

Abstract
Objective
To investigate the status of the basal forebrain cholinergic system in primary progressive aphasia (PPA) as justification for cholinergic therapy.
Methods
A cohort of 36 brains from PPA participants with the neuropathology of Alzheimer disease (PPA-AD, n = 14) or frontotemporal lobar degeneration (PPA-tau, n = 12; PPA-TDP, n = 10) were used for semiquantitative rating of degeneration and gliosis of basal forebrain cholinergic neurons (BFCN). A subpopulation of 5 PPA-AD and 7 control brains underwent detailed analysis of BFCN pathology and cortical cholinergic axonal loss employing immunohistochemical and histochemical methods and stereologic analysis.
Results
Semiquantitatively, 11 (∼80%) PPA-AD participants were rated as having moderate/severe BFCN loss and gliosis, whereas none of the PPA-tau and only 1 (10%) PPA-TDP participant received such a rating. Detailed analysis in the subpopulation of PPA-AD participants revealed substantial tangle formation, loss of BFCN, and degeneration of cortical cholinergic axons. Compared to controls, loss of p75 low affinity neurotrophin receptor-positive BFCN was detected in the PPA-AD participants (p < 0.01). Acetylcholinesterase-positive cholinergic axons in all cortical areas studied displayed loss in PPA-AD (p < 0.005–0.0001). The loss was more severe in the language-dominant left hemisphere and, within the left hemisphere, in language-affiliated cortical areas.
Conclusions
Our results demonstrate prominent depletion of BFCN and cortical cholinergic axons in PPA-AD when compared with normal control or other neuropathologic variants of PPA. The demonstration of cholinergic denervation with an anatomy that fits the clinical picture suggests that cholinergic treatment is justified in patients with PPA who have positive AD biomarkers.
Physicians look for the typical amnestic profile of Alzheimer disease (AD) to prescribe cholinesterase inhibitors (ChEIs) and AD-related clinical trials tend to base inclusion criteria on memory function. However, nearly 40% of patients with primary progressive aphasia (PPA) also have AD neuropathology1,2 but tend to be excluded from AD-related clinical trials and are less likely to be prescribed ChEIs.
Cortical cholinergic innervation originates in the basal forebrain cholinergic neurons (BFCN), mostly in the Ch4 neuronal group of the nucleus basalis of Meynert (nbM-Ch4).3,4 There is early and substantial degeneration of BFCN and cortical cholinergic axons in the typical amnestic variant of AD (AV-AD),5,6 where these lesions are correlated with dementia severity.7,8 Currently, ChEIs, which increase the pool of acetylcholine, represent the major available therapeutic agents in this disorder.9,10
PPA presents with diverse pathologies, including AD (PPA-AD) and frontotemporal lobar degeneration (FTLD) with tauopathy (PPA-Tau) or transactive response DNA binding protein-43 inclusions (PPA-TDP).1,2,11 However, the status of the cholinergic system in PPA has not been investigated in detail. Here, we provide an analysis of the basal forebrain cholinergic system in postmortem PPA brains and report selective neuronal loss and gliosis in PPA-AD when compared with PPA-Tau and PPA-TDP. In a subgroup of PPA-AD participants, we report substantial loss of nbM-Ch4 neurons and their cortically projecting axons with a leftward hemispheric asymmetry. Our results suggest that ChEI therapy in PPA-AD is likely to be as effective as it is in AV-AD.
Methods
Standard protocol approvals, registrations, and patient consents
Patients with PPA had enrolled in the Clinical Core of the Northwestern University Alzheimer's Disease Center where they were followed annually until death and had committed to postmortem brain donation. They had all signed informed consent and the study was approved by the Northwestern University Institutional Review Board.
Participants and tissue processing
Patients were seen at the Neurobehavior and Memory Clinic of the Northwestern Feinberg School of Medicine. The PPA diagnosis was made according to established criteria.12,13
All brains with the clinical diagnosis of PPA in the Northwestern University Alzheimer's Disease Center Brain Bank, in which neuropathologic workup included evaluation of the nbM-Ch4 neurons, were used for semiquantitative rating of neuronal loss and gliosis. This cohort included 36 brains and was used for comparison among PPA-AD (n = 14), PPA-Tau (n = 12), and PPA-TDP (n = 10). The 2 cases with FTLD-TDP type A had GRN mutations. There were no other disease-causing mutations. Two of the AD cases had secondary diagnoses of diffuse Lewy body disease. No other significant secondary neuropathologic diagnoses were noted. Brain tissue from a randomly selected subpopulation of 5 cases from among the 14 PPA-AD participants and 7 elderly controls with no neurologic or psychiatric disorders were used for detailed analysis of nbM-Ch4 tangle burden, neuronal loss, and degeneration of cortical cholinergic axons, including unbiased stereologic methods. Characteristics of participants are listed in tables 1 and 2. In the PPA-AD group selected for quantitative analyses, 3 were of the logopenic variant, 1 was of the mixed variant, and 1 was unclassifiable by the consensus criteria.12
Table 1.
Characteristics of the 36 cases

Table 2.
Characteristics of participants for detailed analysis

Immediately following autopsy, brains were cut into 2–3 cm coronal slabs, fixed in 4% paraformaldehyde or formalin for 30–36 hours at 4°C and taken through sucrose gradients (10%–40% in 0.2 M phosphate buffer, containing 0.02% sodium azide) for cryoprotection and stored at 4°C. In the cohort used for semiquantitative rating of neuronal loss and gliosis (n = 36), a block of tissue containing the nbM-Ch4 was embedded in paraffin, cut at a thickness of 5 μm, and stained with hematoxylin & eosin. Blocks from the brains used for detailed analysis (n = 5 PPA-AD and 7 control) were cut into 40-µm-thick sections on a freezing microtome and stored in 0.1 M phosphate buffer at 4°C until used. Series of 1 in 24 sections spanning regions of interest were mounted on slides, air dried, and stained for Nissl using the cresyl violet stain for determination of neuronal types and delineation of anatomical boundaries, and with thioflavin-S, which binds to β-pleated sheet abnormal protein conformations, to visualize mature plaques and tangles.
Immunohistochemistry
Immunohistochemistry was carried out in a 1 in 24 series of sections spanning the entire extent of the basal forebrain according to the avidin-biotin peroxidase complex method employing the Vecstastain Elite Kit (Vector Laboratories, Burlingame, CA), with diaminobenzidine as chromogen. A monoclonal antibody against the p75 low affinity neurotrophin receptor (p75LNTR, 1/1,000; Millipore Sigma, Burlington, MA), which in the human forebrain is specifically enriched in the BFCN,14,15 was used to visualize these neurons.
Acetylcholinesterase (AChE) histochemistry
We have shown that immunohistochemistry for the specific cholinergic enzyme choline acetyltransferase and the hydrolytic cholinergic enzyme AChE visualize an identical population of cortical cholinergic axons.16 Therefore, we used AChE histochemistry to visualize cortical cholinergic axons in a 1 in 24 series of sections with the help of a highly sensitive method. The principles of this method (incubation in a dilute Karnovsky Roots medium, followed by metal ion-diaminobenzidine intensification) have been described by Hanker et al.17 and Tago et al.18 We have introduced a number of changes in this method, as described elsewhere.19,20
To inhibit butyrylcholinesterase (BuChE, nonspecific cholinesterase), the BuChE inhibitor ethopropazine (2 × 10−4 M; Sigma Chemical Company, St. Louis, MO) was added to the incubation medium. The specific AChE inhibitor BW284C51 (10−4 M; Sigma Chemical Company) was added to demonstrate the specificity of AChE staining.
Analysis of stained sections
Semiquantitative ratings were carried out by an experienced neuropathologist (E.B.). Neuronal loss and gliosis was rated as either normal/mild or moderate/severe. The percentage of participants in each group (PPA-AD, PPA-Tau, and PPA-TDP) that fell in each category was then calculated. The analyses could not be performed in a blinded fashion since the diagnosis was known to the neuropathologist. In the brains used for detailed analysis, the density of BFCN containing various markers and of AChE-positive cortical axons was assessed using careful microscopic examination combined with electronic image documentation, and differences between PPA-AD and controls were noted. Loss of cholinergic neurons was assessed in the entire extent of the nbM-Ch4 complex. Cortical cholinergic axons were examined in 3 regions affiliated with language function (inferior frontal gyrus [IFG], inferior parietal lobule [IPL], and superior temporal gyrus [STG]) and in 2 nonlanguage regions (entorhinal cortex [ENT] and anterior cingulate cortex [ACC]). To assess hemispheric asymmetry of measures, each region was examined bilaterally in PPA-AD participants. Material was available only from one hemisphere in the control cases (primarily left).
Quantitative and statistical analyses
The numbers of p75LNTR immunoreactive nbM-Ch4 neurons (5 PPA-AD and 3 controls) in each hemisphere were quantified using unbiased stereologic methods, employing the optical fractionator probe of the StereoInvestigator Software (MBF Biosciences, MicroBrightfield, Inc., Williston, VT) as previously described.21 Briefly, the top and bottom 2 μm of each section were set as guard height. The region of interest was traced at ×10 magnification, and counting was carried out at ×40. In all cases, enough sections with systematic random sampling of basal forebrain were available to satisfy the requirement for unbiased stereologic estimation of immunoreactive profiles. Stereologic parameters such as dissector spacing, dissector height, and counting frame size were determined through trials to result in coefficients of error equal to or less than 0.1. Numbers of mature tangles in nbM-Ch4 neurons and of mature plaques in the basal forebrain region in which these neurons are located was also determined using unbiased stereology in each hemisphere (2 PPA-AD participants) to obtain a measure of pathologic burden.
To obtain an estimate of the loss of cortical cholinergic axons, the total length of AChE-positive axons was determined (2 PPA-AD, 2 control) in the same cortical areas that were evaluated qualitatively, using the Space Balls probe of StereoInvestigator.
Differences between the total numbers of p75LNTR immunoreactive nbM-Ch4 neurons in the control and each hemisphere of PPA-AD participants were determined using nonparametric Kruskal-Wallis analysis of variance followed by Dunn post hoc tests. Laterality of pathology was expressed as percent difference in tangles and plaques in the 2 hemispheres. Stereologic data obtained per section were used for statistical analysis of cortical cholinergic axonal loss, as previously described.11,22 Density of cortical cholinergic axons was expressed as total length of axons in mm per mm3 cortical volume. Analysis of variance was used to determine the degree of cortical cholinergic axonal loss in PPA-AD compared to normal controls. Bonferroni corrected post hoc tests were employed for pairwise comparisons. The threshold for significance was set at p < 0.05.
Data availability statement
All data generated in this experiment are summarized in this report. De-identified individual data will be provided to qualified investigators upon request.
Results
Semiquantitative rating of neuronal loss and gliosis in nbM-Ch4
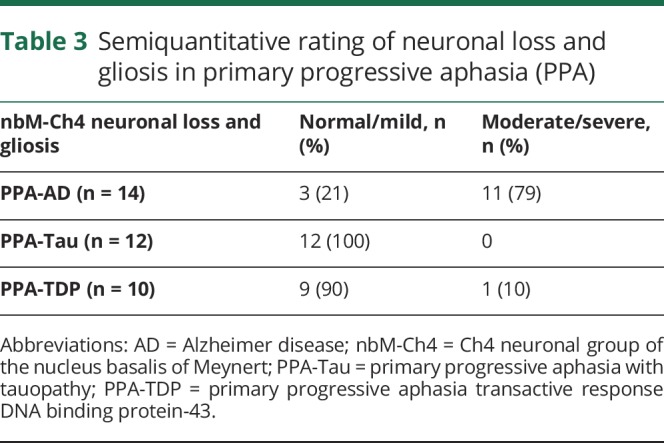
Ratings of the extent of neuronal loss and gliosis in nbM-Ch4 of the 36 PPA brains in the cohort pointed to substantially greater degeneration in PPA-AD when compared with PPA-TDP and PPA-Tau (table 3). Of the 14 PPA-AD participants in this cohort, 11 (∼80%) were rated as having moderate/severe nbM-Ch4 neuronal loss and gliosis. Conversely, of the 12 PPA-Tau and 10 PPA-TDP participants combined, only 1 (a PPA-TDP participant) was rated in the moderate to severe category.
Table 3.
Semiquantitative rating of neuronal loss and gliosis in primary progressive aphasia (PPA)
Alzheimer pathology in nbM-Ch4 of PPA-AD participants
Based on the semiquantitative results showing moderate/severe neuronal loss and gliosis primarily in PPA-AD, we randomly chose 5 PPA-AD brains from our cohort for detailed analysis and compared them with 7 normal aged individuals. Thioflavin-S staining visualized a substantial density of mature tangles within the nbM-Ch4 neurons bilaterally (figure 1, A and B). The estimated numbers of nbM-Ch4 mature tangles in PPA-AD participants was greater in the language-dominant left hemisphere when compared with the right (left = 1939; right = 1,309; left 33% > right). The density of thioflavin-S-positive dense-core/neuritic plaques in the basal forebrain region within which nbM-Ch4 neurons are located was relatively low and displayed only slight hemispheric asymmetry (left = 417; right = 355; left 15% > right).
Figure 1. Substantial accumulation of tangles and loss of Ch4 neuronal group of the nucleus basalis of Meynert (nbM-Ch4) neurons in primary progressive aphasia with Alzheimer disease pathology (PPA-AD).

(A) A substantial density of Thioflavin-S-positive tangles was present in the left nbM-Ch4 neurons. (B) A matching section in the right hemisphere shows fewer nbM-Ch4 tangles. (C) Immunostaining for the p75 low affinity neurotrophin receptor (p75LNTR) reveals a dense collection of nbM-Ch4 neurons in control participants, shown here in the intermediate sector of nbM-Ch4 in the left hemisphere. (D) The number of p75LNTR immunoreactive nbM-Ch4 neurons in PPA-AD displays a substantial decrease, shown here also in the intermediate sector of nbM-Ch4 in the left hemisphere. Magnification ×10.
Quantitative loss of nbM-Ch4 (cholinergic) neurons in PPA-AD
Assessment of p75LNTR immunoreactivity revealed substantial loss of nbM-Ch4 neurons in the basal forebrain of PPA-AD participants when compared with normal controls (figure 1, C and D). There was asymmetry in the loss of cholinergic neurons in PPA-AD such that the density of nbM-Ch4 neurons in the language-dominant hemisphere was visibly and quantitatively lower than that in the nondominant hemisphere (figure 2). Counts of p75LNTR immunoreactive nbM-Ch4 neurons were lower in the left hemisphere of PPA-AD participants when compared with control (figure 2; loss of 73%; p < 0.01). Counts in the right hemisphere were not different from control (p > 0.05). Examination of Nissl-stained tissue corroborated the results obtained from p75LNTR-stained material, demonstrating substantial loss of magnocellular BFCN favoring the language-dominant hemisphere.
Figure 2. Quantitatively determined loss of Ch4 neuronal group of the nucleus basalis of Meynert (nbM-Ch4) neurons and cortical cholinergic axons in primary progressive aphasia with Alzheimer disease pathology (PPA-AD).

(A) The number of p75LNTR immunoreactive nbM-Ch4 neurons in PPA-AD participants was substantially lower when compared with controls. The number of these neurons in the left hemisphere was lower in PPA-AD participants when compared with controls (p < 0.01). The numbers of nbM-Ch4 neurons in the right hemisphere of PPA-AD participants was also lower, but was not significantly different from controls. (B) Total length of acetylcholinesterase-positive cortical cholinergic axons displayed a substantial loss in PPA-AD participants when compared with controls in all areas examined, with greater loss in the left hemisphere. Loss of cortical cholinergic axons was greater in language-related cortical areas (superior temporal gyrus [STG], inferior parietal lobule [IPL], and inferior frontal gyrus [IFG]; 64%–94%, p < 0.05–0.001 compared with control) when compared with nonlanguage regions (anterior cingulate cortex [ACC] and entorhinal cortex [ENT]; 24%–44%, p < 0.05–0.01). C = Control left hemisphere; L = PPA-AD, left hemisphere; R = PPA-AD, right hemisphere.
Qualitative and quantitative loss of cortical cholinergic axons in PPA-AD participants
The AChE histochemical method visualized a dense network of cholinergic axons in all cortical areas in the normal brains. Substantial and regionally selective loss of cortical cholinergic axons was observed in PPA-AD (figures 2 and 3). An initial qualitative assessment was conducted in order to allow a broad survey of all cortical areas that were being investigated. This assessment revealed greater loss of cholinergic axons in the language-related cortical regions (STG, IPL, and IFG) compared to the nonlanguage cortical regions (ACC and ENT).
Figure 3. Regionally selective loss of cortical cholinergic axons in primary progressive aphasia with Alzheimer disease pathology (PPA-AD).

(A) Cortical cholinergic axons in the language-affiliated inferior parietal lobule (IPL) of a control participant. (B) Cortical cholinergic axons in the left IPL of a participant with PPA-AD. Note the virtually complete loss of cholinergic axons. Similar to IPL, the language-affiliated superior temporal gyrus contains a dense network of cholinergic axons (C) that are nearly completely lost in PPA-AD (D). Magnification ×20.
Quantitative assessment in the subset of the 5 cases corroborated the qualitative comparisons, revealing loss of cortical cholinergic axons in all cortical areas investigated in PPA-AD compared to normal controls (p < 0.05–0.0001). Loss of cortical cholinergic axons was greater in language regions (reduced by 64%–94%) compared to nonlanguage regions (reduced by 24%–44%) (figure 2). In all cortical regions of PPA-AD participants, the total length of cholinergic axons in the left hemisphere was smaller than in the right hemisphere. This hemispheric difference was greater in language regions (reduced by 26%–43%) when compared with nonlanguage regions (reduced by 13%–17%). However, the variability in this small sample was such that quantitative comparisons of hemispheric difference in length of axons yielded a p value just shy of threshold (p = 0.062).
Discussion
It is becoming increasingly clear that sporadic AD is clinically heterogeneous. In addition to the most common amnestic multidomain form, AD can present as aphasic, visuospatial, and frontal-type dementias.23 All forms share the common denominator of neuritic β-amyloid plaques and neurofibrillary tangles. However, the distribution of these lesions, especially the tangles, can violate the Braak and Braak24 progression pattern in the nonamnestic clinical presentations. For example, the PPA variant of AD can be associated with tangle counts that are more numerous in left hemisphere language areas and that exceed in density those in hippocampal and entorhinal cortices.2 In visuospatial variant (known as posterior cortical atrophy), the tangles can be most numerous in occipital areas and the superior colliculus25 whereas the frontal-type dementia variant can display tangles that are more numerous in frontal than in medial temporal cortices.26 In addition to differences in tangle distribution, some of these variants may also have atypical molecular associations. For example, APOE4, a robust risk factor for AV-AD, does not have such an association in the PPA variant of AD.27
The current report focuses on the status of the cholinergic system in the atypical form of AD that causes PPA. Historically, the discovery of the cortical cholinergic denervation transformed research on AD from the descriptive stage of plaques and tangles to the neurobiological stage of transmitters and neural networks.28,29 The discoveries that this innervation originates in the BFCN,30 that the BFCN is subject to severe degeneration in AD,31 and that cholinergic antagonists can lead to memory impairments similar to those seen in senile dementia32 rapidly led to the introduction of clinically effective ChEIs.33 Since then, scores of clinical trials on thousands of patients have unequivocally confirmed that ChEIs offer symptomatic improvement at various stages of established AD dementia. The effectiveness of ChEIs at the amnestic mild cognitive impairment (MCI) stages of AD remains controversial. Although the cholinergic degeneration starts very early in the aging–MCI–dementia continuum of AD pathology,34–36 a major clinical trial failed to show efficacy of ChEI in MCI.37 However, the patients entered into the trial had no biomarker confirmation of AD and undoubtedly comprised a highly heterogeneous group with respect to underlying pathology. More recent reviews of the literature suggest that ChEI can be effective at the MCI stages and that they may even have a disease-modifying effect in retarding hippocampal and basal forebrain atrophy.38–40 The cholinergic lesion is one of many components of AD neuropathology so that cholinomimetics can be expected to provide only partial relief.
The status of the cholinergic system in PPA that is caused by AD had not been investigated in any degree of detail. Existing reports have been based mostly on neuroimaging. Structural MRI had shown atrophy of the basal forebrain regions in PPA while PET with an AChE ligand demonstrated a loss of cortical cholinesterase activity selectively in the logopenic variant of PPA.41–43 These in vivo approaches have well-known limitations. For example, the basal forebrain region also contains noncholinergic neural components that are difficult to delineate from the BFCN. Furthermore, cortical ChEs reflect not only presynaptic cholinergic input but also postsynaptic neurons and AChE-positive plaques and tangles.44,45 The results in this report definitively establish the presence of severe and selective BFCN degeneration and the corresponding depletion of cortical cholinergic axons in patients with PPA and AD pathology. In PPA-AD, ∼80% of cases had moderate to severe neuronal loss and gliosis in the BFCN. In contrast, such pathology was seen in only 10% of PPA cases caused by FTLD-TDP and in none of the PPA cases caused by FTLD-Tau. In the PPA patients with AD, the axonal loss was particularly prominent in neocortical areas that encompass the language network. There was also greater loss in the language-dominant left hemisphere. The demonstration of cholinergic denervation with an anatomy that fits the clinical profile suggests that cholinergic treatment should be pursued in these patients.
Reports on the use of cholinomimetics in PPA are mostly anecdotal. One study reported a “trend” toward clinical efficacy of galantamine in a small group of patients with PPA.46 However, the patients were not selected based on AD biomarker positivity. Our results suggest that a rigorous trial of ChEI in patients with PPA with biomarker evidence of AD pathology is indicated. Although the incidence of AD pathology is highest in the logopenic variant of PPA, the relationship is not perfect so that biomarker confirmation will be essential.1,23 Cholinomimetic treatments could conceivably also be combined with speech therapy or with transcranial stimulation modalities.47 Such trials will need to be designed with instruments that are sensitive to the temporal evolution of atrophy in the language network and to the progression of language dysfunction in PPA.48,49 They will also undoubtedly require collaborative recruitment across multiple centers since the aphasic manifestation of AD is far less common than the typical amnestic forms.
Glossary
- ACC
anterior cingulate cortex
- AChE
acetylcholinesterase
- AD
Alzheimer disease
- AV-AD
amnestic variant of Alzheimer disease
- BFCN
basal forebrain cholinergic neurons
- BuChE
butyrylcholinesterase
- ChEI
cholinesterase inhibitor
- ENT
entorhinal cortex
- FTLD
frontotemporal lobar degeneration
- IFG
inferior frontal gyrus
- IPL
inferior parietal lobule
- MCI
mild cognitive impairment
- nbM-Ch4
Ch4 neuronal group of the nucleus basalis of Meynert
- PPA
primary progressive aphasia
- PPA-AD
primary progressive aphasia with Alzheimer disease neuropathology
- PPA-Tau
primary progressive aphasia with tauopathy
- PPA-TDP
primary progressive aphasia with transactive response DNA binding protein-43
- STG
superior temporal gyrus
Appendix. Authors
Study funding
This work was supported by grants from the National Institute of Neurologic Disorders and Stroke (NS085770), National Institute on Deafness and Other Communication Disorders (DC008552), the Louis Family Foundation, the Florane and Jerome Rosenstone Fellowship, an Alzheimer's Disease Center Grant from the National Institute on Aging (AG013854), and a training grant from the National Institute on Aging (T32 AG20506).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer's and frontotemporal pathology in primary progressive aphasia. Brain 2014;137:1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gefen T, Gasho K, Rademaker A, et al. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain 2012;135:1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mesulam MM, Mufson EJ, Levey AI, Wainer BH. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol 1983;214:170–197. [DOI] [PubMed] [Google Scholar]
- 4.Mesulam MM, Geula C. Nucleus basalis (Ch4) and cortical cholinergic innervation in the human brain: observations based on the distribution of acetylcholinesterase and choline acetyltransferase. J Comp Neurol 1988;275:216–240. [DOI] [PubMed] [Google Scholar]
- 5.Rogers JD, Brogan D, Mirra SS. The nucleus basalis of Meynert in neurological disease: a quantitative morphological study. Ann Neurol 1985;17:163–170. [DOI] [PubMed] [Google Scholar]
- 6.Geula C, Mesulam MM. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer's disease. Cereb Cort 1996;6:165–177. [DOI] [PubMed] [Google Scholar]
- 7.Lehéricy S, Hirsch EC, Cervera-Piérot P, et al. Heterogeneity and selectivity of the degeneration of cholinergic neurons in the basal forebrain of patients with Alzheimer's disease. J Comp Neurol 1993;330:15–31. [DOI] [PubMed] [Google Scholar]
- 8.Bierer LM, Haroutunian V, Gabriel S, et al. Neurochemical correlates of dementia severity in Alzheimer's disease: relative importance of the cholinergic deficits. J Neurochem 1995;64:749–760. [DOI] [PubMed] [Google Scholar]
- 9.Bullock R, Touchon J, Bergman H, et al. Rivastigmine and donepezil treatment in moderate to moderately-severe Alzheimer's disease over a 2-year period. Curr Med Res Opin 2005;21:1317–1327. [DOI] [PubMed] [Google Scholar]
- 10.Farlow M. A clinical overview of cholinesterase inhibitors in Alzheimer's disease. Int Psychogeriatr 2002;14(suppl 1):93–126. [DOI] [PubMed] [Google Scholar]
- 11.Kim G, Ahmadian SS, Peterson M, et al. Asymmetric pathology in primary progressive aphasia with progranulin mutations and TDP inclusions. Neurology 2016;86:627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mesulam MM. Primary progressive aphasia. Ann Neurol 2001;49:425–432. [PubMed] [Google Scholar]
- 14.Woolf NJ, Gould E, Butcher LL. Nerve growth factor receptor is associated with cholinergic neurons of the basal forebrain but not the pontomesencephalon. Neuroscience 1989;30:143–152. [DOI] [PubMed] [Google Scholar]
- 15.Mufson EJ, Presley LN, Kordower JH. Nerve growth factor receptor immunoreactivity within the nucleus basalis (Ch4) in Parkinson's disease: reduced cell numbers and co- localization with cholinergic neurons. Brain Res 1991;539:19–30. [DOI] [PubMed] [Google Scholar]
- 16.Mesulam MM, Geula C. Overlap between acetylcholinesterase-rich and choline acetyltransferase-positive (cholinergic) axons in human cerebral cortex. Brain Res 1992;577:112–120. [DOI] [PubMed] [Google Scholar]
- 17.Hanker JS, Thornburg LP, Yates PE, Moore HG. The demonstration of cholinesterases by the formation of osmium blacks at sites of Hatchett's brown. Histochemie 1973;37:223–242. [DOI] [PubMed] [Google Scholar]
- 18.Tago H, Kimura H, Maeda T. Visualization of detailed acetylcholinesterase fiber and neuron staining in rat brain by a sensitive histochemical staining. J Histochem Cytochem 1986;34:1431–1438. [DOI] [PubMed] [Google Scholar]
- 19.Geula C, Mesulam MM. Cortical cholinergic fibers in aging and Alzheimer's disease: a morphometric study. Neurosci 1989;33:469–481. [DOI] [PubMed] [Google Scholar]
- 20.Mesulam MM, Geula C. Chemoarchitectonics of axonal and perikaryal acetylcholinesterase along information processing systems of the human cerebral cortex. Brain Res Bull 1994;33:137–153. [DOI] [PubMed] [Google Scholar]
- 21.Geula C, Bu J, Nagykery N, et al. Loss of calbindin-D28k from aging human cholinergic basal forebrain: relation to neuronal loss. J Comp Neurol 2003;455:249–259. [DOI] [PubMed] [Google Scholar]
- 22.Gliebus G, Bigio EH, Gasho K, et al. Asymmetric TDP-43 distribution in primary progressive aphasia with progranulin mutation. Neurology 2010;74:1607–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogalski E, Sridhar J, Rader B, et al. Aphasic variant of Alzheimer disease: clinical, anatomic, and genetic features. Neurology 2016;87:1337–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braak H, Braak E. Neuropathological staging of Alzheimer's disease. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 25.Hof PR, Archin N, Osmand AP, et al. Posterior cortical atrophy in Alzheimer's disease: analysis of a new case and re-evaluation of a historical report. Acta Neuropathol 1993;86:215–223. [DOI] [PubMed] [Google Scholar]
- 26.Johnson JK, Head E, Kim R, Starr A, Cotman CW. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol 1999;56:1233–1239. [DOI] [PubMed] [Google Scholar]
- 27.Rogalski EJ, Rademaker A, Harrison TM, et al. ApoE E4 is a susceptibility factor in amnestic but not aphasic dementias. Alzheimer Dis Assoc Disord 2011;25:159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mesulam M. The cholinergic lesion of Alzheimer's disease: pivotal factor or side show? Learn Mem 2004;11:43–49. [DOI] [PubMed] [Google Scholar]
- 29.Geula C, Mesulam MM. Cholinergic systems in Alzheimer disease. In: Terry RD, Katzman R, Bick KL, Sisodia SS, eds. Alzheimer Disease, Philadelphia: Lippincott Williams & Wilkins; 1999:269–292. [Google Scholar]
- 30.Mesulam MM, Van Hoesen GW. Acetylcholinesterase-rich projections from the basal forebrain of the rhesus monkey to neocortex. Brain Res 1976;109:152–157. [DOI] [PubMed] [Google Scholar]
- 31.Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol 1981;10:122–126. [DOI] [PubMed] [Google Scholar]
- 32.Drachman DA, Leavitt J. Human memory and the cholinergic system: a relationship to aging? Arch Neurol 1974;30:113–121. [DOI] [PubMed] [Google Scholar]
- 33.Summers WK, Majovski LV, Marsh GM, Tachiki K, Kling A. Oral tetrahydroaminoacridine in long-term treatment of senile dementia, Alzheimer type. N Engl J Med 1986;315:1241–1245. [DOI] [PubMed] [Google Scholar]
- 34.Mesulam M, Shaw P, Mash D, Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol 2004;55:815–828. [DOI] [PubMed] [Google Scholar]
- 35.Geula C, Nagykery N, Nicholas A, Wu CK. Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer disease. J Neuropathol Exp Neurol 2008;67:309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmitz TW, Nathan Spreng R; Alzheimer's Disease Neuroimaging Initiative. Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer's pathology. Nat Commun 2016;7:13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 2005;352:2379–2388. [DOI] [PubMed] [Google Scholar]
- 38.Dubois B, Chupin M, Hampel H, et al. Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer's disease. Alzheimers Dement 2015;11:1041–1049. [DOI] [PubMed] [Google Scholar]
- 39.Cavedo E, Grothe MJ, Colliot O, et al. Reduced basal forebrain atrophy progression in a randomized donepezil trial in prodromal Alzheimer's disease. Sci Rep 2017;7:11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hampel H, Mesulam MM, Cuello AC, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer's disease. Brain 2018, 141:1917–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaeverbeke J, Evenepoel C, Bruffaerts R, et al. Cholinergic depletion and basal forebrain volume in primary progressive aphasia. Neuroimage Clin 2017;13:271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teipel S, Raiser T, Riedl L, et al. Atrophy and structural covariance of the cholinergic basal forebrain in primary progressive aphasia. Cortex 2016;83:124–135. [DOI] [PubMed] [Google Scholar]
- 43.Teipel SJ, Flatz W, Ackl N, et al. Brain atrophy in primary progressive aphasia involves the cholinergic basal forebrain and Ayala's nucleus. Psychiatry Res 2014;221:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mesulam MM, Geula C. Acetylcholinesterase-rich pyramidal neurons in the human neocortex and hippocampus: absence at birth, development during the life span, and dissolution in Alzheimer's disease. Ann Neurol 1988;24:765–773. [DOI] [PubMed] [Google Scholar]
- 45.Geula C, Mesulam MM. Cholinesterases and the pathology of Alzheimer's disease. Alz Dis Assoc Disor 1995;9:23–28. [DOI] [PubMed] [Google Scholar]
- 46.Kertesz A, Morlog D, Light M, et al. Galantamine in frontotemporal dementia and primary progressive aphasia. Dement Geriatr Cogn Disord 2008;25:178–185. [DOI] [PubMed] [Google Scholar]
- 47.Teichmann M, Lesoil C, Godard J, et al. Direct current stimulation over the anterior temporal areas boosts semantic processing in primary progressive aphasia. Ann Neurol 2016;80:693–707. [DOI] [PubMed] [Google Scholar]
- 48.Rogalski E, Cobia D, Harrison TM, Wieneke C, Weintraub S, Mesulam MM. Progression of language decline and cortical atrophy in subtypes of primary progressive aphasia. Neurology 2011;76:1804–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dickerson BC. Quantitating severity and progression in primary progressive aphasia. J Mol Neurosci 2011;45:618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated in this experiment are summarized in this report. De-identified individual data will be provided to qualified investigators upon request.