

Abstract
Bioinformatics approaches are becoming ever more essential in translational drug discovery both in academia and within the pharmaceutical industry. Computational exploitation of the increasing volumes of data generated during all phases of drug discovery is enabling key challenges of the process to be addressed. Here, we highlight some of the areas in which bioinformatics resources and methods are being developed to support the drug discovery pipeline. These include the creation of large data warehouses, bioinformatics algorithms to analyse ‘big data’ that identify novel drug targets and/or biomarkers, programs to assess the tractability of targets, and prediction of repositioning opportunities that use licensed drugs to treat additional indications.
Keywords: computational biochemistry, drug discovery and design, genomics
Introduction
Recent estimates suggest that it takes approximately 13 years and a ‘capitalized’ cost of approximately US$1.8 billion to bring a new drug to the market [1]. This cost includes the development of the licensed drug, and also incorporates the cost of the compounds that failed to make it to the market. Projects can fail in all the different steps of drug discovery process and in particular, during the later stages of development.
Common reasons for this high attrition rate include lack of clinical efficacy of the potential drug (approximately 30%), unexpected toxicities (>20%) as well as the inherent commercial concerns (>20%) of being able to successfully position a new drug within a competitive market [2].
Reducing costs and amount of time required for each of the different steps in the drug discovery pipeline is the key to deliver better drugs to patients in a timely manner [3]. One approach that has the potential to increase the efficiency of the drug discovery process involves maximizing the information acquired from the basic science. Translational drug discovery involves the effective conversion of advances in basic biological and chemical science research into the production of new drugs and treatment options for patients, i.e. the development of new drugs from ‘bench-to-bedside’. Translational approaches also come with the additional benefits of enabling new treatments and research knowledge to reach the patient subpopulations for whom they are intended, inform better clinical trial design, and help to reduce the often severe side effects of treatments. Figure 1 sets out the steps of the process and the bioinformatics techniques that can be brought to bear on them.
Figure 1. Translational bioinformatics opportunities in the drug discovery pipeline.

A schematic diagram of the drug discovery process. Each phase of the drug discovery pipeline (discovery, clinical and postlaunch) is shown as an orange arrow. Underneath the pipeline, shown as blue rectangles, are the types of ‘big data’ that can be generated in each step of the pipeline. Highlighted below the data types are the potential opportunities to improve the pipeline using bioinformatics techniques. For example, during the discovery phase, the focus is on identifying the druggability of potential target proteins. During the clinical trials, phase personalized medicine and patient selection can be used to better sample and categorize subjects while the use of biomarkers can improve efficacy measurements. Finally, at the post-launch phase of a drug’s life cycle drug safety monitoring and disease subtyping can be used to both improve the quality of life for patients as well as help to identify the opportunities for modified interventions that may be more effective for certain subtypes of a given disease. Adapted from [4], Copyright (2011), with permission from Elsevier.
In this review, we illustrate how the recent advances in computational methods, together with ever growing access to publicly available medical big data are revolutionizing translational drug discovery, resulting in the development of better drugs and therapies. This revolution is happening both from the clinical perspective of disease or its pathology the ‘disease-based’ approach and from a chemical perspective, the so-called ‘drug-based’ approach.
Disease-based bioinformatics approaches
Disease-based bioinformatics approaches in translational drug discovery are dependent upon the type of disease under consideration, with different strategies implemented to analyse cancer, genetic and infectious diseases [5].
Cancer cells are characterized by a diverse set of genetic and epigenetic changes, and by chromosomal instability. Bioinformatics approaches can be used to identify the key drivers of cancer in each particular patient. So, they have the potential to enable a more personalized approach to cancer therapy, paving the way for novel and repurposed drugs that target specific proteins, killing or disabling just those cells that are diseased [6,7].
Our genetic makeup affects our likelihood of developing a wide range of diseases, our responses to a variety of drug treatments and the progression of many infectious diseases [8–11]. For genetic diseases, the emphasis of bioinformatics techniques is often on identifying opportunities for gene therapies, as well as identifying noninvasive diagnostic and prognostic tools.
Bioinformatics is also implemented within translational drug discovery in infectious diseases. For instance, the presence of viral or bacterial infection gives rise to specific profiles of gene expression within the cell. Comparing these profiles with those of other diseases and with drug-induced genetic profiles offers repositioning opportunities for existing drugs [12–16].
Target identification in cancer
Since the human genome was first sequenced, genomic, proteomic and metabolomic high-throughput platforms have increasingly allowed analyses of large datasets across many different diseases. Data science, machine learning and/or statistical approaches are used to identify abnormal patterns that correlate with the disease process, often with the ultimate aim of identifying actionable targets that are druggable [3].
Over 200 forms of cancer have been described [17]. Each involves dynamic changes in the genome, including a wide range of different genetic aberrations such as somatic mutations, copy number variations, as well as changes to gene expression profiles, and different epigenetic patterns. Not only do these anomalies vary among cancers, but there is also a significant variation within patient cohorts within the same cancer, with continuing changes as tumours evolve, for instance when tumours develop resistance to specific drugs [18]. The complexity of these changes means that the application of bioinformatics techniques is often critical in identifying the type of cancer presented, with each characterized by a different molecular profile that requires a unique therapeutic strategy.
Within the field of cancer research, there are several large repositories storing multiplatform cancer data including the International Cancer Genomics Consortium (ICGC) [19], the National Cancer Institute Genomics Data Commons (GDC) and The Cancer Gemome Atlas (TCGA) [17]. For example, the GDC [20] provides curated storage for over 14,531 cases previously curated by the TCGA [17], and this is expected to grow to over 30,000 cases with the inclusion of data from Foundation Medicine Inc.. The benefit of such resources is not only the access to raw sequencing data, but also the application of state of the art methods for generating high level data (e.g. mutation calls, structural variants etc.), that allow the first steps of analysis to be standardized for reproducibility as well as clinical data. It provides access to multiple ‘omic’ data types such as mRNA expression, somatic mutations, copy number variation and protein abundance.
Drawing this information together provides a better molecular characterization and understanding of the biological basis for diseases. The use of such ‘big data’ to search for novel drug targets splits into several key elements, including identifying the genes that are driving the cancer, and then determining which of these are actionable.
Identification of genes that may be driving cancer
Within any tumour, only a minority of the genetic changes enable and drive the progression of the disease. The other mutations provide no growth advantage and are often described as passenger mutations. Vogelstein et al. [21] identified approximately 140 potential genes that act as drivers of tumorigenesis. However, as the number of analyses grow so does the list of potentially significant driver genes [22].
A number of methods have developed to separate true driver genes from the more commonly mutated passengers. One approach (e.g. MutSigCV [18]) is to modify the putative mutation background rate to take in account the replication time of the DNA region and incorporating information about gene expression levels. In cancers with particularly high mutation rates, most genetic changes are incidental to the development of the cancer, so it is helpful to assess the functional impact of modifications. There are a number of existing algorithms that can help with this and these are outlined in Table 1. These methods use a variety of approaches to predict the tolerance of amino acid substitutions (or indels) within the protein [23] and several were specifically developed to assess the importance of particular missense mutations within cancer samples [23]. An alternative, powerful approach is to look at the events in a given genetic pathway. Results from such studies can be easier to interpret as they may suggest causal mechanisms relating to concepts such as inflammation or DNA damage response [24–26].
Table 1. Bioinformatics resources to help identify the functional impact of mutations and tools designed to analyse cancer mutations*.
| Tool | Reference | Comments | URL |
|---|---|---|---|
| CHASM* | [27] | Probability that the mutation gives the cells a selective survival advantage | http://wiki.chasmsoftware.org/index.php/Main_Page |
| Condel | [28] | Combines FATHMM, mutation assessor etc. | http://bg.upf.edu/fannsdb/ |
| FATHMM* | [29] | Distinguishes between cancer promoting and ‘neutral’ germline polymorphisms using hidden Markov models | http://fathmm.biocompute.org.uk/about.html |
| Mutation assessor* | [30] | Based on evolutionary conservation of the affected amino acid in protein homologues | http://mutationassessor.org/r3/ |
| Polyphen-2 | [31] | Uses straightforward physical and comparative considerations | http://genetics.bwh.harvard.edu/pph2/ |
| SIFT | [32] | Based on sequence homology and the physical properties of amino acids | http://sift.bii.a-star.edu.sg/ |
| TransFIC* | [23] | Transforms functional impact scores provided by other tools by taking into account the differences in basal tolerance to germline single nucleotide variants (SNVs) of genes that belong to different functional classes | http://bg.upf.edu/transfic/home |
Targeting oncogenes and tumour suppressor genes
Bioinformatics can not only help to identify genes that may drive cancer, but can also help to classify them, according to whether they must be activated (proto-oncogenes) [33] or alternatively inactivated (tumour suppressor genes) before they cause harm. The patterns of mutations seen in these two classes of genes differ considerably and have been used to separate genes between these classes when the biological function of the protein product of the gene in a cancer setting is still unknown [34–36].
Many targeted anticancer drugs work by directly inhibiting activated oncogenes, particularly proteins that contain protein kinase domains or proteins that are nuclear receptors [33,37,38]. For example, dabrafenib has been approved for the treatment of late-stage melanoma, and targets the constitutively activated kinase oncogene BRAF V600E. Whereas cetuximab, panitumumab, gefitinib and erlotinib are the licensed inhibitors of the EGFR tyrosine kinase and crizotinib is an ALK inhibitor, all of which are licensed for the treatment of lung cancer [39–42].
A substantively different approach is needed to provide therapies aimed at controlling the damage done by inactivated tumour suppressor genes. It is not usually feasible to repair the protein products of these genes, if they are inactivated by truncation, although there are on-going attempts to reactivate or restore function to a small subset of p53 missense mutant proteins [43]. While targeting a tumour suppressor gene, it is now becoming common to look for a synthetically lethal partner gene that can be drugged. Two genes are said to be synthetically sensitive or lethal (SSL) if the function of either gene can be disrupted without causing cell death, while alterations in both genes cause cell death [44]. By drugging synthetic lethal partners, it is possible to target only those cells that have the mutation while leaving normal cells viable [45,44].
Genes involved in the DNA damage response are prime candidates for synthetically lethal interactions as there are multiple complementary pathways for repairing DNA [46]. The best example of the therapeutic exploitation of SSLs is the pharmaceutical inhibition of PARP1 [47], a key enzyme in single-strand break repair (SSBR), which is SSL with genetic defects in the BRCA1, BRCA2 or PALB2 homologous recombination (HR) proteins observed in hereditary breast, ovarian, pancreatic and prostate cancers. The furthest progressed PARP inhibitor, olaparib (AZD-2281), was approved by the EMA and the FDA in late 2014 for BRCA-mutated advanced ovarian cancer patients [48] and is in further clinical trials for a variety of other SSBR-deficient cancers.
Targeting genes in genetic disorders
Genetic disorders are generally caused by genetic variants that ultimately induce a detrimental change in protein function within the cell. Genome-wide association studies (GWAS) are undertaken to statistically associate the presence of particular genetic variations with the onset of disease. Early GWAS, based on linear regression models, were successful at identifying Mendelian traits [49–52] and disorders that are highly heritable such as coeliac disease [53] and type-1 diabetes [54,55].
Gene therapies offer a potential way to translate the results from GWAS into new treatments. Early successes were reported in 2000 for a gene therapy to treat X-linked severe combined immunodeficiency (SCID-X1). However, other gene therapy trials were placed on hold following cancers caused by insertional mutagenesis associated with the gene vectors used. More modern vectors have improved safety features, new trials have started [56], and ADA-SCID gene therapy was endorsed by the European Medicines Agency in June 2016 [57].
GWAS are now frequently employed to identify rare variants that contribute to multifactorial diseases, but it is harder to identify the relevance of their significance. This is because there are complex confounding factors in the relationships among individuals, and the relationships among the mutation loci [58,59]. GWAS have not directly identified the existing drug targets for a disease. However, Cao and Moult [60] suggest that new targets will be discovered using GWAS, by combining the techniques with protein interaction network data and machine learning.
Increasingly, the data from GWAS are being used as the starting point for a variety of different machine learning techniques. This work has potential within the clinic to provide noninvasive diagnostic tools. For example, diagnosis of coeliac disease traditionally requires exposure to the allergen gluten. However, Abraham and Inouye [54] used genetic profiling to enable the noninvasive prediction of coeliac disease without recourse to gluten sensitivity testing. Results from 1390 GWAS have been brought together and re-annotated to provide GRASP, a database of over 6.2 million SNP–phenotype associations [61].
Very occasionally, it is also possible to find the deletion of a gene that is associated with unusually good health. The 2010 Longevity Genes Study enabled Barzilai et al. [62] to study the relationship between gene polymorphisms and age. In particular, longevity was found to be associated with a deletion at in the adiponectin (ADIPOQ) gene.
Infectious diseases
There are many infectious diseases that have no effective treatment or where treatment is only effective for a subset of the patient population. Moreover, variants of diseases continue to emerge, threatening the progress already made [63].
Several bioinformatics approaches have been used to stratify the patient populations. For example, GWAS have enabled researchers to identify subpopulations that have genetic variants associated with different patterns in disease progression [63,64]. Alternatively, it is possible to map the gene expression profile that is associated with disease and compare it with pre-existing profiles that are associated with drugs [65].
In 2015, a large GWAS by the Malaria Genomic Epidemiology Network found that approximately 33% protection against severe malaria is provided by genetic variants at a novel genetic locus, which is either in or close to genes encoding the production of glycophorins [66]. Therapies for infections such as HIV have been developed that target host factors [67] and it is now hoped that the same approach can be taken to improve therapies for malaria [68].
As well as enabling identification of differences between patients, ‘omic’ data can be used to identify distinguish related strains of viruses and bacteria, both by looking at evolution of the pathogen genomics, and by looking for changes in the metabolites that they express. For example, the variants of Escherichia coli found in the gut and urinary tract differ in the expression of two small molecules, yersiniabactin and salmochelin, that are known to support bacterial survival. Targeting the metabolic pathways or the strains that produce these molecules may provide a good strategy for preventing recurrent urinary tract infections [69]. Fontana et al. [70] provided a useful overview of this large and growing area.
‘Omic’ data also provide a fast and cheap way of identifying drugs that have potential for repurposing. The publicly available Connectivity Map allows easy comparison of any gene expression profile against the expression profile generation by over 1300 compounds, most of which are drugs that have already been approved for other purposes. The program calculates a connectivity score, an assessment of the positive or negative correlation between gene expression signatures [13]. The later DMAP extends this search to over 289,571 chemical entities [71]. In 2010, Josset et al. [14] identified antiviral agents that are broadly effective against influenza A, a virus noted for its genetic diversity. They reasoned that a viral infection could be treated by manipulating the cell environment away from the optimal conditions required for the viral life cycle [14]. This approach also has the potential to identify candidate small molecules to reverse or prevent the biological responses induced by ZIKV infection, which could have therapeutic benefits for ZIKV-infected individuals [15]. Alternative approaches looked for similarities between two diseases or two drugs by comparing the induced gene expression profiles [16].
Drug-based approaches
Drug repositioning and open source drug discovery
Repositioned drugs, in which the preclinical and safety studies in humans have already been evaluated, enable a faster, cheaper and more efficient translation into the clinic [72]. The use of an existing drug for a new condition is not completely risk free and still requires a drug development phase [72,73]. However, repositioning an already licensed drug can reduce the drug development cycle from 10 to 17 years to as short a time frame as 3–12 years [74].
Iorio et al. [33] mapped cancer-driven alterations on to human cancer cell lines allowed sensitivity testing with 265 existing drugs. The result of this work is the identification of a series of alterations that result in sensitivity and resistance to particular drugs, providing datasets that can act as a resource for researchers looking for therapeutic options for particular cancer subpopulations.
Data sharing, focused around a particular disease or a group of diseases, can also improve the efficacy of drug discovery and allow links to be made to other related conditions. For example, the Malaria Box provides open access to information on safety and effectiveness of compounds that kill malarial parasites in vitro, encouraging collaboration between academia and industry [75]. The resulting drug development programmes suggest that some of the compounds may have much wider therapeutic benefits against other pathogens and have led to the development of a wider initiative–the Pathogen Box [66]. A similar approach was taken by the TDR targets database that provides data and predicted druggability relating to tropical disease pathogens [76].
Target tractability
Bioinformatics techniques are also used to assess whether a target is ‘druggable’. By carrying out such analyses in the early stages of drug discovery, it is possible to reduce the risk of project failure later on in the discovery process [77,78].
Ligand-based druggability
Protein druggability is defined as the protein’s ability to bind small drug-like molecules with high affinity. These interactions depend strongly on both ways in which the protein is folded in space and other physical attributes of the protein such as the distribution of charge. The structure of the small ‘drug-like molecule’ is equally important. An ideal drug should be able to be orally administered in small quantities. Thus, as well as being potent, the drug should successfully cross both the intestinal and cell membranes, be transportable through the blood, diffuse quickly and excreted successfully. Potential drugs that do not have these pharmacokinetic properties are a big factor of overall attrition rates [79]. These properties are well expressed and quantified in Lipinski’s ‘rule of 5’ (see Table 2) [80], and this can be improved by putting in place further restrictions on the polar surface area (PSA) and the number of rotatable bonds [81].
Table 2. Properties of small molecule drug-like compounds.
Introduced in 2002, the concept of the ‘druggable genome’ identified the genes within the human genome that coded for proteins that could be modulated by small drug-like proteins [82]. This bioinformatics analysis evaluated the ‘druggability’ in all human proteins by calculating their sequence identity to known therapeutic targets and predicted that less than 10% of the human proteome was druggable [82]. Of these targets, only 10% are then associated with an FDA-approved drug. The Illuminating the Druggable Genome (IDG) program aims to provide comprehensive access to data on these protein targets in order to stimulate research [38].
The ability of a protein to bind drug-like compounds can be assessed by analysing the chemical qualities of known inhibitors or predicted through virtual screening and docking of inhibitors on these proteins [83]. The CanSAR database [84] provides a ligand-based druggability scores for human proteins estimated from the chemical properties and bioactivity parameters of small molecule compounds deposited in the ChEMBL database [85]. The score is derived from the affinity, diversity, ligand efficiency and other qualities of all compounds tested against both the target and all its family members [84].
Where enzymes have similar ligand-binding profiles, this can indicate that they have similar function. For example, the family of cytochrome P450 enzymes (P450s) play important roles in Mycobacterium tuberculosis. Using fragment screening, Kavanagh et al. [86] identified similarities in the ligand-binding profile of CYP121A1, which is known to be important for M. tuberculosis viability, and the orphan enzyme CYP144A1. An assessment of the similarities and differences in binding between the two enzymes provides insight into both the function of the enzyme and potential inhibitors [86].
Hajduk et al. [87] experimentally showed that the druggability of a binding site is related to its ability to bind small ligands. The same principles were then applied in silico by a virtual screen of over 11,000 fragments on 152 protein-binding sites. This work demonstrated that a small ligand based virtual screen can be effective at predicting druggability of protein-binding sites [88].
Structure-based druggability
Knowing the 3D structure of a target protein greatly assists small molecule drug discovery, enabling analysis of the druggability of each protein pocket, virtual docking with small molecules and comparison of similar proteins [89]. Structure-based druggability calculations starts with a crystallographic or modelled 3D structure. All the ligand-binding sites on the surface of the protein are identified and the probable druggability of each pocket is assessed based on physicochemical parameters such as size, shape and hydrophobicity. Results from these tools correlate well with predictions from NMR screens of fragment libraries [87,90] and drug discovery projects are more likely to fail if they target proteins that have only low scoring pockets [91].
Methods for identifying pockets either assume that the 3D protein structure is static, employ energy-based algorithms or use molecular dynamics simulations [92]. These techniques have been reviewed [93–95] and the main algorithms that search for binding pockets have been summarized by Villoutreix and colleagues [95]. The druggability of each pocket is then assessed by calculating properties such as hydrophobicity, volume, amino acid composition and electrostatics, and then using these features to train a machine learning model on validated drug binding/not drug-binding pockets [90,96–99]. Some of these programmes are automated and score binding pockets on the likelihood of their druggability. An overview of these automated druggability assessment methods is summarized in Table 3. Figure 2 demonstrates the potential to drug the bromodomains BRD1 and TRIM24 using DoGSiteScorer. Although the proteins appear indistinguishable to the eye, nevertheless the analysis identifies that BRD1 has a bromodomain-binding pocket (shown as a mesh) more likely to bind a small molecule.
Table 3. Programs that can be used to calculate structure-based druggability.
| Name | References | Pocket search method | Druggability score | |
|---|---|---|---|---|
| Function | Descriptors | |||
| fPocket | [92] | Geometric criteria based on distance to predetermined points | Partial least square analysis | Hydrophobicity, normalized polarity and local hydrophobicity density |
| DoGSiteScorer | [100] | Geometric criteria based on 3D image enhancement techniques | Support vector machine | Depth, volume and relative number or apolar amino acids |
| SiteMap | [101] | Geometric and energetic criteria on 3D grids | Weighted sum of three descriptors | Hydrophilicity, degree of enclosure, number of site points |
Figure 2. Prediction of druggable pockets in bromodomains.
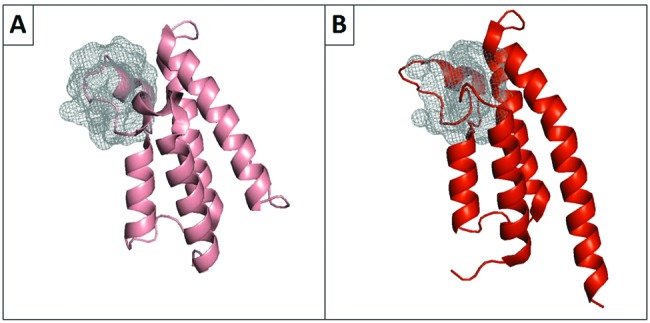
The acetyl-lysine (KAc) binding pockets of two human bromodomains were identified by DoGSiteScorer. (A) shows the non-druggable KAc binding site of the bromodomain from TRIM24 (PDB: 2YYN_A) (druggability score =0.49). (B) shows the druggable KAc binding site of the bromodomain from BRD1 (PDB: 3RCW_A) ( druggability score =0.68). A score greater than 0.50 is indicative of a druggable pocket [102].
Understanding the resemblance between binding pockets can aid in the design of target selective compounds, preventing mistakes in assigning druggability. To aid this understanding, a number of tools have been developed that compare protein-binding sites by representing binding sites through specific features [95,103,104]. Computational druggability investigations have also been undertaken to compare and contrast the druggability of binding sites such as bromodomains that have a similar function [105–108].
Network-based druggability
A number of different networks have been built to represent molecular interactions including drug–target, drug–drug, drug–disease, protein–protein, transcriptional and signalling networks (for an example, see Figure 3). Features from these networks can then be used to train machine learning models with a large number of aims. These range from characterizing drug targets and identifying potential new uses for existing drugs, to predicting the response of patient subpopulations to drug treatments [109–112]. Similarly, Napolitano et al. [113] used machine learning to predict the therapeutic class of FDA-approved compounds with repositioning in mind.
Figure 3. Druggable targets in the DNA damage response.

This illustrates the protein–protein interaction network of proteins (derived from the STRING database [114]) involved in the DNA damage response as described in [46]. Each protein is shown as a node/circle with the interaction described as a connecting line. The network is labelled by some of the DDR processes: HR; UR, ubiquitin response; FA, Fanconi anaemia; NER, nucleotide excision repair, CS, chromosome segregation; CR, chromatin remodelling. Nodes coloured dark green indicate a protein for which these is a licenced drug, light green nodes indicate that the protein is a target of a drug in clinical trials. Pink nodes indicate that a protein is predicted to be druggable as it has the features of a good drug target. Each of these proteins have been predicted to be druggable by the at least two of the druggability methods (ligand, structure and network) provided by the canSAR database [84]. Adapted from Supplementary Information (figure) S15 [46]. .
Computational drug repositioning in this way is only possible because of the breadth of publicly available big data sources that integrate pharmacological, genomic, phenotypic, chemical and clinical information (e.g. Drug Bank [115], ClinicalTrials.gov [116], PharmGKB [117] or PubChem [118]) and contine advances in text mining. Many of these tools and networks rely on the semiautomatic identification of links among genomic data, specific molecular pathways and phenology. The development of gene ontological terms has been of particular importance [119] as have advances in text mining approaches. These have recently been reviewed by Gonzalez et al. [120]. Useful resources for protein–protein and genetic interactions are set out in Table 4.
Table 4. Web-based databases documenting protein and genetic interactions.
| Database | References | Description | URL |
|---|---|---|---|
| BioGRID | Chatr-Aryamontri et al. [121] | Repository of curated genetic and physical interaction data | https://thebiogrid.org |
| STRING | Szklarczyk et al. [122] | Protein–protein interaction data for a wide range of organisms | https://string-db.org |
| IntAct | Hermjakob et al. [123] | Molecular interaction database derived from literature curation or direct user submissions | http://www.ebi.ac.uk/intact/ |
| Syn-lethality database | Li et al. [124] | Cross referenced and annotated resource for synthetic lethal related research | http://ntu.edu.sg/home/zhengjie/software/Syn-Lethality |
| SynLethDB | Guo, Liu and Zheng [125] | Database of genetic interactions focused on selective and sensitive anticancer drug targets | http://histone.sce.ntu.edu.sg/SynLethDB/ |
| Slorth! | Benstead-Hume and Pearl [unpublished] | Genetic interaction data with a focus on orthologues and conserved interactions | http://rails.biochem.susx.ac.uk:4000 |
Patient stratification and personalized medicine
Next generation sequencing and other ‘omic’ technologies are enabling better identification of a wide range of diseases, which will eventually lead to targeted therapies and personalized medicines. Personalized medicine can be used not only in cancer and long-term disorders, but also in infectious diseases.
One of the best known examples of patient stratification currently used in the clinic is the analysis of biomarkers for patients with breast cancer. There already exists tests and endocrine therapies for patients testing positive for elevated levels of HER2, oestrogen or progesterone and these complement chemotherapy, radiation therapy and surgery, the current standard-of-care in cancer treatment [126]. This type of characterzation is also being developed for other cancers enabling the identification of patient cohorts with similar therapeutic needs and potential outcomes. This approach can also reduce the use of aggressive therapies where they are not warranted. Hoadley et al. [127] divided the heterogeneous population of tumours into clinically and biologically meaningful subtypes using the similarity of molecular profiles. Rubio-Perez et al. [128] developed a pan-cancer strategy for therapy based on identifying alterations in driver genes. While only 5.9% of the tumours were treatable using approved drugs following the clinical guidelines, up to 40.2% could benefit from repurposing existing drugs [128]. A number of other teams have focused on specific common cancers [129–131]
Patient stratification can also be applied to infectious diseases, where patient response to treatment can have a strong genetic component. The 2009 GWAS of patient response to treatment for the hepatitis C virus (HCV) found that a genetic polymorphism near the IL28B gene makes a significant difference to patients’ response to pegylated interferon α plus ribavirin [132]. Genotyping people with HCV is now common when determining treatment options [133].
Discussion
The large amount of data generated directly by the drug discovery process that have become publicly available (e.g. ChEMBL), combined with the disease-based data provided by large consortia (e.g. GDC) mean that there has been an explosion of computational approaches linking chemical and disease data. Innovative bioinformatics approaches are already having an impact on the discovery, preclinical and clinical phases of the drug discovery process.
However, the challenges faced by the pharmaceutical industry means that it is becoming crucial to further invest in the bioinformatics resources required to support and expedite translational drug discovery. Approaches include: the development of databases and data warehouses that can archive, maintain and integrate large amounts of drug discovery and biomedical data currently being generated; the development of robust algorithms to enable the analysis of large and complex datasets; development of tools to enable experimental drug discovery for scientists to easily access and interpret these data; formal and informal networking tools such as Biostars that enable bioinformaticians to link up and learn from one another [134].
These type of endeavours will enable a better understanding of how we can use genomics and other ‘omic’ approaches to classify disease, improve diagnoses and inform new approaches to drug repositioning. They will allow us to identify disease biomarkers and genetic variants which correlate well with patient outcomes, and use them to improve therapeutic strategies.
Abbreviations
- DDR
DNA damage response
- GDC
Genomics Data Commons
- GWAS
genome-wide association studies
- HCV
hepatitis C virus
- HR
homologous recombination
- PSA
polar surface area
- SSBR
single-strand break repair
- SSL
synthetically sensitive or lethal
- TCGA
The Cancer Genome Atlas
Competing interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Paul S.M., Mytelka D.S., Dunwiddie C.T., Persinger C.C., Munos B.H., Lindborg S.R. et al. (2010) How to improve R &D productivity: the pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov. 9, 203–214 [DOI] [PubMed] [Google Scholar]
- 2.Kola I. and Landis J. (2004) Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 3, 711–715 [DOI] [PubMed] [Google Scholar]
- 3.Loging W., Harland L. and William-Jones B. (2007) High-throughput electronic biology: mining information for drug discovery. Nat. Rev. Drug Discov. 6, 220–230 [DOI] [PubMed] [Google Scholar]
- 4.Buchan NS., Rajpal DK., Webster Y., Alatorre C., Gudivada RC., Zheng C., Sanseau P. and Koehler J. (2011) The role of translational bioinformatics in drug discovery. Drug Discov. Today 16, 426–34 10.1016/j.drudis.2011.03.002 [DOI] [PubMed] [Google Scholar]
- 5.van Driel M.A. and Brunner H.G. (2006) Bioinformatics methods for identifying candidate disease genes. Hum. Genomics 2, 429–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J., Yang P.L. and Gray N.S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee H., Yoon N.E. and Jung B.H. (2017) Metabolomics study of cancer targeting small molecule kinase inhibitors in cell culture. Drug Metab. Pharmacokinet. 32, S77 [Google Scholar]
- 8.Malaria Genomic Epidemiology Network (2015) A novel locus of resistance to severe malaria in a region of ancient balancing selection. Nature 526, 253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wellcome Trust Case Control Consortium (2007) , Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 10.3410/f.1087106.540084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge D., Fellay J., Thompson A.J., Simon J.S., Shianna K.V., Urban T.J. et al. (2009) Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461, 399–401 [DOI] [PubMed] [Google Scholar]
- 11.Arcanjo A.C., Mazzocco G., de Oliveira S.F., Plewczynski D. and Radomski J.P. (2014) Role of the host genetic variability in the influenza A virus susceptibility. Acta Biochim. Pol. 61, 403–419 [PubMed] [Google Scholar]
- 12.Piñero J., Janet P., Àlex B., Núria Q.-R., Alba G.-S., Jordi D.-P. et al. (2016) DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 45, D833–D839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamb J. (2007) The connectivity map: a new tool for biomedical research. Nat. Rev. Cancer 7, 54–60 [DOI] [PubMed] [Google Scholar]
- 14.Josset L., Textoris J., Loriod B., Ferraris O., Moules V., Lina B. et al. (2010) Gene expression signature-based screening identifies new broadly effective influenza a antivirals. PLoS ONE 5 10.1371/journal.pone.0013169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang F., Hammack C., Ogden S.C., Cheng Y., Lee E.M., Wen Z. et al. (2016) Molecular signatures associated with ZIKV exposure in human cortical neural progenitors. Nucleic Acids Res. 44, 8610–8620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J., Zheng S., Chen B., Butte A.J., Swamidass S.J. and Lu Z. (2016) A survey of current trends in computational drug repositioning. Brief. Bioinform. 17, 2–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomczak K., Czerwińska P. and Wiznerowicz M. (2015) The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp. Oncol. (Pozn.) 19, A68–A77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A. et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J., Baran J., Cros A., Guberman J.M., Haider S., Hsu J. et al. (2011) International Cancer Genome Consortium Data Portal–a one-stop shop for cancer genomics data. Database (Oxford) 2011, bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A. et al. (2016) Toward a shared vision for cancer genomic data. N. Engl. J. Med. 375, 1109–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A. Jr and Kinzler K.W. (2013) Cancer genome landscapes. Science 339, 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamborero D., Gonzalez-Perez A., Perez-Llamas C., Deu-Pons J., Kandoth C., Reimand J. et al. (2013) Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 3, 2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez-Perez A., Deu-Pons J. and Lopez-Bigas N. (2012) Improving the prediction of the functional impact of cancer mutations by baseline tolerance transformation. Genome Med. 4, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akavia U.D., Litvin O., Kim J., Sanchez-Garcia F., Kotliar D., Causton H.C. et al. (2010) An integrated approach to uncover drivers of cancer. Cell 143, 1005–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pe’er D. and Hacohen N. (2011) Principles and strategies for developing network models in cancer. Cell 144, 864–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narayan S., Bader G.D. and Reimand J. (2016) Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome Med. 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong W.C., Kim D., Carter H., Diekhans M., Ryan M.C. and Karchin R. (2011) CHASM and SNVBox: toolkit for detecting biologically important single nucleotide mutations in cancer. Bioinformatics 27, 2147–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.González-Pérez A. and López-Bigas N. (2011) Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 88, 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shihab H.A., Julian G., Cooper D.N., Stenson P.D., Barker G.L.A., Edwards K.J. et al. (2012) Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reva B., Antipin Y. and Sander C. (2011) Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P. et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sim N.-L., Kumar P., Hu J., Henikoff S., Schneider G. and Ng P.C. (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iorio F., Knijnenburg T.A., Vis D.J., Bignell G.R., Menden M.P., Schubert M. et al. (2016) A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baeissa H.M., Benstead-Hume G., Richardson C.J. and Pearl F.M.G. (2016) Mutational patterns in oncogenes and tumour suppressors. Biochem. Soc. Trans. 44, 925–931 [DOI] [PubMed] [Google Scholar]
- 35.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A. Jr and Kinzler K.W. (2013) Cancer genome landscapes. Science 339, 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baeissa H., Benstead-Hume G., Richardson C.J. and Pearl F.M.G. (2017) Identification and analysis of mutational hotspots in oncogenes and tumour suppressors. Oncotarget 8, 21290–21304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shawver L.K., Slamon D. and Ullrich A. (2002) Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell 1, 117–123 [DOI] [PubMed] [Google Scholar]
- 38.Nguyen D.-T., Mathias S., Bologa C., Brunak S., Fernandez N., Gaulton A. et al. (2017) Pharos: collating protein information to shed light on the druggable genome. Nucleic Acids Res. 45, D995–D1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thatcher N., Chang A., Parikh P., Rodrigues Pereira J., Ciuleanu T., von Pawel J. et al. (2005) Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 366, 1527–1537 [DOI] [PubMed] [Google Scholar]
- 40.Shepherd F.A., Rodrigues Pereira J., Ciuleanu T., Tan E.H., Hirsh V., Thongprasert S. et al. (2005) Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 353, 123–132 [DOI] [PubMed] [Google Scholar]
- 41.Stinchcombe T.E. and Socinski M.A. (2008) Gefitinib in advanced non-small cell lung cancer: does it deserve a second chance? Oncologist 13, 933–944 [DOI] [PubMed] [Google Scholar]
- 42.Lindeman N.I., Cagle P.T., Beasley M.B., Chitale D.A., Dacic S., Giaccone G. et al. (2013) Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Mol. Diagn. 15, 415–453 [DOI] [PubMed] [Google Scholar]
- 43.Khoo K.H., Verma C.S. and Lane D.P. (2014) Drugging the p53 pathway: understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 13, 217–236 [DOI] [PubMed] [Google Scholar]
- 44.Hartwell L.H., Szankasi P., Roberts C.J., Murray A.W. and Friend S.H. (1997) Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068 [DOI] [PubMed] [Google Scholar]
- 45.Kaelin W.G., Jr (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 5, 689–698 [DOI] [PubMed] [Google Scholar]
- 46.Pearl L.H., Schierz A.C., Ward S.E., Al-Lazikani B. and Pearl F.M.G. (2015) Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 15, 166–180 [DOI] [PubMed] [Google Scholar]
- 47.Brown J.S., Kaye S.B. and Yap T.A. (2016) PARP inhibitors: the race is on. Br. J. Cancer 114, 713–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim G., Ison G., McKee A.E., Zhang H., Tang S., Gwise T. et al. (2015) FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 21, 4257–4261 [DOI] [PubMed] [Google Scholar]
- 49.Klein R.J. (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamazaki K., McGovern D., Ragoussis J., Paolucci M., Butler H., Jewell D. et al. (2005) Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn’s disease. Hum. Mol. Genet. 14, 3499–3506 [DOI] [PubMed] [Google Scholar]
- 51.Duerr R.H., Taylor K.D., Brant S.R., Rioux J.D., Silverberg M.S., Daly M.J. et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 doi: 10.3410/f.1047044.510892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dubois P.C.A., Trynka G., Franke L., Hunt K.A., Romanos J., Curtotti A. et al. (2010) Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 42, 295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abraham G. and Inouye M. (2015) Genomic risk prediction of complex human disease and its clinical application. Curr. Opin. Genet. Dev. 33, 10–16 [DOI] [PubMed] [Google Scholar]
- 55.Bradfield J.P., Qu H.-Q., Wang K., Zhang H., Sleiman P.M., Kim C.E. et al. (2011) A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 7, e1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ginn S.L., Alexander I.E., Edelstein M.L., Abedi M.R. and Wixon J. (2013) Gene therapy clinical trials worldwide to 2012 - an update. J. Gene Med. 15, 65–77 [DOI] [PubMed] [Google Scholar]
- 57.Ylä-Herttuala S. (2016) ADA-SCID gene therapy endorsed by European medicines agency for marketing authorization. Mol. Ther. 24, 1013–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R. et al. (2014) Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Visscher P.M., Brown M.A., McCarthy M.I. and Yang J. (2012) Five years of GWAS discovery. Am. J. Hum. Genet. 90, 7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cao C. and Moult J. (2014) GWAS and drug targets. BMC Genomics 15, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leslie R., O’Donnell C.J. and Johnson A.D. (2014) GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics 30, i185–i194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barzilai N., Gabriely I., Atzmon G., Suh Y., Rothenberg D. and Bergman A. (2010) Genetic studies reveal the role of the endocrine and metabolic systems in aging. J. Clin. Endocrinol. Metab. 95, 4493–4500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Bakker P.I.W. and Telenti A. (2010) Infectious diseases not immune to genome-wide association. Nat. Genet. 42, 731–732 [DOI] [PubMed] [Google Scholar]
- 64.Cantor R.M., Lange K. and Sinsheimer J.S. (2010) Prioritizing GWAS results: A review of statistical methods and recommendations for their application. Am. J. Hum. Genet. 86, 6–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thiers B.H. (2007) The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Year bk. Dermatol. Dermatol. Surg. 2007, 384–386 [Google Scholar]
- 66.Van Voorhis W.C., Adams J.H., Adelfio R., Ahyong V., Akabas M.H., Alano P. et al. (2016) Open source drug discovery with the malaria box compound collection for neglected diseases and beyond. PLoS Pathog. 12, e1005763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jacobson J.M., Lalezari J.P., Thompson M.A., Fichtenbaum C.J., Saag M.S., Zingman B.S. et al. (2010) Phase 2a study of the CCR5 monoclonal antibody PRO 140 administered intravenously to HIV-infected adults. Antimicrob. Agents. Chemother. 54, 4137–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langhorne J. and Duffy P.E. (2016) Expanding the antimalarial toolkit: targeting host–parasite interactions. J. Exp. Med. 213, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Henderson J.P., Crowley J.R., Pinkner J.S., Walker J.N., Tsukayama P., Stamm W.E. et al. (2009) Quantitative metabolomics reveals an epigenetic blueprint for iron acquisition in uropathogenic Escherichia coli. PLoS Pathog. 5, e1000305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fontana J.M., Alexander E. and Salvatore M. (2012) Translational research in infectious disease: current paradigms and challenges ahead. Transl. Res. 159, 430–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang H., Nguyen T., Ibrahim S., Shantharam S., Yue Z. and Chen J.Y. (2015) DMAP: a connectivity map database to enable identification of novel drug repositioning candidates. BMC Bioinformatics 16 (suppl 13) , S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dudley J.T., Deshpande T. and Butte A.J. (2011) Exploiting drug-disease relationships for computational drug repositioning. Brief. Bioinform. 12, 303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thellung S., Favoni R.E., Würth R., Nizzari M., Pattarozzi A., Daga A. et al. (2016) Molecular pharmacology of malignant pleural mesothelioma: challenges and perspectives from preclinical and clinical studies. Curr. Drug Targets 17, 824–849 [DOI] [PubMed] [Google Scholar]
- 74.Ashburn T.T. and Thor K.B. (2004) Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 3, 673–683 [DOI] [PubMed] [Google Scholar]
- 75.Spangenberg T., Burrows J.N., Kowalczyk P., McDonald S., Wells T.N.C. and Willis P. (2013) The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS ONE 8, e62906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Agüero F., Al-Lazikani B., Aslett M., Berriman M., Buckner F.S., Campbell R.K. et al. (2008) Genomic-scale prioritization of drug targets: the TDR Targets database. Nat. Rev. Drug Discov. 7, 900–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gashaw I., Ellinghaus P., Sommer A. and Asadullah K. (2011) What makes a good drug target? Drug Discov. Today 16, 1037–1043 [DOI] [PubMed] [Google Scholar]
- 78.Surendiran A., Pradhan S.C. and Adithan C. (2008) Role of pharmacogenomics in drug discovery and development. Indian J. Pharmacol. 40, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wenlock M.C., Austin R.P., Barton P., Davis A.M. and Leeson P.D. (2003) A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem. 46, 1250–1256 [DOI] [PubMed] [Google Scholar]
- 80.Lipinski C.A., Franco L., Dominy B.W. and Feeney P.J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1. Adv. Drug Deliv. Rev. 46, 3–26 [DOI] [PubMed] [Google Scholar]
- 81.Veber D.F., Johnson S.R., Cheng H.-Y., Smith B.R., Ward K.W. and Kopple K.D. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623 [DOI] [PubMed] [Google Scholar]
- 82.Hopkins A.L. and Groom C.R. (2002) The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 [DOI] [PubMed] [Google Scholar]
- 83.Svensson F., Norinder U. and Bender A. (2017) Improving screening efficiency through iterative screening using docking and conformal prediction. J. Chem. Inf. Model. 57, 439–444 [DOI] [PubMed] [Google Scholar]
- 84.Bulusu K.C., Tym J.E., Coker E.A., Schierz A.C. and Al-Lazikani B. (2014) canSAR: updated cancer research and drug discovery knowledgebase. Nucleic Acids Res. 42, D1040–D1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gaulton A., Bellis L.J., Bento A.P., Chambers J., Davies M., Hersey A. et al. (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40, D1100–D1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kavanagh M.E., Chenge J., Zoufir A., McLean K.J., Coyne A.G., Bender A. et al. (2017) Fragment profiling approach to inhibitors of the orphan M. tuberculosis P450 CYP144A1. Biochemistry 56, 1559–1572 [DOI] [PubMed] [Google Scholar]
- 87.Hajduk P.J., Huth J.R. and Tse C. (2005) Predicting protein druggability. Drug Discov. Today 10, 1675–1682 [DOI] [PubMed] [Google Scholar]
- 88.Huang N. and Jacobson M.P. (2010) Binding-site assessment by virtual fragment screening. PLoS ONE 5, e10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fauman E.B., Rai B.K. and Huang E.S. (2011) Structure-based druggability assessment–identifying suitable targets for small molecule therapeutics. Curr. Opin. Chem. Biol. 15, 463–468 [DOI] [PubMed] [Google Scholar]
- 90.Hajduk P.J., Huth J.R. and Fesik S.W. (2005) Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 48, 2518–2525 [DOI] [PubMed] [Google Scholar]
- 91.Edfeldt F.N.B., Folmer R.H.A. and Breeze A.L. (2011) Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discov. Today 16, 284–287 [DOI] [PubMed] [Google Scholar]
- 92.Le Guilloux V., Schmidtke P. and Tuffery P. (2009) Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Henrich S., Salo-Ahen O.M.H., Huang B., Rippmann F.F., Cruciani G. and Wade R.C. (2010) Computational approaches to identifying and characterizing protein binding sites for ligand design. J. Mol. Recognit. 23, 209–219 [DOI] [PubMed] [Google Scholar]
- 94.Egner U. and Hillig R.C. (2008) A structural biology view of target drugability. Expert Opin. Drug Discov. 3, 391–401 [DOI] [PubMed] [Google Scholar]
- 95.Pérot S., Sperandio O., Miteva M.A., Camproux A.-C. and Villoutreix B.O. (2010) Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov. Today 15, 656–667 [DOI] [PubMed] [Google Scholar]
- 96.Nayal M. and Honig B. (2006) On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins 63, 892–906 [DOI] [PubMed] [Google Scholar]
- 97.Schmidtke P. and Barril X. (2010) Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 53, 5858–5867 [DOI] [PubMed] [Google Scholar]
- 98.Sheridan R.P., Maiorov V.N., Katharine Holloway M., Cornell W.D. and Gao Y.-D. (2010) Drug-like density: a method of quantifying the “bindability” of a protein target based on a very large set of pockets and drug-like ligands from the Protein Data Bank. J. Chem. Inf. Model 50, 2029–2040 [DOI] [PubMed] [Google Scholar]
- 99.Cheng A.C., Coleman R.G., Smyth K.T., Cao Q., Soulard P., Caffrey D.R. et al. (2007) Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 25, 71–75 [DOI] [PubMed] [Google Scholar]
- 100.Volkamer A., Kuhn D., Grombacher T., Rippmann F. and Rarey M. (2012) Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 52, 360–372 [DOI] [PubMed] [Google Scholar]
- 101.Halgren T.A. (2009) Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 49, 377–389 [DOI] [PubMed] [Google Scholar]
- 102.Vidler L.R., Brown N., Knapp S. and Hoelder S. (2012) Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 55, 7346–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kellenberger E., Schalon C. and Rognan D. (2008) How to measure the similarity between protein ligand-binding sites? Curr. Comput. Aided Drug Des. 4, 209–220 [Google Scholar]
- 104.Schmitt S., Kuhn D. and Klebe G. (2002) A new method to detect related function among proteins independent of sequence and fold homology. J. Mol. Biol. 323, 387–406 [DOI] [PubMed] [Google Scholar]
- 105.Volkamer A., Eid S., Turk S., Jaeger S., Rippmann F. and Fulle S. (2015) Pocketome of human kinases: prioritizing the ATP binding sites of (yet) untapped protein kinases for drug discovery. J. Chem. Inf. Model. 55, 538–549 [DOI] [PubMed] [Google Scholar]
- 106.Radusky L., Defelipe L.A., Lanzarotti E., Luque J., Barril X., Marti M.A. et al. (2014) TuberQ: a Mycobacterium tuberculosis protein druggability database. Database (Oxford) 2014, bau035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Campagna-Slater V., Mok M.W., Nguyen K.T., Feher M., Najmanovich R. and Schapira M. (2011) Structural chemistry of the histone methyltransferases cofactor binding site. J. Chem. Inf. Model. 51, 612–623 [DOI] [PubMed] [Google Scholar]
- 108.Aretz J., Wamhoff E.-C., Hanske J., Heymann D. and Rademacher C. (2014) Computational and experimental prediction of human C-type lectin receptor druggability. Front. Immunol. 5, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Würth R., Thellung S., Bajetto A., Mazzanti M., Florio T. and Barbieri F. (2016) Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov. Today 21, 190–199 [DOI] [PubMed] [Google Scholar]
- 110.Shameer K., Readhead B. and Dudley J.T. (2015) Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr. Top. Med. Chem. 15, 5–20 [DOI] [PubMed] [Google Scholar]
- 111.Mitsopoulos C., Schierz A.C., Workman P. and Al-Lazikani B. (2015) Distinctive behaviors of druggable proteins in cellular networks. PLoS Comput. Biol. 11, e1004597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Menden M.P., Iorio F., Garnett M., McDermott U., Benes C.H., Ballester P.J. et al. (2013) Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS ONE 8, e61318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Napolitano F., Zhao Y., Moreira VM., Tagliaferri R., Kere J., D'Amato M. and Greco D. (2013) Drug repositioning: a machine-learning approach through data integration. J Cheminform 5, 30 10.1186/1758-2946-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J. et al. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y. et al. (2014) DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, D1091–D1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schwartz L.M., Woloshin S., Zheng E., Tse T. and Zarin D.A. (2016) ClinicalTrials.gov and Drugs@FDA: a comparison of results reporting for new drug approval trials. Ann. Intern. Med. 165, 421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thorn C.F., Klein T.E. and Altman R.B. (2013) PharmGKB: the Pharmacogenomics Knowledge Base. Methods Mol. Biol. 1015, 311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim S., Thiessen P.A., Bolton E.E., Chen J., Fu G., Gindulyte A. et al. (2016) PubChem Substance and Compound databases. Nucleic Acids Res. 44, D1202–D1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.The Gene Ontology Consortium (2014) Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gonzalez G.H., Tahsin T., Goodale B.C., Greene A.C. and Greene C.S. (2016) Recent advances and emerging applications in text and data mining for biomedical discovery. Brief. Bioinform. 17, 33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chatr-Aryamontri A., Breitkreutz B.-J., Oughtred R., Boucher L., Heinicke S., Chen D. et al. (2015) The BioGRID interaction database: 2015 update. Nucleic Acids Res. 43, D470–D478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J. et al. (2015) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hermjakob H., Montecchi-Palazzi L., Lewington C., Mudali S., Kerrien S., Orchard S. et al. (2004) IntAct: an open source molecular interaction database. Nucleic Acids Res. 32, D452–D455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li X.-J., Mishra S.K., Wu M., Zhang F. and Zheng J. (2014) Syn-lethality: an integrative knowledge base of synthetic lethality towards discovery of selective anticancer therapies. Biomed. Res. Int. 2014, 196034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guo J., Liu H. and Zheng J. (2015) SynLethDB: synthetic lethality database toward discovery of selective and sensitive anticancer drug targets. Nucleic Acids Res. 44, D1011–D1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cheang M.C.U., Voduc D., Bajdik C., Leung S., McKinney S., Chia S.K. et al. (2008) Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 14, 1368–1376 [DOI] [PubMed] [Google Scholar]
- 127.Hoadley K.A., Yau C., Wolf D.M., Cherniack A.D., Tamborero D., Ng S. et al. (2014) Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158, 929–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rubio-Perez C., Tamborero D., Schroeder M.P., Antolín A.A., Deu-Pons J., Perez-Llamas C. et al. (2015) In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 27, 382–396 [DOI] [PubMed] [Google Scholar]
- 129.Wang C., Machiraju R. and Huang K. (2014) Breast cancer patient stratification using a molecular regularized consensus clustering method. Methods 67, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Riester M., Wei W., Waldron L., Culhane A.C., Trippa L., Oliva E. et al. (2014) Risk prediction for late-stage ovarian cancer by meta-analysis of 1525 patient samples. J. Natl. Cancer Inst. 106 10.1093/jnci/dju048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hofree M., Shen J.P., Carter H., Gross A. and Ideker T. (2013) Network-based stratification of tumor mutations. Nat. Methods 10, 1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Khattab M.A., Abdelghany H.M., Ramzy M.M. and Khairy R.M. (2016) Impact of IL28B gene polymorphisms rs8099917 and rs12980275 on response to pegylated interferon-a/ribavirin therapy in chronic hepatitis C genotype 4 patients. J. Biomed. Res. 30, 40–45 [Google Scholar]
- 133.Urban T., Charlton M.R. and Goldstein D.B. (2012) Introduction to the genetics and biology of interleukin-28B. Hepatology 56, 361–366 [DOI] [PubMed] [Google Scholar]
- 134.Parnell L.D., Lindenbaum P., Shameer K., Dall'Olio G.M., Swan D.C. et al. (2011) BioStar: An Online Question & Answer Resource for the Bioinformatics Community. PLoS Comput. Biol. 7, e1002216 doi: 10.1371/journal.pcbi.1002216 [DOI] [PMC free article] [PubMed] [Google Scholar]