

Abstract
Binding of small inhibitory compounds to human cytochrome P450 3A4 (CYP3A4) could intefere with drug metabolism and lead to drug-drug interactions, the underlying mechanism of which is not fully understood due to insufficient structural information. This study investigated the interaction of recombinant CYP3A4 with a nonspecific inhibitor metyrapone, anti-fungal drug fluconazole, and protease inhibitor phenylmethylsulfonyl fluoride (PMSF). Metyrapone and fluconazole are classic type II ligands that inhibit CYP3A4 with medium strength by ligating to the heme iron, whereas PMSF, lacking the heme-ligating moiety, acts as a weak type I ligand and inhibitor of CYP3A4. High-resolution crystal structures revealed that metyrapone orients similarly but not identically to the previously reported 1W0G model, whereas the flexible fluconazole adapts a conformer markedly different from that observed in the target CYP51 enzymes, which could explain its high potential for cross-reactivity. Besides hydrophobic and aromatic interactions with the heme and active site residues, both drugs establish water-mediated contacts that stabilize the inhibitory complexes. PMSF also binds near the catalytic center, with the phenyl group parallel to the heme. However, it does not displace the water ligand and is held in place via strong H-bonds formed by the sulfofluoride moiety with Ser119 and Arg212. Collectively, our data suggest that PMSF might have multiple binding sites and likely occupies the high-affinity site in the crystal structure. Moreover, its hydrolysis product, phenylmethanesulfonic acid, can also access and be retained in the CYP3A4 active site. Therefore, to avoid experimental artifacts, PMSF should be excluded from purification and assay solutions.
Keywords: CYP3A4, ligand binding, inhibition, metyrapone, fluconazole, PMSF
Graphical Abstract
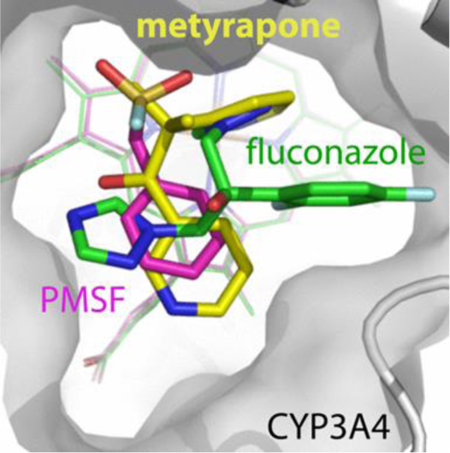
INTRODUCTION
Cytochromes P450 (CYPs) are ubiquitous enzymes that oxidize a wide variety of exogenous and endogenous compounds and play an important role in cellular homeostasis and metabolism.1 Among human CYPs, CYP3A4 is the most abundant and catalytically the most promiscuous isoform that biotransforms the majority of administered pharmaceuticals.2 Having a spacious and malleable active site, CYP3A4 can accommodate and oxidize a wide range of structurally diverse molecules. Moreover, CYP3A4 can simultaneously bind two or more chemicals, whose competing for the catalytic center could lead to atypical (non-Michaelis-Menten) enzyme kinetics and drug-drug interactions (DDIs) in vivo.3–4 Inhibition of CYP3A4 by small pharmaceuticals during multi-drug medication is the major mechanism associated with DDIs. Therefore, a better understanding of the ligand binding mechanism, in general, and the inhibitory complex formation, in particular, is necessary for the development of pharmaceuticals with fewer side effects.
Inhibitors can bind remotely and in the vicinity of the catalytic center or directly coordinate to the heme via the nitrogen heteroatom with a free pair of electrons. Currently, there are only three crystal structures of CYP3A4 bound to inhibitory drugs: metyrapone,5 ketoconazole,6 and ritonavir.7 Unlike ketoconazole and ritonavir, which are large and highly potent inhibitors of CYP3A4, metyrapone (Fig. 1) is a small and nonspecific inhibitor that interferes with drug metabolism.8 Metyrapone preferentially binds to a steroid hydroxylase CYP11B19 and is currently used as a diagnostic drug for testing hypothalamic-pituitary ACTH function and treatment of hypercortisolemia in patients with Cushing’s disease.10 The CYP3A4-metyrapone complex was one of the first to be crystallized (PDB code 1W0G)5 but the resolution was insufficient for resolving solvent molecules that could assist the ligand binding. Also, the 1W0G model has a high Rfree factor (31.8%; an independent quality/reliability measure), which normally should not exceed 30%. Therefore, the interaction of CYP3A4 with metyrapone was revisited by obtaining higher resolution structural data and evaluating the role of Ser119, predicted to be important for metyrapone association/egress by computational studies.11–13
Figure 1.
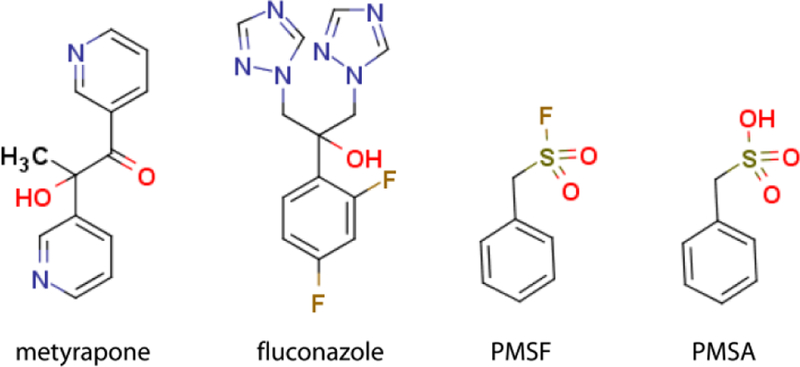
Investigated compounds.
Secondly, we investigated how CYP3A4 interacts with fluconazole (Fig. 1), an antifungal agent implicated in DDIs.14–22 Several fluconazole-bound structures of microbial CYP51 and CYP121 enzymes have been determined 23–27 but none of human CYP. Gaining structural information on azole drug orientation in human CYPs is highly important because it could clarify the cross-reactivity mechanism and help develop new antifungal agents that can be safely used in multi-drug therapy.
Finally, during our purification trials on CYP3A4, we noticed that its chromatographic behavior changes in the presence of phenylmethylsulfonyl fluoride (PMSF; Fig. 1), a protease inhibitor commonly used to prevent protein degradation during purification and functional assays. To better understand this phenomenon, PMSF was included in this study and, for the first time, was identified as a weak type I ligand and inhibitor of CYP3A4. Moreover, its hydrolysis product, phenylmethanesulfonic acid (PMSA; Fig. 1), could also access and be retained in the active site when externally added. Based on these findings, we recommend excluding PMSF from purification and assay solutions to avoid experimental artifacts.
EXPERIMENTAL PROCEDURES
Materials –
Metyrapone, fluconazole, PMSF and PMSA were purchased from Enzo Life Sciences, TCI America, Gold Biotechnology and Sigma-Aldrich, respectively.
Protein expression and purification -
The full-length and Δ3–22 human CYP3A4 were produced as reported previously.28 Δ3–22 CYP3A4 was used for equilibrium titrations and crystallization, whereas the inhibitory assays were conducted with the full-length protein to promote interaction with the redox partner, cytochrome P450 reductase.29 The S119A and R212A mutants of CYP3A4 were prepared as described elsewhere.30–31
Spectral Binding Titrations –
Equilibrium titrations of CYP3A4 were conducted in a Cary 300 spectrophotometer at ambient temperatures in 0.1M phosphate pH 7.4, containing 20% glycerol and 1 mM dithiothreitol (buffer A). Compounds were dissolved in dimethyl sulfoxide (DMSO) and added to a 1.5–2 μM protein solution in small aliquots, with the final solvent concentration less than 2% for metyrapone and fluconazole, and 5% for PMSF and PMSA. Spectral dissociation constants (Ks) were determined from the single-site hyperbolic fits to the plots of the maximal absorbance change (peak-to-trough separations in the difference spectra; ΔA) vs. ligand concentration. For PMSF and PMSA, the long-wavelength (475 or 490 nm) rather than near-UV absorbance peak was used for ΔA calculation to reduce the contribution of light scattering.
Inhibitory Potency Assays –
The inhibitory potency of the investigated compounds on the 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC) O-debenzylation activity of CYP3A4 was evaluated fluorometrically in a soluble, lipid-free reconstituted system as described in detail elsewhere.32 The IC50 values were derived from the [% activity] vs. [inhibitor] plots.
Determination of the X-ray Structures –
CYP3A4 was co-crystallized with metyrapone, fluconazole and PMSF at room temperature by a microbatch method under oil. Protein solution (50 mg/ml) in 100 mM phosphate, pH 7.4, 20% glycerol and 1 mM dithiothreitol was mixed with a 10-fold ligand excess and centrifuged to remove precipitate. The ligand-bound CYP3A4 (0.5–0.8 μl) was mixed with an equal volume of the crystallization solution (10–12% Peg 3350, 50–100 mM malonate, pH 6.8–7.2) and covered with paraffin oil. Crystals were harvested next day and cryoprotected with paratone-N before freezing in liquid nitrogen. X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource beamline 14–1 and the Advanced Light Source beamline 5.0.2. CC1/2 > 0.3 was used as a criterion for the high resolution data cutoff.33 Crystal structures were solved by molecular replacement with PHASER34 and the 5VCC structure as a search model. Ligands were manually fit into electron density with COOT.35 The initial models were refined with PHENIX 36 and rebuilt with COOT. Simulated annealing and polder omit maps were calculated with PHENIX. Data collection and refinement statistics are summarized in Table 1. The atomic coordinates and structure factors for the metyrapone-, fluconazole- and PMSF-bound CYP3A4 were deposited in the Protein Data Bank with the ID codes 6MA6, 6MA7 and 6MA8, respectively.
Table 1.
Data collection and refinement statistics
| Ligand PDB code | metyrapone 6MA6 | fluconazole 6MA7 | PMSF 6MA8 |
|---|---|---|---|
| Data collection statistics | |||
| Space group | I222 | I222 | I222 |
| Unit cell parameters | a = 77 Å, b = 101 Å, c = 127 Å; α, β, γ = 90° | a = 76 Å, b = 102 Å, c = 127 Å; α, β, γ = 90° | a = 77 Å, b = 100 Å, c = 132 Å; α, β, γ = 90° |
| Molecules per asymmetric unit | 1 | 1 | 1 |
| Resolution range (Å) | 39.17–2.18 (2.25–2.18) a | 79.28–2.09 (2.20–2.09) | 66.64–1.83 (1.93–1.83) |
| Total reflections | 147,775 | 189,246 | 216,952 |
| Unique reflections | 26,333 | 29,479 | 45,087 |
| Redundancy | 5.6 (5.5) | 6.4 (6.4) | 4.8 (4.8) |
| Completeness | 99.7 (98.3) | 99.7 (99.5) | 99.8 (100.0) |
| Average I/σI | 10.2 (0.4) | 8.7 (1.2) | 9.0 (0.8) |
| Rmerge | 0.048 (2.959) | 0.089 (1.553) | 0.071 (1.456) |
| Rpim | 0.024 (1.376) | 0.037 (0.644) | 0.035 (0.737) |
| CC ½ | 0.999 (0.781) | 0.997 (0.412) | 0.998 (0.330) |
| Refinement statistics | |||
| R/Rfreeb | 18.6 (26.0) | 19.4 (25.1) | 18.3/22.9 |
| No. of protein atoms | 3845 | 3763 | 3754 |
| No. of ligand atoms | 17 | 22 | 11 |
| No. of water molecules | 81 | 89 | 231 |
| Average B-factor (Å2): | |||
| Protein | 106 | 81 | 52 |
| Heme | 64 | 45 | 37 |
| Ligand | 78 | 46 | 60 |
| Glycerol | 140 | - | - |
| Ethylene glycol | 134 | 103 | 75 |
| DMSO | - | 63 | 94 |
| Solvent | 101 | 71 | 61 |
| Wilson B-value (Å2) | 72 | 55 | 40 |
| r.m.s. deviations: | |||
| Bond lengths, Å | 0.010 | 0.009 | 0.007 |
| Bond angles, ° | 1.136 | 1.003 | 0.964 |
| Ramachandran plotc (residues; %) | |||
| Preferred | 435 (94.6%) | 441 (96.5%) | 440 (96.3%) |
| Allowed | 24 (5.2%) | 14 (3.1%) | 17 (3.7%) |
| Outliers | 1 (0.2%) | 2 (0.4%) | none |
Values in brackets are for the highest resolution shell.
Rfree was calculated from a subset of 5% of the data that were excluded during refinement.
Analyzed with PROCHECK.
RESULTS AND DISCUSSION
To relate the ligand binding affinity, inhibitory potency and association mode, spectral dissociation constants (Ks), half-maximal inhibitory concentrations (IC50) for the BFC debenzylase activity, and co-crystal structures of CYP3A4 with metyrapone, fluconazole and PMSF were determined.
Interaction of CYP3A4 with metyrapone
Binding affinity and inhibitory potency of metyrapone -
During equilibrium titrations, metyrapone induced a red shift in the Soret band (type II spectral changes), indicative of the nitrogen ligation to the heme iron (Fig. 2A). Ks and IC50 derived from the titration and inhibitory plots (Figs. 2A and 3) were 2.4 and 18 μM, respectively, meaning that metyrapone binds and inhibits CYP3A4 with medium strength. The IC50 estimate was 3-fold higher than that for microsomal CYP3A4, but still within the physiologically significant range for DDIs.21 The large difference between the Ks and IC50 values could be due to distinct assay conditions, lower accessibility of metyrapone for the full-length CYP3A4, altered topology of the active site in the presence of the redox partner, cytochrome P450 reductase,37–38 and/or the ability of BFC to weaken the CYP3A4-metyrapone interaction.
Figure 2. Interaction of CYP3A4 with metyrapone.
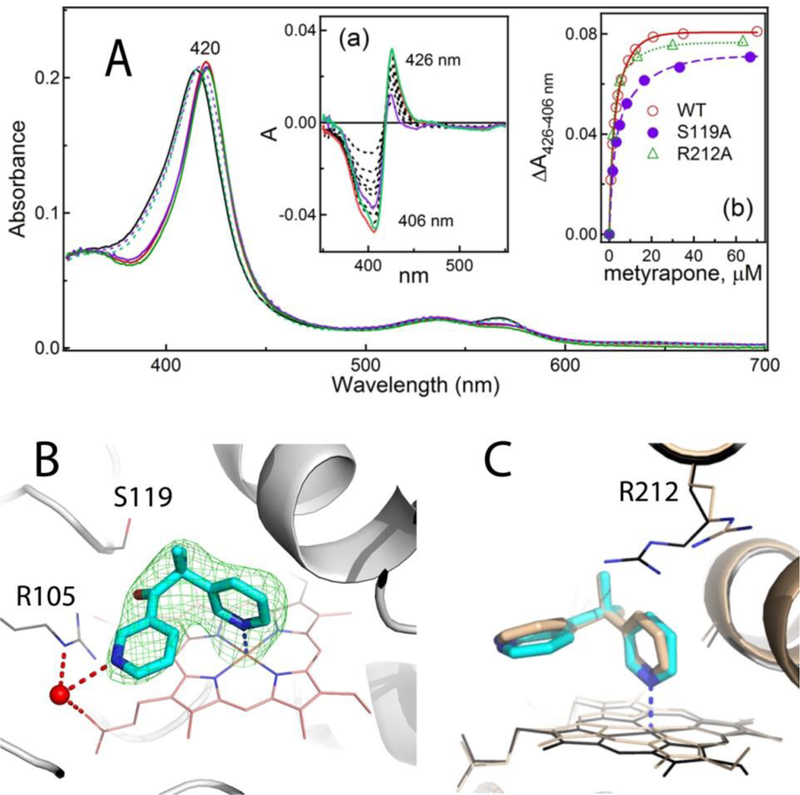
A, Spectral changes induced by metyrapone. Absorbance spectra of the ligand-free and metyrapone-bound WT, recorded at the end of titration, are shown as solid black and red lines, respectively. The corresponding spectra of S119A and R212A CYP3A4 are in dotted/solid violet and green lines, respectively.
Insets a and b, Difference spectra and titration plots with the hyperbolic fittings, respectively. In Inset a, the difference spectra recorded for WT, S119A and R212A CYP3A4 at maximal metyrapone concentrations are in red, purple and green, respectively. Spectral dissociation constants (Ks) derived from three independent experiments are listed in Table 2.
B, Metyrapone binds to CYP3A4 by ligating to the heme iron. A water molecule (red sphere) links the non-ligating pyridine nitrogen to the heme propionate and the Arg105 side group. Hydrogen bonds are depicted as dashed red lines. Green mesh is a simulated annealing omit map contoured at 3σ level. That CYP3A4 preferably binds the depicted conformer of metyrapone is demonstrated in Figure S1.
C, Superposition of 6MA6 and 1W0G structures (in cyan/black and beige, respectively) shows distinct orientations of the pyridine rings and the Arg212 side chain.
Figure 3.
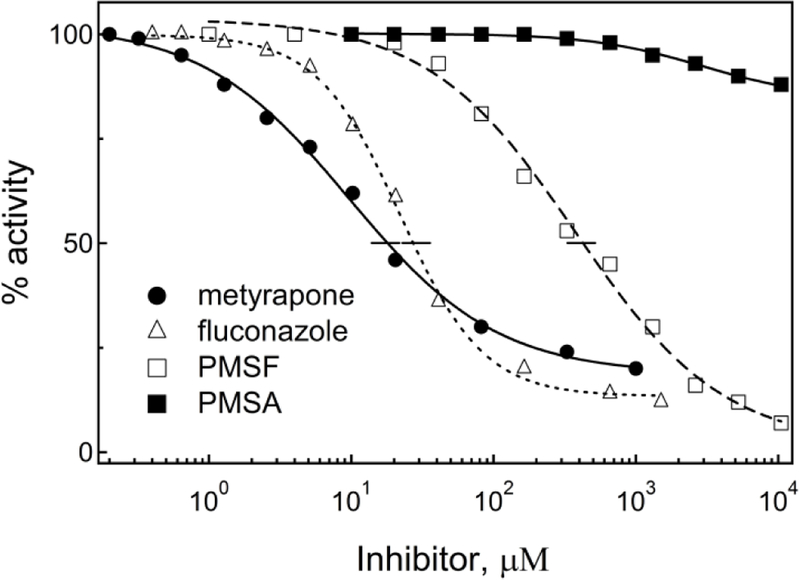
Inhibitory plots for the BFC debenzylase activity of CYP3A4. Each data point represents an average of three measurements, with the standard error <15%. The IC50 values derived from the plots are listed in Table 2.
Crystal structure of the CYP3A4-metyrapone complex –
As mentioned, the previously reported 1W0G model was determined to 2.73 Å resolution, which was too low to resolve solvent molecules that could orient/stabilize the inhibitory complex. We obtained better diffracting crystals of metyrapone-bound CYP3A4 and refined the structure to 2.18 Å and R/Rfree of 18.6/26.0 (Table 1). The ligand fitting was straightforward because, as demonstrated in Figure S1, only one conformer of metyrapone fits into electron density and preferably binds to CYP3A4 (occupancy of ~0.9). This conformer was similar to that modeled in the 1W0G structure, with one pyridine N-heteroatom directly ligated to the heme and the second pyridine ring in a perpendicular orientation and parallel to the heme (Fig. 2B). The Fe-N distance remains nearly the same (2.31 vs. 2.27 Å in 1W0G) but the ring positioning is distinct (Fig. 2C): the heme-ligating pyridine tilts toward the central I-helix by 10° and orients perpendicular to the heme plane, whereas the second ring rotates by 20° to lay parallel to the cofactor. These adjustments optimize the coordination orientation, as well as π-π stacking interactions with the heme macrocycle. One newly identified feature is the water molecule that links the non-ligated pyridine nitrogen to the heme propionate and the Arg105 guanidine group. By strengthening protein-ligand interactions and decreasing the motional freedom of metyrapone, this water-mediated bridge could increase the binding and inhibitory strength.
Ser119 and Arg212 are not critical for the equilibrium binding of metyrapone –
Another structural difference that could influence the binding affinity of metyrapone is conformational variation in the Arg212 side group, lying above and within the van der Waals distance from metyrapone in our structure and swinging aside in 1W0G (Fig. 2C). Ser119, however, is in a similar conformation and does not form direct or water-mediated contacts with metyrapone, which disagrees with theoretical predictions.11–13 To test whether Ser119 and Arg212 contribute to the equilibrium ligand binding, Ks was measured for the S119A and R212A mutants of CYP3A4, which were already available.30–31 Similar to WT, both variants displayed type II spectral changes, consistent with the association of one metyrapone molecule in the active site (Fig. 2A). Variations in the maximal absorbance change (ΔA426–406 nm; Inset b) were, in part, due to differences in the Soret peak (415, 418 and 416 nm for the ligand-free WT, S119A and R212A CYP3A4, respectively), but could also result from an altered coordination and suboptimal Fe-N orbital overlap. Changes in Ks were moderate but bidirectional: 42% increase for S119A and 33% decrease for R212A (Table 2). This suggests that neither residue significantly contributes to the binding affinity of metyrapone but, unlike Arg212, Ser119 assists the inhibitory complex formation to some extent, possibly via transient polar interactions.
Table 2.
Binding affinity and inhibitory potency of the investigated compounds
| Ks (μM) |
IC50 (μM) | |||
|---|---|---|---|---|
| WT | S119A | R212A | ||
| Metyrapone | 2.4± 0.11 | 3.4±0.1 | 1.6±0.1 | 18±2 |
| Fluconazole | 17±3 | 15±1 | 21±2 | 27±3 |
| PMSF | 2650±150 | 12500±1550 | 11500±1150 | 420±30 |
| PMSA | 15380±2060 | ND2 | ND | >20000 |
All values represent an average of three measurements with the standard error.
Not determined because there were no ligand-specific spectral changes.
Interaction of CYP3A4 with fluconazole
Binding affinity and inhibitory potency of fluconazole –
During equilibrium titrations, fluconazole also behaved as a typical type II ligand (Fig. 4A; Table 2), with the red shift in the Soret band increasing continuously with an increase in fluconazole concentration. This is in contrast to the recent study by Godamudunage et al.,39 who observed a high-spin shift in the heme iron (type I spectral change) at low fluconazole concentrations. The Ks value for the CYP3A4-fluconazole complex was in the same range: 22 μM vs. 17 μM reported here and 10 μM for the full-length CYP3A4.40 The IC50 estimate, however, exceeded ours by 10-fold (>200 vs 21 μM, respectively). This discrepancy could arise from differences in the reconstituted system, assay methods and/or CYP3A4 substrates. For microsomal CYP3A4, for instance, the IC50 for fluconazole varied from 12 to 210 μM when midazolam and tacrolimus were used as substrates for the inhibitory assays.21
Figure 4. Interaction of CYP3A4 with fluconazole.
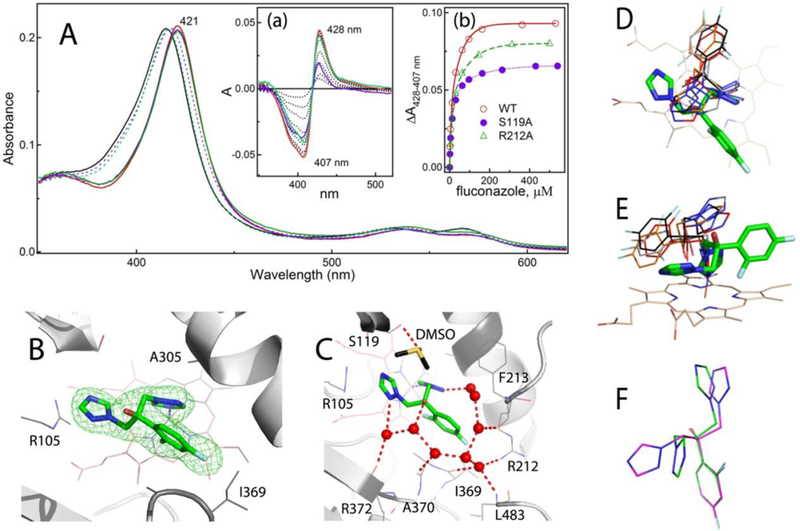
A, Spectral changes induced by fluconazole. Absorbance spectra of the ligand-free and fluconazole-bound WT CYP3A4, recorded at the end of titration, are in solid black and red lines, respectively. The corresponding spectra of the S119A and R212A mutants are in dotted/solid violet and green lines, respectively. Insets a and b, Difference spectra and titration plots with hyperbolic fittings, respectively. In Inset a, the difference spectra recorded for WT, S119A and R212A CYP3A4 at maximal fluconazole concentrations are in red, purple and green, respectively. Ks values are listed in Table 2.
B, View at the fluconazole molecule ligated to the heme via the triazole moiety. Green mesh is a simulated annealing omit map contoured at 3σ level. Additional views at the active site are shown in Figure S2 to demonstrate an unambiguous identification of the ligand binding mode.
C, Solvent network linking fluconazole’s N-heteroatoms and hydroxyl group to the main and side chains of the nearby residues. Water molecules and hydrogen bonds are shown as red spheres and dashed lines, respectively.
D and E, Two views at the overlaid structures of fluconazole-bound CYP3A4 (in green sticks) and CYP51 from Saccharomyces cerevisiae (4WMZ; in red), Mycobacterium tuberculosis (1EA1; in gray), Naegleria fowleri (6AY4; in black), and Tripanosoma brucei (2WV2; in orange). Structures of CYP51 from Trypanozoma cruzi and Leishmania infantum (3KHM and 3L4D, respectively) were not included because of invalid geometry of fluconazole.
F, Superposition of fluconazole molecules bound to CYP3A4 (in green) and CYP51 (6AY4; in magenta).
Generally, the absolute spectra of the ligand-bound CYP (λmax) could reflect electronic properties of the coordinating group, whereas ΔAmax in the difference spectra is more sensitive to the environment/steric properties distant from the heme: the larger the ΔAmax value, the stronger the coordination mode.40 As seen in Figures 3A and 4A, metyrapone and fluconazole induce highly similar spectral changes in CYP3A4. However, although fluconazole has a 7-fold lower affinity, it induces a slightly larger Soret shift and ΔAmax (by 1 nm and ~8%, respectively). Thus, the spectral data alone cannot explain differences in the binding strength.
Crystal structure of the CYP3A4-fluconazole complex –
The crystal structure of the CYP3A4-fluconazole complex, solved to 2.09 Å resolution (Table 1), helped to understand why fluconazole is a weaker ligand than metyrapone. As the equilibrium titrations predicted, only one fluconazole molecule binds to CYP3A4 through the triazole nitrogen coordination, with the Fe-N distance of 2.20 Å (Fig. 4B). Additional views at the heme-bound fluconazole are shown in Figure S2 to demonstrate the quality of the electron density map allowing unambiguous identification of the ligand binding mode. Surprisingly, no conformational change other than swinging of the Arg212 side chain was needed to accommodate the larger and more chemically complex fluconazole (r.m.s.d. between the Cα-atoms of the ligand-free 5VCC and 6MA7 structures is 0.41 Å). However, because of space limitations and conformational restraints, fluconazole cannot adjust positioning of its functional groups as freely as metyrapone does. In particular, the heme-ligating triazole cannot orient perpendicular to the heme (~10° deviation) due to restraints imposed by the adjacent difluorophenyl ring, partially inserted into a narrow cavity formed by Thr309, Ile369 and Ala370. The ortho fluorine atom points toward and is only 3.2 Å from the heme, whereas the para fluorine is trapped between the Arg212 and Ile369 side/main chains (3.3 and 2.8 Å away, respectively), which disallows any lateral or vertical movements. The difluorophenyl ring itself is perpendicular to the heme and, hence, is optimally poised for T-stacking interactions. The non-ligating triazole also lies in the vicinity from the heme (< 4 Å) but with a 35° tilt and cannot adapt a parallel orientation to maximize π-π stacking due to steric clashing with Arg105.
This sub-optimal binding mode is partially compensated by an intricate solvent network that connects fluconazole’s hydroxyl group and triazole nitrogens to the side and main chains of Arg212, Phe213, Ile369, Ala370, Arg372 and Leu483. In addition, the non-ligating triazole makes van der Waals contacts with the DMSO molecule (used to dissolve fluconazole), which sits on top and H-bonds to Ser119 (Fig. 4C). Replacement of Arg212 or Ser119 with alanine had virtually no effect on Ks (Table 2) but led to a more notable decrease in ΔA4max: 10% for R212A and 30% for S119A vs 6% and 9%, respectively, observed for metyrapone (compare Insets b in Figs. 3A and 4A). Again, although Ser119 does not establish any contacts with fluconazole, it seems to affect the ligand coordination to a larger extent than Arg212, part of the polar network. Considering the active site architecture, it is plausible to conclude that Ser119 could modulate the binding of fluconazole not only through transient polar interactions but also by restricting maneuvering space near the heme.
Comparison of the fluconazole binding mode in CYP3A4 and fungal CYP51 -
Unlike drug-metabolizing CYPs, the CYP51 enzymes evolved to perform one specific function, sterol biosynthesis.41 As a result, they have a smaller and more rigid active site, and serve as a target for azole antifungal drugs. Fluconazole inhibits fungal CYP51 very potently (Kd and IC50 for Candida albicans CYP51 are 47 nM and 0.6–1.2 μM, respectively) and with over 500-fold higher selectivity over the human homolog.42–43 One undesired side effect is the ability of fluconazole to bind to drug-metabolizing CYPs tightly enough to cause DDIs during multi-drug therapy.21 To better understand the molecular basis for cross-reactivity, the binding orientations of fluconazole in CYP3A4 and various CYP51 enzymes were compared. As seen from structural overlays (Fig. 4D–F), in order to fit into a larger and differently shaped CYP3A4 active site, fluconazole adapts a distinct conformer and reorients, placing its functional groups in the opposite directions. This shows once again that the binding manner of the antifungal drugs in steroidogenic and nonsteroidogenic CYPs could be profoundly different44 and, thus, accurate predictions for human CYP inhibition cannot be made solely based on the CYP51 models. Novel insights provided by the first-of-its-kind CYP3A4-fluconazole structure could improve in silico screening/computer modeling predictions and facilitate the rational design of pharmaceuticals with fewer off-target activities and lower potential for DDIs.
Interaction of CYP3A4 with PMSF
PMSF is a non-selective protease inhibitor that binds to the catalytic serine of trypsin, chymotrypsin and several other proteases, and is commonly used at 0.1–2 mM concentrations to prevent CYP3A4 proteolysis during purification and functional assays. During our initial purification trials, we noticed that CYP3A4 elutes from the ion-exchange column in a wide, diffused band when PMSF is present in solution, but forms a narrow band in its absence. To better understand the underlying mechanism, we investigated if/how CYP3A4 interacts with PMSF.
PMSF is a weak type I ligand and inhibitor of CYP3A4 –
During equilibrium titrations, PMSF induces type I spectral changes in CYP3A4, meaning that it can access the catalytic site and displace the distal water ligand (Fig. 5A). Compared to other investigated compounds, it took considerably longer (>20 min) to reach equilibrium after each addition of PMSF. Moreover, the ligand saturation could not be fully achieved due to low solubility of PMSF and its instability in aqueous solutions (t1/2 =55 min at pH 7.5).45 To minimize PMSF hydrolysis and correct for DMSO-induced spectral perturbations, titration experiments were repeated to measure the difference absorbance spectra shortly after the ligand addition (<5 min), with equal amounts of solvent added to the reference cuvette. This allowed to identify two spectral phases (Fig. 5A, Inset), indicative of two binding events taking place at low and high PMSF concentrations: ≤ 60 μM and 0.1–8 mM, respectively. Importantly, the first spectral phase, characterized by a small decrease in the Soret band (~5% of total absorbance change), was observed regardless of the length of the time interval between PMSF additions. The second phase was accompanied by the Soret peak decrease and an increase in the near-UV and long-wavelength regions. The Ks estimates for the two binding events (Fig. 5B, C) were 20 μM and 2.65 mM. This could explain why the IC50 value for PMSF, acting as a weak inhibitor of CYP3A4 (Fig. 3), was in the intermediate range (0.42 mM).
Figure 5. Interaction of CYP3A4 with PMSF.
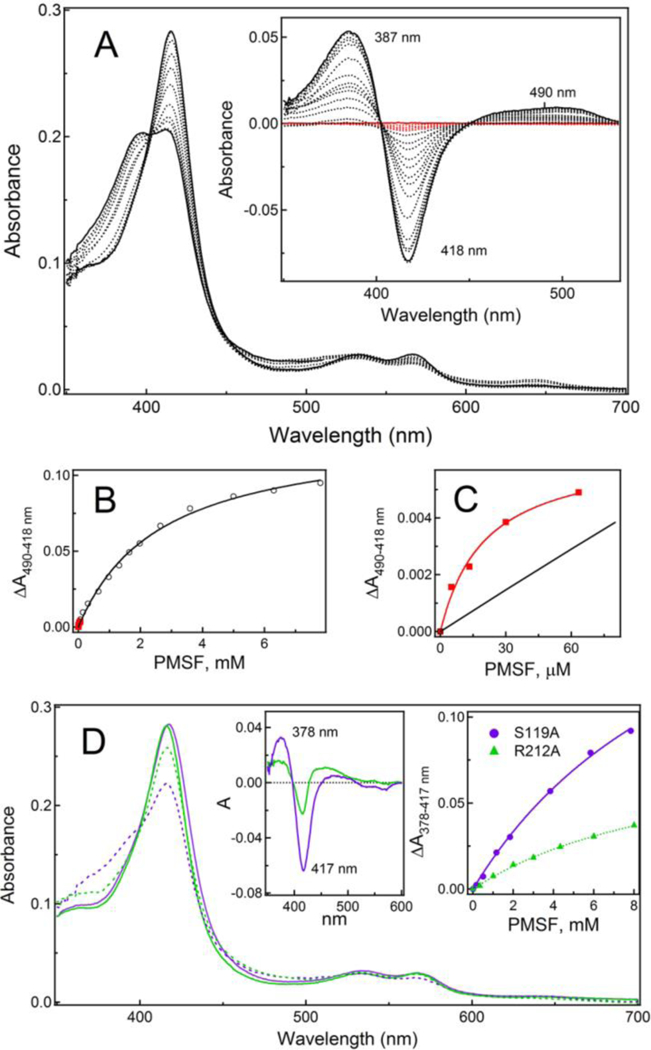
A, Type I spectral changes induced by PMSF in WT CYP3A4 during equilibrium titrations. Inset, Difference spectra recorded in a separate experiment where equal amounts of DMSO were added to the reference cuvette to correct for the solvent-induced spectral perturbations. Absorbance changes occurring during the first spectral phase, accompanied by a small decrease in the Soret band, are highlighted in red.
B and C, Titration plots and hyperbolic fittings for the first and second ligand binding events (shown in red and black, respectively). The Ks estimates for two binding steps are 20 μM and 2.65 mM, respectively.
D, Absorbance spectra of the ligand-free and PMSF-bound S119A and R212A CYP3A4 recorded at the end of titrations (in solid/dashed purple and green lines, respectively). Insets are the respective difference spectra (left) and titration plots with hyperbolic fittings (right). The Ks estimates are listed in Table 2. Spectral data demonstrating two distinct PMSF binding events in both mutants are shown in Figure S3.
PMSF hydrolysis product can also access and be retained in the CYP3A4 active site –
In aqueous solutions, PMSF undergoes hydrolysis to form phenylmethanesulfonic acid (PMSA; Fig. 1). Since PMSA is structurally similar to PMSF but negatively charged, it was of interest to investigate whether it could interact with CYP3A4 in a similar manner. Indeed, during equilibrium titrations, PMSA induced type I spectral changes (Fig. 6A) but its affinity for CYP3A4 was ~6-fold lower (Table 2). The difference absorbance spectra measured in a separate titration experiment with corrections for DMSO-dependent perturbations (Fig. 6A, Inset) allowed to distinguish two spectral phases corresponding to two ligand binding events taking place at low (<0.2 mM) and high (0.3–45 mM) PMSA concentrations (Fig. 6B, C). However, absorbance changes observed during the first phase were distinct from those induced by PMSF (compare Figs. 5C and 6C) and, as the inhibitory assays showed, PMSA only negligibly affects CYP3A4 activity (Fig. 3; Table 1). Thus, the externally added PMSA can access and alter spectral properties of the heme but the negative charge on the sulfonate group decreases the binding affinity and inhibitory potency for CYP3A4.
Figure 6. Interaction of CYP3A4 with PMSA.
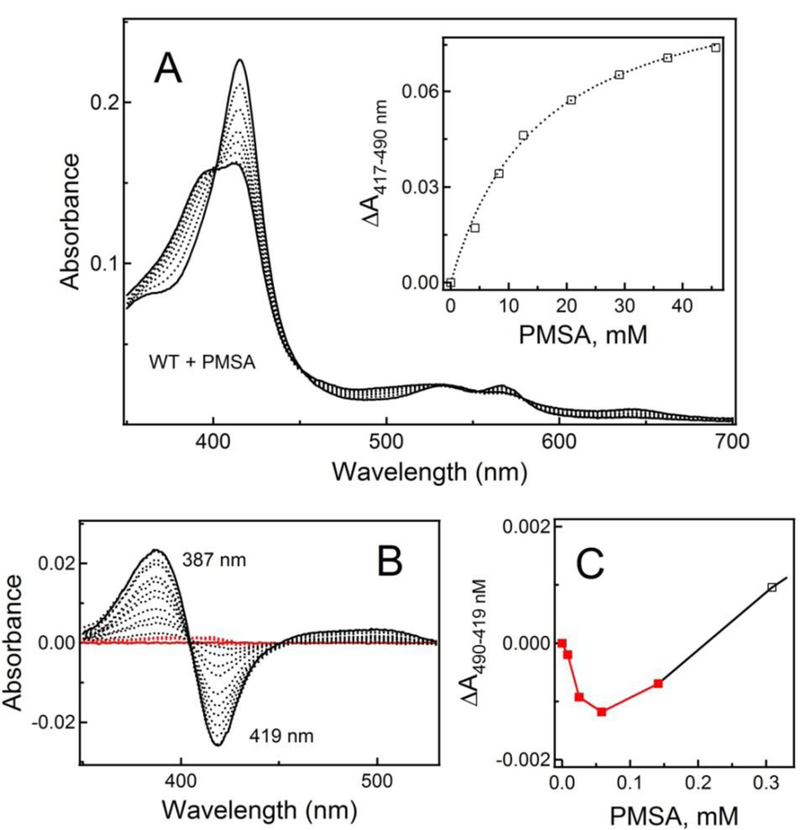
A, Type I spectral changes induced by PMSA in WT CYP3A4 during equilibrium titrations. Inset, Titration plot with hyperbolic fitting. The Ks estimate is given in Table 2.
B, Difference spectra recorded in a separate experiment that included low PMSA concentrations and where equal amounts of DMSO were added to the reference cuvette to correct for the solvent-induced spectral perturbations. Absorbance changes occurring during the first spectral phase, characterized by a rise in the 370–430 nm region, are highlighted in red.
C, The initial lag phase in the titration plot observed at low PMSA concentrations (red data points).
Crystal structure of the CYP3A4-PMSF complex -
To get insights into the association mode and inhibitory mechanism of PMSF, we attempted to obtain its co-crystal structure with CYP3A4. Crystals were grown overnight in the presence of a 10-fold excess of PMSF, and the X-ray structure was solved to 1.83 Å resolution (Fig. 7A; Table 1). There is only one ligand molecule bound in the active site near the catalytic center, with the phenyl ring parallel to and 3.4 Å from the heme. It is not possible to distinguish whether the ligand is PMSF or PMSA, as both compounds fit equally well (Fig. 7B) and the R/Rfree factors do not change upon their substitution. However, since PMSF has a considerably higher affinity for CYP3A4 and its lifetime in the hydrophobic active site could be extended, there is a higher possibility that the structure represents the PMSF-bound complex and, therefore, it will be described as such.
Figure 7. Crystal structure of the CYP3A4-PMSF complex.
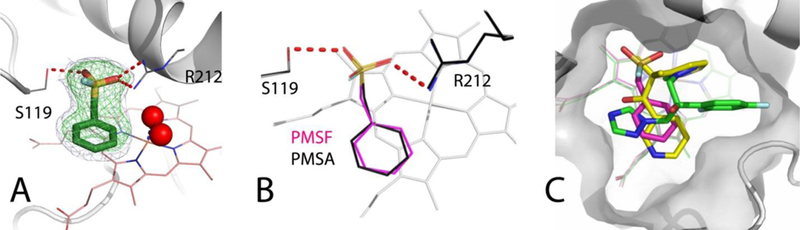
A, View at the active site of PMSF-bound CYP3A4. The sulfofluoride moiety of PMSF forms strong H-bonds (red dashed lines) with the Ser119 and Arg212 side groups. The distal water ligand and the neighboring solvent molecule are depicted as red spheres. Simulated annealing and polder omit maps are shown as green and gray mesh contoured at 3σ and 5σ levels, respectively.
B, PMSA fits equally well into the PMSF binding site, and PMSF-to-PMSA substitution does not affect the refinement statistics.
C, Comparison of the binding modes of metyrapone, fluconazole and PMSF (shown in yellow, green and magenta, respectively).
Superposition of the PMSF-, metyrapone- and fluconazole-bound CYP3A4 shows that the non-ligating aromatic groups of these chemically distinct compounds are clustered above the porphyrin A-ring (Fig. 7C). This space, previously identified as a P2 site,46 seems to be filled by the aromatic groups whenever possible. Besides hydrophobic and π-π stacking interactions, the phenyl moiety of PMSF forms van der Waals contacts with the Arg105 and Ile369 side groups. The sulfofluoride, on the other hand, establishes strong hydrogen bonds (<2.7 Å) with the Ser119 and Arg212 side chains which, like tweezers, keep PMSF in a fixed position (Fig. 7A). These polar contacts are important, as both S119A and R212A mutations decrease the binding affinity of PMSF by 4-fold (Fig. 5D; Table 2). Nonetheless, two spectral phases could be distinguished during titration of both mutants, suggesting the presence of two distinct PMSF binding sites. A substantial high-spin shift was observed only in the S119A variant (~30% of WT; Fig. S3). In contrast, neither mutant exhibits spectral changes in the presence of PMSA (Fig. S4), which suggests that polar/electrostatic interactions are critical for the PMSA association.
Notably, in the crystal structure, PMSF does not displace the distal water ligand, which remains bound to the heme. One reason could be the limited solubility of PMSF, which precludes reaching concentrations sufficient for saturation of both binding sites in crystallization solution. Conformational selection and preferable crystallization of CYP3A4 with one PMSF bound could be another explanation. A nearly full occupancy of the PMSF- and heme-bound water sites (0.94 and 0.96, respectively) excludes the possibility of multiple ligand orientations and a mixed low/high spin state. Therefore, it is plausible that the crystallographic site represents the initial binding site, occupation of which takes place at low ligand concentrations and leads to spectral perturbations comprising the first spectral phase (Fig. 5B, C), whereas association of an additional PMSF molecule(s) is needed to trigger the high spin shift. In other words, the spectral and structural data suggest that PMSF could have multiple binding sites in CYP3A4 and likely occupies the high-affinity site in the crystal structure.
Further, direct engagement of PMSF with Arg212 provides an explanation on how this compound could affect molecular properties of CYP3A4. Arg212 is part of the highly flexible F-G fragment that mediates interactions with the lipid bilayer and promotes protein aggregation.29, 47–48 By H-bonding to Arg212, PMSF could modulate the F-G conformer distribution and, subsequently, alter chromatographic behavior of CYP3A4.
It is unclear how quickly PMSF degrades when bound to CYP3A4. If the life-time could be extended due to hydrophobic environment and limited solvent access, then PMSF would have the ability to co-purify with CYP3A4 or enter and remain in the active site upon de novo addition. If not, hydrolysis of the bound and free PMSF would be harmful as well, because the product could access and/or be retained in the active site via similar interactions. By crowding the active site and limiting an access to the catalytic center, both PMSF and PMSA could perturb spectral properties and catalytic behavior of CYP3A4. PMSF/A are unlikely to efficiently compete with the larger and more hydrophobic molecules, especially those that could ligate to the heme. However, the interference of PMSF/A could be more pronounced during association of small, low affinity substrates. DMSO is another component that could perturb spectral properties of CYP3A4 and, possibly, alter the binding affinity of weak ligands. Therefore, to avoid experimental artifacts, we advise (i) to conduct proper controls for DMSO and other solvents, and (ii) exclude PMSF from purification and assay solutions. In our purification protocol,28 leupeptin is used instead of PMSF and only during bacterial cell lysis, which is sufficient to maintain the integrity of CYP3A4.
SUMMARY
Interaction of CYP3A4 with small inhibitory molecules was investigated using spectral analysis, functional assays, and X-ray crystallography. Metyrapone and fluconazole were found to act as medium-strength type II ligands that inhibit CYP3A4 through coordination to the heme iron. In contrast, PMSF and its hydrolysis product, PMSA, were identified as weak type I ligands, possibly with multiple binding sites, occupation of which could block an access to the catalytic center and alter spectral and functional properties of CYP3A4.
Determination of high-resolution crystal structures allowed to accurately define the ligand binding modes, as well as the water- and protein-mediated polar interactions that stabilize/assist the inhibitory complex formation. No structural rearrangements except the Arg212 side chain reorientation are needed to accommodate the inhibitory compounds. However, in order to fit into the CYP3A4 active site, fluconazole adapts an isomer markedly distinct from that bound to microbial CYP51. This finding confirms the notion that the high flexibility/adaptability of azole drugs underlies their promiscuity and cross-reactivity with human CYPs, and could be used for designing the antifungals with fewer side effects.
Novel results on the CYP3A4-PMSF complex formation, on the other hand, not only suggest that PMSF can co-purify and interfere with the functional assays, but also provide insights on how small pharmaceuticals that lack the heme-ligating moiety could alter the CYP3A4 activity and lead to DDIs.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant ES025767 and involves research carried out at the Stanford Synchrotron Radiation Lightsource and the Advanced Light Source. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. De partment of Energy under Contract No. DE-AC02-05CH11231.
Abbreviations
- CYP3A4
cytochrome P450 3A4
- BFC
7-benzyloxy-4-(trifluoromethyl)coumarin
- DDI
drug-drug interaction
- DMSO
dimethyl sulfoxide
- PMSA
phenylmethanesulfonic acid
- PMSF
phenylmethanesulfonyl fluoride
- WT
wild type
Footnotes
Accession ID
UniProtKB accession ID for human CYP3A4 is P08684
Supporting Information
Two possible orientations of metyrapone, different views at the heme-bound fluconazole, and spectral changes induced by PMSF and PMSA in the S119A and R212A mutants of CYP3A4. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The author declares no competing financial interest.
References
- (1).Danielson PB (2002) The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab 3, 561–597. [DOI] [PubMed] [Google Scholar]
- (2).Guengerich FP (1999) Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol 39, 1–17. [DOI] [PubMed] [Google Scholar]
- (3).Tang W; Stearns RA (2001) Heterotropic cooperativity of cytochrome P450 3A4 and potential drug-drug interactions. Curr. Drug Metab 2, 185–198. [DOI] [PubMed] [Google Scholar]
- (4).Atkins WM (2005) Non-Michaelis-Menten kinetics in cytochrome P450-catalyzed reactions. Annu. Rev. Pharmacol. Toxicol 45, 291–310. [DOI] [PubMed] [Google Scholar]
- (5).Williams PA; Cosme J; Vinkovic DM; Ward A; Angove HC; Day PJ; Vonrhein C; Tickle IJ; Jhoti H (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305, 683–686. [DOI] [PubMed] [Google Scholar]
- (6).Ekroos M; Sjogren T (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U S A 103, 13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sevrioukova IF; Poulos TL (2010) Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. U S A 107, 18422–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Iribarne C; Berthou F; Baird S; Dreano Y; Picart D; Bail JP; Beaune P; Menez JF (1996) Involvement of cytochrome P450 3A4 enzyme in the N-demethylation of methadone in human liver microsomes. Chem. Res. Toxicol 9, 365–373. [DOI] [PubMed] [Google Scholar]
- (9).Liddle GW; Island D; Lance EM; Harris AP (1958) Alterations of adrenal steroid patterns in man resulting from treatment with a chemical inhibitor of 11 beta-hydroxylation. J. Clin. Endocrinol. Metab 18, 906–912. [DOI] [PubMed] [Google Scholar]
- (10).Ciato D; Mumbach AG; Paez-Pereda M; Stalla GK (2017) Currently used and investigational drugs for Cushing s disease. Expert Opin. Investig. Drugs 26, 75–84. [DOI] [PubMed] [Google Scholar]
- (11).Park H; Lee S; Suh J (2005) Structural and dynamical basis of broad substrate specificity, catalytic mechanism, and inhibition of cytochrome P450 3A4. J. Am. Chem. Soc 127, 13634–13642. [DOI] [PubMed] [Google Scholar]
- (12).Li W; Liu H; Luo X; Zhu W; Tang Y; Halpert JR; Jiang H (2007) Possible pathway(s) of metyrapone egress from the active site of cytochrome P450 3A4: a molecular dynamics simulation. Drug Metab. Dispos 35, 689–696. [DOI] [PubMed] [Google Scholar]
- (13).Krishnamoorthy N; Gajendrarao P; Thangapandian S; Lee Y; Lee KW (2009) Probing possible egress channels for multiple ligands in human CYP3A4: a molecular modeling study. J. Mol. Model 16, 607–614. [DOI] [PubMed] [Google Scholar]
- (14).Hsiao SH; Chang HJ; Hsieh TH; Kao SM; Yeh PY; Wu TJ (2016) Rhabdomyolysis caused by the moderate CYP3A4 inhibitor fluconazole in a patient on stable atorvastatin therapy: a case report and literature review. J. Clin. Pharm. Ther 41, 575–578. [DOI] [PubMed] [Google Scholar]
- (15).Nair DR; Morris HH (1999) Potential fluconazole-induced carbamazepine toxicity. Ann. Pharmacother 33, 790–792. [DOI] [PubMed] [Google Scholar]
- (16).Michalets EL; Williams CR (2000) Drug interactions with cisapride: clinical implications. Clin. Pharmacokinet 39, 49–75. [DOI] [PubMed] [Google Scholar]
- (17).Eap CB; Buclin T; Baumann P (2002) Interindividual variability of the clinical pharmacokinetics of methadone: implications for the treatment of opioid dependence. Clin. Pharmacokinet 41, 1153–1193. [DOI] [PubMed] [Google Scholar]
- (18).Davis MW; Wason S; Digiacinto JL (2013) Colchicine-antimicrobial drug interactions: what pharmacists need to know in treating gout. Consult. Pharm 28, 176–183. [DOI] [PubMed] [Google Scholar]
- (19).Skerjanec A (2006) The clinical pharmacokinetics of darifenacin. Clin. Pharmacokinet 45, 325–350. [DOI] [PubMed] [Google Scholar]
- (20).Gibbs MA; Thummel KE; Shen DD; Kunze KL (1999) Inhibition of cytochrome P-450 3A (CYP3A) in human intestinal and liver microsomes: comparison of Ki values and impact of CYP3A5 expression. Drug Metab. Dispos 27, 180–187. [PubMed] [Google Scholar]
- (21).Niwa T; Imagawa Y; Yamazaki H (2014) Drug interactions between nine antifungal agents and drugs metabolized by human cytochromes P450. Curr. Drug Metab 15, 651–679. [DOI] [PubMed] [Google Scholar]
- (22).Saad AH; DePestel DD; Carver PL (2006) Factors influencing the magnitude and clinical significance of drug interactions between azole antifungals and select immunosuppressants. Pharmacotherapy 26, 1730–1744. [DOI] [PubMed] [Google Scholar]
- (23).Seward HE; Roujeinikova A; McLean KJ; Munro AW; Leys D (2006) Crystal structure of the Mycobacterium tuberculosis P450 CYP121-fluconazole complex reveals new azole drug-P450 binding mode. J. Biol. Chem 281, 39437–39443. [DOI] [PubMed] [Google Scholar]
- (24).Podust LM; Poulos TL; Waterman MR (2001) Crystal structure of cytochrome P450 14alpha-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc. Natl. Acad. Sci. U S A 98, 3068–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lepesheva GI; Hargrove TY; Anderson S; Kleshchenko Y; Furtak V; Wawrzak Z; Villalta F; Waterman MR (2010) Structural insights into inhibition of sterol 14alpha-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem 285, 25582–25590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chen CK; Leung SS; Guilbert C; Jacobson MP; McKerrow JH; Podust LM (2010) Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis 4, e651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sagatova AA; Keniya MV; Wilson RK; Monk BC; Tyndall JD (2015) Structural Insights into Binding of the Antifungal Drug Fluconazole to Saccharomyces cerevisiae Lanosterol 14alpha-Demethylase. Antimicrob. Agents Chemother 59, 4982–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sevrioukova IF (2017) High-level production and properties of the cysteine-depleted cytochrome P450 3A4. Biochemistry 56, 3058–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sevrioukova IF; Poulos TL (2015) Anion-dependent stimulation of CYP3A4 monooxygenase. Biochemistry 54, 4083–4096. [DOI] [PubMed] [Google Scholar]
- (30).Sevrioukova IF; Poulos TL (2012) Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a type I ligand. J. Biol. Chem 287, 3510–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sevrioukova IF; Poulos TL (2013) Pyridine-substituted desoxyritonavir is a more potent cytochrome P450 3A4 inhibitor than ritonavir. J. Med. Chem 56, 3733–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Samuels ER; Sevrioukova IF (2018) Inhibition of human CYP3A4 by rationally designed ritonavir-like compounds: Impact and interplay of the side group functionalities. Mol. Pharm 15, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Karplus PA; Diederichs K (2012) Linking crystallographic model and data quality. Science 336, 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Emsley P; Lohkamp B; Scott WG; Cowtan K (2010) Features and development of Coot. Acta Crystallogr. Section D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Section D 66, 213–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yamaguchi Y; Khan KK; He YA; He YQ; Halpert JR (2004) Topological changes in the CYP3A4 active site probed with phenyldiazene: effect of interaction with NADPH-cytochrome P450 reductase and cytochrome b5 and of site-directed mutagenesis. Drug Metab. Dispos 32, 155–161. [DOI] [PubMed] [Google Scholar]
- (38).Modi S; Gilham DE; Sutcliffe MJ; Lian LY; Primrose WU; Wolf CR; Roberts GC (1997) 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine as a substrate of cytochrome P450 2D6: allosteric effects of NADPH-cytochrome P450 reductase. Biochemistry 36, 4461–4470. [DOI] [PubMed] [Google Scholar]
- (39).Godamudunage MP; Grech AM; Scott EE (2018) Comparison of Antifungal Azole Interactions with Adult Cytochrome P450 3A4 versus Neonatal Cytochrome P450 3A7. Drug Metab. Dispos 46, 1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Locuson CW; Hutzler JM; Tracy TS, (2007) Visible spectra of type II cytochrome P450-drug complexes: evidence that “incomplete” heme coordination is common. Drug Metab. Dispos 35, 614–622. [DOI] [PubMed] [Google Scholar]
- (41).Lepesheva GI; Waterman MR (2007) Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 1770, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Warrilow AG; Martel CM; Parker JE; Melo N; Lamb DC; Nes WD; Kelly DE; Kelly SL (2010) Azole binding properties of Candida albicans sterol 14-alpha demethylase (CaCYP51). Antimicrob. Agents Chemother 54, 4235–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Warrilow AG; Parker JE; Kelly DE; Kelly SL (2013) Azole affinity of sterol 14alpha-demethylase (CYP51) enzymes from Candida albicans and Homo sapiens. Antimicrob. Agents Chemother 57, 1352–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Strushkevich N; Usanov SA; Park HW (2010) Structural basis of human CYP51 inhibition by antifungal azoles. J. Mol. Biol 397, 1067–1078. [DOI] [PubMed] [Google Scholar]
- (45).James GT (1978) Inactivation of the protease inhibitor phenylmethylsulfonyl fluoride in buffers. Anal. Biochem 86, 574–579. [DOI] [PubMed] [Google Scholar]
- (46).Sevrioukova IF; Poulos TL (2014) Ritonavir analogues as a probe for deciphering the cytochrome P450 3A4 inhibitory mechanism. Curr. Top. Med. Chem 14, 1348–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Denisov IG; Shih AY; Sligar SG, (2012) Structural differences between soluble and membrane bound cytochrome P450s. J. Inorg. Biochem 108, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Navratilova V; Paloncyova M; Berka K; Otyepka M (2016) Effect of lipid charge on membrane immersion of cytochrome P450 3A4. J. Phys. Chem. B 120, 11205–11213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.