

Abstract
Decades of research reveal that MDM2 participates in cellular processes ranging from macro-molecular metabolism to cancer signaling mechanisms. Two recent studies uncovered a new role for MDM2 in mitochondrial bioenergetics. Through the negative regulation of NDUFS1 (NADH:ubiquinone oxidoreductase 75 kDa Fe-S protein 1) and MT-ND6 (NADH dehydrogenase 6), MDM2 decreases the function and efficiency of Complex I (CI). These observations propose several important questions: (1) Where does MDM2 affect CI activity? (2) What are the cellular consequences of MDM2-mediated regulation of CI? (3) What are the physiological implications of these interactions? Here, we will address these questions and position these observations within the MDM2 literature.
Keywords: apoptosis, bioenergetics, complex I, electron transport chain, MDM2, mitochondria, oncogene, p53, tumor suppressor
Graphical Abstract
1. Introduction
Mitochondria are double membrane organelles responsible for cellular bioenergetics by converting nutrients into energy in the form of adenosine triphosphate (ATP). The outer mitochondrial membrane (OMM) is permeable up to several kilodaltons and allows for equilibration with the cytosol, while the inner mitochondrial membrane (IMM) is impermeable and establishes the electrochemical proton gradient (ΔΨM) necessary for ATP production (Hatefi, 1985; Mitchell, 2011; Saraste, 1999; Smeitink et al., 2001). Central to ATP production is the electron transport chain (ETC), an assembly of five enzymatic complexes within the IMM that convert energy from NADH and/or FADH2 into ATP and H2O via coupled electron (e−) transport and oxidative phosphorylation (OXPHOS) (Figure 2A) (Hatefi, 1985; Letts and Sazanov, 2017; Saraste, 1999; Smeitink et al., 2001). In parallel, and also dependent upon ΔΨM, mitochondria produce metabolic precursors for DNA, protein, and lipid synthesis; and regulate multiple pathways including calcium signaling and reactive oxygen species (ROS) generation.
Figure 2. The electron transport chain mantains mitochondrial respiration and generates the proton-motive gradient.

(A) Schematic diagram of Complexes I-V (CI, CII, CIII, CIV, and CV), which are integrated in the inner mitochondrial membrane (IMM). CI is the entry point for electrons (e−) via the oxidation of NADH to NAD+. These electrons flow through CI and CII to ubiquinone (Q), which is involved in the reduction of oxygen (O2) to free radicals (O2−). CII acts as a secondary electron entry point via the oxidation of FADH2 to FAD+. Collectively, electrons flow from CIII to reduce cytochrome c (Cyto c), which is then utilized by CIV to reduce molecular oxygen (the final electron acceptor) to water (H20). CI, CIII, and CIV generate the proton motive force (ΔψM) by pumping hydrogen ions (H+) from the mitochondrial matrix into the inner mitochondrial space (IMS). In turn, the H+ gradient generated by the electron transport chain (ETC) drives CV (ATPase synthase) to convert adenosine diphosphate and inorganic phosphate (ADP and PO4-, respectively) into ATP. (B) Assembly of CI, CIII, and CIV into supercomplex structures, which promote efficient e− transport, reduced ROS production and optimal ETC capacity.
ATP production requires the biosynthesis, assembly, and catalytic functions of multi-subunit protein complexes that constitute the ETC and ATP synthase. Complexes I, III, IV, and V (CI, CIII, CIV, & CV) are encoded by nuclear and mitochondrial genomes, while Complex II (CII) is exclusively encoded within the nucleus (Letts and Sazanov, 2017; Smeitink et al., 2001; Taanman, 1999). These complexes consist of integral membrane proteins that anchor and support the subunits within the IMM, proteins with intrinsic enzymatic activities to catalyse NADH and FADH2 oxidation, and iron-sulfur (Fe-S) cluster proteins responsible for e movement throughout the complex to eventually reduce e− carriers between the complexes (Figure 2A). The stoichiometric expression and assembly of subunits into large complexes enables efficient e− flow through the ETC, eventually destined for molecular oxygen as the final e− acceptor (Letts and Sazanov, 2017). In addition to e− transport, CI, CIII, and CIV concomitantly pump protons (H+) from the matrix (i.e., the space within the IMM) to the inner membrane space (IMS) to generate ΔΨM, which is utilized by CV (also referred to as ATPase synthase) to convert adenosine diphosphate and inorganic phosphate (ADP and PO4-, respectively) into ATP (Figure 2A) (Letts and Sazanov, 2017).
In addition to the functional and structural subunits that comprise the respiratory complexes within the IMM, there are many essential proteins that assist in the assembly, IMM integration, and maturation of the complexes. For example, CI consists of over forty subunits, which require at least fourteen additional proteins to assemble within the IMM (Letts et al., 2016; Vinothkumar et al., 2014). Of the forty CI subunits, seven are encoded by the mitochondrial genome and translated by mitochondrial ribosomes within the matrix --- with the remaining subunits encoded by the nuclear genome, translated on cytoplasmic ribosomes, and imported into the mitochondrial network (Formosa et al., 2018). Due to the elaborate coordinated nature of multiple genomes and localizations for proteins essential in CI function, the mechanisms of CI assembly remain an active area of investigation; but the current model suggests that CI is assembled through the stoichiometric association of smaller intermediate modules containing core subunits within the IMM (Formosa et al., 2018; Guerrero-Castillo et al., 2017; Lazarou et al., 2007). Given the number of pathways and proteins essential for establishing and maintaining bioenergetics, we likely do not have a clear understanding of all the cellular factors that impact upon individual complex subunits or modules, and how these interactions subsequently alter CI activity and bioenergetics.
Studies from Arena et al. and Elkholi et al. provide new mechanistic insights into how CI assembly and function are regulated by the E3 ubiquitin ligase mouse double minute 2 (MDM2) (Arena et al., 2018; Elkholi et al., 2018). A unifying observation from both studies is that MDM2 negatively regulates CI function. However, these papers describe that MDM2 regulates CI via distinct cellular localizations and mechanisms. Arena et al. offers an interesting insight into MDM2 biology by describing the import of MDM2 into the mitochondrial matrix where it reduces the expression of the mitochondrial encoded gene essential for CI, NADH dehydrogenase 6 (MT-ND6) (Arena et al., 2018). In contrast, Elkholi et al. demonstrates that MDM2 sequesters the CI subunit NADH:ubiquinone oxidoreductase 75 kDa Fe-S protein 1 (NDUFS1) in the cytosol (Elkholi et al., 2018). Both mechanisms result in a reduction of CI activity followed by a decrease in mitochondrial respiration and subsequent increase in ROS. The cellular consequences of MDM2-mediated mitochondrial dysfunction ranges from induction of apoptosis (Elkholi et al., 2018) to increased invasion and migration of cancer cells (Arena et al., 2018). Here, we discuss how these observations fit within the broader scope of MDM2 literature and provide prospective for the role of MDM2 and CI interactions in physiology and disease.
2. MDM2 binding to different components of CI regulates bioenergetics
Wild type MDM2 (MDM2WT) contains several conserved structural domains (Figure 1) that function by regulating MDM2 subcellular localizations and interactions with diverse protein partners (Karni-Schmidt et al., 2016; Nicholson et al., 2014). An example of a highly studied MDM2-interacting protein is the tumor suppressor, p53. This interaction allows for the constitutive degradation of p53 via MDM2’s E3 ubiquitin ligase activity to ensure the absence of p53-dependent effects (e.g., regulation of gene expression, cell cycle arrest, apoptosis, and senescence), which are prevented in the absence of genotoxic stress (Honda et al., 1997). In cancer settings with constitutive genomic stress, p53-dependent effects are also often inherently inhibited by either p53 mutation (i.e., prevents p53-dependent gene expression) or MDM2 amplification, which allows for hyper-constitutive p53 sequestration to block it’s tumor suppressor activities (Bieging and Attardi, 2012; Momand et al., 1992; Wade et al., 2013). One key development in understanding the biological consequences and pharmacological utility of this pathway arose from the development of MDM2 inhibitors (e.g., Nutlin-3A) which liberate p53 from MDM2 to promote p53 mediated responses (Vassilev et al., 2004). As such, several studies have also utilized Nutlin-3A to investigate the MDM2 protein-protein interaction landscape outside the context of p53.
Figure 1: MDM2 gene and protein structure.

The diversity of MDM2 signaling comes from a complex gene and protein structure. Located on the long arm of chromosome 12, Mdm2 contains twelve exons and two promoters (P1 and P2). MDM2WT consists of 491 amino acids that comprise four functional domains essential to its function. They are the amino-terminal p53 binding domain, a central acidic domain, a zinc finger region, and the RING (Really Interesting New Gene) domain, which contains E3 ubiquitin ligase activity. The amino-terminus is commonly referred to as the p53-binding domain because this is its most characterized interaction, but it is also responsible for binding a variety of other proteins (e.g., E2F1 and p73). Following the amino-terminal domain is a linker region containing both a nuclear localization signal and a nuclear export signal (NLS and NES, respectively) that regulate MDM2 subcellular localization. The central acidic domain negatively regulates the p53-signalling pathway by inducing the ubiquitination of p53 or by inducing a conformational change that inhibits p53’s transcriptional activity (Cross et al., 2011). Finally, the E3 ubiquitin ligase activity of the RING domain is responsible for regulating both subcellular localization and degradation of various MDM2 interacting proteins.
For example, a screen conducted by Nicholson et al. discovered that a subset of Nutlin-3A regulated MDM2 interacting proteins are associated with mitochondrial function, including mitochondrial transporters (e.g., TIMM8A, TIMM13) and the CI subunit, NDUFS1 (Nicholson et al., 2014). Elkholi and colleagues corroborated this study and further linked MDM2 to NDUFS1 by discovering that cytosolic MDM2 binds NDUFS1, leading to its cytosolic retention rather than mitochondrial localization (Figure 3) --- moreover, Nutlin-3A also enhanced the binding of MDM2 to NDUFS1(Elkholi et al., 2018). A consequence of MDM2-mediated sequestration of NDUFS1 is a decrease in mitochondrial respiration and increased ROS production, an effect that can be phenocopied by the genetic silencing of NDUFS1 expression (Elkholi et al., 2018).
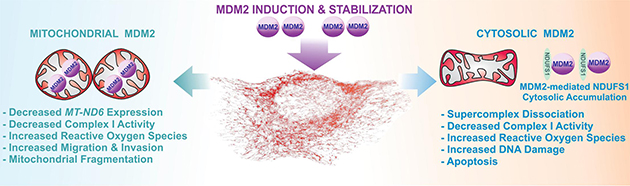
Figure 3. MDM2 over-expression disrupts Complex I activity through two distinct mechanisms.

Mitochondrial MDM2 binds to mitochondrial DNA leading to decreased MT-ND6 expression, Complex I activity, and induced mitochondrial fragmentation and ROS. Conversely, cytosolic MDM2 sequesters NDUFS1 in the cytosol causing supercomplex dissociation, decreased Complex I activity, increased ROS, DNA damage, and apoptosis.
It is also noteworthy that the interaction between MDM2 and NDUFS1 is characteristically akin to the MDM2-p53 interaction (Nicholson et al., 2014). Structural composition analysis comparing the amino termini of p53 and NDUFS1 revealed shared amino acid similarities critical for interacting with MDM2. Pharmacological regulation of MDM2 with its binding proteins can occur either through disruption of the interaction interface (e.g., Nutlin-3A disruption of MDM2 and p53) or by forcing allosteric changes within the MDM2 structure that either enhance or reduce its affinity for interacting proteins; and the enhanced binding of NDUFS1 to MDM2 in the presence of Nutlin-3A appears to support the later (Elkholi et al., 2018). Furthermore, the notion that Nutlin-3A enhances the interaction between MDM2 and NDUFS1 fits with previous observations of Nutlin-3A affecting cell signaling independently of p53 (Fahraeus and Olivares-Illana, 2014; Ye et al., 2017). Taken together, these observations emphasize a broader impact of MDM2 and its pharmacology in cell biology; and more specifically, in CI activity and cellular bioenergetics.
To discuss the potential impact of the MDM2-NDUFS1 interaction, an introduction to NDUFS1 function is necessary. While mechanistic insights into NDUFS1 are limited, NDUFS1 appears to be a critical CI subunit for the entry and efficient transfer of e−, which is based on two observations: (1) NDUFS1 contains three Fe-S clusters that integrate into the redox center in CI and (2) NDUFS1 forms part of the interaction interface between CI and CIII (Lopez-Fabuel et al., 2016; Maio et al., 2017; Wu et al., 2016). The assembly of CI, CIII, and CIV into higher organized structures in the IMM are collectively known as supercomplexes (Figure 2B). According to current opinion, supercomplexes are involved in efficient e− transport, minimized ROS production, stabilization of individual complexes, and optimal respiratory chain capacity (Milenkovic et al., 2017). Ekholi et al. demonstrated that MDM2-mediated decrease in mitochondrial function was not caused by a disassembly of CI itself, but rather through a decrease in CI activity through the dissociation of CI from CIII, suggesting that appropriate import and mitochondrial localization of NDUFS1 are critical to maintain supercomplex assembly and optimal bioenergetic homeostasis (Elkholi et al., 2018). Incidentally, the role of NDUFS1 and supercomplex formation has been compared in primary neuron and astrocyte cultures where bioenergetic differences between these cell types is imposed by NDUFS1-mediated supercomplex assembly (Lopez-Fabuel et al., 2016). Therefore, it stands to reason that MDM2 and Nutlin-3A mediated regulation of NDUFS1 may broadly impact upon a range of energetically demanding cell and tissue types when MDM2 and NDUFS1 are expressed.
Similar to NDUFS1, MT-ND6 is also essential for the assembly and respiratory function of CI (Vartak et al., 2015). Recently, Arena et al. showed that MDM2 can co-localize with transcriptional factor A, mitochondrial (TFAM) in the mitochondrial matrix in a p53-independent manner. In response to ROS production and in hypoxic conditions, MDM2 decreases the binding of TFAM to the light strand promoter (LSP) of the mitochondrial genome, which encodes for MT-ND6, leading to decreased MT-ND6 expression, CI activity, and oxygen consumption (Figure 3) (Arena et al., 2018). Although Arena and colleagues did not investigate if decreased MT-ND6 expression affected the assembly of supercomplexes, other reports have indicated that MT-ND6 is important for the recruitment of ND1–5 into CI, but not necessarily for the assembly of supercomplexes --- however, this later point remains a topic of debate and requires further investigation (Vartak et al., 2015). Collectively, two distinct, but complementary mechanisms are described for MDM2-mediated regulation of CI activity and supercomplex assembly by decreasing MT-ND6 and NDUFS1, respectively.
3. How is MDM2 imported into mitochondria?
Arena and colleagues were the first to demonstrate MDM2 localization to the mitochondrial matrix, an intriguing result considering the majority of literature focuses on the cytosolic and nuclear roles of MDM2. Mitochondria contain nearly two thousand proteins, with the vast majority (>99%) encoded by the nuclear genome, synthesized on cytosolic ribosomes, and imported into the mitochondrial network via a multi-component conserved import machinery (Harbauer et al., 2014; Schmidt et al., 2010). Proteins destined for mitochondrial membranes and matrix are often synthesized as precursors containing a cleavable mitochondrial targeting signal (MTS) within the amino terminus. The MTS is recognized by translocases of the outer membrane (TOM) complex (i.e., TOM20), which facilitate the active import of proteins from the cytosol into the IMS. The translocases of the inner membrane (TIMs, i.e., TIM23) then take control, in a ΔΨM dependent manner, to distribute peptides either laterally across the IMM or completely into the mitochondrial matrix (Harbauer et al., 2014; Schmidt et al., 2010).
As mentioned earlier, MDM2 interacts with multiple mitochondrial transporters (e.g., TIMM8A, TIMM13) in a Nutlin-3A dependent manner (Nicholson et al., 2014), and Arena et al. further expanded upon this interaction repertoire by demonstrating that MDM2 also binds to TOM20 and TIM23 (Arena et al., 2018). Moreover, MDM2 interacts with mitochondrial HSP70, which is potentially required to stabilize mitochondrial MDM2 for efficient binding to mtDNA (Arena et al., 2018). However, analysis of the MDM2 amino acid sequence did not reveal a canonical MTS (Arena et al., 2018), which raises the question of how MDM2 is imported into mitochondria? The authors suggest that mitochondrial import of MDM2 depends on binding to additional MTS-containing proteins (Arena et al., 2018). Curiously, the mechanism for transport of non-MTS containing proteins into mitochondria has been previously proposed for p53 (Ahn et al., 2010). Under hypoxic conditions p53 binds to TID1, a MTS-containing chaperone that assists in the translocation of p53 from the cytosol into the mitochondria to potentially regulate apoptosis. Likewise, MDM2 also interacts with TID1, which gives an indication that MDM2 may share conserved mechanisms (Arena et al., 2018).
Although further analysis is necessary to investigate the regulatory mechanisms of MDM2 import into mitochondria, the presence of MDM2 in different subcellular compartments (i.e., mitochondria, cytosol, and nucleus) and the differing means of regulating CI activity is one of the most interesting observations arising from the Elkholi and Arena studies. These mechanistic differences suggest MDM2 has multiple levels of control over mitochondrial function and bioenergetics in order to assure cellular homeostasis.
4. MDM2 alters mitochondrial dynamics
The above findings position MDM2 into an unexpected mechanism controlling a central core of mitochondrial bioenergetics; yet diverse components of mitochondrial biology (e.g., mitochondrial dynamics, mitochondrial quality control) impact upon bioenergetics, and links between MDM2 and these pathways remain scant. However, MDM2 and mitochondrial biology were recently united via the regulation of mitochondrial morphology (Arena et al., 2018). The mitochondrial network undergoes regulated fusion and fission, which are essential for maintaining mitochondrial homogeneity (e.g., protein, lipid, and nucleoid distribution) within a cell, and the same dynamics also influence ETC function and assembly into supercomplexes (Cogliati et al., 2013). Moreover, reciprocal regulation is also observed as blocking CI function (e.g., pharmacological inhibition with rotenone) affects mitochondrial morphology (Benard et al., 2007).
Mitochondrial fusion is regulated by Mitofusin 1 and 2 (MFN1 & MFN2), whereas fission is mainly mediated by dynamin related protein 1 (DRP1) (Trotta and Chipuk, 2017). Interestingly, MDM2 depletion results in an increase in DRP1 phosphorylation on serine 637 (Arena et al., 2018), a post-translational modification described to inactivate DRP1 (Chang and Blackstone, 2007; Cribbs and Strack, 2007; Trotta and Chipuk, 2017). Nevertheless, the increase in the phosphorylation at serine 637 correlated with an increase in mitochondrial fission --- and not fusion --- an observation that requires further clarification and corroboration using different molecular and cellular tools (Arena et al., 2018). In contrast, over-expression of mitochondrially-localized MDM2 resulted in altered cristae morphology and increased perinuclear clustering of mitochondria --- all of which are indicative that MDM2 may induce mitochondrial fusion and impact on cellular REDOX, potentially linking to CI dysfunction. Indeed, the effects of MDM2 on mitochondrial ROS production can be mitigated by either treating with mitoquinone mesylate (MitoQ) or expression of superoxide dismutase 1 (SOD1) (Elkholi et al., 2018). MDM2-mediated changes to mitochondrial structure could be a response to enforce mitochondrial quality control. For example, mitochondrial fission typically preceds the induction of mitophagy, a form of autophagy that selectively removes dysfunctional mitochondria from the cell (Pickles et al., 2018). Although a direct link between MDM2 and mitochondrial quality control remains unknown, mitochondrial bioenergetics and mitochondrial dynamics mutually influence each other and are regulated by common pathways that include MDM2 signaling. Certainly more investigations into these intersections are critical to fully appreciate the impact of MDM2 on mitochondrial biology.
5. MDM2: a pro-apoptotic and pro-survival signal?
Aside from the bioenergetic and metabolic functions governed by mitochondria, these organelles are also the central mediators and targets of pro-apoptotic signals (Luna-Vargas and Chipuk, 2016). In order for cells to commit themselves to the mitochondrial pathway of apoptosis, a cohort of B-cell lymphoma 2 (BCL-2) proteins must functionally collaborate at the OMM. This activates a process referred to as mitochondrial outer membrane permeabilization (MOMP), which leads to the release of IMS proteins (i.e., cytochrome c, SMAC/Diablo) into the cytoplasm, which induces caspase activation and subsequent commitment to death. The pro-apoptotic proteins BCL-2 antagonist killer (BAK) and BCL-2-associated X (BAX) are responsible for initiating MOMP, however they require interactions with the pro-apoptotic BH3-only proteins (i.e., BIM and BID) to activate and permeabilize the OMM. The anti-apoptotic BCL-2 proteins (e.g., BCL-2, BCL-XL, BCL-W, & MCL-1) preserve OMM integrity by directly inhibiting the pro-apoptotic BCL-2 proteins (Luna-Vargas and Chipuk, 2016).
MDM2-mediated sequestration of NDUFS1 and subsequent CI efficiency is described to cause an accumulation of oxidative stress often leading to apoptosis (Elkholi et al., 2018). An apoptotic program consisting of BIM and BAX are described to promote MDM2-initiated cell death, and indeed molecular studies have previously described a specific role of BIM-mediated BAX activation (Sarosiek et al., 2013). MDM2-induced apoptosis appears to be initiated by the mitochondrial accumulation of BIM-S, the shorter form of BIM, yet the signals linking oxidative stress to BIM-S accumulation remain unknown, but evidence suggests that post-translational mechanisms are likely responsible (Elkholi et al., 2018). As expected, apoptosis induced by MDM2 is also tightly controlled by the anti-apoptotic BCL-2 proteins (e.g., BCL-xL and MCL-1). Collectively, these data are indicative of the mitochondrial pathway of apoptosis being activated following MDM2 overexpression, potentially due to oxidative stress, but what additional cellular components contribute to this response?
Exogenous MDM2 induces a marked production of ROS, which correlates with DNA damage (i.e., measured through the accumulation of nuclear ɣH2AX foci and chromatid breaks), and co-treatment with the antioxidant enzyme superoxide dismutase (SOD1) inhibited MDM2-induced apoptosis (Elkholi et al., 2018). The likely source underlying ROS accumulation and apoptosis is likely mediated by MDM2’s sequestration of NDUFS1. Indeed, RNAi of NDUFS1 phenocopied many of the cell stress and death effects induced by MDM2. Linking the direct MDM2-NDUFS1 interaction to apoptosis, Elkholi and colleagues discovered a glycine residue at position 58 (MDM2G58) that is essential for the interaction with NDUFS1 and apoptosis (Elkholi et al 2018). Mutation of this glycine residue to isoleucine (MDM2G58I) reduced MDM2 binding to NDUFS1, ROS production, and apoptosis compared to MDM2WT (Elkholi et al., 2018).
A consequence of supercomplex disassembly and loss of CI activity is inefficient e− transport and accumulation of ROS (Lopez-Fabuel et al., 2016), which appears to be sufficient to provoke DNA damage and apoptosis. In contrast, mitochondrial-targeted MDM2 variants investigated by Arena et al do not induce apoptosis, despite using the same lung cancer cell line (H1299) and observing decreased CI activity and increased ROS levels. Perhaps in this setting, MDM2 mediated decreases in CI are useful to reduce cellular bioenergetics to preserve key nutrients while transiently disrupting REDOX. This discrepancy may also be partially explained by differences in the amount of MDM2 expressed, timing of experiments (e.g., Elkholi et al. notes that MDM2-induced apoptosis requires at least two days), or culture conditions impacting upon MDM2 and NDUFS1 expression and localization. Indeed, cytosolic localization of MDM2 is critical for apoptosis, not nuclear MDM2, yet this observation does not exclude a role for nuclear MDM2 in stress signaling or cell death commitment. Taken together, the contrasting yet complementary observations reveal that additional investigations are necessary to understand how signaling and setting influence MDM2-mediated cellular phenotypes. As the majority of observations are based on exogenous expression of MDM2, more clear insights may come from studies that investigate which physiological scenarios promote MDM2’s regulation of bioenergetics.
6. Physiological impact of MDM2-mediated CI regulation
The development of Mdm2 transgenic animal models have been vital to understanding the significance of MDM2 in normal physiology and cancer settings. The first literature describing genetic regulation of Mdm2 expression in vivo was the creation of Mdm2 null mice, which exhibit embryonic lethality, unless trp53 is conjointly deleted (Itahana et al., 2007; Jones et al., 1995; Montes de Oca Luna et al., 1995). Interestingly, this lethality phenotype was also observed in a murine model where the MDM2 E3 ligase activity was disrupted by mutating the MDM2 RING domain (MDM2C462A), indicating that constitutive MDM2-p53 regulation is critical for murine development (Itahana et al., 2007).
Around the same time that Mdm2 null mice were created, fundamental observations across a wide variety of human tumor specimens established that MDM2 was frequently amplified, suggesting an oncogenic function (Biernat et al., 1997; Higashiyama et al., 1997; Jones et al., 1998; Leite et al., 2001; McCann et al., 1995; Nakayama et al., 1995). However, initial gain-of-function mouse models were challenging to generate as mice expressing Mdm2 two-fold over physiological levels succumbed to lethality in utero, suggesting a tumor suppressor (potentially pro-apoptotic) role (Jones et al., 1998). Using conditional alleles allowing for specific expression in breast epithelial cells of WT and trp53 null mice, Mdm2 exhibited growth inhibitory characteristics: including induction of S-phase cell cycle arrest, increased polyploidy, and abnormal mammary gland development --- indicating that MDM2 has tumor suppressor activity that was not fully dependent upon trp53 (Alkhalaf et al., 1999).
Recent findings point to an evolutionary conserved function of MDM2 to regulate apoptosis, potentially through CI, in Drosophila melanogaster (Elkholi et al., 2018). This in vivo model offers an interesting alternative to Mdm2 mouse models for three main reasons: (1) components of cell stress and caspase-dependent cell death pathways are conserved between Drosophila and mammals (Tittel and Steller, 2000), (2) the Drosophila p53 pathway has no known MDM2 homolog, and (3) Drosophila p53 is not regulated by mammalian MDM2 (Folberg-Blum et al., 2002). Specific overexpression of MDM2 in either the eye or imaginal wing disc, resulted in increased mitochondrial ROS and DNA damage. This oxidative stress resulted in apoptotic cell death and tissue ablation in an MDM2 dose-dependent manner. All of these apoptotic phenotypes can be effectively reversed in flies genetically engineered to disable cellular commitment to apoptosis (i.e., deletion of reaper/grim/hid prevent apoptosis from initiating or crossing with DIAP1 or p35 transgenic animals to suppress caspase activation). Interestingly, many of the transgenic MDM2 effects observed were phenocopied when the Drosophila homologue of NDUFS1 (i.e., ND-75) was silenced by RNAi, reinforcing the direct links between MDM2, mitochondrial biology, and apoptosis (Elkholi et al., 2018). These results are particularly interesting to consider because MDM2 has no effect on yeast mitochondrial bioenergetics, probably because NADH oxidase activity is mediated by a single protein, NDI1 (Marres et al., 1991). These observations potentially suggest that the evolution of CI into a multi-subunit, highly regulated assembled holoenzyme may have occurred simultaneously with a series of regulatory proteins (i.e, MDM2) as a mean of adding further complexity to bioenergetics.
In stark contrast to MDM2’s ability to induce cell death is a large body of literature describing its potent oncogenic function. Transgenic Mdm2 mouse models expressing four-fold over normal levels developed tumors, with a tumor spectrum that differs from trp53 null mice (Jones et al., 1998). Moreover, mice that express two copies of the Mdm2 gene have a greater tumor burden compared with mice that only express one copy of the transgene, indicating a dose-dependent susceptibility to MDM2 over-expression (Jones et al., 1998). Indeed, MDM2 over-expression has been correlated with poorer patient prognosis (Reifenberger et al 1993, Matsumura et al 1996). Interestingly, several tumor types (e.g., liposarcoma) are characterized by the amplification of MDM2, decreased mitochondrial function, and high glycolytic rates (Nakayama et al., 1995), suggesting potential mechanistic links between MDM2 overexpression and metabolic reprogramming (i.e., the Warburg effect). Indeed, sarcoma cells are sensitive to glycolytic inhibitors like 2-deoxy-D-glucose, but not CI inhibitors, such as metformin (Issaq et al., 2014). From these findings one can speculate that MDM2 amplification in sarcoma results in mitochondrial dysfunction through decreased supercomplex formation and CI activity, which contributes changes to cellular metabolism and sensitivity to CI inhibitors. Similarly, mitochondrial localization of MDM2 promotes tumor progression by increasing the migration and invasion capacity of lung cancer cells in vitro and in vivo (Arena et al., 2018). These cellular changes are also accompanied by an increase in the expression of genes that control epithelial to mesenchymal transition, a key phenotype of metastasising cancer cells. Intriguely, many cancer cells contain loss-of-function mutations within MT-ND6 that concomitantly occur with increased expression of metastasis genes (Koshikawa et al., 2017; Yuan et al., 2015). For example, primary lung cancer cytoplasmic hybrid (i.e., cybrid) cells expressing nonsense or missense MT-ND6 mutations possessed higher migratory and invasion capacity compared to wild type MT-ND6 cells (Yuan et al., 2015). Based on these observations it appears that inhibition of MT-ND6 expression, CI activity, and decreased mitochondrial activity, either through mitochondrial MDM2 or acquisition of MT-ND6 mutations is sufficient to alter gene expression profiles to promote tumor metastasis.
Beyond the effects mitochondrial MDM2 has on tumorigenesis, Arena et al. also provide interesting mechanistic insight into how MDM2 over-expression disrupts normal cellular homeostasis. For example, the relevance of the MDM2-mediated regulation of CI was analysed in vivo in murine skeletal muscle cells (Arena et al., 2018). Under hypoxic conditions, MDM2 levels were increased in the mitochondria and this correlated with a decrease in MT-ND6 levels. When Mdm2 was silenced in striated skeletal muscles from trp53 null mice, MT-ND6 expression and subsequent CI activity increased under normoxia and hypoxia conditions compared to mice expressing Mdm2. Notably, the physical endurance of Mdm2 knockout mice under hypoxic conditions was higher when compared to control mice, suggesting that mitochondrial MDM2 may be a key regulator of the energetic output of skeletal muscles, which may impact on cellular survival in these cell types. Altogether, the regulation of CI by MDM2 has broad physiological effects in different tissues and cell types that range from the control of bioenergetic requirements of highly activity tissues to the regulation of cell death and survival of cancer cells.
7. Future directions
Two recent articles have shown that MDM2 regulates CI, revealing a new field of research in the complex interplay between mitochondrial bioenergetics and the MDM2 pathway in physiology and disease. The identification and characterization of new MDM2 interacting partners affecting mitochondrial function propose multiple areas that require further attention. There is evidence suggesting MDM2 enters mitochondria under hypoxic conditions to affect MT-ND6 expression, but the molecular mechanisms underlying MDM2 transport into mitochondria remain enigmatic. It has been suggested to occur through mitochondrial transporters, but because MDM2 has no MTS, further analysis is required to completely elucidate this mechanism. Similarly, the cellular conditions and upstream signals that promote the interaction between MDM2 and NDUFS1 remain undefined. It will be interesting to evaluate the MDM2-NDUFS1 interaction and the potential role of MDM2-mediated regulation of CI in pathological conditions where mitochondrial respiration is compromised, such as additional cancers and neuropathies. Also, a broader understanding of MDM2-mitochondrial signaling axes is necessary to uncover the potential clinical applications of MDM2 modulation in disease.
Acknowledgements
We thank everyone in the Chipuk Laboratory and the Department of Oncological Sciences. We thank Joshua R. Kaminetsky for mitochondrial imaging. This work was supported by: NIH grants R01 CA157740 (J.E.C.) and R01 CA206005 (J.E.C.); the JJR Foundation, the William A. Spivak Fund, the Fridolin Charitable Trust, an American Cancer Society Research Scholar Award, a Leukemia & Lymphoma Society Career Development Award, and an Irma T. Hirschl / Monique Weill- Caulier Trust Research Award. This work was also supported in part by two research grants (5-FY11–74 and 1-FY13–416) from the March of Dimes Foundation, the Developmental Research Pilot Project Program within the Department of Oncological Sciences at the Icahn School of Medicine at Mount Sinai, and the Tisch Cancer Institute Cancer Center Support Grant (P30 CA196521).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no conflict of interest.
References
- Ahn BY, Trinh DL, Zajchowski LD, Lee B, Elwi AN, and Kim SW (2010). Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene 29, 1155–1166. [DOI] [PubMed] [Google Scholar]
- Alkhalaf M, Ganguli G, Messaddeq N, Le Meur M, and Wasylyk B (1999). MDM2 overexpression generates a skin phenotype in both wild type and p53 null mice. Oncogene 18, 1419–1434. [DOI] [PubMed] [Google Scholar]
- Arena G, Cisse MY, Pyrdziak S, Chatre L, Riscal R, Fuentes M, Arnold JJ, Kastner M, Gayte L, Bertrand-Gaday C, et al. (2018). Mitochondrial MDM2 Regulates Respiratory Complex I Activity Independently of p53. Mol Cell 69, 594–609 e598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, and Rossignol R (2007). Mitochondrial bioenergetics and structural network organization. J Cell Sci 120, 838–848. [DOI] [PubMed] [Google Scholar]
- Bieging KT, and Attardi LD (2012). Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol 22, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernat W, Kleihues P, Yonekawa Y, and Ohgaki H (1997). Amplification and overexpression of MDM2 in primary (de novo) glioblastomas. J Neuropathol Exp Neurol 56, 180–185. [DOI] [PubMed] [Google Scholar]
- Chang CR, and Blackstone C (2007). Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282, 21583–21587. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs JT, and Strack S (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8, 939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross B, Chen L, Cheng Q, Li B, Yuan ZM, and Chen J (2011). Inhibition of p53 DNA binding function by the MDM2 protein acidic domain. J Biol Chem 286, 16018–16029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkholi R, Abraham-Enachescu I, Trotta AP, Rubio-Patiño C, Mohammed JN, Luna-Vargas MP, Gelles JD, Kaminetsky JR, Serasinghe MN, Zou C, et al. (2018). MDM2 integrates cellular respiration and apoptotic signaling through NDUFS1 and the mitochondrial network In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahraeus R, and Olivares-Illana V (2014). MDM2’s social network. Oncogene 33, 4365–4376. [DOI] [PubMed] [Google Scholar]
- Folberg-Blum A, Sapir A, Shilo BZ, and Oren M (2002). Overexpression of mouse Mdm2 induces developmental phenotypes in Drosophila. Oncogene 21, 2413–2417. [DOI] [PubMed] [Google Scholar]
- Formosa LE, Dibley MG, Stroud DA, and Ryan MT (2018). Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin Cell Dev Biol 76, 154–162. [DOI] [PubMed] [Google Scholar]
- Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, and Nijtmans L (2017). The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab 25, 128–139. [DOI] [PubMed] [Google Scholar]
- Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, and Meisinger C (2014). The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab 19, 357–372. [DOI] [PubMed] [Google Scholar]
- Hatefi Y (1985). The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem 54, 1015–1069. [DOI] [PubMed] [Google Scholar]
- Higashiyama M, Doi O, Kodama K, Yokouchi H, Kasugai T, Ishiguro S, Takami K, Nakayama T, and Nishisho I (1997). MDM2 gene amplification and expression in non-small-cell lung cancer: immunohistochemical expression of its protein is a favourable prognostic marker in patients without p53 protein accumulation. Br J Cancer 75, 1302–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, and Yasuda H (1997). Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420, 25–27. [DOI] [PubMed] [Google Scholar]
- Issaq SH, Teicher BA, and Monks A (2014). Bioenergetic properties of human sarcoma cells help define sensitivity to metabolic inhibitors. Cell Cycle 13, 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, Bhat KP, Godfrey VL, Evan GI, and Zhang Y (2007). Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 12, 355–366. [DOI] [PubMed] [Google Scholar]
- Jones SN, Hancock AR, Vogel H, Donehower LA, and Bradley A (1998). Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci U S A 95, 15608–15612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, and Bradley A (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208. [DOI] [PubMed] [Google Scholar]
- Karni-Schmidt O, Lokshin M, and Prives C (2016). The Roles of MDM2 and MDMX in Cancer. Annu Rev Pathol 11, 617–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshikawa N, Akimoto M, Hayashi JI, Nagase H, and Takenaga K (2017). Association of predicted pathogenic mutations in mitochondrial ND genes with distant metastasis in NSCLC and colon cancer. Sci Rep 7, 15535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, McKenzie M, Ohtake A, Thorburn DR, and Ryan MT (2007). Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol Cell Biol 27, 4228–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite KR, Franco MF, Srougi M, Nesrallah LJ, Nesrallah A, Bevilacqua RG, Darini E, Carvalho CM, Meirelles MI, Santana I, et al. (2001). Abnormal expression of MDM2 in prostate carcinoma. Mod Pathol 14, 428–436. [DOI] [PubMed] [Google Scholar]
- Letts JA, Fiedorczuk K, and Sazanov LA (2016). The architecture of respiratory supercomplexes. Nature 537, 644–648. [DOI] [PubMed] [Google Scholar]
- Letts JA, and Sazanov LA (2017). Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat Struct Mol Biol 24, 800–808. [DOI] [PubMed] [Google Scholar]
- Lopez-Fabuel I, Le Douce J, Logan A, James AM, Bonvento G, Murphy MP, Almeida A, and Bolanos JP (2016). Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc Natl Acad Sci U S A 113, 13063–13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Vargas MPA, and Chipuk JE (2016). Physiological and Pharmacological Control of BAK, BAX, and Beyond. Trends Cell Biol 26, 906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maio N, Kim KS, Singh A, and Rouault TA (2017). A Single Adaptable Cochaperone-Scaffold Complex Delivers Nascent Iron-Sulfur Clusters to Mammalian Respiratory Chain Complexes I-III. Cell Metab 25, 945–953 e946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marres CA, de Vries S, and Grivell LA (1991). Isolation and inactivation of the nuclear gene encoding the rotenone-insensitive internal NADH: ubiquinone oxidoreductase of mitochondria from Saccharomyces cerevisiae. Eur J Biochem 195, 857–862. [DOI] [PubMed] [Google Scholar]
- McCann AH, Kirley A, Carney DN, Corbally N, Magee HM, Keating G, and Dervan PA (1995). Amplification of the MDM2 gene in human breast cancer and its association with MDM2 and p53 protein status. Br J Cancer 71, 981–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic D, Blaza JN, Larsson NG, and Hirst J (2017). The Enigma of the Respiratory Chain Supercomplex. Cell Metab 25, 765–776. [DOI] [PubMed] [Google Scholar]
- Mitchell P (2011). Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. 1966. Biochim Biophys Acta 1807, 1507–1538. [DOI] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, and Levine AJ (1992). The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, and Lozano G (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Toguchida J, Wadayama B, Kanoe H, Kotoura Y, and Sasaki MS (1995). MDM2 gene amplification in bone and soft-tissue tumors: association with tumor progression in differentiated adipose-tissue tumors. Int J Cancer 64, 342–346. [DOI] [PubMed] [Google Scholar]
- Nicholson J, Scherl A, Way L, Blackburn EA, Walkinshaw MD, Ball KL, and Hupp TR (2014). A systems wide mass spectrometric based linear motif screen to identify dominant in-vivo interacting proteins for the ubiquitin ligase MDM2. Cell Signal 26, 1243–1257. [DOI] [PubMed] [Google Scholar]
- Pickles S, Vigie P, and Youle RJ (2018). Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M (1999). Oxidative phosphorylation at the fin de siecle. Science 283, 1488–1493. [DOI] [PubMed] [Google Scholar]
- Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, and Letai A (2013). BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 51, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, and Meisinger C (2010). Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11, 655–667. [DOI] [PubMed] [Google Scholar]
- Smeitink J, van den Heuvel L, and DiMauro S (2001). The genetics and pathology of oxidative phosphorylation. Nat Rev Genet 2, 342–352. [DOI] [PubMed] [Google Scholar]
- Taanman JW (1999). The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410, 103–123. [DOI] [PubMed] [Google Scholar]
- Tittel JN, and Steller H (2000). A comparison of programmed cell death between species. Genome biology 1, Reviews0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotta AP, and Chipuk JE (2017). Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci 74, 1999–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartak R, Deng J, Fang H, and Bai Y (2015). Redefining the roles of mitochondrial DNA-encoded subunits in respiratory Complex I assembly. Biochim Biophys Acta 1852, 1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Vinothkumar KR, Zhu J, and Hirst J (2014). Architecture of mammalian respiratory complex I. Nature 515, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M, Li YC, and Wahl GM (2013). MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Gu J, Guo R, Huang Y, and Yang M (2016). Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell 167, 1598–1609 e1510. [DOI] [PubMed] [Google Scholar]
- Ye C, Tang H, Zhao Z, Lei CT, You CQ, Zhang J, Gao P, He FF, Chen S, Wang YM, et al. (2017). MDM2 mediates fibroblast activation and renal tubulointerstitial fibrosis via a p53-independent pathway. Am J Physiol Renal Physiol 312, F760–F768. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Wang W, Li H, Yu Y, Tao J, Huang S, and Zeng Z (2015). Nonsense and missense mutation of mitochondrial ND6 gene promotes cell migration and invasion in human lung adenocarcinoma. BMC Cancer 15, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]