

Abstract
Striated muscle contraction is regulated by the actin-associated proteins tropomyosin and troponin. The extent of activation of myosin ATPase activity is lowest in the absence of both Ca2+ and activating cross-bridges (i.e., S1-ADP or rigor S1). Binding of activating species of myosin to actin at a saturating Ca2+ concentration stabilizes the most active state (M state) of the actin–tropomyosin–troponin complex (regulated actin). Ca2+ binding alone produces partial stabilization of the active state. The extent of stabilization at a saturating Ca2+ concentration depends on the isoform of the troponin subunits, the phosphorylation state of troponin, and, in the case of cardiac muscle, the presence of hypertrophic cardiomyopathy-producing mutants of troponin T and troponin I. Cardiac dysfunction is also associated with mutations of troponin C (TnC). Troponin C mutants A8V, C84Y, and D145E increase the Ca2+ sensitivity of ATPase activity. We show that these mutants change the distribution of regulated actin states. The A8V and C84Y TnC mutants decreased the inactive B state distribution slightly at low Ca2+ concentrations, but the D145E mutants had no effect on that state. All TnC mutants increased the level of the active M state compared to that of the wild type, at a saturating Ca2+ concentration. Troponin complexes that contained two mutations that stabilize the active M state, A8V TnC and Δ14 TnT, appeared to be completely in the active state in the presence of only Ca2+. Because Ca2+ gives full activation, in this situation, troponin must be capable of positioning tropomyosin in the active M state without the need for rigor myosin binding.
Graphical Abstract:

Hypertrophic cardiomyopathy is a cardiac disease in which the left ventricle becomes stiff, thus compromising muscle relaxation during diastole.1 Missense and frame-shift mutations in sarcomeric proteins are the most common cause of this clinical presentation.1 On average, 75% of the hypertrophic cardiomyopathy patients screened for sarcomeric mutations are found to be positive for at least a single gene mutation, including those encoding proteins of the troponin complex.2–5 However, the molecular basis for the poor relaxation kinetics of the heart and diastolic dysfunction in these patients remains obscure. We investigated the effects of mutations in the regulatory apparatus to understand how regulation is altered in hypertrophic cardiomyopathy.
Cardiac troponin C (TnC) is a Ca2+ binding protein of the thin filament that confers Ca2+ sensitivity to the actin-activated ATPase activity of myosin.6,7 At low resting levels of intracellular Ca2+, the single functional site of the N-terminal domain, site II, is free of bound Ca2+, while high-affinity sites III and IV in the C-terminal domain are bound to Ca2+ and/or Mg2+.8 When Ca2+ is released from the sarcoplasmic reticulum into the cytoplasm, it binds to site II of TnC. This promotes a series of conformational changes in the troponin–tropomyosin complex that culminates in faster actomyosin cycling, thus generating contraction.9–12 Troponin is a ternary complex that in addition to TnC is comprised of troponin I (TnI), the subunit known to inhibit contraction,13 and troponin T (TnT), which connects the ternary complex to tropomyosin and directly modulates the rate of actomyosin ATPase activity.14,15 The ability of troponin to control actin activation of ATPase activity is known to be altered by hypertrophic cardiomyopathy-associated mutations in troponin.16
The ability of the actin–tropomyosin–troponin complex to stimulate the ATPase activity of myosin S1 depends on the level of Ca2+ bound to troponin C and S1 or S1-ADP bound to actin. Many features of actin activation of ATPase activity can be simulated by assuming that these actin states fall into two categories: those that activate ATP hydrolysis and those that do not.17 The activating and nonactivating species of actin were assumed to bind to S1 during steady state ATP hydrolysis as suggested by experimental evidence.18–21 Several lines of evidence suggest three states of regulated actin are in rapid equilibrium with each other at a saturating Ca2+ concentration.22–25 Despite the fact that two of these states are nonactivating, the transitions between them are important because these transitions are affected by disorders such as hypertrophic cardiomyopathy.16
The inactive state is heavily populated in the absence of either bound Ca2+ or activating cross-bridges. This state is often called the blocked state (B state) because tropomyosin appears to block the binding site of activating cross-bridges (rigor S1 or S1-ADP). Myosin–ATP can bind to actin in this state, however.18,26–28 Solution studies show that the VMax, rather than the KM, is most effected by an increase in the Ca2+ concentration.19,28–30 At a saturating Ca2+ concentration, ∼60% of the actin is in an intermediate state that is often called the closed or calcium state (C state). That state also has low activity with respect to stimulating myosin ATPase activity.25 The active state, sometimes called the open or myosin state (M state), is stabilized by high-affinity myosin binding31,32 and by some reports requires both Ca2+ and myosin binding.33 The ATPase activity of actin–myosin is approximately defined by the fraction of actin in the active state. Note that in some earlier publications we used the terms I, X, and A for the inactive, intermediate, and active forms of regulated actin, respectively.
Actin–tropomyosin–troponin complex-activated ATPase activity is a function of the cooperative transition from the inactive to the active states that occurs with an increase in the Ca2+ concentration32,34,35 and the absolute activities at the limits of zero Ca2+ and saturating Ca2+ concentrations. These limiting activities are altered by the troponin structure; several mutants of TnT and TnI changed these limiting activities by changing the distribution of states of regulated actin at the extremes of Ca2+.30,36–38 We wished to determine if mutations in TnC function in the same manner.
Three mutations in the TnC gene have been linked to hypertrophic cardiomyopathy in humans.39 Recent reviews describe these and other TnC mutations.40–42 Incorporation of the TnC mutants A8V, C84Y, and D145E into skinned fibers increased the Ca2+ sensitivity of contraction, which was also recapitulated in a reconstituted actomyosin ATPase assay.4,39 Furthermore, A8V TnC knock-in mice develop diastolic dysfunction.43 The changes caused by these TnC mutations might be linked to an increase in the level of the M state at a saturating Ca2+ concentration.
We now show that the cardiomyopathy mutations A8V, C84Y, and D145E in TnC stabilize the active state of regulated actin to various extents. Furthermore, actin filaments containing both A8V TnC and Δ14 TnT, another activating mutation36,38 that is associated with hypertrophic cardiomyopathy,44,45 were fully activated by Ca2+ without high-affinity binding of myosin to actin. This demonstrates that troponin can actively direct tropomyosin to the activating position as well as perform its established function of stabilizing the inhibitory position.
EXPERIMENTAL PROCEDURES
Proteins.
Actin was prepared from rabbit back muscle.46 Pyrene-labeled actin was prepared by reacting F-actin with N-(1-pyrenyl) iodoacetamide.47 Myosin was prepared from rabbit back muscle.48 Skeletal myosin S1 was made from myosin digested with chymotrypsin.49 N-Ethylmaleimide-modified S1 (NEM-S1) was prepared by reacting 33 μM S1 with 495 μM N-ethylmaleimide for 30 min at 25 °C in a buffer containing 100 mM KCl and 50 mM tris(hydroxyethyl)aminomethane hydrochloride (pH 8.0).50 The reaction was quenched with 5 mM dithiothreitol, and NEM-S1 was dialyzed exhaustively against 100 mM KCl and 10 mM MOPS, frozen in a dry ice/ethanol bath, and stored at −80 °C.
Bovine cardiac left ventricles were used to prepare tropomyosin.51 Acrylodan-labeled cardiac muscle tropomyosin was prepared using a 10:1 acrylodan:tropomyosin molar ratio.38 These conditions gave >70% labeling using an extinction coefficient of 14400 M−1 cm−1 at 372 nm for acrylodan.52
Human cardiac troponin TnT (isoform 2) in pSBETA and human cardiac TnI in pET17b were purified after expression.36 The preparation of wild-type and mutant human cardiac TnC in pET3d was described previously.53 Troponin subunits were reconstituted following the previously described protocol54 with modifications. TnT, TnI, and TnC were mixed in a 1:1:1.2 molar ratio, and following dialysis, the intact troponin complex was isolated by chromatography on a Protein Pak DEAE 15HR column (Waters, Milford, MA). The start buffer consisted of 0.1 M NaCl, 20 mM Tris-HCl (pH 8.0), and 5 mM MgCl2. Troponin was eluted with a 0.1 to 1.0 M NaCl gradient in the same buffer.
Protein concentrations were determined using the following extinction coefficients (ε0.1%) for 280 nm: 1.15 for actin and 0.75 for myosin S1. Concentrations of tropomyosin and troponin subunits were determined by the Lowry protein assay with a bovine serum albumin standard. Protein molecular weights were assumed to be 42000 for actin, 120000 for myosin S1, 68000 for tropomyosin, 24000 for troponin I, 35923 for troponin T, and 18400 for troponin C.
ATPase Assays.
Rates of ATP hydrolysis were measured by the time course of liberation of 32Pi from [γ−32P]ATP.19 The total reaction volume was generally 0.2 mL. A 0.05 mL aliquot of the reaction mixture was analyzed at each of three or four time intervals to ensure that Pi release was linear over the time period in this cooperative system. Data were fitted with a linear least-squares program to determine the initial rate. Measurements were taken at 25 °C with 0.1 μM S1, 10 μM F-actin, 2.2 μM tropomyosin, and various concentrations of troponin. The buffer was 1 mM ATP, 3 mM MgCl2, 34 mM NaCl, 10 mM MOPS, 1 mM dithiothreitol, 2 mM EGTA, or 0.1 mM CaCl2 (pH 7.0).
With two populations of regulated actin, one inactive (B and C states) and one active (M state), the observed rate, vobs, is equal to (1 − fM)(VMax‑ BC[A])/(KM‑ BC + [A]) + f M(VMax‑M[A])/(KM‑M + [A]). The fraction of actin in the active M state is f M, and [A] is the actin concentration. Because [A] < KM and because the M state is much more active than the B and C states, the rates are proportional to fMVMax/KM.
Determination of f M, from values of vobs, requires knowledge of the minimum, vmin, and maximum, vmax, values of ATPase activity under these same conditions of actin concentration, ATP concentration, temperature, and ionic strength. The fraction of regulated actin in the active M state is given by (vobs − vmin)/(vmax − vmin). The lowest actin activated ATPase activity that we have measured was at low Ca2+ with a protein kinase C phosphomimetic mutation, S45E TnI.37 Actin filaments containing that troponin had a rate in EGTA that was 0.61 times that of filaments containing wild-type troponin. We assume that represents the fully inactive state and use that value for vmin. Not to be confused with VMax at a saturating actin concentration, vmax is rate at the actin concentration used to measure vobs when actin is fully stabilized in the M state.
Stabilization of the M state was achieved by high-affinity binding of myosin to regulated actin. The cooperative binding of ATP-free myosin to actin55 enhances the ability of actin to stimulate myosin S1 ATPase activity9,31,56 and maximizes the phosphate release step.33 Myosin S1 that is modified with N-ethylmaleimide has a very low ATPase activity but still retains the ability to bind regulated actin and stabilize the active state.57–59 To maintain a constant free regulated actin concentration in the presence of a competitive inhibitor of binding of S1-ATP to actin, the concentration of the actin− tropomyosin–troponin complex was increased by an amount equal that of the added NEM-S1.59 ATPase rates were corrected for the rate of hydrolysis of S1 in the absence of actin [0.06 μM s−1 (μM S1)−1] and for the rate of hydrolysis of the added NEM-S1 in the presence of the actin–tropomyosin− troponin complex [0.02 μM s−1 (μM NEM-S1)−1].
Rapid Kinetic Measurements.
Binding of rigor S1 to the actin–tropomyosin–troponin complex stabilizes the active state. Rapid dissociation of S1 with ATP in EGTA causes the regulated actin to relax to the inactive B state after transiently passing through the intermediate C state.60 Skeletal S1 detaches more rapidly than cardiac S1 and is preferred for these assays.38
A mixture of actin, troponin, tropomyosin, and S1 (protein concentrations and buffers are given in the figure legends) was rapidly mixed with a high concentration of ATP at 10 °C. The kinetics of the transition from the active state to the inactive state were measured with a SF20 sequential mixing stopped-flow spectrometer (Applied Photophysics, Leatherhead, U.K.). The temperature was maintained with a circulating water bath. Acrylodan was excited through a monochromator set at 370 nm (below the optimum of 391 nm to reduce light scattering) and a slit width of 0.5 mm. The fluorescence was measured through a Schott (Duryea, PA) GG 475 long-pass filter.
The reaction is shown diagrammatically in Scheme 1, where T is ATP, S is myosin S1, and the states of actin are shown in bold. The active (M) and inactive (B) states are characterized by high acrylodan–tropomyosin fluorescence.61
Scheme 1.

The ATP concentration used to dissociate the actin–myosin complex was chosen to make the formation of free actin (in the active M state) occur sufficiently fast that the first two processes in Scheme 1 do not affect the kinetics of the acrylodan tropomyosin changes.61 The transition from actin in the active M state to actin in the C state occurs with a rate constant that is close to the limit of the stopped-flow device. The observed rate constant for that process is essentially k5 + k6. This was confirmed by direct fitting of the differential equations describing Scheme 1 to the data.61 The observed rate constant for the slow increase is essentially equal to k7 + k8. Because k7 + k8 was much slower than the preceding reactions, this slow increase appeared as a monoexponential function.
The equilibrium constants describing the transitions among actin states are KB and KT, where KB = [C]/[B] = k8/k7 and KT = [M]/[C] at equilibrium.22 Apparent rate constants were obtained by exponential fits to the data using the software provided by Applied Photophysics. Estimates of true rate constants were determined by solving the set of differential equations61 using KinTek Explorer.62,63 Rate constants k1−k6 were estimated earlier as follows: k1 = 10 μM−1 s−1, k3 = 700 s−1, k5 = 200 s−1, k6 = 15.4 s−1, and k2 and k4 were set to zero.61 Values of k7 + k8 were equal to the apparent rate constants obtained from exponential fits.
The amplitude of the slow fluorescence increase, corresponding to k7 + k8, is a measure of the fraction of actin in the inactive B state.38,61 Wild-type regulated actin filaments are approximately 87% in the inactive B state at low Ca2+ concentrations (see Table 2) with the rest primarily in the intermediate low-fluorescence C state. Thus, the amplitude of the fluorescence increase is proportional to the occupancy of the inactive B state.
Table 2.
Distribution of Regulated Actin States at Low Ca2+ Concentrations for Several Types of Troponin
| KB(pyrene)a | Bpyreneb | MATPasec | Facr/Facr‑wtd | Bacrylodane | |
|---|---|---|---|---|---|
| WT | 0.15 | 0.87 | 0.01 | 1 | 0.87(column 2) |
| C84Y TnC | nd | ndf | 0.02 | 0.85 | 0.74 |
| D145E TnC | nd | ndf | 0.01 | 1.03 | 0.9 |
| A8V TnC | 0.2 | 0.83 | 0.01 | 0.78–0.87 | 0.68–0.76 |
| Δ14 TnT | ndf | ndf | 0.04 | ≈0 | ≈0 |
| A8V TnC and Δ14 TnT | 2.1f,g | 0.31f,g | 0.05 | ≈0 | ≈0 |
From ref 64: KB = 1/(kcalcium/kEGTA − 1).
B =1 − M − C. C/B = KB. B = (1 − M)/(1 + KB).
From ATPase rate measurements normalized to filaments stabilized in the M state (Figures 2 and 4). ATPase measurements were taken at ionic strengths lower than those used for fluorescence measurements.
Fluorescence amplitudes of acrylodan relative to the wild type give the B state relative to the wild type.
B state estimated from column 4 assuming the wild-type value from pyrene–actin studies.
There is no assurance that KT is small in these systems as required for calculation of KB from S1 binding kinetics.
KB calculated using kcalcium as the average from wild-type and A8V TnC cases. Using kcalcium from the A8V TnC and Δ14 TnT pair would decrease all values of KB.
Equilibrium constant KB can also be determined from the ratio of the observed rate of binding of S1 to pyrene–actin in EGTA (kEGTA) to that under saturating Ca2+ conditions (kcalcium):64 KB is 1/(kcalcium/kEGTA − 1). This equation is valid when KT is small compared to KB. The process was assumed to follow Scheme 2, where the states of actin are in bold and an asterisk indicates a high-fluorescence state. The assumptions are that Ca2+ alone gives the maximal rate of binding of ATP-free S1, that the reduced rate of binding in EGTA is due to the reduction in the number of actin sites available for S1 binding in the inactive or blocked state, and that the affinity of actin for S1 is the same for the C and M states. That is, if the B state binds to rigor S1 but with a reduced rate constant,65 then rate constants k8′ and k7′ are not necessarily identical with k8 and k7 measured with acrylodan–tropomyosin.
Scheme 2.
Pyrene actin binding kinetics were measured in the stopped flow using excitation at 365 nm with emission measured through a 384 nm long pass filter with a midpoint of 400 nm. Slit widths of 0.5 or 1 mm were used. Pyrene-labeled actin was stabilized with a 1:1 complex of phalloidin (Sigma-Aldrich, St. Louis, MO). An excess of phalloidin-stabilized pyrene-labeled actin–tropomyosin–troponin complex was rapidly mixed with nucleotide-free myosin S1 in the stopped flow. Ratios of initial rates of fluorescence decreases (low Ca2+ to high Ca2+) were used to estimate the population of the B state.
The curve for the fractional change in rate as a function of the total troponin concentration was determined by fitting a quadratic binding equation to the data using Mathematica 10.4 (Wolfram Research, Inc. Champaign, IL). The fraction of change in rate was assumed to be proportional to the amount of troponin bound: fractional change = {ATotal/7 + TTotal + KD − [(ATotal/7 + TTotal + KD)2 − (4ATotal)/(7TTotal)]1/2}/2. The dissociation constant for binding of troponin to the actin− tropomyosin complex is KD; ATotal and TTotal are the total concentrations of actin and troponin, respectively. The factor of 7 was to convert the total actin concentration into the concentration of possible troponin binding sites. Student’s t tests were performed with either SigmaPlot (Systat Software, San Jose, CA) or SPSS (IBM Corp., Armonk, NY).
RESULTS
ATPase measurements in solution can define changes in the functional state of the actin–tropomyosin–troponin complex that occur with disease-associated mutations. However, changes in ATPase rates reflect changes in distribution only if two conditions are met. First, the ATPase rates must be compared at comparable levels of saturation of actin filaments with each type of troponin complex. Second, the ATPase rates must be normalized to the rates of the inactive and fully active states of regulated actin under the same conditions.
Figure 1 shows the actin-activated myosin S1 ATPase activity, at a low Ca2+ concentration, as a function of the concentration of each type of troponin used. There are small differences in activity among the troponin types. In particular, the C84Y mutant was slightly less inhibitory than wild type with a p value of 0.008. However, the TnC mutations do not appear to have significantly changed the affinities of troponin for actin–tropomyosin. The inset of Figure 1 shows the ATPase data normalized to the highest and lowest rates observed with each troponin. This shows more clearly that the ATPase rate has a similar concentration dependency for each troponin type. That is, the mutations did not cause a significant change in the affinity of troponin for actin–tropomyosin.
Figure 1.
ATPase rates as a function of the ratio of concentrations of total troponin to actin. The troponin contained wild-type TnI and TnT. The TnC was the wild type (○), the A8V mutant (□), the D145E mutant (◇), or the C84Y mutant (△). The figure also shows data of skeletal troponin for comparison (●). The inset shows the ATPase rates normalized to the range of 0–1 for comparison among the troponin types. Measurements were taken at 25 °C and pH 7.0 in solutions containing 1 mM ATP, 3 mM MgCl2, 34 mM NaCl, 10 mM MOPS, 1 mM dithiothreitol, and 2 mM EGTA. The concentrations of S1, actin, and tropomyosin were 0.1, 10, and 2.2 μM, respectively. Data are plotted with the standard deviation.
The ATPase activities, in the absence of Ca2+ at the high troponin concentrations of Figure 1, are compared with those of other types of actin filaments in Figure 2. The concentration of ATP was saturating, but the actin concentration was well below the KM. Actin was most active as a cofactor in the absence of tropomyosin and troponin. Tropomyosin reduced the activity to 0.63 of the unregulated rate. Addition of wild-type troponin reduced the rate to 0.17 of the unregulated rate. The three TnC mutants produced minor changes in the degree of inhibition. The largest effect was seen with C84Y TnC where the activity increased 20% compared with that of wild-type troponin. For comparison, Δ14 TnT, which is known to eliminate the B state of regulated actin,38 doubled the ATPase activity. Interestingly, the double mutant of the troponin complex containing A8V TnC and Δ14 TnT increased the rate 2.4-fold compared to that of wild-type troponin containing actin filaments. Although the average activity of filaments containing the double mutant exceeded that of filaments containing Δ14 TnT, the difference between them was not statistically significant (p = 0.67).
Figure 2.
Effect of different regulatory complexes on ATPase activities in the absence of Ca2+. Three TnC mutants, one TnT mutant (Δ14), and one double mutant are compared with the wild type (WT), bare actin filaments (actin), and actin-tropomyosin (actin-Tm). The conditions were the same as those described for Figure 1, except that the troponin concentration was fixed at 2.8 μM. Data are plotted with the standard deviation. Of the TnC mutants, only the C84Y mutant was significantly different from the wild type with a p value of 0.008.
Under saturating Ca2+ conditions (Figure 3), the wild-type actin–tropomyosin–troponin complex was approximately twice as effective as actin alone at stimulating myosin ATPase activity. The activities of actin filaments containing the C84Y, D145E, and A8V mutants of TnC were all greater than those of wild-type regulated actin filaments. All of these changes were significant with p values of 0.026, 0.015, and 0.004, respectively. Regulated actin filaments containing A8V TnC had 3.7 times the activity of pure actin. The activity produced by actin filaments containing A8V TnC approached that observed with the Δ14 TnT mutant (4.4 times that of pure actin). The Δ14 TnT mutant produced higher levels of activation with Ca2+ alone than any troponin type that we have investigated.36,38 Hybrid troponin that contained both A8V TnC and Δ14 TnT produced levels of activation of myosin S1 ATPase rates higher than that of either mutant alone. The double mutant had 1.8 times the activity of A8V alone (p = 0.001) and 1.5 times the activity of Δ14 TnT alone (p = 0.019). Filaments containing the double mutant produced ATPase rates that were 6.5 times those of pure actin.
Figure 3.
Effect of different regulatory complexes on ATPase activities at a saturating Ca2+ concentration. Three TnC mutants, one TnT mutant (Δ14), and one double mutant are compared with the wild type (WT), bare actin filaments (actin), and actin-tropomyosin (actin-Tm). The conditions were the same as those described for Figure 1, except that the EGTA was replaced with 0.1 mM CaCl2 and the troponin concentrations were fixed at 2.8 μM. The activities of actin filaments with the TnC mutants were significantly higher than that of wild type with p values of 0.026, 0.015, and 0.004 for the C84Y, D145E, and A8V TnC mutants, respectively. The actin and actin-tropomyosin data are the same as those from Figure 2 and are shown for comparison. Data are plotted with the standard deviation.
To determine the maximal actin-activated ATPase activity at this actin–tropomyosin–troponin complex concentration, we measured activities at a saturating Ca2+ concentration in the presence of NEM-S1. Actin filaments containing Δ14 TnT troponin were particularly prone to form the M state and were employed as a control to ensure that we had reached full activation. Figure 4 shows that the activity of bare actin filaments was unaffected by the presence of NEM-S1. Actin filaments containing tropomyosin, and wild-type TnC, C84Y TnC, D145E TnC, or A8V TnC containing troponin, were more active in the presence of NEM-S1. Actin filaments containing tropomyosin and both A8V TnC and Δ14 TnT had the same activity as in the presence of Ca2+ but in the absence of NEM-S1 (see Figure 3). Therefore, these actin filaments were fully activated by Ca2+ without the need for high-affinity myosin binding. Table 1 gives values of the fraction of regulated actin in the active M state using ATPase values from the figures.
Figure 4.
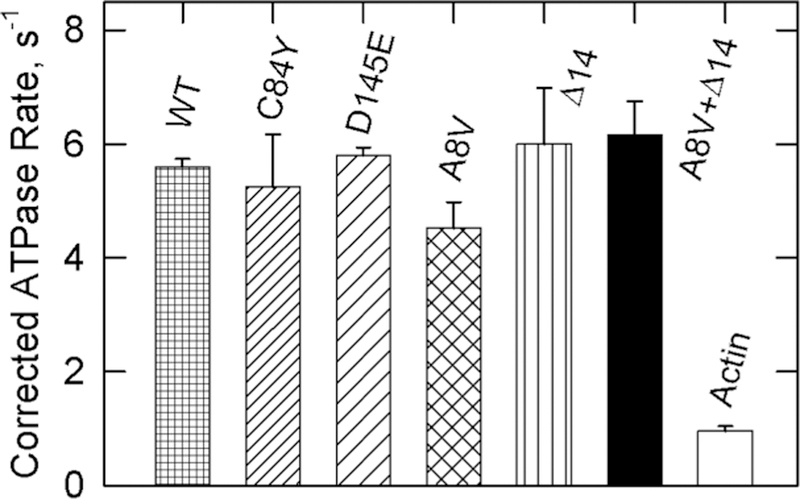
Effect of different regulatory complexes on ATPase activities at a saturating Ca2+ concentration in the presence of NEM-S1. Three TnC mutants, one TnT mutant (Δ14), and one double mutant are compared with the wild type (WT) and bare actin filaments (actin). The actin data are the same as those from Figure 2 and are included for comparison. The conditions were the same as those described for Figure 1, except that the EGTA was replaced with 0.1 mM CaCl2. NEM-S1 (10 μM) was included in each assay to fully stabilize the active state. Furthermore, the concentrations of actin, tropomyosin, and troponin were increased to 20, 4.4, and 5.7 μM, respectively, to maintain 10 μM free actin as in Figures 1–3. None of the TnC mutants had activities significantly different from that of the wild type (p > 0.33). Data are plotted with the standard deviation.
Table 1.
Fractions of the Actin–Tropomyosin–Troponin Complex in the Active M State under the Conditions Described for Figsures 1–4
| wild type | C84Y TnC | D145E TnC | A8V TnC | A8V TnC and Δ14 TnT | |
|---|---|---|---|---|---|
| fraction MEGTA | 0.01 | 0.02 | 0.01 | 0.01 | 0.05 |
| fraction MCa2+ | 0.31 | 0.4 | 0.41 | 0.42 | 1.0 |
Δ14 TnT increases the population of the M state and eliminates the B state. We wished to determine if activating TnC mutants also eliminated or reduced the B state. Measurements of ATPase activity are inaccurate for distinguishing the B and C states because both states have little ability to stimulate ATPase activity. We showed earlier that the fluorescence of acrylodan-labeled tropomyosin increases when the C state undergoes its transition to the B state,38,61 giving a qualitative indication of the presence of the B state. Furthermore, the ratio of amplitudes to a standard gives the occupancy of the B state relative to that standard, in this case wild-type troponin.
Figure 5 shows time courses of acrylodan–tropomyosin fluorescence changes as actin underwent its transition from the active M state following rapid detachment of S1-ATP. The average results of two experiments are shown. The experiment that yielded the data presented in panel B had a higher free troponin concentration and a lower ionic strength compared to those used for panel A and was performed with different preparations of proteins. Similar results were obtained under both conditions.
Figure 5.
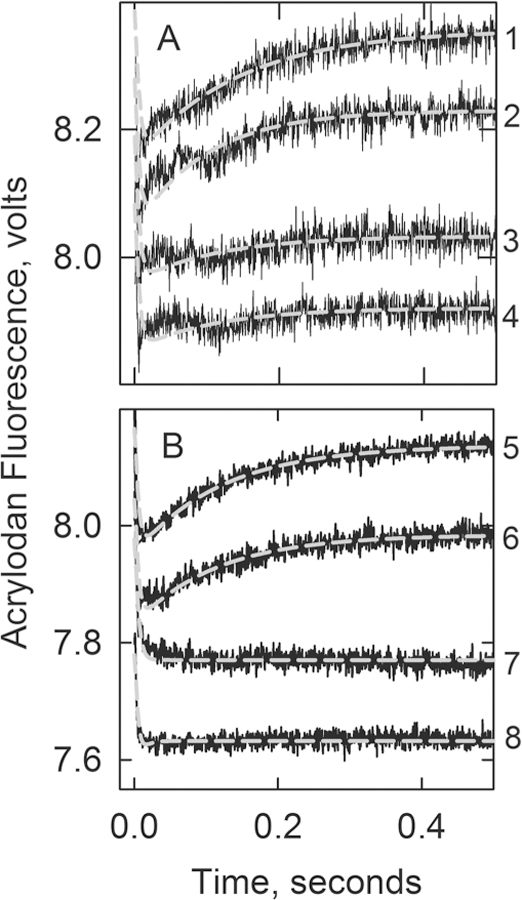
Acrylodan-labeled tropomyosin fluorescence changes following the rapid detachment of myosin S1 in the absence of Ca2+ at 10 °C: curve 1, actin filaments with wild-type troponin; curve 2, A8V TnC; curve 3, A8V TnC and Δ14 TnT; curve 4, Δ14 TnT. The gray lines are best fits of differential equations describing Scheme 1 to the data (see Experimental Procedures). Panel A included 4 μM actin, 0.86 μM tropomyosin, 0.86 μM troponin, and 2.5 μM S1 in 20 mM MOPS, 152 mM KCl, 4 mM MgCl2, 1 mM dithiothreitol, 2 mM EGTA rapidly mixed with 4 mM ATP, 20 mM MOPS, 132 mM KCl, 8 mM MgCl2, 1 mM dithiothreitol and 2 mM EGTA. Values of k7 + k8: 7.8 s−1 for the wild type and 7.7 s−1 for A8V. Amplitudes: 0.17 ± 0.05 V for the wild type, 0.15 ± 0.03 V for A8V, and <0.02 for Δ14 TnT and the double mutant. Panel B included 2 μM actin, 0.86 μM troponin, 0.86 μM tropomyosin, 2 μM S1 in 90 mM KCl, 20 mM MOPS buffer (pH 7), 4 mM MgCl2, 2 mM EGTA, and 1 mM dithiothreitol rapidly mixed with 2 mM ATP in 90 mM KCl, 20 mM MOPS buffer (pH 7), 4 mM MgCl2, 2 mM EGTA, and 1 mM dithiothreitol at 10 °C. Values of k7 + k8: 8.1 s−1 for the wild type and 7.7 s−1 for A8V. Amplitudes: 0.17 ± 0.02 V for the wild type and 0.13 ± 0.01 V for A8V.
The rapid fluorescence decrease, completed within 50 ms (near the limit of detection), results from the transition from the active M state to the intermediate C state. Actin filaments containing the wild type (curve 1), A8V (curve 2), C84Y (not shown), or D145E (not shown) exhibited a subsequent fluorescence increase as the inactive B state became populated. The average values for k7 + k8 were 7.8 and 7.7 s−1 for the filaments containing the wild type and A8V TnC, respectively (see Scheme 2). Actin filaments containing A8V troponin had 87 and 78% of the amplitude of filaments containing wild-type troponin in panels A and B, respectively. Actin filaments containing C84Y TnC exhibited an amplitude of ∼85% of that of the wild type, while those with D145E TnC had an amplitude of acrylodan tropomyosin fluorescence that was indistinguishable (1.03 times) from that of the wild type.
Table 2 lists the fluorescence amplitudes relative to the wild type (Facr/Facr‑wt) for actin filaments containing various troponin species. The last column of Table 2 gives the estimated fraction of actin in the B state with the assumption that the occupancy of wild-type filaments is 87% B state.
Regulated actin filaments containing Δ14 TnT or both Δ14 TnT and A8V TnC showed little or no fluorescence increase characteristic of the B state. There was a possible increase of ≤12% for these systems in Figure 5A. However, no such increase was seen in Figure 5B or in our earlier work with Δ14 TnT.38 Taken together, the results of Figure 5 show that while the Δ14 TnT mutation appeared to eliminate the B state at low Ca2+ concentrations, the A8V TnC mutation alone decreased the probability of forming the B state marginally but D145E TnC had no effect.
The population of the B state was also estimated by the rate of binding of S1 to pyrene-labeled actin filaments in the absence of a nucleotide.22 Figure 6A shows time courses for binding of S1 to the actin–tropomyosin–troponin complex at very low free Ca2+ concentrations. Actin filaments containing A8V TnC alone (curve 2; kapp = 0.12 s−1) exhibited similar rates of binding to wild-type regulated actin filaments (curve 1; kapp = 0.1 s−1). S1 bound to actin filaments containing both A8V TnC and Δ14 TnT (curve 3; kapp = 0.5 s−1) at a significantly faster rate. Because the rate of binding to the B state is slower than that to the C and M states, this indicates a decrease in the B state population for the double mutant.
Figure 6.
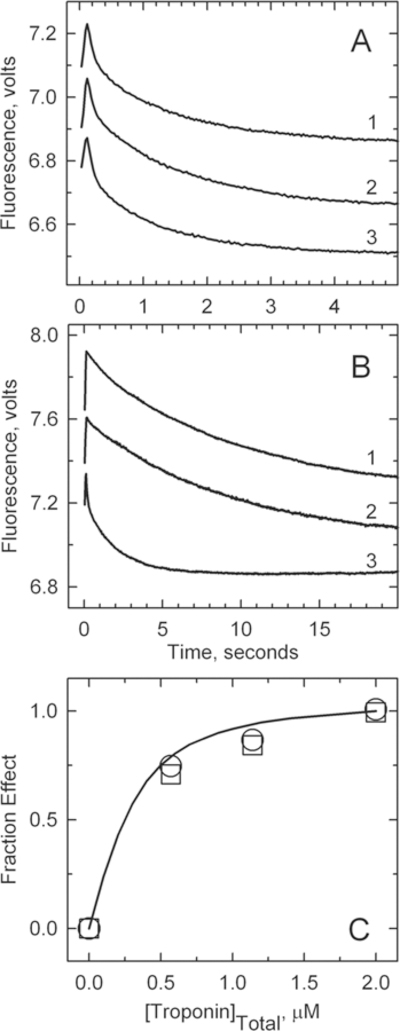
Rate of binding of S1 to pyrene-labeled actin filaments containing tropomyosin and troponin in the absence of ATP. For this experiment, 4 μM pyrene–actin (40% labeled), 0.86 μM tropomyosin, and 2 μM troponin were rapidly mixed with 0.4 μM myosin S1 in a buffer containing 152 mM KCl, 2 mM EGTA, 20 mM MOPS buffer (pH 7), 4 mM MgCl2, and 1 mM dithiothreitol at 10 °C. (A) Low Ca2+ concentrations. Curve 1: wild type, kapp = 0.10 ± 0.01 s−1. Curve 2: A8V, k = 0.12 ± 0.02 s−1. Curve 3: A8V TnC and Δ14 TnT, kapp = 0.5 ± 0.02 s−1. Solid lines are monoexponential fits to each curve. (B) High Ca2+ concentrations. Curve 1: wild type, kapp = 0.77 ± 0.14 s−1. Curve 2: A8V, kapp = 0.70 ± 0.05 s−1. Curve 3: A8V TnC and Δ14 TnT, kapp = 0.91 ± 0.15 s−1. (C) Fraction of the maximal effect on the binding kinetics at low Ca2+ concentrations vs the free troponin concentration: wild type (○) and A8V (□). The highest point corresponds to the conditions used for the main figure. The fitted curve is for a dissociation constant of 0.12 μM.
Figure 6B shows time courses for binding of S1 to the actin− tropomyosin–troponin complex at a saturating Ca2+ concentration. Again, the rate of binding of S1 to actin filaments containing A8V TnC alone (curve 2; kapp = 0.7 s−1) was similar to that for wild-type filaments (curve 1; kapp = 0.77 s−1). S1 bound to actin filaments containing both A8V TnC and Δ14 TnT (curve 3; kapp = 0.91 s−1) at a significantly faster rate. This might be an indication of previously unidentified differences in binding kinetics between the C and M states.
Figure 6C shows the fractional change in the rate of binding of S1 to actin−tropomyosin as a function of the concentration of free troponin (2 μM total troponin gave 1.4 μM free troponin, and 1.1 μM total troponin gave 0.62 μM free troponin). The affinity of A8V TnC troponin for actin− tropomyosin was indistinguishable from that of wild-type troponin as both exhibited the same concentration dependence. Actin filaments containing both A8V TnC and Δ14 TnT were also at a plateau of activity at the same troponin concentration (data not shown). Data shown in panels A and B of Figure 6 were from the highest troponin concentration shown in Figure 6C.
Estimates of the fraction of actin filaments in the B state were made from ratios of binding rates at a saturating Ca2+ concentration to those at a very low Ca2+ concentration. Column 2 of Table 2 shows that A8V TnC increased the value of KB slightly, leading to a 5% reduction in the level of the B state (column 3). This change was smaller than that calculated from acrylodan−tropomyosin studies. The value of KB for the system containing A8V TnC and Δ14 TnT was elevated by 14-fold, leading to a large reduction in the level of the B state. However, the reduction in the level of the B state was not as large as that observed in the acrylodan–tropomyosin studies.
DISCUSSION
Disease-associated mutations in human cardiac TnT and TnI lead to changes in the distribution of states of regulated actin.16,30 We show here that some disease-associated TnC mutations alter the distribution of states of regulated actin at high and low free Ca2+ concentrations (Tables 1 and 2). The D145E mutant produced the greatest effect in our earlier ATPase assays under different conditions,53 so that mutant was also included in some of the more detailed analyses. However, the focus of the work presented here was the A8V TnC mutant that tended to produce larger effects in our current assays. The A8V TnC mutation decreased the level of the B state to a small degree under relaxing conditions but increased the level of the active M state under activating conditions. The effects were not as dramatic as observed earlier with the Δ14 TnT mutation, which eliminated the B state under low-Ca2+ conditions and stabilized the M state under high-Ca2+ conditions to a greater extent.36,38
Changes in the distribution of actin states may be a normal regulatory function of troponin. For instance, introduction of negative charges at the protein kinase C phosphorylation sites of TnI (S45E, S43E/S45E, and S43E/S45E/T144E) changed the actin state distribution to favor the inactive state.37 An important function of the troponin complex is to modulate the orientation of tropomyosin on actin filaments, and it appears that the magnitude of this function may be altered by disease or by regulatory changes.
It may be surprising that changes in the structure of TnC or TnT increased the stability of the M state. The active M state is named after myosin because it is the state that is stabilized by high-affinity myosin cross-bridges. This comes from early steady state measurements of ATP hydrolysis that showed an acceleration of activity with time as the amount of S1-ADP bound to actin increased.17,31,56 However, as shown in Figure 3, troponin promotes the M state of regulated actin even in the absence of high-affinity cross-bridge binding. That is, wild-type cardiac troponin–tropomyosin increased the activity relative to that of pure actin. An earlier study showed a more pronounced effect when skeletal troponin–tropomyosin was added to amoeba actin.66 In all of these cases, the rates of ATP hydrolysis were greater than those measured with actin having neither tropomyosin nor troponin bound. This clearly showed that the actin–tropomyosin–troponin complex is a more effective cofactor in ATP hydrolysis than is actin. This concept is discussed in more detail elsewhere.67
Although wild-type cardiac troponin partially stabilized the M state, in the absence of strong myosin binding (Figures 2 and 3), the three TnC mutants (C84Y, D145Y, and A8V) gave an additional 1.3-fold stabilization of that state (Figure 3). This result is consistent with earlier reports showing troponin containing A8V or D145E TnC to be more activating than reconstituted wild-type troponin.53,68,69 The Δ14 TnT mutant has been shown previously to stabilize the M state ∼2-fold relative to the wild type.36,38
We show here that troponin containing both A8V TnC and Δ14 TnT produced virtually full occupancy of the M state at a saturating Ca2+ concentration in the absence of a high-affinity S1 species (Figures 3 and 4 and Table 1). We conclude that cardiac troponin, with Ca2+ bound, has been designed to position tropomyosin in the M state but only to a limited degree. Mutations in troponin can remove that limitation. The advantage of this self-limitation may be to allow high-affinity myosin binding to give further activation. Some mutations in troponin circumvent the system and give greater activation with Ca2+, leading to progressive cardiac failure. Rigor-type binding of myosin S1 to actin occupies the B and C state sites for tropomyosin binding, so that tropomyosin is forced into the actin helical groove (M state). Troponin may produce the same end by an alternative mechanism.
The ability of the C84Y, D145E, and A8V TnC mutants to increase the population of the M state of regulated actin at a saturating Ca2+ concentration may be related to the increased affinity of troponin for Ca2+ produced by these mutants.53,70 Binding of rigor-type cross-bridges to regulated actin increases Ca2+ affinity,70,71 so it follows that an increasing Ca2+ affinity is linked with an increase in the M state. The D145E and A8V TnC mutants had different effects on binding of Ca2+ to the nonregulatory C domain sites of isolated TnC.72 The greatest effect was seen with the D145E TnC mutant, where the Ca2+ binding affinity was greatly weakened. At present, it is unknown how alterations in the C domain affect binding of Ca2+ to the regulatory sites.
Because the C84Y, D145E, and A8V TnC mutants stabilize the M state of regulated actin, they make the actin filaments more sensitive to activators such as Ca2+ and rigor-type cross-bridges. However, actin filaments containing any type of troponin seem to have the same limiting ATPase activity. As this limit is approached, the effect of activators is blunted.
The A8V TnC mutant produced a small decrease in the B state (Table 2). It was shown earlier that A8V TnC has an increased α-helical content in the presence and absence of divalent metals.53 The A8V mutation is located in the N helix of TnC where it induces a more open conformation of the N-terminal domain in the presence and absence of Ca2+.73 That observation was substantiated by a demonstration that TnI binds more tightly to A8V TnC than to wild-type TnC under low-Ca2+ conditions.68
We observed earlier that the Δ14 TnT mutant appeared to eliminate the B state.38 Similarly, we show here the actin filaments containing Δ14 TnT or Δ14 TnT and A8V TnC had little or no occupancy of the B state as judged from acrylodan− tropomyosin fluorescence changes. The kinetics of binding of S1 to pyrene-labeled actin also showed a reduction in the B state but to a lesser degree. Whereas the acrylodan− tropomyosin study showed 0% occupancy of the B state, S1 binding kinetics showed 31% occupancy of the B state. This difference may, in part, result from the assumptions used to estimate the B state from acrylodan tropomyosin fluorescence changes and S1 binding kinetics to pyrene actin. Acrylodan− tropomyosin fluorescence gives changes in the B state relative to the wild type. The absolute B state was estimated by assuming that wild type was 87% in the B state (determined from S1 binding kinetics). The level of agreement between the methods diverged as the change in the B state increased.
Interpretation of the pyrene–actin study is complicated by the observation that the rate of binding of S1 to filaments containing the double mutant was higher than that of the wild type at a saturating Ca2+ concentration, yet it has generally been assumed that binding to wild-type filaments at a saturating Ca2+ concentration is at its maximum; i.e., the kinetics of binding to the C and M states is identical. If the maximal rate of S1 binding is actually that of the double mutant, then all values in column 1 of Table 2 are overestimates and the differences between the methods would be even greater. Perhaps the assumptions for estimating KB from S1 binding kinetics are not valid for the A8V TnC and Δ14 TnT double mutant. Calculation of KB by this method requires that KT be small; that may not be true in the case of the A8V TnC and Δ14 TnT double mutant. Rationalizing these differences will require additional investigation. However, both measurements gave similar qualitative changes: the TnC mutants had much smaller effects on the B state under low-Ca2+ conditions compared with the effects of Δ14 TnT and the combined Δ14 TnT and A8V TnC mutants.
The different methods required to measure occupancies of the M and B states required different experimental conditions. ATPase measurements were taken at an ionic strength of ≈50 mM to obtain sufficient actin activation to accurately determine changes in the M state. Measurements of the B state with binding of S1 to pyrene-labeled actin were taken at an ionic strength of ≈180 mM but in the absence of ATP; pyrene fluorescence reveals the transition to the high-affinity actin− myosin interaction. Acrylodan–tropomyosin measurements of the B state were taken at ionic strengths of ≈122 and ≈190 mM with similar results. The differences in conditions could result in errors in the estimation of the C state in Table 2 because the C state was determined by the conservation of mass. However, because the M state was sparsely populated, the errors are likely to be small.
Each of the three TnC mutants examined here favored the active M state (increased KT) at a saturating Ca2+ concentration. The C84Y and A8V mutations of TnC also favored the C state (increased KB) at low Ca2+ concentrations. The other activating mutant that we investigated, Δ14 TnT, had a greater stabilizing effect on the M state and appeared to eliminate the B state altogether.36,38 This means that the two equilibrium constants controlling the distribution of regulated actin states may be independently affected by mutations. This could be an important consideration when attempting to manage disorders of the regulatory apparatus.
Comparison of Figures 3 and 4 shows that the effects of A8V TnC and Δ14 TnT were additive; other examples of additive effects of troponin mutants have been reported.38,74 The implication of additivity is that the deleterious effects of a mutant might be mitigated by altering the structure of troponin (i.e., by drug binding or modification of an amino acid residue) at a site remote from the mutation.
We use the term B state here for the inactive state as this is now common usage. However, we do not imply that the binding of myosin S1 to actin is blocked during steady state ATP hydrolysis as there is evidence that such binding occurs.18,20,27,28,33 Tropomyosin does partially block the binding of rigor S1 and S1-ADP to regulated actin at low free Ca2+ concentrations. However, it seems reasonable that this provides a mechanism for a high myosin concentration to influence the activity of regulated actin just as Ca2+ does.75 The results presented here are consistent with the view that tropomyosin controls the activity of actin rather than acting simply as a blocker/unblocker of myosin binding. Both binding of S1 to actin and, in the case of these mutants, binding of Ca2+ to troponin appear to move tropomyosin deep into the actin groove where full activation can occur. At low Ca2+ concentrations, and in the virtual absence of S1-ADP or rigor S1, troponin positions tropomyosin in the inactivating position where actin is an ineffective activator of myosin ATPase activity. While it has been appreciated for some time that troponin can stabilize tropomyosin in an inactivating position on actin, it now seems that troponin can also stabilize tropomyosin in an activating position even without high-affinity binding of myosin to actin.
ACKNOWLEDGMENTS
The authors thank Dr. Li Zhu for assistance with some of the rapid kinetic measurements.
Funding
This research was supported by the National Heart, Lung and Blood Institute of the National Institutes of Health (Grant HL128683 to J.R.P. and Grant ARO35216 to J.M.C.) and a Brody Brothers grant to J.M.C.
ABBREVIATIONS
- acrylodan
6-acryloyl-2-dimethylaminonaphthalene
- EGTA
ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- NEM
N-ethylmaleimide
- S1
myosin subfragment 1
- MOPS
3-(N-morpholino)propanesulfonic acid
- TnC
troponin C
- TnI
troponin I
- TnT
troponin T
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, Anderson ME, Boineau R, Byrne BJ, Cappola TP, Kalluri R, LeWinter MM, Maron MS, Molkentin JD, Ommen SR, Regnier M, Tang WH, Tian R, Konstam MA, Maron BJ, and Seidman CE (2010) Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation 122, 1130–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Marian A (2008) Utilities and limitations of genetic testing for hypertropic cardiomyopathy. Expert Opin. Med. Diagn 2, 539–546. [DOI] [PubMed] [Google Scholar]
- (3).Konno T, Chang S, Seidman JG, and Seidman CE (2010) Genetics of hypertrophic cardiomyopathy. Curr.Opin. Cardiol 25, 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, and Potter JD (2010) Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol 48, 882–892. [DOI] [PubMed] [Google Scholar]
- (5).Maron BJ, and Maron MS (2013) Hypertrophic cardiomyopathy. Lancet 381, 242–255. [DOI] [PubMed] [Google Scholar]
- (6).Greaser ML, and Gergely J (1971) Reconstitution of troponin activity from three protein components. J. Biol. Chem 246, 4226–4233. [PubMed] [Google Scholar]
- (7).Farah CS, and Reinach FC (1995) The troponin complex and regulation of muscle contraction. FASEB J 9, 755–767. [DOI] [PubMed] [Google Scholar]
- (8).Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, and Potter JD (1980) The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J. Biol. Chem 255, 11688–11693. [PubMed] [Google Scholar]
- (9).Chalovich JM (1992) Actin mediated regulation of muscle contraction. Pharmacol. Ther 55, 95–148. [DOI] [PubMed] [Google Scholar]
- (10).Chalovich JM (2002) Regulation of striated muscle contraction: a discussion. J. Muscle Res. Cell Motil 23, 353–361. [DOI] [PubMed] [Google Scholar]
- (11).Leavis PC, Gergely J, and Szent-Gyorgyi AG (1984) Thin filament proteins and thin filament-linked regulation of vertebrate muscle contraction. Crit. Rev. Biochem. Mol. Biol 16, 235–305. [DOI] [PubMed] [Google Scholar]
- (12).Gordon AM, Homsher E, and Regnier M (2000) Regulation of contraction in striated muscle. Physiol. Rev 80, 924. [DOI] [PubMed] [Google Scholar]
- (13).Perry SV, Cole HA, Head JF, and Wilson FJ (1973) Localization and mode of action of the inhibitory protein component of the tropomyosin complex. Cold Spring Harbor Symp. Quant. Biol 37, 251–262. [Google Scholar]
- (14).Potter JD, Sheng Z, Pan BS, and Zhao J (1995) A direct regulatory role for troponin T and a dual role for troponin C in the Ca 2+ regulation of muscle contraction. J. Biol. Chem 270, 2557–2562. [DOI] [PubMed] [Google Scholar]
- (15).Malnic B, Farah CS, and Reinach FC (1998) Regulatory properties of the NH2- and COOH-terminal domains of troponin T. ATPase activation and binding to troponin I and troponin C. J. Biol. Chem 273, 10594–10601. [DOI] [PubMed] [Google Scholar]
- (16).Chalovich JM (2012) Disease causing mutations of troponin alter regulated actin state distributions. J. Muscle Res. Cell Motil 33, 493–499. [DOI] [PubMed] [Google Scholar]
- (17).Hill TL, Eisenberg E, and Chalovich JM (1981) Theoretical models for cooperative steady-state ATPase activity of myosin subfragment-1 on regulated actin. Biophys. J 35, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chalovich JM, Chock PB, and Eisenberg E (1981) Mechanism of action of troponin-tropomyosin: inhibition of actomyosin ATPase activity without inhibition of myosin binding to actin. J. Biol. Chem 256, 575–578. [PMC free article] [PubMed] [Google Scholar]
- (19).Chalovich JM, and Eisenberg E (1982) Inhibition of actomyosin ATPase activity by troponin-tropomyosin without blocking the binding of myosin to actin. J. Biol. Chem 257, 2432–2437. [PMC free article] [PubMed] [Google Scholar]
- (20).Kraft T, Chalovich JM, Yu LC, and Brenner B (1991) Weak cross-bridge binding is essential for force generation. Evidence at near physiological conditions. Biophys. J 59, 375a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Brenner B, Schoenberg M, Chalovich JM, Greene LE, and Eisenberg E (1982) Evidence for cross-bridge attachment in relaxed muscle at low ionic strength. Proc. Natl. Acad. Sci. U. S. A 79, 7288–7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McKillop DFA, and Geeves MA (1993) Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J 65, 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lehman W, Hatch V, Korman V, Rosol M, Thomas L, Maytum R, Geeves MA, Van Eyk JE, Tobacman LS, and Craig R (2000) Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol 302, 593–606. [DOI] [PubMed] [Google Scholar]
- (24).Pirani A, Xu C, Hatch V, Craig R, Tobacman LS, and Lehman W (2005) Single particle analysis of relaxed and activated muscle thin filaments. J. Mol. Biol 346, 761–772. [DOI] [PubMed] [Google Scholar]
- (25).Mathur MC, Kobayashi T, and Chalovich JM (2009) Some cardiomyopathy causing troponin I mutations stabilize a functional intermediate actin state. Biophys. J 96, 2237–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kraft T, Chalovich JM, Yu LC, and Brenner B (1995) Parallel inhibition of active force and relaxed fiber stiffness by caldesmon fragments at physiological ionic strength and temperature conditions: Additional evidence that weak cross-bridge binding to actin is an essential intermediate for force generation. Biophys. J 68, 2404–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Houmeida A, Heeley DH, Belknap B, and White HD (2010) Mechanism of Regulation of Native Cardiac Muscle Thin Filaments by Rigor Cardiac Myosin-S1 and Calcium. J. Biol. Chem 285, 32760–32769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tobacman LS, and Adelstein RS (1986) Mechanism of Regulation of Cardiac Actin-Myosin Subfragment 1 by Troponin-Tropomyosin. Biochemistry 25, 798–802. [DOI] [PubMed] [Google Scholar]
- (29).El-Saleh SC, and Potter JD (1985) Calcium-insensitive binding of heavy meromyosin to regulated actin at physiological ionic strength. J. Biol. Chem 260, 14775–14779. [PubMed] [Google Scholar]
- (30).Johnson D, Mathur MC, Kobayashi T, and Chalovich JM (2016) The Cardiomyopathy Mutation, R146G Troponin I, Stabilizes the Intermediate ″C″ State of Regulated Actin under High- and Low-Free Ca2+ Conditions. Biochemistry 55, 4533–4540. [DOI] [PubMed] [Google Scholar]
- (31).Bremel RD, Murray JM, and Weber A (1973) Manifestations of cooperative behavior in the regulated actin filament during actin-activated ATP hydrolysis in the presence of calcium. Cold Spring Harbor Symp. Quant. Biol 37, 267–275. [Google Scholar]
- (32).Hill TL, Eisenberg E, and Greene LE (1980) Theoretical model for the cooperative equilibrium binding of myosin subfragment 1 to the actin-troponin-tropomyosin complex. Proc. Natl. Acad. Sci. U. S. A 77, 3186–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Heeley DH, Belknap B, and White HD (2006) Maximal Activation of Skeletal Muscle Thin Filaments Requires Both Rigor Myosin S1 and Calcium. J. Biol. Chem 281, 668–676. [DOI] [PubMed] [Google Scholar]
- (34).Geeves MA, and Lehrer SS (2002) Modeling thin filament cooperativity. Biophys. J 82, 1677–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Aboelkassem Y, Bonilla JA, McCabe KJ, and Campbell SG (2015) Contributions of Ca2+-Independent Thin Filament Activation to Cardiac Muscle Function. Biophys. J 109, 2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Gafurov B, Fredricksen S, Cai A, Brenner B, Chase PB, and Chalovich JM (2004) The Δ14 mutation of human cardiac troponin T enhances ATPase activity and alters the cooperative binding of S1-ADP to regulated actin. Biochemistry 43, 15276–15285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mathur MC, Kobayashi T, and Chalovich JM (2008) Negative Charges at Protein Kinase C Sites of Troponin I Stabilize the Inactive State of Actin. Biophys. J 94, 542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Franklin AJ, Baxley T, Kobayashi T, and Chalovich JM (2012) The C-Terminus of Troponin T Is Essential for Maintaining the Inactive State of Regulated Actin. Biophys. J 102, 2536–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Landstrom AP, Parvatiyar MS, Pinto JR, Marquardt ML, , Bos JM, Tester DJ, Ommen SR, Potter JD, and Ackerman MJ (2008) Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol 45, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cheng Y, and Regnier M (2016) Cardiac troponin structure-function and the influence of hypertrophic cardiomyopathy associated mutations on modulation of contractility. Arch. Biochem. Biophys 601, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kalyva A, Parthenakis FI, Marketou ME, Kontaraki JE, and Vardas PE (2014) Biochemical characterisation of Troponin C mutations causing hypertrophic and dilated cardiomyopathies. J. Muscle Res. Cell Motil 35, 161–178. [DOI] [PubMed] [Google Scholar]
- (42).Li MX, and Hwang PM (2015) Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 571, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Martins AS, Parvatiyar MS, Feng HZ, Bos JM, Gonzalez-Martinez D, Vukmirovic M, Turna RS, Sanchez-Gonzalez MA, Badger CD, Zorio DA, Singh RK, Wang Y, Jin JP, Ackerman MJ, and Pinto JR (2015) In Vivo Analysis of Troponin C Knock-In (A8V) Mice: Evidence that TNNC1 Is a Hypertrophic Cardiomyopathy Susceptibility Gene. Circ.: Cardiovasc. Genet 8, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, and Seidman CE (1995) Mutations in the genes for cardiac troponin T and à-tropomyosin in hypertrophic cardiomyopathy. N. Engl. J. Med 332, 1058–1064. [DOI] [PubMed] [Google Scholar]
- (45).Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seldman JG, and Seidman CE (1994) à-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 77, 701–712. [DOI] [PubMed] [Google Scholar]
- (46).Spudich JA, and Watt S (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem 246, 4866–4871. [PubMed] [Google Scholar]
- (47).Kouyama T, and Mihashi K (1981) Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labeled F-actin: local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem 114, 33–38. [PubMed] [Google Scholar]
- (48).Kielley WW, and Harrington WF (1960) A model for the myosin molecule. Biochim. Biophys. Acta 41, 401–421. [DOI] [PubMed] [Google Scholar]
- (49).Weeds AG, and Taylor RS (1975) Separation of subfragment-1 isozymes from rabbit skeletal muscle myosin. Nature 257, 54–56. [DOI] [PubMed] [Google Scholar]
- (50).Chalovich JM, Lutz E, Baxley T, and Schroeter MM (2011) Acrylodan-labeled smooth muscle tropomyosin reports differences in the effects of troponin and caldesmon in the transition from the active state to the inactive state. Biochemistry 50, 6093–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Smillie LB (1982) Preparation and identification of alpha- and beta-tropomyosins. Methods Enzymol 85, 234–241. [DOI] [PubMed] [Google Scholar]
- (52).Hibbs RE, Talley TT, and Taylor P (2004) Acrylodan-conjugated Cysteine Side Chains Reveal Conformational State and Ligand Site Locations of the Acetylcholine-binding Protein. J. Biol. Chem 279, 28483–28491. [DOI] [PubMed] [Google Scholar]
- (53).Pinto JR, Parvatiyar MS, Jones MA, Liang J, Ackerman MJ, and Potter JD (2009) A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. J. Biol. Chem 284, 19090–19100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kobayashi T, and Solaro RJ (2006) Increased Ca2+ Affinity of Cardiac Thin Filaments Reconstituted with Cardiomyopathy-related Mutant Cardiac Troponin I. J. Biol. Chem 281, 13471–13477. [DOI] [PubMed] [Google Scholar]
- (55).Greene LE, and Eisenberg E (1980) Cooperative binding of myosin subfragment-1 to the actin-troponin-tropomyosin complex. Proc. Natl. Acad. Sci. U. S. A 77, 2616–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Eisenberg E, and Kielley W (1970) Native tropomyosin: effect on the interaction of actin with heavy meromyosin and subfragment-1. Biochem. Biophys. Res. Commun 40, 50–56. [DOI] [PubMed] [Google Scholar]
- (57).Pemrick S, and Weber A (1976) Mechanism of inhibition of relaxation by N-ethylmaleimide treatment of myosin. Biochemistry 15, 5193–5198. [DOI] [PubMed] [Google Scholar]
- (58).Meeusen RL, and Cande ZW (1979) N-ethylmaleimide-modified heavy meromyosin. A probe for actomyosin interactions. J. Cell Biol 82, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Williams DL, Greene LE, and Eisenberg E (1988) Cooperative Turning on of Myosin Subfragment-1 Adenosine-Triphosphatase Activity by the Troponin Tropomyosin Actin Complex. Biochemistry 27, 6987–6993. [DOI] [PubMed] [Google Scholar]
- (60).Trybus KM, and Taylor EW (1980) Kinetic studies of the cooperative binding of subfragment 1 to regulated actin. Proc. Natl. Acad. Sci. U. S. A 77, 7209–7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Borrego-Diaz E, and Chalovich JM (2010) Kinetics of regulated actin transitions measured by probes on tropomyosin. Biophys. J 98, 2601–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Johnson KA, Simpson ZB, and Blom T (2009) FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem 387, 30–41. [DOI] [PubMed] [Google Scholar]
- (63).Johnson KA, Simpson ZB, and Blom T (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem 387, 20–29. [DOI] [PubMed] [Google Scholar]
- (64).Head JG, Ritchie MD, and Geeves MA (1995) Characterization of the equilibrium between blocked and closed states of muscle thin filaments. Eur. J. Biochem 227, 694–699. [DOI] [PubMed] [Google Scholar]
- (65).Chen Y, Yan B, Chalovich JM, and Brenner B (2001) Theoretical kinetic studies of models for binding myosin subfragment-1 to regulated actin: Hill model versus Geeves model. Biophys. J 80, 2338–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Eisenberg E, and Weihing RR (1970) Effect of skeletal muscle native tropomyosin on the interaction of amoeba actin with heavy meromyosin. Nature 228, 1092–1093. [DOI] [PubMed] [Google Scholar]
- (67).Chalovich JM, and Johnson D (2016) Commentary: Effect of Skeletal Muscle Native Tropomyosin on the Interaction of Amoeba Actin with Heavy Meromyosin. Front. Physiol 7, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Zot HG, Hasbun JE, Michell CA, Landim-Vieira M, and Pinto JR (2016) Enhanced troponin I binding explains the functional changes produced by the hypertrophic cardiomyopathy mutation A8V of cardiac troponin C. Arch. Biochem. Biophys 601, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Albury AN, Swindle N, Swartz DR, and Tikunova SB (2012) Effect of hypertrophic cardiomyopathy-linked troponin C mutations on the response of reconstituted thin filaments to calcium upon troponin I phosphorylation. Biochemistry 51, 3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Pinto JR, Reynaldo DP, Parvatiyar MS, Dweck D, Liang J, Jones MA, Sorenson MM, and Potter JD (2011) Strong cross-bridges potentiate the Ca2+ affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments: a fast kinetic approach. J. Biol. Chem 286, 1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Hofmann PA, and Fuchs F (1987) Effect of length and cross-bridge attachment on Ca2+ binding to cardiac troponin C. Am. J. Physiol 253, C90–C96. [DOI] [PubMed] [Google Scholar]
- (72).Swindle N, and Tikunova SB (2010) Hypertrophic cardiomyopathy-linked mutation D145E drastically alters calcium binding by the C-domain of cardiac troponin C. Biochemistry 49, 4813–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Cordina NM, Liew CK, Gell DA, Fajer PG, Mackay JP, and Brown LJ (2013) Effects of calcium binding and the hypertrophic cardiomyopathy A8V mutation on the dynamic equilibrium between closed and open conformations of the regulatory N-domain of isolated cardiac troponin C. Biochemistry 52, 1950–1962. [DOI] [PubMed] [Google Scholar]
- (74).Liu B, Lee RS, Biesiadecki BJ, Tikunova SB, and Davis JP (2012) Engineered troponin C constructs correct disease-related cardiac myofilament calcium sensitivity. J. Biol. Chem 287, 20027–20036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Resetar AM, Stephens JM, and Chalovich JM (2002) Troponin-tropomyosin: an allosteric switch or a steric blocker? Biophys. J 83, 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]