg for 30 min at 4 °C.
Transfer the centrifuge tube back into the anaerobic chamber and decant the supernatant into a 0.25 L bottle and cap the bottle tightly.
Remove the sealed bottle from the glove box and place in a water bath set to 80 °C for 30 min.
Transfer the bottle into an ice bath for 15 min.
Reintroduce the bottle back into the anaerobic chamber and transfer the lysate to centrifuge tubes. Cap the lid tightly, and centrifuge at 18,000 xg for 30 min at 4 °C.
Reintroduce the centrifuge bottle back into the anaerobic chamber and transfer the clarified lysate to a measuring cylinder and record the volume.
Transfer the clarified lysate to a clean beaker containing a stir bar.
Slowly add solid AmSO4 to the stirring lysate to a final concentration of 1 M and stir to completely dissolve.
All column chromatography steps are carried out using a AKTAprime plus instrument with a UV detector set to 280 nm.
Manually pack a butyl-Sepharose FF column (11 × 3 cm) (GE Healthcare)and equilibrate with butyl column buffer.
Load the lysate onto the equilibrated column and wash with the loading buffer until the absorbance has returned to baseline. This typically takes 2–3 column volumes.
Elute adsorbed protein with a step gradient to 100% lysis buffer and collect brown fractions.
Using the AKTAprime plus equilibrate a 5 mL HisTrap chealting HP column (GE Healthcare) charged with nickel chloride according to manufacturer’s directions with HisTrap loading buffer.
Load the pooled fractions from the butyl column directly onto the equilibrated HisTrap chelating column. When all lysate is loaded wash the column with 2–3 column volumes of HisTrap loading buffer until the absorbance has returned to baseline.
Elute adsorbed protein with a step gradient to 100% HisTrap elution buffer, collect and pool the protein that elutes.
Equilibrate a Bio-Gel P-6 DG gravity column (8 × 3 cm) with 100 mL of desalting buffer.
Load up to 35 mL of protein onto the equilibrated column at one time. Start collecting protein at 14 mL and stop collecting at 50 mL. If more than 35 mL of protein needs to be desalted, re-equilibrate the column with 100 mL of desalting buffer and repeat.
At this point the His6-TYW1 can be flash frozen and stored at −80 °C, or be treated with TEV protease to remove the purification tag.
2.3. Cleavage of the His6 tag with TEV protease
Expression of TYW1 from the pAY613 plasmid places a TEV protease site between the His6 affinity tag and the protein. Following cleavage, native TYW1 can be obtained by chromatography of the sample on a HisTrap column to remove the tag, as well as uncleaved protein, and TEV protease. Expression and purification of the TEV protease are described in Section 3.
The buffer to perform the cleavage should contain DTT, which was introduced in the previous desalting step. The TEV protease cleavage is carried out overnight in the anaerobic chamber with stirring. Following chromatography on the HisTrap column, the protein is desalted to remove imidazole and place the protein in buffer that is compatible with either freezing or proceeding to the chemical reconstitution of the iron-sulfur clusters.
Protocol for TEV protease cleavage of the His6 tag.
In an anaerobic chamber prepare the following buffers using water, salts, and concentrated buffer stocks that have been stirred in the chamber for 24 hours: HisTrap loading buffer – 50 mM Kpi (pH 7.4) containing 0.5 M KCl and 50 mM imidazole; desalting buffer – 50 mM PIPES-NaOH (pH 7.4) containing 2 mM DTT.
Combine His6-TYW1 purified as described in Section 2.2 in a beaker with a stir bar and ~ 1 mL of TEV protease (~ 120 µM) and stir overnight at room temperature.
Equilibrate a 5 mL HisTrap chealting HP column (GE Healthcare) charged with nickel chloride according to manufacturer’s directions with HisTrap loading buffer using the AKTAprime plus.
Load the TYW1 TEV mixture onto the equilibrated column and collect the protein that flows through the column.
When all the protein has been loaded, wash the column with HisTrap loading buffer until the absorbance returns to baseline, collecting all protein that elutes during the wash.
Pool all protein that did not bind to the column.
Equilibrate a Bio-Gel P-6 DG gravity column (8 × 3 cm) with 100 mL of desalting buffer.
Load up to 35 mL of protein onto the equilibrated column at one time. Start collecting protein at 14 mL and stop collecting at 50 mL. If more than 35 mL of protein needs to be desalted, re-equilibrate the column with 100 mL of desalting buffer and repeat.
Protein eluting from the desalting column can be flash frozen at −80 °C or undergo chemical reconstitution.
2.4. Chemical Reconstitution of the iron-sulfur Clusters
TYW1 as isolated is inactive and requires chemical reconstitution to obtain cofactor-replete protein. We have determined empirically using the increase of absorbance at 420 nm that 4 h is the optimal reconstitution time for TYW1. At longer incubation times, we find significantly more aggregated protein upon gel filtration, suggesting that the protein may undergo irreversible loss of activity when reconstitution time is extended.
Chemical reconstitution of iron sulfur clusters is performed in a reducing anaerobic environment in the presence of excess iron and sulfide. We have found that an 10-fold molar excess of FeCl3 and Na2S over the concentration of TYW1 is ideal. To determine the protein concentration a Bradford assay is performed. A correction factor, for the Bradford assay, is determined by amino acid analysis, we use UC Davis Genome Center Molecular Structure Facility. Following anaerobic reconstitution, any precipitate is removed by centrifugation and the protein is concentrated and subjected to size exclusion chromatography, which separates the aggregated protein from monomeric TYW1.
Protocol for Chemical Reconstitution of Iron-sulfur clusters
In an anaerobic chamber prepare the following buffer using water, salts, and concentrated buffer stocks that have been stirred in the chamber for 24 hours: gel filtration buffer – 50 mM PIPES-NaOH (pH 7.4) containing 150 mM KCl, and 2 mM DTT.
To determine the quantity of Fe and S to use in the reconstitution, an accurate protein concentration is essential. We routinely use a Bradford assay (Boston BioProducts) with BSA as standard to quantify TYW1. An empirically-determined correction factor of 0.32 (Young et al, 2015), which is based on the absolute amino acid content of the protein preparation, is applied to the Bradford assays.
Weigh out solid iron(III)chloride and sodium sulfide into separate glass vials and cycle into the anaerobic chamber.
Add enough anaerobic water to obtain solutions of iron(III)chloride and sodium sulfide at final concentration of 1 M.
Transfer the protein solution to a beaker with a stir bar and add 10-fold molar excess of iron(III)chloride, followed by sodium sulfide.
Stir the reconstitution mixture at room temperature for 4 h.
Aliquot the reconstitution mixture into multiple Eppendorf tubes and remove precipitated protein by centrifuging for 10 min at 16,000 xg at room temperature.
Combine the supernatants from the centrifugation.
Equilibrate a Bio-Gel P-6 DG gravity column (8 × 3 cm) with 100 mL of gel filtration buffer.
Load up to 35 mL of protein onto the equilibrated column at one time. Start collecting protein at 14 mL and stop collecting at 50 mL. If more than 35 mL of protein needs to be desalted, re-equilibrate the column with 100 mL of gel filtration buffer and repeat.
Concentrate the protein using an Amicon concentrator with a YM-10 membrane (Millipore) under nitrogen gas to a volume of ~ 1 mL. This is readily accomplished by bringing in nitrogen gas through one of the nipple ports in the glove box. We find that this allows for rapid protein concentration without exposing the protein to oxygen.
Equilibrate a Sephacryl 16/60 S-200 column (GE Healthcare) in gel filtration buffer.
Desalt TYW1 in 1 mL aliquots over the equilibrated column.
Pool and concentrate the brown fractions that elute at 65 mL, corresponding to monomeric protein, using an Amicon concentrator with a YM-10 membrane under nitrogen gas.
Aliquot, flash freeze, and store the concentrated protein at −80°C. Typical yield of homogeneous TYW1 is 1 mg / g wet cell paste. Fig. 6 shows a sample of TYW1 on a SDS-PAGE gel purified in this manner
Figure 6. SDS-PAGE gel showing purified TYW1.
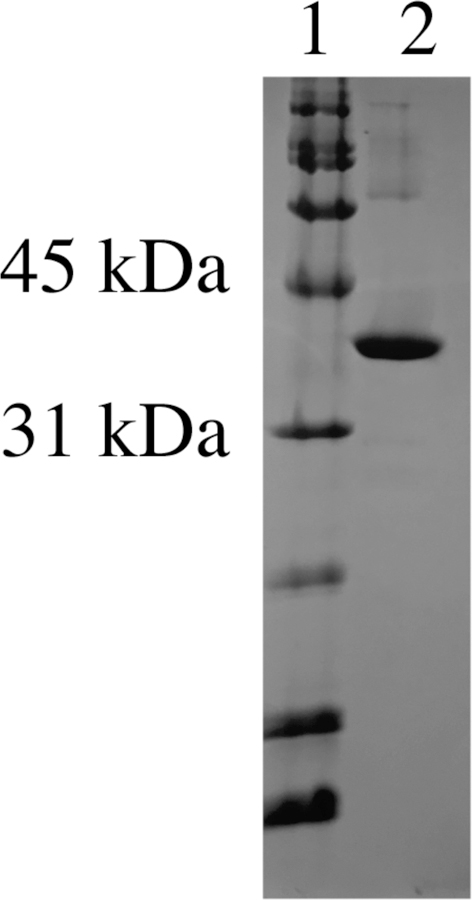
A gel showing TYW1 with BioRad broad range standard (cat # 161–0317) in lane 1 and TYW1 in lane 2.
3. Expression and Purification of Tobacco Etch Virus (TEV) Protease
The TEV protease recognizes the sequence ENLYFQ/G (Carrington & Dougherty, 1988; Kapust, Tözsér, Copeland & Waugh, 2002). TYW1 is cloned into pET28t, which is a modified pET28a vector containing the TEV protease cut site in place of thrombin cut site (Thoden et al, 20005), such that cleavage would lead to amino acids GH on the N-terminus of TYW1. The expression vector for TEV protease (prTEV) and the cell line used for expression of TEV protease (SG13000009) was a gift from Professor Hazel Holden at the University of Wisconsin-Madison.
3.1. Expression of TEV Protease
Expression of TEV protease is carried out in an E. coli cell line SG13000009 containing pLacIRARE. The pLacIRARE plasmid already in the strain has chloramphenicol (Cam) resistance, while the prTEV plasmid has Amp resistance. Expression of TEV protease is carried out in LB with cells initially grown at 37 °C and then the temperature is decreased to 16 °C when protein expression is induced by addition of IPTG.
Protocol for Expression of Tobacco Etch Virus (TEV) Protease
Make LB (Lennox) agar plates as described in Section 2.1.
Transform E. coli cell line SG1300009 pLacIRARE with prTEV and plated transformants on a LB agar plate containing 34 µg/mL Cam (dissolved in ethanol) and 100 µg/mL Amp. Incubate plates at 37 °C overnight.
Make 1 L of LB broth as described in Section 2.1. Use a single colony to inoculate a 125 mL of LB broth in a 250 mL Erlenmeyer flask containing 34 µg/mL Cam and 100 µg/mL Amp. Flasks are grown at 37 °C shaking at 225 rpm overnight.
Make 6 L of LB broth and distribute among six 2.8 L Fernbach flasks. Autoclave for 30 min and allow to cool.
Add 34 µg/mL Cam and 100 µg/mL Amp to each flask and inoculate with 10 mL of the overnight culture.
Allow the cultures to shake at 175 rpm and at 37 °C to OD600 ~ 0.6.
Add 1 mL of a 1 M stock solution of IPTG (1 mM final concentration) to induce expression of the protein. At this point, reduce the temperature to 16 °C and allow the cultures to grow for a further 18 h.
Harvest cells by centrifugation at 5,000 xg and flash freeze cell pellet in liquid nitrogen. Typical yield from this protocol is ~5 g / L media.
3.2. Purification of TEV Protease
The TEV protease used to cleave His6-TYW1 is readily purified using a His6 affinity tag as described below. The presence of the His6 affinity tag allows TEV protease to be easily purified from E. coli and it also provides a method for purification of the protease when it is used to cleave the tag from a protein of interest.
The purification procedure involves lysing the cells and then passing the clarified lysate over a HisTrap column charged with nickel. Bound protein is eluted via a gradient and the fractions containing concentrated protein are dialyzed against HisTrap loading buffer. Finally, glycerol is added and the protein is stored at −20 °C.
Protocol for Purification of TEV Protease
Prepare the following buffers using water at 4 °C: lysis buffer – 50 mM potassium phosphate (Kpi) (pH 7.4) containing 0.5 M KCl, 50 mM imidazole, and 20% glycerol (v/v); elution buffer – 50 mM Kpi (7.4) containing 0.5 M KCl, 0.5 M imidazole, and 20% glycerol (v/v).
Place 30 g of frozen cells, a stir bar, and solid PMSF in a stainless steel beaker and add 0.15 L of lysis buffer.
Place beaker in a crystallization dish with ice surrounding the beaker on all sides. Place crystallization dish on a stir-plate and adjust speed so that the cells are stirring.
Disrupt cells by 48 bursts of sonication for 15 seconds at 50% power, followed by 60 second breaks to avoid heating the samples.
Transfer lysate to a centrifuge tube that has a lid with an O-ring, cap the lid tightly, and centrifuge at 18,000 xg for 30 min at 4°C.
Using an AKTA FPLC at 4 °C, equilibrate a 5 mL HisTrap chelating HP column (GE Healthcare) charged with nickel chloride, according to manufacturer’s directions, with lysis buffer.
Transfer clarified lysate from centrifuge tube to a clean beaker.
Load clarified lysate onto the equilibrated column. When all lysate is loaded wash the column with lysis buffer (10 column volumes) until the absorbance has returned to baseline.
Elute adsorbed protein with a gradient to 100% elution buffer over 50 mL.
Identify TEV protease containing fractions by SDS-PAGE and pool the more concentrated fractions.
Dialyze pooled TEV protease against 4 L of lysis buffer.
Change dialysis buffer twice after at least 4 h.
Add glycerol to the pooled TEV protease to a final concentration of 50% (v/v), aliquot, and flash freeze in liquid nitrogen. Store aliquots at −20°C.
4. Expression and Purification of TRM5
The first step in the biosynthesis of wybutosine is methylation of the G at position 37 of tRNA Phe (Björk et al, 2001; Droogmans et al, 1987; Noma et al, 2010). The synthesis of a tRNA substrate for TYW1 using in vitro transcription requires a methyl transferase capable of methylating the tRNA. In our studies, we have routinely employed TRM5 from M. jannaschii. This section will describe expression and purification of MjTRM5.
4.1. Expression of MjTRM5 gene
The gene for MjTRM5, which was codon optimized for expression in E. coli, was synthesized (GenScript) and cloned between the NdeI and HindIII sites of pET29a vector to create pAY423 (see Fig. 7 for optimized gene sequence). This vector contains a T7 promotor that is inducible with IPTG and a Kan resistance marker. The protein is expressed in LB media at 37 °C.
Figure 7.

Codon optimized gene for MjTRM5 and the corresponding protein sequence. Cloning sites are shown in italic.
Protocol for Expression of TRM5 gene
Make LB (Lennox) agar plates containing 34 µg/mL Kan as described in Section 2.1.
Transform E. coli cell line BL21(DE3) with pAY423, which contains codon optimized TRM5 gene in pET29a vector. Plate transformants on LB agar plates containing 34 µg/mL Kan and incubate at 37 °C overnight.
Make 1 L of LB broth as described in Section 2.1.
Use a single colony from the plate to inoculate a 125 mL of LB broth containing 34 µg/mL Kan in a 250 mL Erlenmeyer flask. Cells were grown overnight at 37 °C with shaking speed set to 200 rpm.
Make 6 L of LB broth as described in Section 2.1.
Inoculate 1 L of LB broth containing 34 µg/mL Kan in a 2.8 fernbach flask with 10 mL of the overnight culture.
Grow the cells at 37 °C with shaking set to 200 rpm.
At OD600 ~ 0.6 add 100 µL of IPTG from a 1 M stock to make a final concentration of 100 µM.
Allow cells to grow for ~18 h.
Harvest cells by centrifugation at 5,000 xg and flash freeze cell paste in liquid nitrogen. Store at −80°C.
4.2. Purification of TRM5
The purification of TRM5 involves a heat step to remove the majority of E. coli proteins. A hydrophobic column is then used to remove heat stable E. coli proteins and nucleic acids that co-purify with TYW1. The final step in the purification is dialysis to remove the AmSO4 used in the hydrophobic chromatography step, resulting in pure protein capable of methylating the tRNAPhe transcript.
Protocol for Purification of TRM5
Prepare the following buffers using water at 4°C: Lysis buffer – 20 mM Tris-HCl (pH 8), 2 mM DTT; Butyl column buffer – 20 mM Tris-HCl (pH 8), 1.5 M ammonium sulfate, 2 mM DTT; Butyl column elution buffer – 20 mM Tris-HCl (pH 8), 2 mM DTT; Dialysis buffer – 20 mM Tris-HCl (pH 8), 50 mM KCl, 2 mM DTT.
Place 12 g of frozen cells, a stir bar, solid PMSF, and add 0.15 L of lysis buffer into a stainless steel beaker.
Place beaker into a crystallization dish surrounded on all sides by ice.
While stirring the frozen cells in buffer, sonify with 48 bursts of 15 sec with sonifier at 50% amplitude, with 60 sec rest to allow the solution to remain cold.
Transfer lysate to a centrifuge tube that has a lid with an O-ring, cap the lid tightly, and centrifuge at 18,000 xg for 30 min at 4°C.
Transfer the clarified lysate from the centrifuge tube to a glass bottle and seal the lid tightly.
Place the bottle in an 80 °C water bath and incubate for 30 min.
Transfer the bottle to an ice water bath and incubate for 15 min.
Transfer the lysate to a centrifuge tube, and centrifuge for 30 min at 18,000 xg at 4°C.
Transfer the clarified lysate to a measuring cylinder and determine the volume.
Add the lysate to a beaker containing a stir bar and stir over a stir plate while slowly adding solid ammonium sulfate to a final concentration of 1.5 M over the course of 20 min.
Manually pack a butyl-Sepharose FF column (GE Healthcare) (11 × 3 cm) and equilibrate with butyl column buffer using an AKTA FPLC at 4°C.
Load the lysate onto the equilibrated column and wash with the loading buffer until the absorbance has returned to baseline. This typically takes 2–3 column volumes.
Elute adsorbed protein with a gradient to 100% butyl column elution buffer over 0.4 L.
Identify fractions containing pure TRM5 by SDS-PAGE.
Pool fractions with highest purity protein and transfer to dialysis tubing with a 10 kDa molecular weight cut off.
Dialyze against 4 L of dialysis buffer for at least 4 hours with three changes.
Concentrate the protein to a minimal volume using an Amicon stirred cell with a YM10 membrane (Millipore).
Aliquot and flash freeze with liquid nitrogen. Store frozen aliquots at −80°C.
5. Synthesis of Substrate for TYW1 via in vitro transcription
One method utilized to prepare substrate from TYW1 is in vitro transcription using a DNA oligonucleotide as the template with T7 RNA polymerase. A DNA oligonucleotide complementary to M. jannaschii MJ-t16 (phenylalanine tRNA gene) with the T7 promoter at the 3′ end is combined with an oligonucleotide consisting of the T7 promoter sequence. The two primers are annealed and then used as a template for recombinant T7 polymerase. This reaction is carried out at 37 °C in the presence of inorganic pyrophophatase, which hydrolyzes pyrophosphate into phosphate in order to stop it from binding MgCl2 and forming a precipitate. The final step is incubation with DNase I to remove the template oligonucleotides. This section describes in vitro transcription reaction. Expression and purification of the T7 RNA polymerase used in these studies is described in Section 6.
Protocol for synthesis of tRNAPhe
Prepare diethyl pyrocarbonate (DEPC) treated water to be used in the synthesis of tRNA and the purification of S. cerevisiae tRNA in the next section as follows: add 1 mL of diethyl pyrocarbonate to 1 L of water. Incubate for 2 h at 37 °C and then autoclave for 15 min.
Obtain the following oligonucleotides nucleotides: 5′-TGGTGCCGCGGGGGTGATTTGAACACCCGACAACTGGATCTTCAGTCCAGCGTTCTCCCAGGCTGAACTACCGCGGCTATAGTGAGTCGTATTAGGATCC-3′ (MJ-t16) and 5′-GGATCCTAATACGACTCACTATAG-3′ (T7 promoter) and re-suspend them in DEPC treated water.
Prepare the following buffer using water that has been DEPC treated: 5X transcription buffer – 200 mM Tris-HCl (pH 7.9), 30 mM magnesium chloride (MgCl2), 50 mM DTT, 50 mM NaCl, 10 mM spermidine.
Anneal 10 µM each of T7 promotor primer with (Mj-t16) primer using a thermocycler with the following conditions in a volume of 200 µL: 95°C for 20 seconds with a −0.5 °C change in temperature for 34 cycles.
Setup a reaction containing the following in a 500 µL volume: 1X transcription buffer, 0.75 µM annealed primers, 10 mM DTT, 10 mM MgCl2, 4 mM each of ribonucleotide triphospahtes, 0.5 units inorganic pyrophosphatase (Sigma), and 10% (v/v) recombinant T7 RNA polymerase.
Incubate at 37 °C for 3 h.
Add 20 units of RNase free DNase I.
Incubate at 37 °C for 30 min.
Split reaction into two tubes with 260 µL in each tube.
Freeze or purify further as described below in Section 9.3.
6. Expression and Purification of T7 RNA polymerase
A glycerol stock containing a Rosetta (DE3) E. coli strain transformed with a pET42a plasmid containing T7 RNA polymerase (constructed by Bevilacqua Lab at Penn State) was a generous gift from Prof. Squire Booker at The Penn State University. T7 RNA polymerase recognizes the T7 promoter and has a low error rate. The T7 RNA polymerase expressed from this plasmid contains a His tag which aids in purification.
6.1. Expression of T7 RNA polymerase
The T7 RNA polymerase gene is cloned downstream of a IPTG inducible promoter on a pET42a plasmid. The pET42a plasmid contains a gene for Amp resistance, while the Rosetta (DE3) strain has Cam resistance. The protein is expressed in LB at 37 °C for 3 h.
Protocol for the expression of T7 RNA polymerase
Make LB (Lennox) agar plates containing 34 µg/mL Cam and 100 µg/mL Amp as described in Section 2. Streak out a loop of T7 RNA polymerase in Rosetta DE3 glycerol stock and incubate overnight at 37 °C.
Make 1 L of LB broth as described in Section 2.
Use a single colony from the plate to inoculate a 125 mL of LB broth, containing 34 µg/mL Cam and 100 µg/mL Amp, in a 250 mL Erlenmeyer flask. Cells were grown overnight at 37 °C with shaking speed set to 200 rpm.
Make 10 L of LB broth as described in Section 2 and distribute among ten 2.8 L Fernbach flask. Autoclave for 30 min and allow to cool.
Add 34 µg/mL Cam and 100 µg/mL Amp to each flask and inoculate with 10 mL of the overnight culture.
Grow the cells at 37 °C with shaking set to 200 rpm.
At OD600 ~ 0.6 add 0.5 mL of 1 M IPTG to a final concentration of 500 µM.
Allow cells to grow for ~3 h.
Harvest cells by centrifugation at 5,000 xg and flash freeze cell paste in liquid nitrogen. Store at −80°C.
Typical yield is 3.4 g per L of culture.
6.2. Purification of T7 polymerase
The T7 RNA polymerase expressed from this plasmid has a His6 affinity tag which greatly aids the purification. Protein is purified in a very similar manner to that used to purify TEV protease in Section 3.2 with a HisTrap chelating HP column charged with nickel utilized.
Protocol for purification of T7 polymerase
Prepare the following buffers using water at 4 °C: Lysis buffer – 50 mM Tris-HCl (pH 8) buffer containing 100 mM NaCl, 5 mM DTT, 10 mM imidazole, and 5% (v/v) glycerol; Elution buffer – 50 mM Tris-HCl (pH 8) buffer containing 100 mM NaCl, 5 mM DTT, 400 mM imidazole, and 20% (v/v) glycerol; Desalting buffer – 50 mM Tris-HCl (pH 8) buffer containing 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 10 mM DTT, and 50% (v/v) glycerol.
Place 34 g of frozen cells, a stir bar, solid PMSF, and add 0.2 L of lysis buffer into a stainless steel beaker.
Place beaker into a crystallization dish surrounded on all sides by ice.
While stirring the frozen cells in buffer, sonify with 48 bursts of 15 sec with sonifier at 50% amplitude, with 60 s rest to allow the solution to remain cold.
Transfer lysate to a centrifuge tube that has a lid with an O-ring, cap the lid tightly, and centrifuge at 18,500 xg for 30 min at 4°C.
Using an AKTA FPLC, equilibrate a 5 mL HisTrap chelating HP column charged with nickel chloride, according to manufacturer’s directions, with lysis buffer.
Transfer clarified lysate from centrifuge tube to a clean beaker.
Load clarified lysate onto the equilibrated column. When all lysate is loaded wash the column with lysis buffer (20 column volumes) until the absorbance has returned to baseline.
Elute adsorbed protein with a gradient to 100% elution buffer over 100 mL.
Identify T7 polymerase containing fractions by SDS-PAGE and pool the fractions.
Concentrate to 3 mL using a Amicon stirred cell with a YM10 membrane (Millipore).
Equilibrate a BioRad Econo-Pac 10 DG desalting column by filling the column to the top with desalting buffer and allowing it to flow through.
Add the 3 mL of concentrated protein to the desalting column and allow it to enter the column.
Elute the protein by adding 4 mL of desalting buffer and collecting the flow through.
Aliquot the protein and store at −80 °C.
7. Isolation of Substrate for TYW1 from S. cerevisiae Deletion Strain
As an alternative to the synthetic substrate, TYW1 can be assayed with tRNA enriched for tRNAPhe obtained from a deletion strain of Saccharomyces cerevisiae, where TYW1 has been deleted. The sequence of the genes encoding tRNAPhe from S. cerevisiae and M. jannaschii are shown in Fig. 8. The tRNAPhe isolated from the TYW1 (YPL207W) deletion strain will be fully modified with the exception of G37, which will contain a m1G residue instead of the expected wybutosine.
Figure 8. Alignment of M. jannaschii and S. cerevisiae tRNAPhe.

The sequence of the S. cerevisiae tRNAPhe is similar to that of M. jannaschii, allowing it to substituted as substrate TYW1 from M. jannaschii. The site of modification is shown in bold.
7.1. Growth of S. cerevisiae deletion strain
S. cerevisiae is grown in YPD broth at 30 °C in order to generate cell mass. Cells are grown to an high optical density, harvested by centrifugation, and flash frozen with liquid nitrogen.
Protocol for Growth of S. cerevisiae YPL207W deletion strain
Make 1 L of YPD broth by combining 20 g of peptone, 10 g yeast extract, and 20 g glucose with 1 L of water in a 1 L bottle. Autoclave for 30 min and allow to cool. Transfer 0.1 L to an autoclaved 250 mL Erlenmeyer flask aseptically.
Inoculate a 0.1 L YPD broth culture inoculated with a loop of S. cerevisiae YPL207W deletion strain (YSC1–21-547084 Thermo Scientific).
Grow at 30 °C with shaking at 200 rpm for approximately 48 h.
Make 6 L of YPD broth and distribute among six 2.8 L Fernbach flasks. Autoclave for 30 min and allow to cool. Inoculate each with 10 mL of the culture from step 3.
Grow at 30 °C until the OD600 reaches ~10. This typically takes 72 h.
Harvest by centrifugation at 5,000 xg and flash freeze cell pellet with liquid nitrogen. This typically yields 20 g per L of culture.
7.2. Purification of soluble RNA from S. cerevisiae
Soluble RNA is extracted from S. cerevisiae via a phenol extraction. Phenol permeabilzes the cell membrane and wall releasing the cytoplasmic contents. The aqueous layer is extracted, followed by an ethanol precipitation to collect RNA. The pellet is washed with 1 M NaCl to remove high molecular weight RNA (von Ehrenstein, 1967), followed by an ethanol precipitation of the supernatant.
Protocol for purifying soluble RNA from S. cerevisiae YPL207W deletion strain
Prepare DEPC water as described in Section 5.1.
Prepare the following buffers using DEPC treated water: resuspension buffer - 10 mM Tris-HCl (pH 8) containing 10 mM MgCl2 and 150 mM NaCl; sodium acetate solution - 3 M sodium acetate (pH 5.2); sodium chloride solution – 1 M NaCl.
Add resuspension buffer to cells (1 mL / 1 g of wet cell paste) in a beaker containing a stir bar and stir for 2 h in order to re-suspend the cells.
Add liquified phenol (pH 4.3) (Fisher) (1 mL / 1 g of wet cell paste) and stir the mixture 1 h.
Transfer to a centrifuge bottle and centrifuge at 1,500 xg at room temperature for 10 min.
Transfer supernatant aqueous layer to a clean beaker and add liquefied phenol (pH 4.3) (1 mL / 1 g of wet cell paste) and mix by inversion.
Transfer to a centrifuge bottle and centrifuge at 15,000 xg at 4°C for 30 min.
Extract the upper layer and determine the volume
Add 0.1 volume of sodium acetate solution and 3 volumes of ethanol.
Incubate at −20 °C for at least 2 h.
Transfer to a centrifuge bottle and centrifuge at 15,000 xg for 30 min at 4 °C.
Discard the supernatant and re-suspend the pellet in sodium chloride solution.
Centrifuge at 7,000 xg for 30 min at 4 °C.
Discard the supernatant.
Re-suspend the pellet in DEPC-treated water, aliquot, and freeze.
Determine the concentration using A260 (ε = 0.025 (µg/mL)−1cm−1)
8. Synthesis of S-adenosyl-l-methionine
While SAM is commercially available, it is a mixture of isomers where only one is biologically active. In order to prepare enantiomerically pure SAM, a cell free extract from cells overexpressing SAM synthethase (Markham, Hafner, Tabor & Tabor, 1980) DM22-(pK8) (generous gift from Prof. Doug Markham) is utilized. SAM synthetase catalyzes the reaction between Met and ATP to generate SAM. SAM is subsequently purified on a carboxymethyl cellulose column and frozen in small aliquots for subsequent assays (Iwig & Booker, 2004).
8.1. Growth of E. coli strain overexpressing SAM synthetase
The E. coli strain DM22-(pK8) is used in the cell free assays to synthesize SAM. The strain is grown for 12 h at 37 °C and has oxytetracycline resistance.
Protocol for growth of E. coli strain overexpressing SAM synthetase
Make 1 L of LB broth as described in Section 2.
Inoculate a 0.125 L LB broth culture containing 35 µg/mL oxytetracycline (in ethanol) with a loop of E. coli strain DM22-(pK8).
Grow culture overnight at 37 °C with shaking at 225 rpm.
Make 6 L of LB broth as described in Section 2 and distribute equally between six 2.8 L Fernbach flasks. Autoclave for 30 min and allow to cool.
Add 35 µg/mL oxytetracycline to each flask and inoculate with 10 mL of the overnight culture.
Grow at 37 °C with shaking at 225 rpm for 12 h.
Harvest by centrifugation 5,000 xg and flash freeze cell paste in liquid nitrogen. Store at −80°C. The typical yield is 4 g of cell paste from each L of growth.
8.2. Preparation of cell lysates for synthesis
The method used to produce SAM enzymatic involves lysate from the E. coli strain DM22-(pK8). The cells are lysed using a sonifier and then dialyzed against buffer in order to remove small molecules such as ATP and Met that may interfere with synthesis of isotopically enriched SAM.
Protocol for cell lysis
Prepare the following buffer using water at 4 °C: cell lysis buffer – 20 mM Tris-HCl (pH 8), 1 mM EDTA and 50 µg/mL lysozyme.
Place 17 g of frozen cells and a stir bar in a stainless steel beaker and add 0.04 L of cell lysis buffer.
Stir at room temperature for 30 min.
Add solid PMSF to give a final concentration of 1 mM (7 mg)
Place beaker in a crystallization dish with ice surrounding the beaker on all sides. Place crystallization dish on a stir-plate and adjust speed so that the cells are stirring.
Disrupt cells by 40 bursts of sonication for 30 seconds at 50% power, followed by 60 second breaks to avoid heating the samples.
Transfer lysate to a centrifuge tube that has a lid with an O-ring, cap the lid tightly, and centrifuge at 18,000 xg for 30 min at 4°C.
Transfer the clarified lysate to a dialysis bag and dialyze against 4 L of cell lysis buffer at 4 °C for a minimum of 4 h with 2 changes.
Aliquot the lysate in 15 mL aliquots, flash freeze with liquid nitrogen, and store at −80 °C.
8.3. Synthesis of SAM
Enantiomerically pure SAM is synthesized from ATP and Met using a cell free system. DM22-(pK8) cell lysate is incubated at room temperature for 5 h. in the presence of ATP and methionine allowing for the incorporation of specific isotopes creating enriched SAM. Acetonitrile is included in the reaction mixture to prevent product inhibition.
Following the synthesis reaction SAM is purified on a cation exchange column, to which the positively charged SAM binds. SAM is eluted with a gradient to 0.2 M HCl. Fractions containing SAM are identified with HPLC analysis, pooled, and lyophilized.
Protocol for synthesizing SAM
Prepare the following buffers using cold water: loading buffer – 1 mM sodium acetate (pH 5); elution buffer – 200 mM HCl.
Set up the synthesis reaction in a reaction containing 0.1 M Tris-HCl (pH 8), 50 mM KCl, 1 mM EDTA, 20% acetonitrile (v/v), 26 mM magnesium chloride, 13 mM ATP, 10 mM Met, and 12 mL of cell lysate, in a total volume of 0.1 L.
Incubate for 5 h with stirring at room temperature.
To quench the reaction add 12.1 M HCl dropwise to the reaction till the pH decreases to 5.
Place the mixture on ice for 15 min.
Transfer the reaction to a centrifuge tube and centrifuge at 26,500 xg for 30 min at 4°C.
Transfer the clarified reaction to a 1 L beaker and add 0.9 L of cold loading buffer.
Manually pack a column (5 × 18 cm) containing CM-52 column resin (Whatman).
Equilibrate a column (5 × 18 cm) containing CM-52 resin (Whatman) column with loading buffer using a AKTA FPLC.
Load the solution from step 7 onto the equilibrated column. Wash the column with the loading buffer until the absorbance returns to baseline using a AKTA FPLC.
Elute SAM material with a gradient to 100% elution buffer over 0.8 L.
We typically determine fractions containing SAM by HPLC analysis as follows. A 50 µL aliquot from each fraction is injected onto a Agilent 1100 HPLC. The fractions are analyzed using a Eclipse XDB-C18 reverse phase column (250 X 2.6 mm) equilibrated in 0.1% TFA:water (v/v) with a gradient to 10% acetonitrile:0.1% TFA (v/v) over 20 min with a flow rate of 0.75 mL/min with monitoring at 260 nm.
Pool fractions containing pure SAM and lyophilize.
Resuspend the lyophilized product in a minimal volume of water and determine concentration by UV-vis spectroscopy, using the extinction coefficient of adenosine at 260 nm (15.4 mM−1cm−1).
9. Assay conditions for TYW1
The conversion of m1G to imG-14 catalyzed by TYW1 is best monitored by LC-MS. We typically include methyl viologen in the assays since early experiments on the system indicated that it appears to be required in vitro to mediate reduction of the protein with dithionite(Young et al, 2011).
The large size of the substrate makes direct detection of the modification difficult. However, the RNA can be extracted, digested to the nucleoside level by treating with P1 nuclease, phosphodiesterase, and alkaline phosphatase (Miles, McCarty, Molnar & Bandarian, 2011; Pomerantz & McCloskey, 1990). The nucleosides are separated on a C-18 HPLC column and detected by in-line mass spectrometer. The modified base is often present in concentrations that are far below the canonical base. However, it is readily observed by extracted ion analysis of the mass spectral data.
There are two different methods that can be used to provide substrate for TYW1. One of the methods uses tRNA produced via in vitro transcription, which is then modified to the m1G substrate by TRM5. The other method uses tRNA purified from a S. cerevisiae TYW1 deletion strain, which contains all the normal modifications found in tRNAPhe in S. cerevisiae, except for wybutosine. The protocol for both and subsequent analysis via LC-MS are described below.
9.1. Assay using in vitro transcript as substrate
The tRNA substrate prepared via in vitro transcription contains only the four standard RNA bases. In order to be a substrate for TYW1, G37 needs to be methylated using a SAM dependent methyltransferase. TRM5 is employed to do this and can be used to make the substrate in situ in the presence of TYW1 and all the other components required for the assay.
Protocol for TYW1 assay using in vitro transcript substrate
All steps should be carried out in an anaerobic chamber. We carry out the assays in the same chambers used to purify the protein described above.
All reagents should be dissolved in oxygen free water when possible. Typical assay should mixtures contain 0.1 M Tris-HCl (pH 8), 0.1 M KCl, 4 mM DTT, 2 mM MgCl2, 2 mM SAM, 20 µM tRNA, 20 µM TRM5, 1.5 mM methyl viologen, 10 mM sodium dithionite, 2 mM pyruvate, and 20 µM TYW1.
Incubate the reactions mixtures at 50 °C for desired period of time.
When the incubation time is over, the assays can be either frozen or the RNA can be extracted using the procedure described in Section 9.3.
9.2. Assay using S. cerevisiae tRNA as substrate
The other source of substrate is from a S. cerevisiae knockout strain. In this assay all the soluble RNA extracted from a S. cerevisiae YPL207w (TYW1) deletion strain is incubated in the presence of TYW1, reductant etc. Following incubation the RNA is extracted and digested to the nucleoside level. The high level of RNA in the assay causes there to be a high background of nucleotides when they are analyzed via LC-MS, however imG-14 is easily detected by looking at the extracted ion chromatograms at m/z 322.
Protocol for TYW1 assay using S. cerevisiae tRNA
All steps should be carried out in an anaerobic chamber.
All reagents should be dissolved in oxygen free water when possible. Typical assay should mixtures contain 0.1 M Tris-HCl (pH 8), 4 mM DTT, 1.5 mM methyl viologen, 10 mM sodium dithionite, 2 mM SAM, 2 mM pyruvate, 2 µg yeast tRNA, and 20 µM TYW1.
Incubate the reactions mixtures at 50 °C for desired period of time.
When the incubation time is over, the assays can be either frozen or the RNA can be extracted using the procedure described in Section 8.3.
9.3. Extraction of nucleic acids from reaction mixtures
In order to analyze the product made in a TYW1 reaction the RNA is extracted from the reaction mixture using Qiazol, HiBind RNA minicolumns, and other Qiagen buffers. In this procedure the RNA is extracted using Qiazol and chloroform, followed by an ethanol precipitation. The precipitating RNA is loaded onto a HiBind RNA minicolumn and subsequently washed. Finally, purified RNA is eluted using DEPC treated water, which can then be analyzed or used as required.
Protocol for RNA Extraction
Add 250 µL of Qiazol (Qiagen) to reaction and vortex.
Add 50 µL of chloroform to reaction and vortex.
Centrifuge for 5 min at 21,000 xg at room temperature.
Extract upper clear layer and combine with 400 µL of cold (−20°C) ethanol in a new tube.
Transfer contents of tube (including precipitate if present) to a HiBind RNA minicolumn (Omega bio-tek) inside a collection tube.
Centrifuge at 21,000 xg for 1 minute at room temperature.
Discard flow through in collection tube and add 700 µL RWT buffer (Qiagen) to the minicolumn.
Centrifuge at 21,000 xg for 1 min at room temperature.
Discard flow through in collection tube and add 500 µL RPE buffer (Qiagen) to the minicolumn.
Centrifuge at 21,000 xg for 1 min at room temperature.
Discard flow through in collection tube and add 500 µL RPE buffer (Qiagen) to the minicolumn.
Centrifuge at 21,000 xg for 1 min at room temperature.
Discard flow through in collection tube and centrifuge empty minicolumn for 2 min at 21,000 xg at room temperature.
Discard collection tube and transfer minicolumn to a clean 1.5 mL tube. Add 50 µL of DEPC-treated water to the minicolumn and incubate at room temperature for 5 min.
Centrifuge at 21,000 xg for 1 min at room temperature.
Discard minicolumn and store the RNA sample at −20°C until needed.
9.4. Digestion of RNA to the nucleoside level
In order to detect imG-14 it is necessary to digest the RNA to the nucleoside level, creating a sample that can be analyzed on a C18 column. P1 nuclease and phosphodiesterase I are used to cleave the RNA backbone to form mononucleotides. Alkaline phosphatase is then used to dephosphorylate the mononucleotides, forming nucleosides that are detected via reverse phase chromatography coupled with mass spectrometry. This protocol is adapted from that of Crain (Crain, 1990).
Protocol for Digestion to the Nucleoside level
Prepare the following buffers: P1 buffer – 0.1 M ammonium acetate (pH 5.3); PD buffer – 1 M ammonium bicarbonate; AP buffer – 10 mM Tris-HCl (pH 7.5) containing 10 mM MgCl2.
Add 5 µL of P1 buffer to 50 µL of sample.
Add 1 µL of P1 nuclease (from Penicillium citrinum) (Fisher) (1.4 U/µL) to the reaction.
Incubate for 6 h at 45 °C.
Add 5 µL of PD buffer to the sample.
Add 3 µL of PD (from Crotalus adamanteus) (Sigma) (2 U/µL) to the reaction.
Incubate for 2 h at 37 °C.
Add 100 µL of AP buffer to the sample.
Add 3 µL of fast AP (Thermo) (150 U/µL) to the sample.
Incubate for 2 hours at 37 °C.
Centrifuge the reaction for 20 min at 8,000 xg with a YM-10 centrifugal concentrator to remove enzymes.
9.5. LC-MS analysis of imG-14
Nucleosides are analyzed using reverse phase chromatography (c18) on a Thermo Vanquish UHPLC interfaced with a Thermo LTQ OrbiTrap XL. The absorbance is monitored at 260 nm, with the mass range m/z 250–350 scanned. The mobile phase consists of a volatile ammonium acetate buffer, along with acetonitrile.
Protocol for analyzing samples via LC-MS
Prepare the following buffers using Optima grade (Fisher) water and acetonitrile: Buffer A – 50 mM ammonium acetate (pH 5); Buffer B – 40% acetonitrile (v/v), 60% water (v/v).
We typically separate analytes at 0.2 mL/min on a Thermo Vanquish UHPLC using a Thermo Hypersil gold C18 column (150 × 2.1 mm) 1.9 µ particle size. The UHPLC is interfaced with a Thermo LTQ OrbiTrap XL.
Preequilibrate the C18 column in Buffer A. Set up the following separation program: 0–3 min, 0% buffer B; 3–3.25 min, 0–0.2% buffer B; 3.25–3.5 min, 0.2–0.8% buffer B; 3.5–3.75 min, 0.8–3.2% buffer B; 3.75–4 min, 3.2–5% buffer B; 4–7 min, 5–25% buffer B; 7–10 min, 25–50% buffer B; 10–12 min, 50–75% buffer B; 12–12.1 min, 75–100% buffer B; 12.1–15 min, 100% buffer B; 15–15.1 min, 100–0% buffer B; 15.1–18 min, 0% buffer B.
The LTQ OrbiTrap XL is operated in positive ion mode with the FT analyzer set to 100,000 resolution. Typical settings are: capillary temperature 275 °C, sheath gas flow 35, auxiliary gas flow 12, and a source voltage of 3 kV.
A 50 µL aliquot of the sample is typically injected.
Under these conditions, TYW1 elutes at 12 min and is detected in extracted ion chromatogram at m/z 322. A representative chromatogram using S. cerevisiae RNA as substrate is shown in Fig. 9.
Figure 9. The UV-vis and extracted ion chromatogram from a typical TYW1 assay.
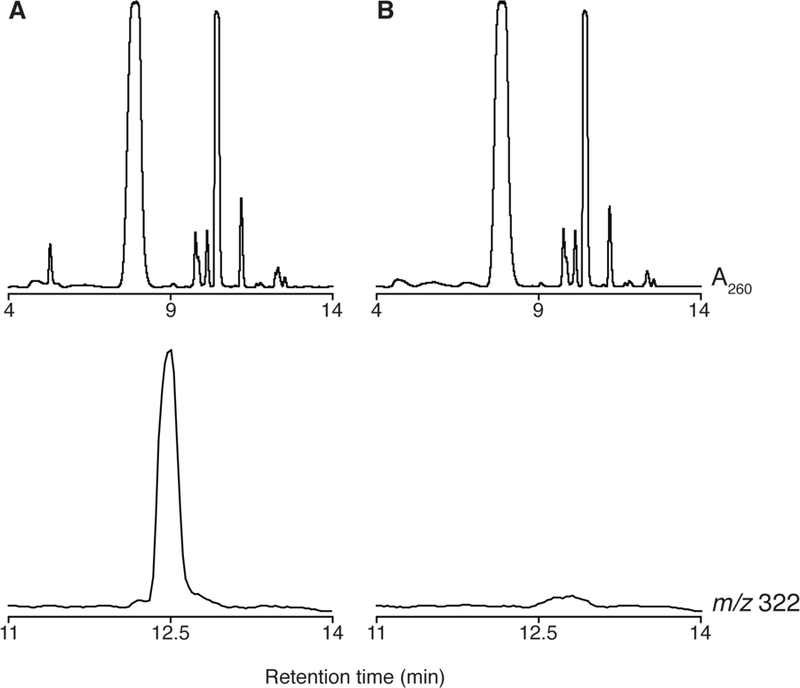
(A) The UV-vis absorbance trace at 260 nm (upper trace) and the extracted ion chromatogram at m/z 322 (lower trace) obtained when all components of the reaction are present. (B) UV-vis absorbance trace at 260 nm (upper trace) and the extracted ion chromatogram at m/z 322 (lower trace) when TYW1 is not present in the reaction. ImG-14 elutes at 12.5 minutes and is only observed when all reaction components are present.
10. Conclusions
The discovery of pyruvate as the source of the two carbon atoms that are required for the biosynthesis of the imidazoline ring of imG-14 has made it possible for mechanistic studies of this TYW1. The outline of the reaction has now been established by stable isotope studies with pyruvate (Young et al, 2011; Young et al, 2015). The methods that are described in this chapter should allow detailed studies aimed at understanding the mechanism of this complex radical-mediated transformation.
Acknowledgments
This work was supported by the National Institutes of Health (GM072623 and GM120638).
Abbreviations:
- AmSO4
ammonium sulfate
- Amp
ampicillin
- ATP
adenosine 5’-triphosphate
- dAdo•
5’-deoxyadenosyl radical
- DEPC
diethylpyrocarbonate
- DTT
dithiothrietol
- EDTA
ethylenediaminetetraacetic acid
- imG-14
4-demethylwyosine
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- Kan
kanamycin
- Kpi
potassium phosphate
- LB
lysogeny broth
- m1G
N-methylguanosine
- PIPES
piperazine-N,N’-bis(2-ethanesulfonic acid)
- PMSF
phenylmethane sulfonyl fluoride
- RS
radical SAM
- SAM
S-adenosyl-l-methionine
- tRNA
transfer RNA
Contributor Information
Anthony P. Young, University of Utah, Department of Chemistry, 315 S 1400 E, Salt Lake City, UT 84112
Vahe Bandarian, University of Utah, Department of Chemistry, 315 S 1400 E, Salt Lake City, UT 84112.
References
- Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, & Drennan CL (2004). Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science, 303, 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk GR, Jacobsson K, Nilsson K, Johansson MJO, Byström AS, & Persson OP (2001). A primordial tRNA modification required for the evolution of life? EMBO J, 20, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobstein SH, Grunberger D, Weinstein IB, & Nakanishi K (1973). Isolation and structure determination of the fluorescent base from bovine liver phenylalanine transfer ribonucleic acid. Biochemistry, 12, 188–193. [DOI] [PubMed] [Google Scholar]
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, et al. (2011). The RNA modification database, RNAMDB: 2011 update. Nucl. Acids Res, 39, D195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington JC, & Dougherty WG (1988). A viral cleavage site cassette: identification of amino acid sequences required for tobacco etch virus polyprotein processing. Proc. Natl. Acad. Sci. U.S.A, 85, 3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain PF (1990). Preparation and enzymatic hydrolysis of DNA and RNA for mass spectrometry. Methods in Enzymology, 193, 782–790. [DOI] [PubMed] [Google Scholar]
- de Crécy-Lagard V, Brochier-Armanet C, Urbonavičius J, Fernandez B, Phillips G, Lyons B, et al. (2010). Biosynthesis of wyosine derivatives in tRNA: an ancient and highly diverse pathway in archaea. Mol. Biol. Evol, 27, 2062–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demick JM, & Lanzilotta WN (2011). Radical SAM activation of the B12-independent glycerol dehydratase results in formation of 5’-deoxy-5’-(methylthio)adenosine and not 5’-deoxyadenosine. Biochemistry, 50, 440–442. [DOI] [PubMed] [Google Scholar]
- Dowling DP, Bruender NA, Young AP, McCarty RM, Bandarian V, & Drennan CL (2013). Radical SAM enzyme QueE defines a new minimal core fold and metal-dependent mechanism. Nature Chemical Biology, 10, 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droogmans L, & Grosjean H (1987). Enzymatic conversion of guanosine 3’ adjacent to the anticodon of yeast tRNAPhe to N1-methylguanosine and the wye nucleoside: dependence on the anticodon sequence. EMBO J, 6, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ehrenstein G (1967). Isolation of sRNA from intact Escherichia coli cells. Methods in Enzymology, 12, 588–596. [Google Scholar]
- Frazzon J, Fick JR, & Dean DR (2002). Biosynthesis of iron-sulphur clusters is a complex and highly conserved process. Biochem. Soc. Trans, 30, 680–685. [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ishii R, Ito T, Shibata R, Fusatomi E, Sekine SI, et al. (2007). Structure of an archaeal TYW1, the enzyme catalyzing the second step of wye-base biosynthesis. Acta Crystallogr. D Biol. Crystallogr, 63, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Iwig DF, & Booker SJ (2004). Insight into the polar reactivity of the onium chalcogen analogues of S-adenosyl-l-methionine. Biochemistry, 43, 13496–13509. [DOI] [PubMed] [Google Scholar]
- Kapust RB, Tözsér J, Copeland TD, & Waugh DS (2002). The P1’ specificity of tobacco etch virus protease. Biochemical and Biophysical Research Communications, 294, 949–955. [DOI] [PubMed] [Google Scholar]
- Kasai H, Goto M, Ikeda K, Zama M, Mizuno Y, Takemura S, et al. (1976). Structure of wye (Yt base) and wyosine (Yt) from Torulopsis utilis phenylalanine transfer ribonucleic acid. Biochemistry, 15, 898–904. [DOI] [PubMed] [Google Scholar]
- Kasai H, Yamaizumi Z, Kuchino Y, & Nishimura S (1979). Isolation of hydroxyl-Y base from rat liver tRNAPhe. Nucl. Acids Res, 6, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathirvelu V, Perche-Letuvée P, Latour J-M, Atta M, Forouhar F, Gambarelli S, et al. (2017). Spectroscopic evidence for cofactor-substrate interaction in the radical-SAM enzyme TYW1. Dalton Trans, 11, 134–139. [DOI] [PubMed] [Google Scholar]
- Kato M, Araiso Y, Noma A, Nagao A, Suzuki T, Ishitani R, et al. (2011). Crystal structure of a novel JmjC-domain-containing protein, TYW5 involved in tRNA modification. Nucl. Acids Res, 39, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, McCarty RM, Ogasawara Y, Liu Y-N, Mansoorabadi SO, LeVieux J, et al. (2013). GenK-catalyzed C-6′ methylation in the biosynthesis of gentamicin: isolation and characterization of a cobalamin-dependent radical SAM enzyme. J. Am. Chem. Soc, 135, 8093–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HJ, Nakanishi K, Grunberger D, & Weinstein IB (1973). Biosynthetic studies of the Y base in yeast phenylalanine tRNA. Incorporation of guanine. Biochemical and Biophysical Research Communications, 55, 818–823. [DOI] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, et al. (2013). MODOMICS: a databse of RNA modification pathways-2013 update. Nucl. Acids Res, 41, D262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta N, Fedoseyenko D, Dairi T, & Begley TP (2013). Menaquinone biosynthesis: formation of aminofutalosine requires a unique radical SAM enzyme. J. Am. Chem. Soc, 135, 15318–15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham GD, Hafner EW, Tabor CW, & Tabor H (1980). S-adenosylmethionine synthetase from Escherichia coli. J. Biol. Chem, 256, 9082–9092. [PubMed] [Google Scholar]
- McCloskey JA, Crain PF, Edmonds CG, Gupta R, Hashizume T, Phillipson DW, et al. (1987). Structure determination of a new fluorescent tricyclic nucleoside from archaebacterial tRNA. Nucl. Acids Res, 15, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles ZD, McCarty RM, Molnar G, & Bandarian V (2011). Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proc. Natl. Acad. Sci. U.S.A, 108, 7368–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Kirino Y, Ikeuchi Y, & Suzuki T (2006). Biosynthesis of wybutosine, a hyper-modified nucleoside in eukaryotic phenylalanine tRNA. EMBO J, 25, 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Ishitani R, Kato M, Nagao A, Nureki O, & Suzuki T (2010). Expanding role of the jumonji C domain as an RNA hydroxylase. J. Biol. Chem, 285, 34503–34507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perche-Letuvée P, Kathirvelu V, Berggren G, Clemancey M, Latour J-M, Maurel V, et al. (2012). 4-Demethylwyosine synthase from Pyrococcus abyssi is a radical S-adenosyl-l-methionine enzyme with an additional [4Fe-4S]+2 cluster that interacts with the pyruvate co-substrate. J. Biol. Chem, 287, 41174–41185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz SC, & McCloskey JA (1990). Analysis of RNA hydrolyzates by liquid chromatography-mass spectrometry. Methods in Enzymology, 193, 796–824. [DOI] [PubMed] [Google Scholar]
- RajBhandary UL, Chang SH, Stuart A, Faulkner RD, Hoskinson RM, & Khorana HG (1967). Studies on polynucleotides, LXVIII. The primary structure of yeast phenylalanine transfer RNA. Proc. Natl. Acad. Sci. U.S.A, 57, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RajBhandary UL, Stuart A, Hoskinson RM, & Khorana HG (1968). Studies on polynucleotides, LXXVIII. Yeast phenylalanine transfer ribonucleic acid: terminal sequences. J. Biol. Chem, 214, 565–574. [PubMed] [Google Scholar]
- RajBhandary UL, & Chang SH (1968). Studies on polynucleotides LXXXII. Yeast phenylalanine transfer ribonucleic acid: partial digestion with ribonuclease T1 and derivation of the total primary structure. J. Biol. Chem, 243, 598–608. [PubMed] [Google Scholar]
- Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, & Miller NE (2001). Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucl. Acids Res, 29, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Noma A, Suzuki T, Senda M, Senda T, Ishitani R, et al. (2007). Crystal structure of the radical SAM enzyme catalyzing tricyclic modified base formation in tRNA. J. Mol. Biol, 372, 1204–1214. [DOI] [PubMed] [Google Scholar]
- Thoden JB, & Holden HM (2005). The molecular architecture of human N-acetylgalactosamine kinase. J. Biol. Chem, 280, 32784–32791. [DOI] [PubMed] [Google Scholar]
- Umitsu M, Nishimasu H, Noma A, Suzuki T, Ishitani R, & Nureki O (2009). Structural basis of AdoMet-dependent aminocarboxypropyl transfer reaction catalyzed by tRNA-wybutosine synthesizing enzyme, TYW2. Proc. Natl. Acad. Sci. U.S.A, 106, 15616–15621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waas WF, de Crécy-Lagard V, & Schimmel P (2005). Discovery of a gene family critical to wyosine base formation in a subset of phenylalanine-specific transfer RNAs. J. Biol. Chem, 280, 37616–37622. [DOI] [PubMed] [Google Scholar]
- Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, et al. (2002). Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active [4Fe−4S]+ cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc, 124, 3143–3151. [DOI] [PubMed] [Google Scholar]
- Young AP, & Bandarian V (2015). Mechanistic studies of the radical S-Adenosyl-l-methionine enzyme 4-demethylwyosine synthase reveal the site of hydrogen atom abstraction. Biochemistry, 54, 3569–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AP, & Bandarian V (2011). Pyruvate is the source of the two carbons that are required for formation of the imidazoline ring of 4-demethylwyosine. Biochemistry, 50, 10573–10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, et al. (2010). Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme. Nature, 465, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]